

Abstract
The Us2 gene encodes a tegument protein that is conserved in most members of the Alphaherpesvirinae. Previous studies on the pseudorabies virus (PRV) Us2 ortholog indicated that it is prenylated, associates with membranes, and spatially regulates the enzymatic activity of the MAP (mitogen-activated protein) kinase ERK (extracellular signal-related kinase) through direct binding and sequestration of ERK at the cytoplasmic face of the plasma membrane. Here we present an analysis of the herpes simplex virus 2 (HSV-2) Us2 ortholog and demonstrate that, like PRV Us2, HSV-2 Us2 is a virion component and that, unlike PRV Us2, it does not interact with ERK in yeast two-hybrid assays. HSV-2 Us2 lacks prenylation signals and other canonical membrane-targeting motifs yet is tightly associated with detergent-insoluble membranes and localizes predominantly to recycling endosomes. Experiments to identify cellular proteins that facilitate HSV-2 Us2 membrane association were inconclusive; however, these studies led to the identification of HSV-2 Us2 as a ubiquitin-interacting protein, providing new insight into the functions of HSV-2 Us2.
INTRODUCTION
The Alphaherpesvirinae are a large group of viruses that include the common human pathogens herpes simplex virus 1 (HSV-1), HSV-2, and varicella-zoster virus, as well as numerous important veterinary viruses, including the swine pathogen pseudorabies virus (PRV) (1). All herpesvirus virions have a common structure (2): an icosahedral nucleocapsid containing a linear double-stranded DNA genome, surrounded by a lipid envelope embedded with numerous glycoprotein spikes. Between the nucleocapsid and the envelope lies a proteinaceous compartment called the tegument.
The initial stages of herpesvirus assembly take place in the nucleus, where newly replicated virus genomes are packaged into preformed capsids. Although some controversy exists, strong biochemical and genetic evidence indicates that DNA-containing capsids gain access to the cytoplasm by first acquiring a primary envelope at the inner nuclear membrane by budding into the perinuclear space (3). Perinuclear virions are subsequently de-enveloped through fusion of the primary virion envelope with the outer nuclear membrane, thereby releasing the capsid into the cytoplasm (4). The tegument is formed through the recruitment of tegument proteins to capsid components, through interactions between tegument proteins, and through interactions between tegument proteins and the cytoplasmic tails of membrane proteins destined for the envelopes of mature virions. The most widely accepted model of herpesvirus egress posits that the virion acquires its final envelope through the budding of capsid-tegument complexes into membranes derived from the trans-Golgi network (TGN), or possibly late endosomes (LE), in a process referred to as secondary envelopment (5–8). The TGN- or LE-derived vesicles containing infectious enveloped virus then traffic to and fuse with the plasma membrane (PM) of the cell, releasing virus into the extracellular environment. Little is known about the molecular and cellular biology of the trafficking of these virus-containing vesicles to the cell surface. A general assumption has been that these vesicles follow a default, constitutive, secretory pathway to reach the cell surface. However, evidence supporting the idea that this process is regulated by both viral and cellular components is accumulating.
Rémillard-Labrosse and colleagues have reported that the cellular serine-threonine kinase protein kinase D (PKD) facilitates the transport of TGN-derived vesicles containing HSV-1 virions to the PM (9). This finding strongly supports the notion that the transport of virions from the site of secondary envelopment to the cell surface is regulated. Previous studies have indicated that the alphaherpesvirus tegument protein Us2 is involved in the release of enveloped viruses from infected cells. The Us2 gene is found in the genomes of many alphaherpesviruses, including those of human, equine, canine, bovine, avian, and porcine origin (10–16). Although it is highly conserved, Us2 has been found to be nonessential for virus replication in many cultured cells, and aside from the ability of PRV Us2 to regulate ERK (extracellular signal-related kinase) activity spatially, no specific function has been assigned to the protein (17, 18, 19, 20). Wagenaar and colleagues have reported that mature enveloped virions accumulate inside cytoplasmic vesicles in primary cultures of porcine nasal mucosa epithelial cells infected with PRV mutants with deletions of Us2 (21), and previous work from our laboratory has demonstrated that PRV Us2-null mutants accumulate infectious virus inside cells (22). Possible interpretations of these findings include the hypothesis that Us2 activity is required for the efficient fusion of virion-containing vesicles with the PM or, alternatively, that Us2 functions in the transport of the vesicles to the cell periphery. In support of the latter hypothesis, Kramer and colleagues recently identified PRV Us2 as part of a complex containing the tail-anchored type II viral membrane protein Us9 and the microtubule motor KIF1A, which are required for axonal sorting and efficient anterograde transport of virion-containing vesicles within axons (23).
In this study, we sought to characterize the Us2 ortholog encoded by HSV-2. The results indicate that although HSV-2 Us2 lacks canonical membrane-targeting motifs, a large fraction of HSV-2 Us2 is found in detergent-resistant membranes and localizes to recycling endosomes and the PM. We also discovered that HSV-2 Us2 interacts with ubiquitin-conjugated proteins, suggesting a role for Us2 in the trafficking of endosomal membranes in HSV-2-infected cells.
MATERIALS AND METHODS
Cells and viruses.
Vero and 293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) in a 5% CO2 environment. HSV-1 (17+) and HSV-2 (HG52) were generously provided by A. Dolan and D. McGeoch, formerly of the MRC Virology Unit, Institute of Virology, Glasgow, Scotland. HSV-1 and HSV-2 strains were propagated and titers determined on Vero cells.
The recombinant HSV-2 strain encoding Flag-tagged Us2 was constructed by a two-step Red-mediated mutagenesis procedure (24) utilizing the HSV-2 (186) bacterial artificial chromosome (BAC) pYEbac373 in Escherichia coli strain GS1783 (25). GS1783 was a kind gift of G. Smith, Northwestern University. Primers 5′-GTGAGTGAAGATCGGACCACGGGCCTAATATACCGACATGGATTACAAGGATGACGACGATAAG-3′ and 5′-CGTTGGTCTAGGAGGGTAACCACACTTACAACAACAACGCCCTTATCGTCGTCATCCTTGTAATCCATGTCCAACCAATTAACCAATTCTGATTAG-3′ were used to amplify a PCR product from pEP-Kan-S2, a kind gift of N. Osterrieder, Freie Universität Berlin, which was then used to introduce a Flag epitope onto the N terminus of HSV-2 Us2. An HSV-2 Us2-null virus was constructed using primers 5′-ACAGGCGAGTGAGTGAAGATCGGACCACGGGCCTAATATACCGACATGGGCTGATGAGTTAGGATGACGACGATAAGTAGGG-3′ and 5′-TCGTTGGTCTAGGAGGGTAACCACACTTACAACTCATCAGCCCATGTCGGTATATTAGGCCAACCAATTAACCAATTCTGATTAG-3′. The repaired HSV-2 Us2-null strain was constructed using primers 5′-ACAGGCGAGTGAGTGAAGATCGGACCACGGGCCTAATATACCGACATGGGCGTTGTTGTTAGGATGACGACGATAAGTAGGG-3′ and 5′-TCGTTGGTCTAGGAGGGTAACCACACTTACAACAACAACGCCCATGTCGGTATATTAGGCCAACCAATTAACCAATTCTGATTAG-3′. Using an HSV-1 (F) BAC, pYEbac102 (26) (kindly provided by Y. Kawaguchi, The University of Tokyo), an HSV-1 recombinant carrying a Flag-tagged Us2 gene was constructed using primers 5′-GTGCCCCAAATCGGACACGGGCCTGTAATATACCAACATGGATTACAAGGATGACGACGATAAG-3′ and 5′-TCTGGTCAAGGAGGGTCATTACGTTGACGACAACAACGCCCTTATCGTCGTCATCCTTGTAATCCATGTTCAACCAATTAACCAATTCTGATTAG-3′.
Virus reconstitution.
To produce HSV-2 (186) and HSV-1 (F) strains that lacked the BAC DNA sequences, we cotransfected wild-type (WT) or recombinant pYEbac373 or pYEbac102 and the NLS (nuclear localization signal)-Cre-expressing plasmid pOG231 (27) into Vero cells. Briefly, Vero cells were trypsinized and were resuspended in DMEM–10% FBS (fetal bovine serum) containing 10 mM BES [N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid] (pH 7.2) to a concentration of 4 × 107 cells/ml. pYEbac373, or pYEbac102, and pOG231 (1 μg each) were added to a 250-μl cell suspension, which was then transferred to an electroporation cuvette (gap, 0.4 cm; Fisher Scientific, Ottawa, Ontario, Canada). Electroporation was carried out at settings of 210 V, 950 μF, and 200 Ω by using a BTX ECM 630 electroporator. Cells and DNA were immediately plated onto 100-mm dishes, and the infection was allowed to proceed for as long as 3 days. Supernatants were collected, and the WT or recombinant HSV strains were plaque purified twice on Vero cells.
Immunological reagents.
Monoclonal antibodies against HSV-2 gG, gD, ICP5, ICP8, and ICP27 were purchased from Virusys (Taneytown, MD) and were used for Western blotting at the following dilutions: anti-gG, 1:5,000; anti-gD, 1:10,000; anti-ICP5, 1:3,000; anti-ICP8, 1:4,000; anti-ICP27, 1:1,000. An anti-GFP monoclonal antibody (Clontech, Mountain View, CA) was used for Western blotting at a dilution of 1:1,000; horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG and rabbit anti-rat IgG (Sigma, St. Louis, MO) were used for Western blotting at dilutions of 1:10,000 and 1:80,000, respectively. Alexa Fluor 488-conjugated donkey anti-rat IgG, Alexa Fluor 568-conjugated donkey anti-rat IgG, and Alexa Fluor 568-conjugated donkey anti-mouse IgG (Invitrogen, Burlington, Ontario, Canada) were used for indirect immunofluorescence microscopy at a dilution of 1:500. The GM1 ganglioside was detected by Western blotting by use of biotinylated cholera toxin B subunit (Sigma, St. Louis, MO) diluted to 1 μg/ml, followed by incubation with an HRP-streptavidin conjugate (Invitrogen, Burlington, Ontario, Canada) diluted at 1:1,000. An antibody against mono- and polyubiquitin (FK2) was purchased from Enzo Life Sciences (Burlington, Ontario, Canada).
Insoluble GST (glutathione S-transferase)-Us2 fusion proteins derived from HSV-1 and HSV-2 were isolated from IPTG (isopropyl-β-d-thiogalactopyranoside)-induced E. coli Rosetta (DE3) using the B-Per bacterial protein extraction reagent (Pierce, Rockford, IL). The partially purified proteins were electrophoresed on preparative 10% SDS-PAGE gels. Slabs of polyacrylamide containing the GST-Us2 fusion proteins were excised from the preparative gels and were sent to Cedarlane Laboratories (Burlington, Ontario, Canada) for the production of polyclonal antisera in Wistar rats. The resulting antisera were used for Western blotting at a dilution of 1:500 and for indirect immunofluorescence microscopy at a dilution of 1:200.
Construction of Us2 expression plasmids.
To construct the HSV-2 Us2 expression plasmid pHSV2Us2, the full-length HSV-2 Us2 DNA sequence flanked by EcoRI sites was obtained by PCR amplification using forward primer 5′-CCGAATTCATGGCCTGTCGTAAGTTCTGTGG-3′ and reverse primer 5′-CCGAATTCTCACTTAGGGTGAAATAGCG-3′ from the pATHSV-2HindIIIL template (a kind gift from A. Dolan and D. McGeoch), which contains a HindIII fragment encompassing the majority of the unique short region of the HSV-2 genome (10). The amplified fragment was subcloned into the pCR-Blunt II-TOPO vector (Invitrogen, Burlington, Ontario, Canada) by using the manufacturer's protocols. All plasmids utilizing PCR in their construction were sequenced to ensure that no spurious mutations had been introduced in the process. An EcoRI fragment encompassing the amplified sequences was excised from this intermediate vector, purified, and then ligated to a similarly digested pCINeo vector (Promega, Madison, WI).
To fuse the red fluorescent protein monomeric Cherry (mCherry) (28) to the N terminus of HSV-2 Us2, the mCherry gene was amplified by PCR with forward primer 5′-CGCGCTAGCATGGTGAGCAAGGGCGAGG-3′ and reverse primer 5′-CGCCTCGAGTCTTGTACAGCTCGTCCATGC-3′ from the pRSETb mCherry template, a kind gift from R. Tsien, University of California at San Diego, and the amplified fragment was cloned directly into the pCR-Blunt II TOPO vector. An NheI/XhoI fragment containing the amplified sequences was excised from this intermediate vector, purified, and ligated to similarly digested pHSV2Us2.
A plasmid encoding Flag-tagged HSV-2 Us2 was constructed by amplifying the HSV-2 Us2 gene using forward primer 5′-ATCGATATCGATGGGCGTTGTTGTTGTAAGTGTGG-3′ and reverse primer 5′-GTAGTCGACTTAGAGGTTGGTGATTGGATAGC-3′, and the amplified fragment was cloned directly into the pCR-Blunt II TOPO vector. Next, an EcoRV/SalI fragment containing the amplified sequences was excised from this intermediate vector, purified, and ligated to similarly digested pFlag-CMV-2 (Sigma, St. Louis, MO). A yeast expression plasmid encoding HSV-2 Us2 fused to GAL4-DBD was constructed by amplifying the HSV-2 Us2 gene using forward primer 5′-GATCGAATTCATGGGCGTTGTTGTTGTAAGTGTGG-3′ and reverse primer 5′-GTAGTCGACTTAGAGGTTGGTGATTGGATAGC-3′, and the amplified fragment was cloned directly into the pCR-Blunt II TOPO vector. Next, an EcoRI/SalI fragment containing the amplified sequences was excised from this intermediate vector, purified, and ligated to similarly digested pGBKT7 (Clontech, Mountain View, CA).
Analysis of Us2 expression kinetics.
Confluent monolayers of Vero cells growing in 6-well dishes were infected with HSV-2 at a multiplicity of infection (MOI) of 10. At 0, 2, 4, and 6 h postinfection, the medium was removed, and the cells were washed three times with phosphate-buffered saline (PBS). Cells were scraped into 200 μl of lysis buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 1% NP-40, 1% Na-deoxycholate) containing protease inhibitors (Roche, Laval, Quebec, Canada) and were transferred to a 1.5-ml microcentrifuge tube. Lysates were kept on ice for 30 min with intermittent mixing and were then centrifuged at 10,000 × g for 5 min. For Western blot analysis, 10 to 20 μl of the cellular extract was mixed with SDS-PAGE sample buffer, heated to 100°C for 5 min, electrophoresed through 12.5% SDS-PAGE gels, and analyzed by Western blotting with a polyclonal antiserum to HSV-2 Us2 and monoclonal antibodies to gG, ICP5, or ICP8.
PAA treatment.
Confluent monolayers of Vero cells growing in 6-well plates were used for experiments with phosphonoacetic acid (PAA). At 1 h prior to infection, the cell culture medium was replaced with DMEM–10% FCS either with or without 200 μg/ml PAA (Sigma, St. Louis, MO). Cells were infected with HSV-2 at an MOI of 10 and were incubated in the presence or absence of 200 μg/ml PAA for 6 h. Cell extracts were prepared as described above and were electrophoresed through a 12.5% SDS-PAGE gel. Western blotting was performed with a polyclonal antiserum to Us2 or a monoclonal antibody to gG, ICP5, or ICP8.
Virion purification.
Virions were purified essentially as described previously (29). Briefly, three confluent 150-mm dishes of Vero cells were infected with HSV-2 at an MOI of 10. At 16 h postinfection, the medium was collected and was centrifuged three times in succession at 600 × g to pellet detached cells and cellular debris. The clarified supernatant was layered onto a 30% sucrose (wt/vol, in PBS) cushion and was centrifuged in a Beckman SW28 rotor at 23,000 rpm for 3 h. The pelleted virions were resuspended in 1 ml of PBS, layered onto a 4-ml 30% sucrose (wt/vol, in PBS) cushion, and centrifuged in a Beckman SW55 Ti rotor at 28,000 rpm for 90 min. Pelleted virions were resuspended in 200 μl of PBS, snap-frozen in liquid nitrogen in 100-μl aliquots, and stored at −80°C.
Protease treatment of virions.
Isolated virions were treated with 10 μg of proteinase K (PK) (Fisher Scientific, Ottawa, Ontario, Canada) per ml in either the presence or the absence of 1% NP-40 (Igepal; Sigma, St. Louis, MO). After incubation for 60 min at room temperature, phenylmethylsulfonyl fluoride (Sigma, St. Louis, MO) was added to each sample to a final concentration of 2 mM to inhibit further proteolysis. Samples were immediately separated on 10% SDS-PAGE gels, and proteins were transferred to membranes for Western blot analysis.
Transfections.
293T cells were transfected for the purpose of preparing cellular extracts by using the calcium phosphate coprecipitation method (30). Vero cells were transfected for the purpose of microscopic analyses by using FuGene 6 (Roche, Laval, Quebec, Canada) according to the manufacturer's instructions.
Indirect immunofluorescence microscopy.
Cells for microscopic analyses were grown either on glass coverslips or on glass-bottom dishes (MatTek, Ashland, MA). Cells were then transfected or infected as described above. In some experiments, cells were incubated with 3 μg/ml brefeldin A (BFA) (Epicentre Biotechnologies, Madison, WI) or with 0.3% ethanol (vehicle). Cells were fixed in 4% paraformaldehyde–PBS for 10 min at room temperature. Fixed cells were washed 3 times with PBS containing 1% BSA (bovine serum albumin) (PBS-BSA) and were permeabilized for 3 min at room temperature with PBS-BSA containing 0.1% Triton X-100 (TX-100). Cells were then washed 3 times with PBS and once with PBS-BSA, and 100 μl of primary antiserum diluted appropriately in PBS-BSA was applied for 45 min at room temperature. Cells were washed 3 times with PBS-BSA, and 100 μl of an Alexa Fluor-conjugated secondary antibody diluted appropriately in PBS-BSA was applied for 30 min at room temperature. Cells were then washed 3 times with PBS-BSA. To visualize nuclei, cells were incubated with Hoechst 33342 (Sigma, St. Louis, MO) diluted to 0.5 μg/ml in PBS for 7 min at room temperature. Stained cells on coverslips were washed 3 times in PBS and were mounted in PBS containing 50% (vol/vol) glycerol on glass slides; stained cells in glass-bottom dishes were washed 3 times in PBS and were stored under PBS-BSA. Images were captured by using an Olympus FV1000 laser scanning confocal microscope with a 60× objective (numerical aperture [NA], 1.42) and a digital zoom factor of 2 to 4 and were analyzed by FluoView CS3 software. Composites of representative images were prepared using Adobe Photoshop CS3 software.
Live-cell imaging.
Vero cells growing on glass-bottom dishes were transfected with an mCherry–HSV-2 Us2 expression plasmid. At 24 h after transfection, the medium was replaced with DMEM lacking phenol red and containing 10% FCS, and the dish was mounted on a confocal microscope contained within a humidified 37°C, 5% CO2 environment. Images were captured every 2.2 s by using an Olympus FV1000 laser scanning confocal microscope with a 60× objective (numerical aperture [NA], 1.42) and FluoView CS3 software. Composite images of the time course were prepared using Adobe Photoshop CS3 software.
Transferrin uptake assays.
Vero cells growing on glass coverslips were transfected with an mCherry–HSV-2 Us2 expression plasmid or were infected with HSV-2 at an MOI of 1. At 24 h posttransfection or 8 h postinfection, cells were incubated with Alexa Fluor 647-conjugated transferrin (Invitrogen, Burlington, Ontario, Canada) for 30 min at 4°C in DMEM. The cells were then washed; fresh medium was added; and the cells were transferred to 37°C for the indicated times. Samples were taken at different time points, fixed, and, in the case of HSV-2-infected cells, stained for Us2. Images were then collected by confocal microscopy. Measurement of the colocalization of Us2-labeled vesicles and fluorescent transferrin was facilitated by FluoView CS3 software. Briefly, regions of interest (ROIs) containing individual Us2-labeled vesicles were selected, and the average fluorescence intensity of the Us2 and transferrin signals was measured. An ROI was scored positive for transferrin if the average fluorescence intensity of the ROI was twice that of the background fluorescence in the transferrin channel. Background fluorescence was defined as the averaged fluorescence intensity of 5 ROIs per field that contained no discernible transferrin-labeled puncta.
Membrane flotation assays.
Methods for the analysis of membrane-associated proteins by sucrose density gradient flotation were used essentially as described by Brignati and colleagues (31), with the following modifications. Three subconfluent 150-mm-diameter dishes of 293T cells were cotransfected with HSV-2 Us2 and EGFP (enhanced green fluorescent protein) expression plasmids. Cells were washed and were harvested using trypsin at 48 h posttransfection. Cells were pelleted at 220 × g for 7 min at 4°C and were washed once with cold PBS and once with homogenization buffer (0.25 M sucrose, 10 mM KCl, 10 mM Tris [pH 7.4], 1.5 mM MgCl2). Cells were then suspended by gentle vortexing in 0.5 ml of homogenization buffer supplemented with a protease inhibitor cocktail (Roche, Laval, Quebec, Canada). Following a 30-min incubation on ice, cells were passed repeatedly through a 26-gauge syringe needle in order to homogenize the cells while leaving the nuclei intact. Cell homogenates were centrifuged at 600 × g for 10 min at 4°C to remove unbroken cells and nuclei. Then 0.3 ml of the postnuclear supernatant (PNS) was mixed with 2.7 ml of 85% (wt/vol) sucrose in NTE buffer (100 mM NaCl, 10 mM Tris [pH 7.4], 1 mM EDTA) (final sucrose concentration, 71.5%) and was placed in the bottom of an SW41 centrifuge tube. Next, 6 ml of 65% sucrose in NTE buffer, followed by 3 ml of 10% sucrose in NTE buffer, was layered onto the PNS. This sucrose step gradient was centrifuged at 100,000 × g for 18 h at 4°C in a Beckman SW41 rotor. Twelve 1-ml fractions were collected from the top of the tube. Fraction 13 represented the material that pelleted at the bottom of the tube. Aliquots (500 μl) of fractions 1 to 12 were diluted with 500 μl of NTE buffer plus protease inhibitor cocktail, placed in an MLA-130 centrifuge tube, and centrifuged for 15 min at 100,000 rpm in a Beckman MLA-130 rotor to pellet the membranes. Supernatants were discarded, and pelleted material was resuspended in 60 μl of PBS; 60 μl of 2× SDS-PAGE sample buffer was added, and the samples were boiled. Fraction 13 was boiled in 120 μl of 1× SDS-PAGE sample buffer.
For further analysis, the membrane-associated material at the interface of the 10%–65% sucrose gradient was collected, and aliquots were treated with either 1 M NaCl, 0.2 M Na2CO3 (pH 11), 1% Triton X-100, or 0.01% digitonin for 1 h at 4°C and were subsequently centrifuged for 15 min at 100,000 rpm in a Beckman MLA-130 rotor to pellet the membranes. Supernatants were collected in a new tube, and pelleted material was resuspended in 30 μl of SDS-PAGE sample buffer. Proteins in the supernatants were precipitated with trichloroacetic acid (TCA), pelleted, washed three times with cold acetone, dried, and resuspended in SDS-PAGE sample buffer.
Isolation of detergent-insoluble membranes.
Detergent-insoluble membranes were isolated by flotation using OptiPrep (Sigma, St. Louis, MO) as described by Lyman and colleagues (32). Briefly, Vero cells cultured in 150-mm dishes were infected with HSV-2 at a multiplicity of infection of 5. At 12 h postinfection (hpi), cells were washed with cold DMEM followed by cold PBS. Cells were lysed with 1 ml of lysis buffer consisting of 1% TX-100 in TNE buffer (25 mM Tris-HCl [pH 6.8], 150 mM NaCl, 5 mM EDTA) with a protease inhibitor cocktail (Roche, Laval, Quebec, Canada). The lysate was homogenized by passage through an 18-gauge needle and was mixed with 2 ml of ice-cold 60% OptiPrep. The mixture was placed at the bottom of a Beckman SW41 ultracentrifuge tube and was overlaid sequentially with 5 ml of ice-cold 30% OptiPrep in TNE buffer and 4 ml of ice-cold 5% OptiPrep in TNE buffer. Samples were centrifuged at 200,000 × g and 4°C for 20 h. Twelve 1-ml fractions were collected from the top to the bottom of the tube. Proteins and gangliosides in fractions 3 through 11 were precipitated with TCA and were analyzed by Western blotting.
Coimmunoprecipitation.
To identify HSV-2 Us2-interacting partners, a plasmid expressing Flag-HSV-2 Us2 was transfected into 293T cells grown in 150-mm dishes. At 24 h posttransfection, cells were collected to purify membrane fractions by using the membrane flotation assay described above. Membranes were incubated with an anti-Flag M2 affinity gel overnight at 4°C. Anti-Flag beads were washed three times with Tris-buffered saline (TBS). Precipitated protein complexes were subjected to SDS-PAGE and were stained using SimplyBlue SafeStain according to the manufacturer's instructions (Invitrogen, Burlington, Ontario, Canada). Bands were excised and were sent for LC (liquid chromatography)–MS-MS (tandem mass spectrometry) analysis to the University of Victoria Genome BC Protein Centre.
To determine whether HSV-2 Us2 could interact with ubiquitin-conjugated proteins in infected cells, Vero cells growing in 150-mm dishes were infected with HSV-2 WT strain 186, an HSV-2 Us2-null strain, or an HSV-2 Us2 repair strain at an MOI of 5. At 6 hpi, cells were harvested in lysis buffer and were centrifuged at 14,000 × g for 20 min at 4°C. Lysates were incubated with an anti-Flag M2 affinity gel overnight at 4°C. Anti-Flag beads were washed three times with TBS. Precipitated protein complexes were subjected to Western blotting. To determine whether HSV-2 Us2 could be pulled down by ubiquitin-conjugated proteins, lysates were incubated with an antibody against mono- and polyubiquitin (FK2; Enzo Life Sciences, Burlington, Ontario, Canada) overnight at 4°C. Protein G agarose was added to the antigen/antibody complexes, and samples were nutated at 4°C for 2 h. Protein G agarose was pelleted, washed three times with TBS, resuspended in 1× SDS-PAGE sample buffer, boiled for 5 min, and analyzed by Western blotting.
Ubiquitin interaction assay.
To determine whether HSV-2 Us2 interacts with ubiquitin, Vero cells grown in 150-mm dishes were infected with HSV-2 at an MOI of 5 for 6 h. Cells were collected in lysis buffer and were centrifuged at 14,000 × g for 20 min at 4°C. Ubiquitin-conjugated beads (Boston Biochem, Cambridge, MA) were washed twice with 25 mM HEPES, pH 7.5, before use. Lysates were incubated with monoubiquitin-conjugated beads or with K48-linked or K63-linked tetraubiquitin-conjugated beads overnight at 4°C. Beads were washed three times with wash buffer (10 mM Tris [pH 7.4], 100 mM NaCl, 0.1% NP-40), resuspended with 1× SDS-PAGE sample buffer, boiled for 5 min, and analyzed by Western blotting.
To determine the ability of Us2 to interact with ubiquitin in the absence of other viral proteins, a plasmid expressing Flag-tagged HSV-2 Us2 was transfected into 293T cells growing on 150-mm dishes. At 24 h posttransfection, cells were harvested in 800 μl of lysis buffer per dish and were centrifuged at 14,000 rpm for 15 min. Supernatants were incubated with an anti-Flag M2 affinity gel (Sigma, St. Louis, MO) overnight at 4°C. The immune complexes were washed three times with TBS and were then subjected to competitive elution by using a 3×Flag peptide (Sigma, St. Louis, MO) according to the manufacturer's instructions. Eluates from two dishes were pooled and were incubated with monoubiquitin-conjugated beads overnight at 4°C. Beads were washed three times with wash buffer, resuspended in 1× SDS-PAGE sample buffer, boiled for 5 min, and analyzed by Western blotting.
RESULTS
Expression of HSV-2 Us2.
To enable the detection of HSV-2 Us2, a rat polyclonal antiserum was raised against a GST-Us2 fusion protein. This antiserum detected a protein of the predicted molecular mass for HSV-2 Us2 (∼33 kDa) in 293T cells transfected with an HSV-2 Us2 or mCherry-fused HSV-2 Us2 (∼60-kDa) expression construct (Fig. 1A), as well as in extracts prepared from Vero cells infected with HSV-2 (HG52) (Fig. 1B). This antiserum was unable to detect HSV-1 Us2 (Fig. 1B). Consistent with the findings of Jiang and colleagues, working with HSV-2 strain 186 (20), we found that Us2 encoded by strain HG52 was first detected in cell lysates by Western blotting by 4 h after infection (Fig. 1C). The viral proteins gG and ICP5 were also detected by 4 h after infection, and the ICP8 protein was detected by 2 h postinfection. Herpesvirus genes are grouped into four kinetic classes: immediate early, early, late, and leaky late. Late gene expression is dependent on viral DNA replication, whereas leaky-late genes are expressed poorly if viral DNA synthesis is inhibited (2). As was seen with ICP5 and gG, expression of HSV-2 Us2 was barely detectable in the presence of the viral DNA synthesis inhibitor PAA, whereas the expression of the early gene product ICP8 was unaffected (Fig. 1C). These findings are consistent with the notion that HSV-2 Us2 is expressed with late or leaky-late kinetics, like the equine herpesvirus-1 (EHV-1) Us2 protein (18) but in contrast to the PRV Us2 protein, which is expressed with early kinetics (33). To determine whether HSV-2 Us2 was packaged into virions, we purified extracellular virions from infected-cell supernatants. Purified virions and cell lysates were probed for the presence of Us2 and the nonstructural virus protein ICP27 by Western blotting (Fig. 1D). While ICP27 was readily detected in the infected-cell lysate, it was not detected in the virion preparation. Us2 was detected in both the virion preparation and the infected-cell lysate. These data suggest that Us2 protein is a component of HSV-2 particles. To determine whether Us2 in the virion preparation was contained within the virion envelope, as would be expected for a tegument protein, a protease protection assay was performed (Fig. 1E). Whereas the ectodomain of the virion transmembrane protein gD was susceptible to protease digestion in the absence of detergent (NP-40), Us2 was resistant to protease in the absence of NP-40 but not in its presence, indicating that Us2 is protected by the virion envelope.
Fig 1.
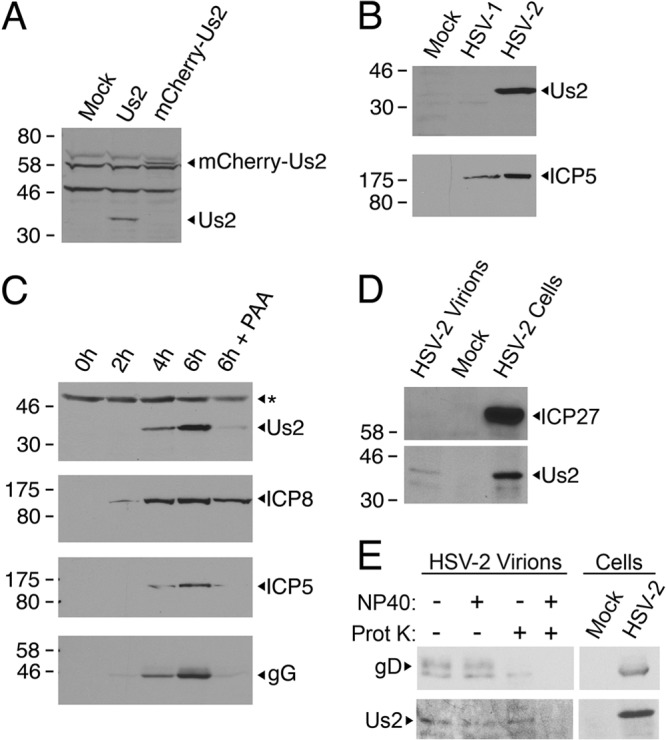
Analysis of HSV-2 Us2 expression and virion localization. (A) A polyclonal antiserum from rats immunized with GST–HSV-2 Us2 specifically detects HSV-2 Us2. Equal volumes of cellular extracts prepared from 293T cells either mock transfected or transfected with a plasmid encoding HSV-2 Us2 or mCherry–HSV-2 Us2 were analyzed by Western blotting. Note the shift in the mobility of the band detected in cells transfected with the plasmid encoding the mCherry–HSV-2 Us2 fusion protein. (B) The anti-HSV-2 Us2 polyclonal antiserum reacts with HSV-2 Us2 but not with HSV-1 Us2. Equal volumes of cellular extracts prepared from uninfected, HSV-1 17+-infected, or HSV-2 HG52-infected Vero cells were analyzed by Western blotting. (Top) The gel was probed with a rat polyclonal anti-HSV-2 Us2 antiserum. (Bottom) The infection control gel was probed with anti-HSV-2 ICP5, which also cross-reacts with ICP5 from HSV-1. (C) Kinetics of Us2 synthesis in HSV-2-infected cells. At the indicated times postinfection, the expression of Us2, ICP8, ICP5, and gG in cell lysates was analyzed by Western blotting. Additionally, the expression of Us2, ICP8, ICP5, and gG in the presence of the viral DNA synthesis inhibitor PAA was analyzed at 6 h postinfection. The asterisk at the top indicates the position of a nonspecific cross-reacting band that serves as an internal loading control. (D) Us2 is incorporated into HSV-2 virions. At 16 h after the infection of Vero cells with HSV-2 HG52, virions were purified from the cell medium and were analyzed alongside HSV-2-infected and mock-infected cell lysates for the presence of ICP27 and Us2 by Western blotting. Molecular size markers (in kilodaltons) are indicated on the left. (E) Protease protection assay. (Left) Purified virions were treated with either PBS (−), NP-40, PK (Prot K), or both NP-40 and PK and were then analyzed for the presence of the virion transmembrane protein gD and Us2. Note that gD is degraded by PK in the absence of NP-40, whereas Us2 remains intact. (Right) Migration of gD and Us2 from an infected-cell lysate.
Interactions of HSV-2 Us2 with ERK.
Previous studies have indicated that PRV Us2 binds specifically to ERK and recruits it to cellular membranes (22, 34). To determine whether this property is shared with HSV-2 Us2, we analyzed the interaction of HSV-2 Us2 with ERK in a yeast two-hybrid assay. As expected, PRV Us2 interacted strongly with ERK2, whereas even weak interactions between HSV-2 Us2 and ERK1 or ERK2 could not be detected in these assays (data not shown). These data suggest that HSV-2 interacts with a different subset of proteins than PRV Us2.
HSV-2 Us2 is tightly associated with cellular membranes.
In transfected Vero cells, HSV-2 Us2 localized to punctate cytoplasmic structures, to small vesicles that often appeared associated with the PM, and to the PM itself (Fig. 2A). In infected Vero cells, HSV-2 Us2 localized primarily to cytoplasmic vesicles at 6 h postinfection and could be detected at the PM by 10 h postinfection (Fig. 2Bi and ii). At these times, a fraction of Us2 was also localized diffusely throughout the cell. At 18 h postinfection, HSV-2 Us2 was found predominantly at the PM, in cytoplasmic vesicles, and also localized diffusely throughout the cytoplasm (Fig. 2Biii). Because HSV-2 Us2 lacks any obvious signals or motifs predicated to target it to membranes, we investigated the nature of Us2 membrane association further.
Fig 2.
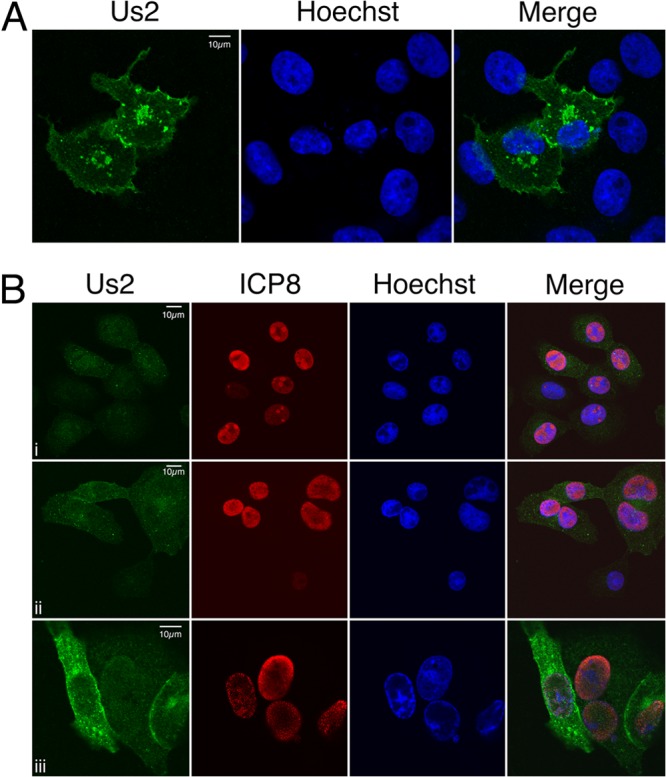
Subcellular localization of HSV-2 Us2. (A) HSV-2 Us2 localizes to membranes in transfected Vero cells. Shown is a representative image of Vero cells transfected with a plasmid encoding HSV-2 Us2 and stained with a polyclonal antiserum specific for HSV-2 Us2 (green) at 24 h posttransfection. Nuclei were detected using the DNA stain Hoechst 33342. (B) Kinetics of HSV-2 Us2 expression and localization in infected Vero cells. Shown are representative images of Vero cells infected with HSV-2. Cells were fixed at 6 h (i), 10 h (ii), or 18 h (iii) after infection and were stained for Us2 (green) or the HSV-2 nuclear protein ICP8 (red). Nuclei were stained with Hoechst 33342 (blue). Images of stained cells were captured by confocal microscopy.
A membrane flotation assay was performed to determine whether HSV-2 Us2 was capable of physically associating with membranes. 293T cells were cotransfected with HSV-2 Us2 and EGFP expression plasmids, and membrane fractions were isolated as described in Materials and Methods. As expected, EGFP was not enriched in the membrane-containing fractions (Fig. 3B). In contrast, Us2 floated with cellular membranes (Fig. 3A, fractions 3 and 4), in agreement with the idea that Us2 is membrane associated.
Fig 3.

HSV-2 Us2 physically associates with membranes. (A and B) 293T cells were cotransfected with an HSV-2 Us2 and EGFP expression plasmid. At 24 h after transfection, membrane fractions were isolated by membrane flotation on discontinuous sucrose step gradients as described in Materials and Methods. Fractions from the top (10% sucrose) to the bottom (71.5% sucrose) of the step gradient were analyzed by Western blotting for the presence of Us2 (A) or EGFP as a negative control (B). (C through E) Membrane fractions were treated with either 1 M NaCl (C), 0.2 M Na2CO3 or 1% Triton X-100 (D), or 0.01% digitonin (E) for 1 h at 4°C prior to the pelleting of membranes by centrifugation. Pelleted membranes and protein from supernatant fractions that had been concentrated by TCA precipitation were analyzed for Us2 contents by Western blotting.
Analysis of the amino acid sequence of HSV-2 Us2 indicates that it lacks a canonical N-terminal signal sequence, has no putative membrane-spanning domain or signals for lipid modifications, and has an isoelectric point of 8.44. These features, combined with the data indicating that Us2 is incorporated into the virion tegument (18, 20, 33), suggested that HSV-2 Us2 is a peripheral membrane protein that associates with the cytoplasmic faces of membranes, possibly through electrostatic interactions. It was expected that, if Us2 associated with membranes through electrostatic interactions, treatment of membranes with 1 M NaCl or 0.2 M Na2CO3 (pH 11) would strip Us2 from the membranes (35, 36). The data shown in Fig. 3C and D indicate that treatment with these agents did not liberate Us2 from the membrane fraction. Treatment of Us2-containing membrane fractions with the cholesterol-sequestering agent digitonin (37) also failed to release Us2 from the membranes, suggesting that Us2 does not associate with membranes by binding cholesterol (Fig. 3E). In contrast, treatment of membranes with 1% Triton X-100, a nonionic detergent, solubilized a portion of Us2 (Fig. 3D). However, the finding that a significant portion of Us2 remained in the pelleted membranes after Triton X-100 exposure suggested that a fraction of Us2 associates with detergent-resistant membranes (38).
To explore this idea further, detergent-resistant membranes were isolated from HSV-2-infected cells and were probed for the presence of Us2, the lipid raft-associated molecules caveolin-1 and ganglioside GM1, and the non-raft-associated membrane protein transferrin receptor (Fig. 4). These experiments indicate that in virus-infected cells, the bulk of Us2 associates with detergent-resistant membranes and cofractionates with caveolin-1 and GM1.
Fig 4.
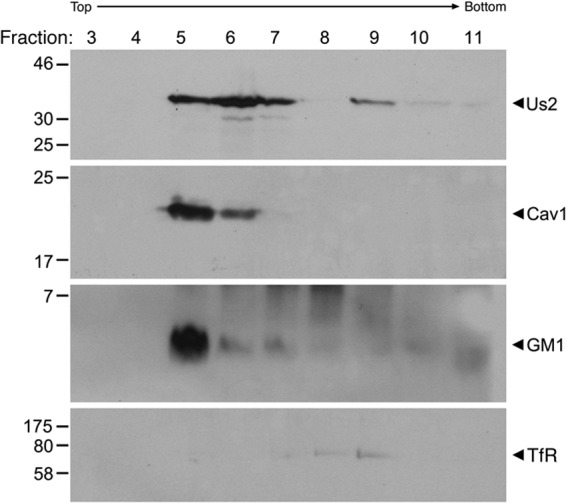
HSV-2 Us2 fractionates with detergent-insoluble membranes. Vero cells infected with HSV-2 were harvested at 12 h after infection, and detergent-insoluble membranes were isolated by flotation through an OptiPrep step gradient. Gradient fractions 3 through 11 were analyzed by Western blotting for Us2, caveolin-1 (Cav1), ganglioside GM1, or the transferrin receptor (TfR). Detergent-insoluble membranes float to the interface between the 5% and 30% OptiPrep portions of the step gradient, which is collected in fraction 5. The migration positions of molecular size markers (in kilodaltons) are shown on the left of each blot.
Us2 localization after BFA treatment.
The data presented so far suggest that Us2 is associated with detergent-insoluble membranes and localizes predominantly to the PM and cytoplasmic vesicles. To determine whether Us2-containing cytoplasmic vesicles represent components of the biosynthetic secretory pathway, cells were exposed to the fungal metabolite BFA, which prevents anterograde trafficking from the endoplasmic reticulum (ER) through the Golgi apparatus, with the net result that Golgi components are relocalized to the ER (39–41). As expected, the Golgi apparatus was reversibly disrupted by BFA treatment, as indicated by the staining of cells for the Golgi apparatus-resident protein giantin (Fig. 5A, B, and C). In contrast to that of giantin, the localization of Us2 appeared to be unaffected by BFA treatment; however, under these conditions, a fraction of Us2 appeared to colocalize with giantin in what might represent cytoplasmic Golgi remnants (42) (Fig. 5B). To determine whether Us2 localization to the PM was dependent on ER/Golgi trafficking, we treated cells with BFA immediately after the transfection of cells with a Us2 expression plasmid (Fig. 5D). While the architecture of the Golgi apparatus was perturbed, as expected, Us2 localization to the PM was not affected by extended BFA treatment. These findings raise the possibility that Us2 does not travel through the biosynthetic secretory pathway en route to the PM.
Fig 5.
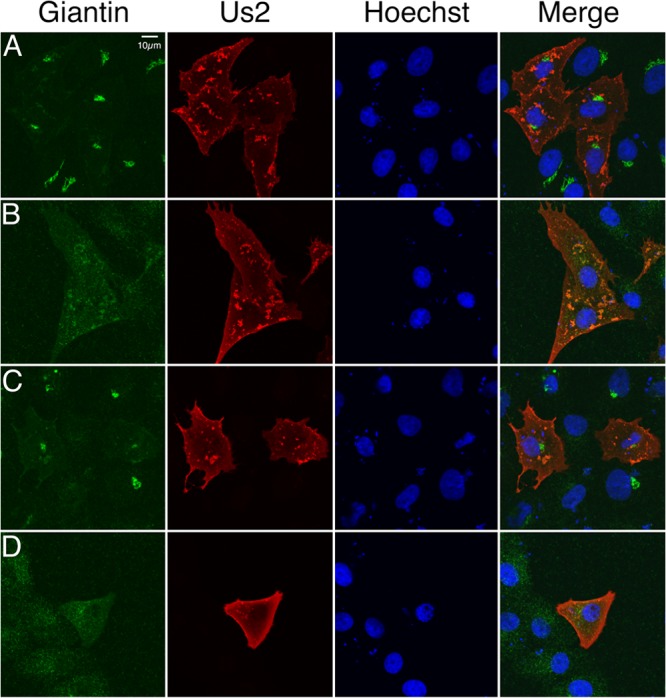
Effect of BFA treatment on Us2 localization. (A to C) Twenty-four hours after transfection with an HSV-2 Us2 expression plasmid, Vero cells either were treated with vehicle (0.3% ethanol) (A) or with 3 μg/ml BFA (B) for 1 h or were treated with BFA for 1 h, washed, and cultured for an additional 2 h in a medium lacking BFA (C). Cells were fixed and immunostained for the Golgi apparatus-resident protein giantin (green) and Us2 (red). Nuclei were detected using the DNA stain Hoechst 33342 (blue). (D) Alternatively, cells were treated with 3 μg/ml BFA 6 h after transfection and were maintained in BFA for 24 h prior to fixing and staining. Images of stained cells were captured by confocal microscopy. Representative images are shown.
Us2-containing vesicles share the properties of recycling endosomes.
Imaging of Us2 dynamics in live Vero cells transiently transfected with mCherry–HSV-2 Us2 expression constructs revealed that Us2-containing cytoplasmic vesicles are highly dynamic, frequently forming long, filamentous tubules, a hallmark of recycling endosomal compartments (Fig. 6; see also movie S1 in the supplemental material) (43, 44). These results prompted us to examine the association of Us2 with endosomes more closely.
Fig 6.
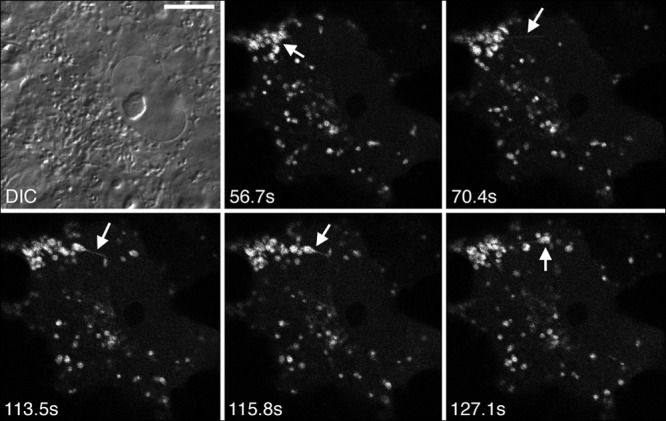
Dynamics of Us2-containing vesicles. Vero cells were transfected with a plasmid encoding mCherry–HSV-2 Us2. At 24 h after transfection, live cells were imaged over 134 s at a rate of 2.2 frames per second by using a confocal microscope. Static images demonstrating the dynamics of Us2 in a representative cell are shown. The signal is mCherry fluorescence. Arrows highlight an example of a Us2-containing vesicle emitting an extended membrane tubule. The full data set is provided as movie S1 in the supplemental material. DIC, differential inference contrast.
Upon binding to its cell surface receptor, diferric transferrin is internalized into clathrin-coated vesicles and is delivered to an early endosomal compartment. Two Fe3+ atoms are released from transferrin in the acidic environment of the early endosome, and the resultant apotransferrin, still bound to its receptor, traffics to an endocytic recycling compartment, where it is sorted back to the PM (44). This movement through the endocytic pathway can be monitored using fluorescently conjugated diferric transferrin. Vero cells were transfected with an mCherry–HSV-2 Us2 expression plasmid. At 24 h posttransfection, cells were incubated with Alexa Fluor 647-conjugated transferrin on ice for 30 min. Unbound transferrin was washed away, and the cells were transferred to 37°C for 0, 5, 12, or 30 min before fixation and analysis by confocal microscopy (Fig. 7). After 5 min at 37°C, the majority of the transferrin localized near the cell surface, and 15% of HSV-2 Us2-containing vesicles colocalized with transferrin (Fig. 7A and D). By 12 min after the shift to 37°C, the majority of the transferrin had entered the cells, and 49% of Us2-containing vesicles colocalized with transferrin (Fig. 7B and D). At 30 min after incubation at 37°C, more-extensive colocalization of transferrin and Us2 was observed: 64% of Us2-containing vesicles colocalized with transferrin (Fig. 7C and D). To examine the association of Us2 with the endocytic pathway in the context of virus infection, the kinetics of transferrin association with Us2-containing vesicles was analyzed at 8 h after HSV-2 infection of Vero cells (Fig. 7E). After 5 min at 37°C, 11% of Us2-containing vesicles colocalized with transferrin, compared to 26% and 40% at 12 and 30 min after the temperature shift, respectively. The kinetics of transferrin-Us2 colocalization in transfected and infected cells are consistent with the localization of Us2 to an endocytic recycling compartment (45, 46).
Fig 7.

A subset of Us2 colocalizes with endosomal vesicles. (A to C) Vero cells transfected with an mCherry–HSV-2 Us2 expression plasmid were incubated with Alexa Fluor 647-conjugated transferrin for 30 min at 4°C, washed, and shifted to 37°C for 5 min (A), 12 min (B), or 30 min (C) prior to fixation. The transferrin signal is shown in green. The Us2 signal is red. Nuclei were detected using the DNA stain Hoechst 33342 (blue). Representative images are shown. (D) Quantification of transferrin colocalization with Us2 puncta in cells transfected with mCherry–HSV-2 Us2. The data are presented as percentages of Us2-containing puncta that are also positive for transferrin. A total of 12 fields of cells and 298 Us2 puncta were analyzed. Error bars represent the standard errors of the means observed for differences between different fields of cells. (E) Quantification of transferrin colocalization with Us2 puncta in HSV-2-infected cells at each time point. The data are presented as percentages of Us2-containing puncta that are also positive for transferrin. Us2 puncta were identified by indirect immunofluorescence confocal microscopy using a polyclonal antiserum against Us2 and an Alexa Fluor 588-conjugated secondary antibody. A total of 30 fields of cells and 938 Us2 puncta were analyzed. Error bars represent the standard errors of the means observed for differences between different fields of cells at each time point.
To identify Us2-containing endosomal membranes more precisely, cells were transfected with plasmids encoding either the early endosomal marker Rab5-GFP, the late endosomal marker Rab7-GFP, or the recycling endosomal marker Rab11-GFP, along with a Us2 expression plasmid. The cells were then examined by confocal microscopy, and colocalization was quantified (Fig. 8). In agreement with the transferrin trafficking data shown above, 25% of Us2-containing vesicles colocalized with Rab5-GFP, 10% with Rab7-GFP, and 63% with Rab11-GFP (Fig. 8D). Taken together, these findings strongly suggest that Us2 localizes predominantly to recycling endosomes.
Fig 8.

Us2 colocalizes predominantly with Rab11, a marker of recycling endosomes. (A to C) Vero cells were cotransfected with plasmids encoding HSV-2 Us2 and either Rab5-GFP (A), Rab7-GFP (B), or Rab11-GFP (C). Plasmids encoding Rab5-, Rab7-, and Rab11-GFP fusion proteins were a kind gift from C. Parish, Cornell University. Cells were fixed at 24 h posttransfection and were stained for Us2. Images of stained cells were captured by confocal microscopy. Rab signals are EGFP fluorescence and are displayed in green. The Us2 signal is red. Nuclei were detected using the DNA stain Hoechst 33342 (blue). Arrowheads in panel C highlight the colocalization of Us2 and Rab11 signals. Representative images are shown. (D) Quantification of the colocalization of Us2-labeled puncta with Rab5, Rab7, and Rab11. The data are presented as percentages of Us2-containing puncta that are also positive for Rab5, Rab7, or Rab11. A total of 12 fields of cells and 249 Us2 puncta were analyzed. The analysis was performed in a manner similar to that for the quantification of Us2-transferrin colocalization, for which results are shown in Fig. 7D and E. Error bars represent the standard errors of the means observed for differences between different fields of cells for each Rab protein analyzed.
HSV-2 Us2 binds to ubiquitinated proteins.
In an attempt to identify cellular proteins that facilitate Us2 interactions with membranes, a Flag–HSV-2 Us2 expression plasmid was transfected into 293T cells. At 24 h posttransfection, cells were collected, and membranes were purified by membrane flotation as described above for Fig. 3A. Purified membranes were incubated with an anti-Flag affinity gel to immunoprecipitate protein complexes. Protein species specifically binding to Flag-Us2 were excised from SDS-PAGE gels and were identified by LC–MS-MS analysis. While a number of proteins were identified in this analysis, including the membrane proteins erlin-1, erlin-2, and calnexin, these proved to have nonspecific interactions with HSV-2 Us2 (data not shown). Strikingly, a large proportion of peptides identified in our LC–MS-MS analysis were derived from ubiquitin.
To investigate the interaction of Us2 with ubiquitin in infected cells, Vero cells were infected with either WT HSV-2, HSV-2 ΔUs2, or HSV-2 with Flag-tagged Us2. Cell lysates were prepared at 6 hpi and were incubated with an anti-Flag affinity gel. Precipitated protein complexes were subjected to Western blotting and were probed with monoclonal antibody FK2, which is reactive against mono- and polyubiquitinated proteins (Fig. 9A). The results showed that Flag-tagged Us2 could specifically pull down ubiquitin-conjugated proteins. Similar results were obtained for lysates of cells infected with a recombinant HSV-1 strain with Flag-tagged Us2 (Fig. 9B). Despite the observation that Flag-tagged HSV-1 Us2 was not detected at high levels in infected-cell lysates, immunoaffinity purification of HSV-1 Us2 clearly demonstrated the copurification of ubiquitin-modified proteins.
Fig 9.

Us2 is a ubiquitin-interacting protein. (A and B) Vero cells were infected with WT HSV-2, HSV-2 ΔUs2, or recombinant HSV-2 with Flag-tagged Us2 (A) or with WT HSV-1 or recombinant HSV-1 with Flag-tagged Us2 (B). At 6 hpi, cell lysates were prepared, and proteins were immunoprecipitated (IP) using an anti-Flag affinity gel. Immunoprecipitated proteins were detected using antibodies against HSV-1 Us2, HSV-2 Us2, or mono- and polyubiquitin (FK2). (C) HSV-2 Us2 coimmunoprecipitates with ubiquitin-conjugated proteins from infected cells. Vero cells were infected with WT HSV-2 or a ΔUs2 virus for 6 h. Cells were collected, and lysates were incubated overnight with a monoclonal antibody against mono- and polyubiquitin (FK2). Proteins were pulled down using protein G agarose and were subjected to Western blotting for detection of the presence of Us2, ICP5, ICP27, UL21, and gD. Asterisks indicate the Ig heavy chain. (D) HSV-2 Us2 interacts directly with ubiquitin. Vero cells were infected with WT HSV-2 for 6 h. Cell lysates were prepared and were incubated overnight with ubiquitin-conjugated beads. Interacting proteins were subjected to Western blotting and were probed with an antibody against Us2. G, protein G agarose (negative control); mono, monoubiquitin-conjugated agarose; K48, K48-linked tetraubiquitin-conjugated agarose; K63, K63-linked tetraubiquitin-conjugated agarose. (E) HSV-2 Us2 interacts with ubiquitin in the absence of other viral proteins. An empty vector (pFlag-CMV2) or N-terminally Flag-tagged Us2 was transfected into 293T cells. At 24 h posttransfection, cells were collected in lysis buffer, and lysates were incubated overnight with anti-Flag beads at 4°C. Immunoprecipitated proteins were eluted using 3×Flag peptides. Eluates were incubated overnight with either protein G agarose or monoubiquitin-conjugated beads at 4°C. Us2 bound to the beads was detected by Western blotting using anti-Flag antibodies. G, protein G agarose; mono, monoubiquitin-conjugated beads.
HSV-2 Us2 is coprecipitated by ubiquitin-conjugated proteins.
To confirm the interaction between HSV-2 Us2 and ubiquitinated proteins, Vero cells were either mock infected or infected with WT HSV-2 or HSV-2 ΔUs2 for 6 h. Cell lysates were incubated overnight with antibody FK2, which is reactive against mono- and polyubiquitin. Antigen/antibody complexes were precipitated using protein G agarose and were analyzed by Western blotting (Fig. 9C). The results show that Us2 could be pulled down using antibody FK2, confirming the interaction between HSV-2 Us2 and ubiquitin-conjugated proteins. As a specificity control, viral proteins, including the major capsid protein ICP5, the tegument protein UL21, the regulatory protein ICP27, and glycoprotein gD, were tested for their abilities to interact with ubiquitin-conjugated proteins. None of these viral proteins were coprecipitated with the FK2 antibody, suggesting that the interaction of Us2 with ubiquitin-conjugated proteins is specific.
HSV-2 Us2 interacts with ubiquitin.
The data obtained so far suggested that Us2 either was capable of binding to multiple ubiquitin-conjugated proteins or could interact with ubiquitin specifically. To determine whether HSV-2 Us2 interacts with ubiquitin, rather than with multiple ubiquitinated species, Vero cells were infected with WT HSV-2. At 6 hpi, cell lysates were prepared and were incubated with either protein G agarose, as a negative control, or monoubiquitin-conjugated agarose. Precipitated complexes were washed and were analyzed by Western blotting for Us2 (Fig. 9D). The results showed that Us2 could be pulled down by monoubiquitin-conjugated agarose but not by protein G agarose, suggesting that Us2 interacts specifically with ubiquitin.
To determine whether the interaction between HSV-2 Us2 and ubiquitin requires other viral proteins, N-terminally Flag-tagged Us2 was transfected into 293T cells. Due to the low levels of Us2 expression in transfected cells, Us2 was first immunoprecipitated using an anti-Flag affinity gel and then eluted using a Flag peptide that competes for binding with anti-Flag antibodies. The eluate containing purified Us2 was then incubated with monoubiquitin-conjugated agarose beads or with protein G agarose as a negative control. The ability of Us2 to interact with ubiquitin was determined by Western blotting (Fig. 9E). The results showed that HSV-2 Us2 interacted with ubiquitin purified from transfected cells, suggesting that the interaction does not require other viral proteins.
Ubiquitin binding proteins can interact either with ubiquitin molecules or with the linkages between conjugated ubiquitin molecules. The preference of binding toward a specific ubiquitin linkage is associated with the function of ubiquitin binding proteins. We hypothesized that if HSV-2 Us2 was involved in the proteasomal degradation pathway, it would interact with K48-linked polyubiquitin chains with higher affinity. Alternatively, if Us2 was involved in endocytosis or lysosomal degradation, it might be predicted to interact preferentially with K63-linked polyubiquitin chains. To study the Us2-ubiquitin interaction in more detail, HSV-2-infected cell lysates were incubated with either K48-linked or K63-linked tetraubiquitin-conjugated agarose, and Us2 interactions were determined by Western blotting (Fig. 9D). The results showed that HSV-2 Us2 interacted with both K48-linked tetraubiquitin and K63-linked tetraubiquitin, with similar efficiencies.
DISCUSSION
Jiang and coworkers reported previously that HSV-2 Us2 was a virus structural component that was expressed with late kinetics (20). The data presented here are consistent with those findings (Fig. 1). However, the previous study suggested that HSV-2 Us2 localized primarily to discrete granules within the nucleus in infected cells and diffusely throughout the cytoplasm of transfected cells (20). Our findings are in stark contrast to those results, since we have demonstrated that Us2 localizes predominantly to recycling endosomes and to the PM. Differences in specificity between the anti-HSV-2 Us2 antisera reported here and that described by Jiang and colleagues may be responsible for this discrepancy; however, it is noteworthy that the localizations of Flag-tagged Us2, mCherry-Us2, and nonfused WT Us2 were indistinguishable in our studies (data not shown).
Although HSV-2 Us2 lacks canonical membrane association motifs, membrane flotation experiments revealed that it physically associates with membranes (Fig. 3A). Previous reports have shown that the EHV-1 Us2 protein localized to the PM despite lacking a classical N-terminal signal sequence, a putative transmembrane domain, or signals for lipid modifications (18). Likewise, PRV Us2 also associates with the PM; however, PRV Us2 PM localization can be explained, in part, by a prenylation motif that directs the addition of a farnesyl moiety to the C terminus of the protein (33). Interestingly, PRV Us2 mutants that cannot be prenylated maintain the ability to associate with vesicular membranes, indicating that additional membrane-targeting information is contained within Us2 (34). How then does HSV-2 Us2 associate with membranes? Based on the subcellular localization of Us2 and its ability to be incorporated into the virion tegument, we and others (18) hypothesized that Us2 is a peripheral membrane protein that associates with the cytoplasmic face of the membrane. Accordingly, we predicted that treatment with 1 M NaCl or 0.2 M Na2CO3 (pH 11) would strip HSV-2 Us2 from membranes, as has been observed with other herpesvirus tegument proteins that associate with membranes (31). Surprisingly, treatment of purified membrane fractions with these agents failed to release Us2 from membranes (Fig. 3C and D). Additionally, exposure of membranes to 0.01% digitonin failed to liberate Us2 from the membrane, suggesting that Us2 does not associate with membranes via a direct interaction with cholesterol (Fig. 3E). Collectively, these findings suggest that a portion of Us2 may be inserted into the membrane. It is unlikely that HSV-2 Us2 is a membrane-spanning protein, because, as has been reported for PRV Us2 (33), in the absence of detergent, virion-associated HSV-2 Us2 is protected from proteolytic digestion (Fig. 1E). A conserved stretch of hydrophobic amino acids between residues 121 and 137 of HSV-2 Us2 may be involved in anchoring Us2 to membranes.
Us2 membrane localization was largely restricted to endocytic vesicles and the PM. Most notably, Us2 did not colocalize with the Golgi apparatus or the ER, nor was its localization sensitive to BFA. At late times postinfection, Us2 was also observed to localize diffusely throughout the cytoplasm (Fig. 2B), suggesting that its synthesis was not occurring on ER-associated ribosomes. Taken together, these findings suggest that Us2 is not a component of the biosynthetic secretory pathway and that its association with membranes may occur posttranslationally. Us2 demonstrated substantial colocalization with Rab11, a marker of recycling endosomes (Fig. 8C and D). The finding that Us2 was associated with detergent-insoluble membranes (Fig. 4) is also consistent with its localization in recycling endosomes. Endocytic recycling compartments are rich in cholesterol, a major component of detergent-insoluble membranes, and it has been proposed that detergent-insoluble membranes play a role in the sorting of proteins within the recycling endosome (47, 48). Rab11 functions both in exocytic membrane traffic from the TGN to the PM (49) and in endocytic trafficking from early endosomes to the TGN (50). Consistent with these activities, imaging of Us2 in living cells suggested that Us2 is highly dynamic, and vesicles bearing Us2 exhibited rapid exchange between other Us2-containing vesicles and the PM. Moreover, the elaboration of tubular membrane projections from Us2 vesicles (Fig. 6; see also movie S1 in the supplemental material) and the kinetics of colocalization of Us2 with internalized transferrin (Fig. 7) suggested that Us2 localized to compartments that recycle membrane lipids and/or membrane proteins (43, 44). A recent ultrastructural study demonstrated that HSV-1 capsids are enveloped at the membranes of endocytic tubules in primary human fibroblasts and that Rab5 and Rab11, which are required for the formation of these endocytic tubules, are required for efficient virus production (51). The localization of Us2 to Rab11-positive membranes is consistent with the idea that Us2 is incorporated into nascent virions at endocytic tubules.
A number of enveloped viruses utilize Rab11-containing membranes for their assembly and egress (52). After their synthesis in the nucleus, the viral ribonucleoprotein complexes (vRNPs) of influenza virus A are exported into the cytoplasm and must traffic to the PM for incorporation into budding virions. The transport of these vRNPs from a perinuclear compartment to the PM is dependent on Rab11. It has been hypothesized that the vRNPs are tethered to the cytoplasmic faces of Rab11-containing vesicles en route to the PM (53, 54). A similar mechanism has been proposed to explain the trafficking of Sendai virus vRNPs from their site of synthesis in the cytoplasm to the PM (55). Andes virus, a hantavirus that acquires its envelope by budding into Golgi apparatus-derived membranes, relies on Rab11/Rab8-bearing vesicles for the transport of vesicles containing enveloped virions from the Golgi apparatus to the cell surface (56). Since Rab11 vesicle cargos are directed to the apical surfaces of polarized cells, it has been suggested that the engagement of these RNA viruses with Rab11 vesicles promotes the budding and/or release of virus from the apical surfaces of polarized cells (52). Because alphaherpesviruses, such as HSV-2, infect a variety of polarized cell types during natural infections, including mucosal epithelial cells and neurons of the peripheral nervous system, it may be that Us2 interactions with Rab11 vesicles regulate the trafficking of virions or subvirion components to apical surfaces.
An unresolved question is how Us2 achieves specificity in its membrane association. A number of motifs, typically found in transmembrane proteins, have been identified that can facilitate the localization of proteins to endocytic membranes and the TGN (57). These include the tyrosine-based YXXØ (where Y is tyrosine, X is any amino acid, and Φ is any bulky hydrophobic amino acid) motif and dileucine motifs that interact with the mu subunits of the clathrin adapters AP-1 and AP-2, which are involved in selecting cargos from the TGN and PM, respectively. Acidic clusters of amino acids, which additionally contain serine or threonine residues capable of being phosphorylated, have also been shown to localize proteins to the TGN. Notable alphaherpesvirus proteins that contain TGN-targeting acidic clusters include varicella-zoster virus glycoprotein gE and the HSV-1 tegument protein VP22 (58, 59). HSV-2 Us2 does not contain an acidic cluster motif; however, it does contain both YXXØ and dileucine motifs. The positions of these motifs in the Us2 sequence are not widely conserved among Us2 orthologs, so it is not clear if they are functional. It may be that the specificity of Us2 membrane localization is imparted through interaction with one or more cellular proteins.
To identify membrane-associated interacting partners of HSV-2 Us2, Flag-Us2 was transfected into mammalian cells, and protein complexes were immunoprecipitated and identified by LC–MS-MS analysis. Although membrane proteins identified in this analysis that also localize to detergent-resistant membranes (erlin-1 and erlin-2 [60]) were confirmed to be false positives, conspicuous amounts of ubiquitin were present in the precipitated protein complexes, suggesting that HSV-2 Us2 might interact with ubiquitin. The interaction between HSV-2 Us2 and ubiquitin was further confirmed by reciprocal immunoprecipitations and ubiquitin binding assays using Us2 derived from both infected- and transfected-cell lysates (Fig. 9). Together, these results indicate that Us2 interacts specifically with ubiquitin and can do so in the absence of other viral proteins; however, the possibility remains that the interaction between Us2 and ubiquitin is indirect and is mediated by a cellular ubiquitin-binding molecule that bridges the interaction. It has been estimated that more than 150 cellular proteins contain one or more ubiquitin binding domains (UBDs), and more than 20 distinct UBDs have been identified thus far (61). HSV-2 Us2 does not appear to contain any previously recognized UBD.
Ubiquitin and ubiquitination play important roles in virtually every cellular process. The role of ubiquitination in the endocytic pathway is a subject of intense investigation (62). Mono- or polyubiquitination of cell surface transmembrane proteins, usually by a K63 linkage, is sufficient to promote their rapid internalization. Enzymatic removal of ubiquitin by deubiquitinases can direct the internalized proteins back to the PM via recycling endosomes, while failure to remove the ubiquitin moiety from the internalized protein promotes interaction with ESCRT (endosomal sorting complex required for transport) components, incorporation into multivesicular bodies (MVBs), and, ultimately, degradation upon fusion of the MVB with lysosomes. A number of alphaherpesvirus glycoproteins undergo endocytosis from the PM, and some, but not all, bear well-defined motifs that promote their internalization (63). Additionally, at least two HSV-1 membrane proteins, Us9 and gB, are modified by ubiquitination (64, 65). Calistri and colleagues have suggested that K63 ubiquitination of gB promotes the trafficking of gB to modified MVB membranes that could serve as sites of final virion envelopment (65). HSV-1 Us9 is a tail-anchored type II membrane protein that lacks lysine residues, to which ubiquitin molecules are most frequently attached (64, 66, 67). HSV-1 Us9 is nevertheless conjugated to ubiquitin (64), presumably through the addition of a cysteine to the N terminus of the protein (68), as is seen in the modification of E2 ubiquitin-conjugating enzymes by E1 ubiquitin-activating enzymes (69), or by a nonconventional mechanism, such as that mediated by select E3 ligases on serine and threonine residues (70). The consequences of HSV-1 Us9 ubiquitination are not presently understood, nor is it known whether Us9 orthologs from other alphaherpesviruses are modified by ubiquitin. It is intriguing, however, that PRV Us2 was identified in a complex containing Us9 and the microtubule motor KIF1A, both of which are required for axonal sorting and efficient anterograde transport of virion-containing vesicles within neurons (23). These findings raise the possibility that Us2 interacts with, and modulates the activity of, the Us9/KIF1A vesicular sorting complex via its interaction with ubiquitin.
Defining the precise role of Us2 in the regulation and trafficking of ubiquitin-modified proteins requires further study. The importance of ubiquitin modifications to herpesvirus infections is underscored by the conservation of proteins containing E3 ubiquitin ligase activity or deubiquitinase activity throughout the Herpesviridae (71–73). While the functions of these activities are not fully understood at present, future investigation of the role of Us2-ubiquitin interactions during herpesvirus infections promises to advance our understanding of key virus-cell interactions and reveal new targets for antiviral intervention strategies for this group of important human and veterinary pathogens.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Canadian Institutes of Health Research operating grant 93804, Natural Sciences and Engineering Council of Canada Discovery Grant 418719, Canada Foundation for Innovation award 16389, and an award from the Violet E. Powell Research Fund to B.W.B. M.-H.K. was supported in part by R. Samuel McLaughlin and Robert John Wilson fellowships from Queen's University and by an Ontario Graduate Scholarship.
We thank R. Tsien (UCSD/HHMI), C. Parish (Cornell University), and N. Osterrieder (Freie Universität Berlin) for providing plasmids, A. Dolan and D. McGeoch (MRC Virology Unit, University of Glasgow) for providing plasmids and viruses, and G. Smith (Northwestern University) and Y. Kawaguchi (The University of Tokyo) for providing bacterial strains used in this study. Special thanks to Sina Kachoei for initial characterization of mCherry–HSV-2 Us2 fusion constructs. We acknowledge members of the Banfield laboratory for helpful comments on the manuscript and M. Lyman for helpful discussions.
Footnotes
Published ahead of print 19 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00994-13.
REFERENCES
- 1.Ben-Porat T, Kaplan AS. 1985. Molecular biology of pseudorabies virus, p 105–173 In Roizman B. (ed), The herpesviruses. Plenum Press, New York, NY [Google Scholar]
- 2.Roizman B, Knipe DM. 2001. Herpes simplex viruses and their replication, p 2399–2459 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 3.Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234 [DOI] [PubMed] [Google Scholar]
- 4.Wisner TW, Wright CC, Kato A, Kawaguchi Y, Mou F, Baines JD, Roller RJ, Johnson DC. 2009. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J. Virol. 83:3115–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones F, Grose C. 1988. Role of cytoplasmic vacuoles in varicella-zoster virus glycoprotein trafficking and virion envelopment. J. Virol. 62:2701–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turcotte S, Letellier J, Lippe R. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 79:8847–8860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whealy ME, Card JP, Meade RP, Robbins AK, Enquist LW. 1991. Effect of brefeldin A on alphaherpesvirus membrane protein glycosylation and virus egress. J. Virol. 65:1066–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394 [DOI] [PubMed] [Google Scholar]
- 9.Rémillard-Labrosse G, Mihai C, Duron J, Guay G, Lippe R. 2009. Protein kinase D-dependent trafficking of the large herpes simplex virus type 1 capsids from the TGN to plasma membrane. Traffic 10:1074–1083 [DOI] [PubMed] [Google Scholar]
- 10.Dolan A, Jamieson FE, Cunningham C, Barnett BC, McGeoch DJ. 1998. The genome sequence of herpes simplex virus type 2. J. Virol. 72:2010–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haanes EJ, Tomlinson CC. 1998. Genomic organization of the canine herpesvirus US region. Virus Res. 53:151–162 [DOI] [PubMed] [Google Scholar]
- 12.Leung-Tack P, Audonnet JC, Riviere M. 1994. The complete DNA sequence and the genetic organization of the short unique region (US) of the bovine herpesvirus type 1 (ST strain). Virology 199:409–421 [DOI] [PubMed] [Google Scholar]
- 13.McGeoch DJ, Dolan A, Donald S, Brauer DH. 1986. Complete DNA sequence of the short repeat region in the genome of herpes simplex virus type 1. Nucleic Acids Res. 14:1727–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Telford EA, Watson MS, McBride K, Davison AJ. 1992. The DNA sequence of equine herpesvirus-1. Virology 189:304–316 [DOI] [PubMed] [Google Scholar]
- 15.van Zijl M, van der Gulden H, de Wind N, Gielkens A, Berns A. 1990. Identification of two genes in the unique short region of pseudorabies virus; comparison with herpes simplex virus and varicella-zoster virus. J. Gen. Virol. 71(Part 8):1747–1755 [DOI] [PubMed] [Google Scholar]
- 16.Sakaguchi M, Urakawa T, Hirayama Y, Miki N, Yamamoto M, Hirai K. 1992. Sequence determination and genetic content of an 8.9-kb restriction fragment in the short unique region and the internal inverted repeat of Marek's disease virus type 1 DNA. Virus Genes 6:365–378 [DOI] [PubMed] [Google Scholar]
- 17.Longnecker R, Roizman B. 1987. Clustering of genes dispensable for growth in culture in the S component of the HSV-1 genome. Science 236:573–576 [DOI] [PubMed] [Google Scholar]
- 18.Meindl A, Osterrieder N. 1999. The equine herpesvirus 1 Us2 homolog encodes a nonessential membrane-associated virion component. J. Virol. 73:3430–3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weber PC, Levine M, Glorioso JC. 1987. Rapid identification of nonessential genes of herpes simplex virus type 1 by Tn5 mutagenesis. Science 236:576–579 [DOI] [PubMed] [Google Scholar]
- 20.Jiang YM, Yamada H, Goshima F, Daikoku T, Oshima S, Wada K, Nishiyama Y. 1998. Characterization of the herpes simplex virus type 2 (HSV-2) US2 gene product and a US2-deficient HSV-2 mutant. J. Gen. Virol. 79(Part 11):2777–2784 [DOI] [PubMed] [Google Scholar]
- 21.Wagenaar F, Pol JM, Peeters B, Gielkens AL, de Wind N, Kimman TG. 1995. The US3-encoded protein kinase from pseudorabies virus affects egress of virions from the nucleus. J. Gen. Virol. 76(Part 7):1851–1859 [DOI] [PubMed] [Google Scholar]
- 22.Lyman MG, Randall JA, Calton CM, Banfield BW. 2006. Localization of ERK/MAP kinase is regulated by the alphaherpesvirus tegument protein Us2. J. Virol. 80:7159–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kramer T, Greco TM, Taylor MP, Ambrosini AE, Cristea IM, Enquist LW. 2012. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 12:806–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634:421–430 [DOI] [PubMed] [Google Scholar]
- 25.Le Sage V, Jung M, Alter JD, Wills EG, Johnston SM, Kawaguchi Y, Baines JD, Banfield BW. 2013. The herpes simplex virus 2 UL21 protein is essential for virus propagation. J. Virol. 87:5904–5915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Gorman S, Fox DT, Wahl GM. 1991. Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science 251:1351–1355 [DOI] [PubMed] [Google Scholar]
- 28.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22:1567–1572 [DOI] [PubMed] [Google Scholar]
- 29.Lyman MG, Demmin GL, Banfield BW. 2003. The attenuated pseudorabies virus strain Bartha fails to package the tegument proteins Us3 and VP22. J. Virol. 77:1403–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graham FL, van der Eb AJ. 1973. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52:456–467 [DOI] [PubMed] [Google Scholar]
- 31.Brignati MJ, Loomis JS, Wills JW, Courtney RJ. 2003. Membrane association of VP22, a herpes simplex virus type 1 tegument protein. J. Virol. 77:4888–4898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lyman MG, Curanovic D, Enquist LW. 2008. Targeting of pseudorabies virus structural proteins to axons requires association of the viral Us9 protein with lipid rafts. PLoS Pathog. 4:e1000065. 10.1371/journal.ppat.1000065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clase AC, Lyman MG, del Rio T, Randall JA, Calton CM, Enquist LW, Banfield BW. 2003. The pseudorabies virus Us2 protein, a virion tegument component, is prenylated in infected cells. J. Virol. 77:12285–12298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang MH, Banfield BW. 2010. Pseudorabies virus tegument protein Us2 recruits the mitogen-activated protein kinase extracellular-regulated kinase (ERK) to membranes through interaction with the ERK common docking domain. J. Virol. 84:8398–8408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subramaniam VN, bin Mohd Yusoff AR, Wong SH, Lim GB, Chew M, Hong W. 1992. Biochemical fractionation and characterization of proteins from Golgi-enriched membranes. J. Biol. Chem. 267:12016–12021 [PubMed] [Google Scholar]
- 36.Cockrell AS, Muggeridge MI. 1998. Herpes simplex virus 2 UL45 is a type II membrane protein. J. Virol. 72:4430–4433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harder T, Kellner R, Parton RG, Gruenberg J. 1997. Specific release of membrane-bound annexin II and cortical cytoskeletal elements by sequestration of membrane cholesterol. Mol. Biol. Cell 8:533–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hooper NM. 1999. Detergent-insoluble glycosphingolipid/cholesterol-rich membrane domains, lipid rafts and caveolae. Mol. Membr. Biol. 16:145–156 [DOI] [PubMed] [Google Scholar]
- 39.Doms RW, Russ G, Yewdell JW. 1989. Brefeldin A redistributes resident and itinerant Golgi proteins to the endoplasmic reticulum. J. Cell Biol. 109:61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD. 1989. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56:801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y. 1986. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J. Biol. Chem. 261:11398–11403 [PubMed] [Google Scholar]
- 42.Ulmer JB, Palade GE. 1991. Effects of brefeldin A on the Golgi complex, endoplasmic reticulum and viral envelope glycoproteins in murine erythroleukemia cells. Eur. J. Cell Biol. 54:38–54 [PubMed] [Google Scholar]
- 43.Bonifacino JS, Rojas R. 2006. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 7:568–579 [DOI] [PubMed] [Google Scholar]
- 44.Maxfield FR, McGraw TE. 2004. Endocytic recycling. Nat. Rev. Mol. Cell Biol. 5:121–132 [DOI] [PubMed] [Google Scholar]
- 45.Ghosh RN, Maxfield FR. 1995. Evidence for nonvectorial, retrograde transferrin trafficking in the early endosomes of HEp2 cells. J. Cell Biol. 128:549–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sönnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. 2000. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J. Cell Biol. 149:901–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hao M, Mukherjee S, Sun Y, Maxfield FR. 2004. Effects of cholesterol depletion and increased lipid unsaturation on the properties of endocytic membranes. J. Biol. Chem. 279:14171–14178 [DOI] [PubMed] [Google Scholar]
- 48.Mayor S, Sabharanjak S, Maxfield FR. 1998. Cholesterol-dependent retention of GPI-anchored proteins in endosomes. EMBO J. 17:4626–4638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen W, Feng Y, Chen D, Wandinger-Ness A. 1998. Rab11 is required for trans-Golgi network-to-plasma membrane transport and a preferential target for GDP dissociation inhibitor. Mol. Biol. Cell 9:3241–3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilcke M, Johannes L, Galli T, Mayau V, Goud B, Salamero J. 2000. Rab11 regulates the compartmentalization of early endosomes required for efficient transport from early endosomes to the trans-Golgi network. J. Cell Biol. 151:1207–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hollinshead M, Johns HL, Sayers CL, Gonzalez-Lopez C, Smith GL, Elliott G. 2012. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J. 31:4204–4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bruce EA, Stuart A, McCaffrey MW, Digard P. 2012. Role of the Rab11 pathway in negative-strand virus assembly. Biochem. Soc. Trans. 40:1409–1415 [DOI] [PubMed] [Google Scholar]
- 53.Amorim MJ, Bruce EA, Read EK, Foeglein A, Mahen R, Stuart AD, Digard P. 2011. A Rab11- and microtubule-dependent mechanism for cytoplasmic transport of influenza A virus viral RNA. J. Virol. 85:4143–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eisfeld AJ, Kawakami E, Watanabe T, Neumann G, Kawaoka Y. 2011. RAB11A is essential for transport of the influenza virus genome to the plasma membrane. J. Virol. 85:6117–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chambers R, Takimoto T. 2010. Trafficking of Sendai virus nucleocapsids is mediated by intracellular vesicles. PLoS One 5:e10994. 10.1371/journal.pone.0010994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rowe RK, Suszko JW, Pekosz A. 2008. Roles for the recycling endosome, Rab8, and Rab11 in hantavirus release from epithelial cells. Virology 382:239–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bonifacino JS, Traub LM. 2003. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72:395–447 [DOI] [PubMed] [Google Scholar]
- 58.Zhu Z, Hao Y, Gershon MD, Ambron RT, Gershon AA. 1996. Targeting of glycoprotein I (gE) of varicella-zoster virus to the trans-Golgi network by an AYRV sequence and an acidic amino acid-rich patch in the cytosolic domain of the molecule. J. Virol. 70:6563–6575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O'Regan KJ, Brignati MJ, Murphy MA, Bucks MA, Courtney RJ. 2010. Virion incorporation of the herpes simplex virus type 1 tegument protein VP22 is facilitated by trans-Golgi network localization and is independent of interaction with glycoprotein E. Virology 405:176–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Browman DT, Resek ME, Zajchowski LD, Robbins SM. 2006. Erlin-1 and erlin-2 are novel members of the prohibitin family of proteins that define lipid-raft-like domains of the ER. J. Cell Sci. 119:3149–3160 [DOI] [PubMed] [Google Scholar]
- 61.Dikic I, Wakatsuki S, Walters KJ. 2009. Ubiquitin-binding domains—from structures to functions. Nat. Rev. Mol. Cell Biol. 10:659–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clague MJ, Liu H, Urbe S. 2012. Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev. Cell 23:457–467 [DOI] [PubMed] [Google Scholar]
- 63.Brideau AD, Enquist LW, Tirabassi RS. 2000. The role of virion membrane protein endocytosis in the herpesvirus life cycle. J. Clin. Virol. 17:69–82 [DOI] [PubMed] [Google Scholar]
- 64.Brandimarti R, Roizman B. 1997. Us9, a stable lysine-less herpes simplex virus 1 protein, is ubiquitinated before packaging into virions and associates with proteasomes. Proc. Natl. Acad. Sci. U. S. A. 94:13973–13978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Calistri A, Sette P, Salata C, Cancellotti E, Forghieri C, Comin A, Gottlinger H, Campadelli-Fiume G, Palu G, Parolin C. 2007. Intracellular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J. Virol. 81:11468–11478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brideau AD, Banfield BW, Enquist LW. 1998. The Us9 gene product of pseudorabies virus, an alphaherpesvirus, is a phosphorylated, tail-anchored type II membrane protein. J. Virol. 72:4560–4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lyman MG, Kemp CD, Taylor MP, Enquist LW. 2009. Comparison of the pseudorabies virus Us9 protein with homologs from other veterinary and human alphaherpesviruses. J. Virol. 83:6978–6986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ciechanover A, Ben-Saadon R. 2004. N-terminal ubiquitination: more protein substrates join in. Trends Cell Biol. 14:103–106 [DOI] [PubMed] [Google Scholar]
- 69.Pickart CM, Eddins MJ. 2004. Ubiquitin: structures, functions, mechanisms. Biochim. Biophys. Acta 1695:55–72 [DOI] [PubMed] [Google Scholar]
- 70.Shimizu Y, Okuda-Shimizu Y, Hendershot LM. 2010. Ubiquitylation of an ERAD substrate occurs on multiple types of amino acids. Mol. Cell 40:917–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Everett RD, Boutell C, McNair C, Grant L, Orr A. 2010. Comparison of the biological and biochemical activities of several members of the alphaherpesvirus ICP0 family of proteins. J. Virol. 84:3476–3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ohmura-Hoshino M, Goto E, Matsuki Y, Aoki M, Mito M, Uematsu M, Hotta H, Ishido S. 2006. A novel family of membrane-bound E3 ubiquitin ligases. J. Biochem. 140:147–154 [DOI] [PubMed] [Google Scholar]
- 73.Kattenhorn LM, Korbel GA, Kessler BM, Spooner E, Ploegh HL. 2005. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol. Cell 19:547–557 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.