

Abstract
Legionella pneumophila, the etiological agent for Legionnaires' disease, is ubiquitous in the aqueous environment, where it replicates as an intracellular parasite of free-living protozoa. Our understanding of L. pneumophila pathogenicity is obtained mostly from study of derivatives of several clinical isolates, which employ almost identical virulent determinants to exploit host functions. To determine whether environmental L. pneumophila isolates interact similarly with the model host systems, we analyzed intracellular replication of several recently isolated such strains and found that these strains cannot productively grow in bone marrow-derived macrophages of A/J mice, which are permissive for all examined laboratory strains. By focusing on one strain called LPE509, we found that its deficiency in intracellular replication in primary A/J macrophages is not caused by the lack of important pathogenic determinants because this strain replicates proficiently in two protozoan hosts and the human macrophage U937 cell. We also found that in the early phase of infection, the trafficking of this strain in A/J macrophages is similar to that of JR32, a derivative of strain Philadelphia 1. Furthermore, infection of these cells by LPE509 caused extensive cell death in a process that requires the Dot/Icm type IV secretion system. Finally, we showed that the cell death is caused neither by the activation of the NAIP5/NLRC4 inflammasome nor by the recently described caspase 11-dependent pathway. Our results revealed that some environmental L. pneumophila strains are unable to overcome the defense conferred by primary macrophages from mice known to be permissive for laboratory L. pneumophila strains. These results also suggest the existence of a host immune surveillance mechanism differing from those currently known in responding to L. pneumophila infection.
INTRODUCTION
Microorganisms in the Legionella genus are Gram-negative motile rod-shaped bacteria, of which at least 58 species have been described (1, 2). These bacteria are ubiquitous in freshwater habitats, where they exist as parasites of protozoa. These protists are the natural hosts of Legionella and thus are believed to provide the primary evolutionary pressure for the acquisition and maintenance of virulence factors necessary for the intracellular lifestyle of these bacteria. Several species of these bacteria, Legionella pneumophila in particular, are an important opportunistic pathogen for humans. Infection by L. pneumophila occurs when susceptible individuals inhale contaminated aerosols. Subsequent replication of the bacteria in alveolar macrophages could lead to Legionnaires' disease, a severe form of pneumonia characterized by inflammation and tissue damage (1).
The Dot/Icm type IV secretion system is essential for successful intracellular replication of L. pneumophila (3). Building with appropriately 26 proteins, the structure of the transporter is believed to traverse the two bacterial membranes and the phagosomal membrane, thus allowing the translocation of protein substrates into the host cell (4). One unique feature of the Dot/Icm system is its large cohort of substrate proteins, which comprise almost 10% of the protein coding capacity of the bacterial genome (5–7). These virulence factors, also called effectors, re-orchestrate diverse cellular signaling cascades involved in various host processes such as innate immunity and vesicle trafficking to create a niche permissive for bacterial replication (8, 9). It is well established that a set of effectors function to prevent the delivery of the bacterial phagosome into the lysosomal network by actively remodeling its membrane composition and biochemical properties (8, 9). These effectors employ sophisticated mechanisms to hijack the activities of some small GTPases critical for the formation and transport of vesicles between the endoplasmic reticulum and the Golgi apparatus (10). Another set of effectors interfere with the host cell death pathways to ensure that the infected cell will not trigger the programmed cell death pathway to eliminate the infection before the completion of bacterial replication (11). Other cellular processes, such as autophagy and those regulated by phosphatidylinositol, are targeted by highly specific and effective bacterial virulence factors to make the intracellular environment of phagocytes conducive for bacterial replication (12, 13).
In addition to the understanding of their roles in bacterial virulence, detailed biochemical characterization of these effectors has led to the discovery of enzymes of novel modes of action, thus revealing previously unrecognized mechanisms of signal transduction in the host. For example, the bacterium regulates the activity of the small GTPase Rab1 by reversible AMPylation (14–16) and phosphorylcholination (17, 18) with specific effectors. It is believed that these posttranslational modifications and signaling mechanisms also occur in host cells to regulate functions in cells of some as-yet-unrecognized functions or under special conditions (19).
Robust immune response of a healthy individual normally is sufficient to clear L. pneumophila, making legionellosis largely a disease of people with a compromised immune system (20). Experiments with the mouse model have revealed that immune cells are able to recognize classic L. pneumophila PAMP molecules such as lipopolysaccharide, flagellin, and nucleic acids to mount a robust response (21). In addition, in infection model using primary bone-derived macrophages, inflammatory cell death or pyroptosis has been recognized as an effective mechanism for macrophages to recognize and respond to intracellular L. pneumophila. Flagellin delivered into the macrophage cytosol presumably as a noncognate substrate of the Dot/Icm transporter, dictates the susceptibility of different mouse lines to L. pneumophila infection (22, 23). Further analyses of this mouse strain-specific susceptibility have revealed that direct sensing of flagellin by the NOD family protein NAIP5 leads to the activation of the NLRC4 inflammasome and subsequent clearance of bacterial infection (24, 25). These studies thus have provided a clear molecular explanation for the susceptibility of A/J mice, whose allele of NAIP5 harbors a number of polymorphisms to resistant mouse lines such as C57BL/6 (26). Primary mouse macrophages also respond robustly to activities conferred by the Dot/Icm system independent of the pathways triggered by the Toll-like receptors (TLRs) or intracellular flagellin. Infection of macrophages by wild-type L. pneumophila but not a mutant deficient in Dot/Icm induces the mitogen-activated protein (MAP) kinase pathway, as well as the NF-κB pathway (27–29). Interestingly, protein synthesis inhibition conferred by five Dot/Icm substrates is part of the signals sensed by both the NF-κB and the MAP kinase pathways (30, 31). Prolonged activation of the NF-κB pathway is achieved by the delayed resynthesis of IκB, the labile inhibitory protein of NF-κB (31). A similar mechanism was postulated to be involved in the activation of MAP kinase pathway (31). However, the labile molecule akin to IκB involved in such activation has not yet been identified. Similarly, the induction of type I interferon by macrophages only occurs when the cells are challenged with bacterial strains harboring a functional Dot/Icm machinery, but the biochemical property of the ligand(s) responsible for the activation of these immune responses remains elusive (32). Thus, activity conferred either by the components of the transporter or by functions of its protein substrates can be recognized by the host immune surveillance machineries. The detection of activities specific to virulent microorganisms may allow immune cells to distinguish pathogen from nonpathogen, a mechanism that complements the function of classic receptors which sense ligands present in both pathogen and nonpathogen (33).
Because environmental bacteria are considered the primary source of outbreaks of Legionnaires' disease, many studies have been directed to analyze L. pneumophila isolated from natural or manufactured water systems (34, 35). Although some of these studies have examined intracellular bacterial growth of these environmental isolates in cultured human macrophages, few of them have determined the response of different hosts such as primary murine macrophages to challenge of these bacteria (35–37). In our study to analyze the virulence of several serogroup 1 strains recently isolated from the environment, we found that none of these strains can replicate in mouse bone marrow-derived macrophages permissive for commonly used laboratory strains under standard experimental conditions. We showed that infection of primary murine macrophages by one such strain, LPE509 leads to extensive cytotoxicity in a process that requires the Dot/Icm transporter but not the flagellin or inflammatory responses mediated by caspase-1 or caspase-11. Our results suggest that strain LPE509 codes for one or more factors capable of inducing a pyroptosis-like cell death once delivered into mouse primary macrophages by the Dot/Icm transporter.
MATERIALS AND METHODS
Bacterial strains, plasmids, media.
The bacterial strains and plasmids used in the present study were listed in Table 1. The environmental strains were isolated from water distribution systems in Shanghai hospitals, the serogroup of which was determined by an Oxoid Legionella latex test kit (DR0800) (38). The strain JR32 was a derivative of Legionella pneumophila Philadelphia 1, which is resistant to streptomycin and deficient in the restriction system (39). L. pneumophila was cultured on charcoal-yeast extract (CYE) plates or in ACES-buffered yeast extract (AYE) broth (40). Bacteria used for infection were grown in AYE broth to post-exponential phase based on both the optical value (OD600) and bacterial motility (41). Escherichia coli strains were grown on L-agar plates or in L broth, and antibiotics were used at the following concentrations: for E. coli, ampicillin (Amp) was used at 100 μg/ml, kanamycin (Km) was used at 30 μg/ml, and streptomycin was used at 50 μg/ml, and for L. pneumophila, Km was used at 20 μg/ml and Amp was used at 100 μg/ml. To express His6-RavZ in LPE509, we inserted the ravZ gene into pZL507 (42), which allows the expression of His6-tagged protein in L. pneumophila to give pHis6-RavZ. The in-frame deletion mutant of flaA(lpg1340) in strain JR32 and strain LPE509 was constructed using the standard method (43). Briefly, 1.2-kbp fragments upstream and downstream of lpg1340 were amplified from genomic DNA of JR32, digested with appropriate restriction enzymes, and ligated to the pir protein-dependent suicide plasmid pSR47s (44) to produce pZLΔflaA. The primer pairs used were as follows (restriction sites are underlined): 5′-CTGGAGCTCTATCATATCAATATGTCCAT-3′/5′-CTGGGATCCCCCGAAACACCCAAATTAC G-3′ and 5′-CTGGGATCCCAGGTACAGCGATGTTGGCA-3′/5′-CTGGTCGACCCCAATCT CTGCTTTCTTTA-3′. Because the sequence of this locus in JR32 and LPE509 is identical (45), pZLΔflaA was used to introduce the deletion in both strains. Similarly, plasmid pZLΔdotA (46) was used to delete dotA (lpg2686), the gene essential for the activity of the Dot/Icm transporter from both strains JR32 and LPE509. To delete icmW, the locus from strain Lp01ΔicmW (47) was amplified with the primers 5′-GGGAGCTCCAATAACCCTTGCCTGTAC-3′ and 5′-CCGTCTAGACCATTGTTGGTTTGGTTGTC-3′ and was inserted into pSR47s to give pΔicmW. Plasmids were introduced into L. pneumophila strains by triparental mating (43) or by electroporation, transconjugants, or transformants were streaked onto the CYE plate supplemented with 5% sucrose, and mutants were screened by colony PCR with primers flanking the genes of interest.
Table 1.
Bacterial strains used in this study
| Strain or plasmid | Genotype and relevant markersa | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α(λpir) | supE44 dlacU169(ϕ80lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 pir tet::Mu recA | Our collection |
| XL1-Blue | recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′ proAB lacIqZM15 Tn10(Tetr)] | Stratagene |
| L. pneumophila | ||
| LPE509 | Environmental wild-type strain | 38 |
| LPEG1 | Environmental wild-type strain | 38 |
| LPE602 | Environmental wild-type strain | 38 |
| LPE422 | Environmental wild-type strain | 38 |
| JR32 | r− m+ Strr derivative of strain Philadelphia 1 | 39 |
| JR32ΔflaA | ΔflaA mutant of JR32 | This study |
| JR32ΔdotA | ΔdotA mutant of JR32 | This study |
| LPE509ΔflaA | ΔflaA mutant of LPE509 | This study |
| LPE509ΔdotA | ΔdotA mutant of LPE509 | This study |
| LPE509ΔicmW | ΔicmW mutant of LPE509 | This study |
| D. discoideum AX4 | Wild-type strain for intracellular growth analysis | 72 |
| A. castellanii | ATCC 30234 | ATCC |
| Plasmids | ||
| pZLΔdotA | dotA in-frame deletion construct on pSR47s | 46 |
| pZLΔflaA | flaA in-frame deletion construct on pSR47s | This study |
| pZLdotA | Derivative of pJB908 expressing DotA | 46 |
| pSdhA | Derivative of pJB908 expressing SdhA | 57 |
| pHis6-RavZ | Derivative of pZL507 expressing His6-RavZ | This study |
| pΔplaA | plaA in-frame deletion construct on pSR47s | 55 |
| pΔicmW | icmW in-frame deletion construct on pSR47s | This study |
Tetr, tetracycline resistance; Strr, streptomycin resistance.
Mouse macrophage preparation, cell culture and infection.
A/J, C56BL/6, caspase 3−/− mice were purchased from the Jackson laboratory (Bar Harbor, ME). Caspase 1/11−/− (48) and the corresponding C56BL/6 control mice were generously supplied by R. E. Vance (UC Berkeley). Bone marrow-derived macrophages were prepared from 6- to 10-week-old female mice with L-supernatant as described previously (49). Macrophages were seeded in 24-well plate the day before infection. Cell density of 4 × 105/well was used for intracellular bacterial growth, and 2 × 105/well was used for immunofluorescence assay and lactate dehydrogenase (LDH) release assay. U937 cells were grown in RPMI cell culture medium supplemented with heat-inactivated 10% fetal bovine serum. Cells were induced by phorbol myristate acetate at a final concentration of 10 ng/ml for 36 h as described previously (50). Dictyostelium discoideum was grown and maintained at 21°C in HL-5 medium supplemented with penicillin and streptomycin (100 U/ml; Invitrogen, Carlsbad, CA) as previously described (51). Acanthamoeba castellanii (ATCC 30234) was grown at 28°C in proteose peptone-yeast extract-glucose medium (52). A total of 5 × 105 cells were seeded in each well of 24-well plate with MB medium and incubated at room temperature for 4 h before infection. Infection of host cells was performed according to established protocols (22, 41). For intracellular bacterial growth, a multiplicity of infection (MOI) of 0.05 was used to allow analysis of multiple time points. For cytotoxicity assays and single cell-based analyses such as LAMP-1 staining, infection center, and evaluation of LDH release, moderate MOIs (1 or 2) were used.
Intracellular bacterial growth.
To determine bacterial intracellular growth, we infected host cells plated seeded in 24-well plates at an MOI of 0.05. At 2 h after adding the bacteria, we synchronized the infection by washing the cells with phosphate-buffered saline (PBS) for three times. Infected macrophages were incubated at 37°C with 5% CO2, whereas infected D. discoideum and A. castellanii were incubated at 25°C. At the desired time points, the cells were lysed with 0.02% saponin, diluted lysate was plated on CYE plates, and CFU were determined from triplicate infections of each strain. When appropriate, the apoptosis inhibitor z-VAD (53), the necrosis inhibitor necrostatin (59), and the autophagy inhibitor 3-methyladenine (54) was added to final concentrations of 20 μM, 30 μM, and 10 mM, respectively.
Antibodies, immunostaining, and immunoblotting.
For immunostaining, mouse macrophages were infected with L. pneumophila at an MOI of 2. Samples were collected 1 h after infection for LAMP-1 staining, 2 and 6 h after infection for Rab1 staining. After fixation, samples were first stained for bacteria using anti-L. pneumophila antibodies (42) at a dilution of 1:10,000. Extracellular and intracellular bacteria were distinguished using secondary antibodies conjugated to distinct fluorescent dyes. LAMP-1 was labeled with antibody clone IDB4 (Development Studies Hybridoma Bank, Iowa City, IA) at a dilution of 1:10. Rab1 was labeled with antibody (Santa Cruz Biotechnology, catalog no. sc-599) at a 1:200 dilution. Processed coverslips were mounted on glass slides with an antifade reagent (Vector Laboratories, CA). For infection center, mouse macrophages were infected at an MOI of 1; extracellular bacteria were removed by washing the cells with PBS for three times. Samples were collected at 14 h after infection. After fixation, extracellular and intracellular Legionella were stained with an anti-Legionella antibody followed by secondary antibody conjugated with fluorescein isothiocyanate and Texas Red, respectively. Samples were examined by visual inspection with an Olympus IX-81 fluorescence microscope. Infection centers were classified into three categories as small (1 to 3 bacteria), medium (4 to 9 bacteria), and large (≥10 bacteria). The average distribution of the infection centers was determined from triplicate infections of each bacterial strain.
For immunoblot, the His tag antibody from Sigma (catalog no. H1029) was used at 1:40,000; the SdhA (55) was used as 1:5,000 and the flagellin antibody (56) was used at 1:5,000. To detect the protein of interest in L. pneumophila, bacteria grown in AYE broth to the indicated OD600 or post-exponential phase ready for infection were collected by centrifugation and lysed by boiling in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. SDS-PAGE-separated proteins were transferred to nitrocellulose membranes and were incubated with appropriately diluted primary antibodies in PBS before incubating with IRDye-conjugated secondary antibodies. The signals were detected by a Li-Cor's Odyssey system (Li-Cor Biosciences, Lincoln, NE).
TUNEL staining.
A total of 2 × 105 mouse macrophages were infected with an MOI of 1 and then incubated for 14 h after infection. Samples were fixed and stained with intracellular and extracellular bacteria as described above, and then stained using TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling) with an in situ cell death detection kit (Roche Diagnostics, Indianapolis, IN). A 50-μl TUNEL reaction mixture was added to each coverslip and incubated at room temperature for 30 min. After three times washes with PBS, coverslips were mounted with antifade reagent.
PI and Hoechst 33342 staining.
A total of 2 × 105 mouse macrophages were infected with an MOI of 1 and incubated for 2 h after infection. After centrifugation at 200 × g for 5 min, the culture medium was removed, and 0.5 ml of the dye solution containing 500 nM propidium iodide (PI) and 1:10,000 Hoechst was then added into each well. The plate was incubated in the dark for 30 min before image acquisition using an Olympus IX-81 fluorescence microscope.
LDH release assay and glycine protection.
LDH release during infection was determined using the CytoTox 96 Assay (Promega, WI). Macrophages were infected at an MOI of 1 on 24-well plates and incubated in 37°C for 2 or 4 h. After centrifugation at 200 × g for 5 min, 50-μl supernatant of each well was transferred to a new 96-well enzymatic assay plate. Then, 50 μl of reconstituted substrate mix was added to each well of the plate and incubated in dark at room temperature for 30 min. The enzymatic reaction was stopped by adding 50-μl stop solution to each well. The absorbance 490 was measured using a Bio-Tek microplate reader. Total LDH release was determined by complete lysis of the cells using a lysis solution provided by the kit, and spontaneous LDH release was determined by using the supernatant of cells without infection. For glycine protection, glycine was added to tissue culture medium at a final concentration of 20 mM for 1 h prior to infection and was kept in infection samples throughout the experimental duration. The percentage of LDH release was calculated with the following formula: LDH release (%) = (experimental LDH release − spontaneous LDH release)/(total LDH release − spontaneous LDH release)·100.
Statistical analyses.
Statistical significance for relevant data was calculated using the unpaired Student t test.
RESULTS
Several environmental L. pneumophila strains are unable to replicate in A/J macrophages.
L. pneumophila can be readily isolated from aquatic niches, but few studies have investigated how environmental isolates interact with mammalian hosts (35–37). We thus initiated a study to analyze the interactions between mouse bone marrow macrophages and several serogroup 1 L. pneumophila strains isolated from hospital water distribution systems in Shanghai, China (38). Infection of the A/J macrophages with strain JR32, a derivative of the Philadelphia 1 strain (39), at an MOI of 0.05, led to robust intracellular bacterial growth (Fig. 1). As expected, a mutant of JR32 defective in the Dot/Icm type IV transporter failed to replicate in these cells. Surprisingly, although the number of internalized bacteria was very similar to strain JR32, under this low-MOI condition, infections with four environmental isolates did not yield productive bacterial replication throughout the experimental duration (Fig. 1). Moreover, whereas the Dot/Icm mutant was cleared slowly by the macrophages, the bacterial counts of the environmental strains dropped drastically in 24 h postinfection and continued to decrease throughout the experimental duration (Fig. 1). These results indicate under the infection condition used, none of these four environmental L. pneumophila strains can replicate in mouse macrophages permissive for standard laboratory strains.
Fig 1.
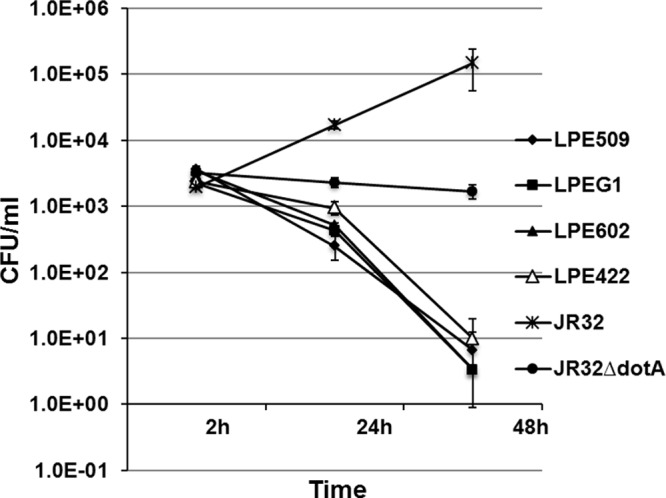
Intracellular replication of several L. pneumophila environmental isolates in A/J mouse macrophages. Bone marrow-derived macrophages were challenged with indicated bacterial strains grown to post-exponential phase at an MOI of 0.05. Infections were synchronized 2 h after adding the bacteria. At the indicated time points, total bacterial counts (CFU) were determined by plating appropriately diluted saponin-solubilized infected cells onto bacteriological medium. Infections were performed in triplicate, and the data shown were from one representative experiment of three independent experiments with similar results.
Strain LPE509 replicates proficiently in two amoebae hosts and differentiated human monocytes.
To understand the mechanism that restricts the growth of these environmental strains in A/J mouse macrophages, we chose one strain, LPE509 for further analysis because of the availability of its genome sequence information (45). We first sought to determine whether such growth deficiency is due to the absence of some essential virulence factors by examining its ability to replicate in four different hosts. Bacteria grown to post-exponential phase were used to challenge D. discoideum, A. castellanii, human macrophages derived from the U937 cell and primary A/J macrophages, respectively. As expected, the laboratory strain JR32 grew robustly in all four hosts (Fig. 2). Importantly, D. discoideum, A. castellanii, and U937 cells support the growth of strain LPE509 at rates comparable to those of JR32 (Fig. 2A to C). Consistent with our earlier observation, strain LPE509 was unable to productively replicate in A/J macrophages (Fig. 2D). We next determined whether intracellular growth of LPE509 requires the Dot/Icm transporter. Deletion of dotA abolished the ability of LPE509 to replicate in U937 cells and such defects can be fully complemented by a plasmid expressing dotA of the Philadelphia 1 strain (see Fig. S1 in the supplemental material). Therefore, similar to strain JR32, the Dot/Icm system is essential for the pathogenicity of strain LPE509 in permissive hosts. These results indicate that the intracellular growth defects exhibited by strain LPE509 in A/J macrophage is host specific.
Fig 2.

Intracellular growth of strain LPE509 in three different hosts. Differentiated human D. discoideum (A), A. castellanii (B), U937 macrophage (C), or bone marrow-derived macrophages (D) from A/J mouse were infected with indicated bacterial strains grown to post-exponential phase at an MOI of 0.05. Infections were synchronized 2 h after adding the bacteria to the cells. At the indicated time points, infected samples were lysed with saponin, and appropriately diluted cell lysates were plated onto bacteriological charcoal yeast extract medium. All infections were performed in triplicate, and the data shown were from one representative experiment of four experiments with highly similar results.
Vacuoles containing LPE509 are targeted properly in A/J macrophage.
To determine the mechanism underlying the growth defect of LPE509 in A/J macrophage, we analyzed the trafficking of its phagosome in this host. Cells infected with LPE509, its dotA mutant and appropriate control strains for 1 h were stained for the lysosomal marker LAMP-1. Similar to wild-type JR32, positive staining signals for LAMP-1 were absent in ca. 82% of the vacuoles formed by strain LPE509 (Fig. 3A). Deletion of dotA led to acquisition of this marker by the bacterial vacuoles (Fig. 3A). We also examined the recruitment of Rab1, the small GTPase important for L. pneumophila infection (10) to vacuoles of LPE509. About 60% of phagosomes containing LPE509 stained positively for Rab1, which was abolished by the deletion of the dotA (Fig. 3B). Thus, vacuoles containing strain LPE509 are not delivered into the lysosomal network, and its defect in intracellular growth is not caused by its inability to manipulate the cellular processes such as the recruitment of Rab1.
Fig 3.
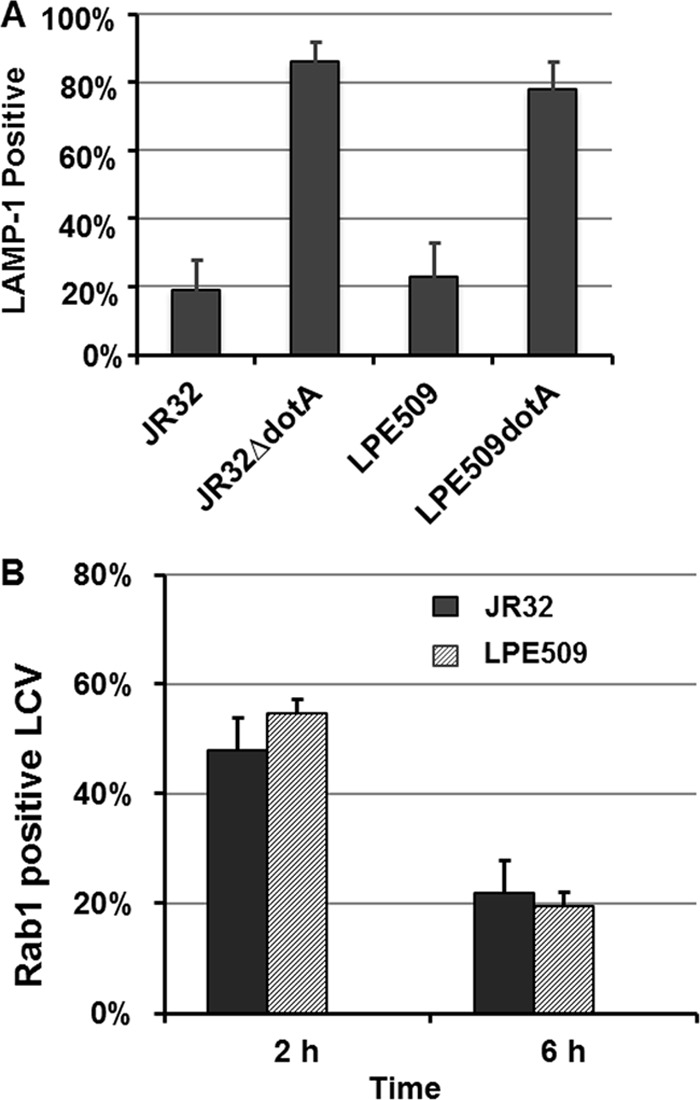
Intracellular trafficking of strain LPE509 in A/J macrophages. Bone marrow-derived macrophages seeded on glass coverslips were infected with properly grown bacterial strains at an MOI of 2. One hour after infection, samples were washed with medium and were fixed at the indicated time points. After labeling extracellular and intracellular bacteria differently by immunostaining, the host proteins LAMP-1 (A) and Rab1 (B) were stained with specific antibodies. The data were collected using an IX-81 Olympus microscope by inspecting bacterial vacuoles for the association of the protein staining signals. All infections were performed in triplicate, and at least 100 bacterial phagosomes were scored for each sample. The data shown were the averages of two independent experiments.
A fraction of internalized LPE509 bacteria establish larger vacuoles in A/J macrophages.
The observation that phagosomes of LPE509 were properly targeted in macrophages indicates that some of them may develop into large vacuoles containing multiple bacteria when infection proceeds to later phase. We thus determined the distribution of vacuoles formed by this strain 14 h after uptake. In infections using strain JR32, >63% of the vacuoles were large phagosomes containing ≥10 bacteria. The fractions of small (1 to 3 bacteria/vacuole) and medium-size (4 to 9 bacteria/vacuole) phagosomes were ca. 19 and 17%, respectively (Fig. 4). In contrast, the majority (∼58%) of the LPE509 vacuoles at this time point contained 1 to 3 bacteria; ca. 22% of them were intermediate vacuoles with 4 to 9 bacteria, and only ca. 18% were large vacuoles containing more than 10 bacteria (Fig. 4). Thus, strain LPE509 was able to form replicative vacuoles in A/J macrophages, but further development of most of these vacuoles did not occur.
Fig 4.
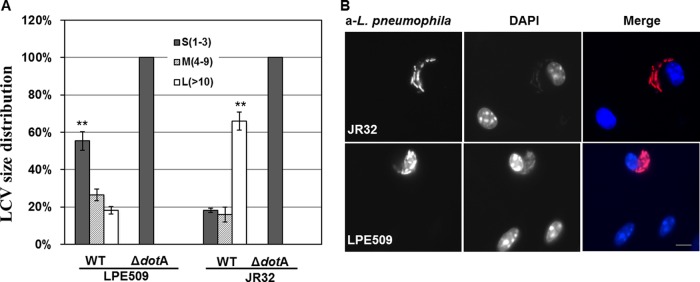
Formation of replicative vacuoles by strain LPE509 in macrophages. A/J mouse macrophages on glass coverslips were infected with appropriately grown bacterial strains at an MOI of 1. Infections were synchronized 1 h after bacterial challenge and were allowed to proceed for an additional 13 h. Samples were then fixed and subjected to immunostaining to label intracellular and extracellular L. pneumophila with different fluorescent colors. When necessary, samples were further staining with DAPI (4′,6′-diamidino-2-phenylindole) to highlight the nuclei of host cells. The bacterial phagosomes were scored under a fluorescence microscope according to the number of bacteria they harbored. Phagosomes containing more than 10 bacteria were categorized as large; those with 4 to 9 bacteria were called medium and those with 1 to 3 bacteria were classified as small vacuoles. (A) The distributions of vacuole sizes of strain JR32 and LPE509 and their derivatives defective in the Dot/Icm transporter. **, P < 0.001 for values of big or small vacuoles between strain JR32 and LPE509. (B) Representative images of large vacuoles by both bacterial strains. Note the condensed nucleus of the cell infected by strain LPE509. Bar, 10 μm.
LPE509 flagellin is not responsible for its intracellular replication restriction in A/J macrophages.
The phenotypes associated with the interactions between strain LPE509 and A/J macrophage are reminiscent of those seen in infections of C57BL/6 macrophages with strain Philadelphia 1, in which sensing of flagellin by NAIP5 leads to pyroptosis and termination of intracellular bacterial replication (24, 25). To examine whether the defects were caused by the recognition of LPE509 flagellin by NAIP5 of A/J mice, we constructed LPE509ΔflaA, a mutant that no longer expressed flagellin (Fig. 5A). Challenge of A/J macrophages with this mutant at a low MOI (0.05) did not lead to productive intracellular bacterial growth (Fig. 5B). These results indicate that the lack of intracellular growth of LPE509 in A/J macrophages was not due to variations in flagellin that can be recognized by NAIP5 of A/J to cause pyroptosis.
Fig 5.
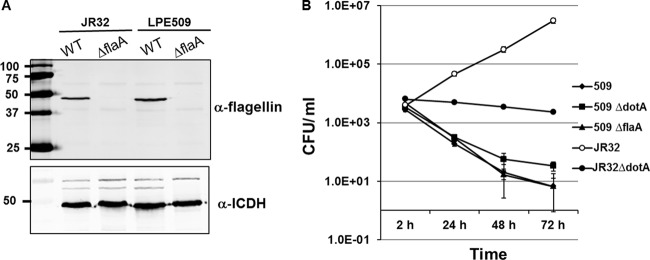
Intracellular growth of a mutant of strain LPE509 lacking flagellin in A/J macrophages. (A) Expression of flagellin by the testing bacterial strains. Cells of indicated strains grown to post-exponential phase were lysed with Laemmli sample buffer and total protein was resolved by SDS-PAGE. Flagellin was detected by a specific antibody and the isocitrate dehydrogenase (ICDH) was probed as a loading control (lower panel). (B) Testing bacterial strains grown in AYE broth to post-exponential phase were used to infect bone marrow-derived macrophages from A/J mice at an MOI of 0.05. Infections synchronized 2 h after adding bacteria were lysed with saponin at indicated time points, and total bacterial counts were determined by plating appropriate diluted lysates onto bacteriological medium. Infections were performed in triplicate, and similar results were obtained in three independent experiments.
LPE509 causes extensive membrane damage in A/J macrophages.
To determine the phenotypic changes associated with infection of A/J macrophages by LPE509, we examined the morphology of these cells infected with a moderate MOI (2). Under this condition, strain JR32 did not cause discernible change in cell morphology (Fig. 6A, top panel). However, challenge with LPE509 led to extensive cell rounding and no such phenotype was detected in infections with the strain LPE509ΔdotA (Fig. 6A, middle and lower panels, respectively). We further examined the integrity of plasma membrane of macrophages infected with these strains. Infected macrophages were stained simultaneously by PI, which is membrane impermeable and generally excluded from viable cells and by Hoechst 33342, which allows the staining of nuclei of both live and dead cells. In samples receiving LPE509 at an MOI of 1, ca. 45% of macrophages became permeable to PI. In infections using strain JR32, only ca. 6% of the cells were stained positively by PI (Fig. 6B and C). The dotA mutants of neither strain caused detectable membrane damage under this infection condition (Fig. 6B and C).
Fig 6.

Infection by strain LPE509 damages macrophage morphology and membrane integrity. Bone marrow derived macrophages from A/J mice were challenged with indicated bacterial strains grown to post-exponential phase at an MOI of 1 for 2 h. (A and B) Cell morphology and membrane integrity of infected samples was examined under a microscope. Similarly infected samples were simultaneously stained with PI and Hoechst 33342 and were analyzed by imaging with a fluorescence microscope. Bar, 10 μm. (C) Quantitation of membrane damaged cells. Membrane damage was determined by positive staining by PI under a microscope, at least 500 cells were scored in each sample for infections done in triplicate. The data shown are from one representative experiment from three independent experiments with similar results.
At 14 h postinfection, a portion of vacuoles containing more than 10 bacteria were established but total bacterial counts at 24 h actually decreased, suggesting that these bacteria were cleared by macrophages (Fig. 1, 2, and 4). We thus examined whether these cells were undergoing cell death by TUNEL staining, which is an effective way for macrophages to eliminate internalized and replicating bacteria. At this time point, ca. 40% of cells infected by strain LPE509 stained positively for TUNEL and deletion of flaA did not significantly reduce the rate of TUNEL-positive cells (Fig. 7A and B). For cells harboring JR32, <20% were TUNEL positive (Fig. 7A and B). Disruption of the Dot/Icm transporter abolished the ability of both strains to induce cell death (Fig. 7A and B).
Fig 7.

Infection by LPE509 caused extensive cell death on A/J macrophages. (A and B) Bone marrow-derived macrophages from A/J mice were infected by indicated L. pneumophila strains at an MOI of 1 for 14 h, and infected cells were immunostained to identify extracellular and intracellular bacteria, followed by TUNEL staining to obtain representative images of the nuclei of infected cells. The label of host nuclei by the TUNEL reagent was inspected and scored under a fluorescence microscope. Bar, 10 μm. (C) Cells infected with the similar bacterial strains at an MOI of 1 were assayed for the release of LDH. Each experiment was performed in triplicate, and similar results were obtained in four independent experiments.
We further examined the cytotoxicity of LPE509 to A/J macrophages by measuring membrane integrity of infected cells using LDH release as an indicator. At an MOI of 1, LDH release induced by strain LPE509 was significantly higher than that caused by strain JR32 (Fig. 7C). Deletion of flaA abolished the ability of strain JR32. However, although deletion of flaA in strain LPE509 detectably reduced the cytotoxicity, this mutant still significantly caused LDH release when infection was performed with an MOI of 1 (Fig. 7C). Again, the type IV secretion system in both strains is essential for the cytotoxicity (Fig. 7C). Taken together, these results indicate that strain LPE509 is more cytotoxic than strain JR32 on A/J macrophages. Importantly, the cytotoxicity of strain LPE509 on these macrophages requires the Dot/Icm transporter but not flagellin.
Cell death induced by LPE509 is independent of several known bacterial or host factors involved in cell death.
In A/J macrophages, the Dot/Icm substrate SdhA functions to prevent infected cells from cell death by protecting the integrity of the bacterial phagosomal membranes (55). We thus determined the expression of SdhA in strain LPE509, similar amount of SdhA was detected in strain LPE509 and JR32 (see Fig. S2 in the supplemental material). Furthermore, a plasmid carrying sdhA capable of complementing the Lp02ΔsdhA strain (57) did not rescue the growth defect of strain LPE509 (see Fig. S2 in the supplemental material). Finally, deletion of the phospholipase gene plaA, the loss of which is known to suppress the intracellular growth defect of the sdhA mutant (55), did not detectably rescue the growth defect or cytotoxicity of LPE509 (see Fig. S2 in the supplemental material). Thus, the growth defect exhibited by LPE509 in primary murine macrophages is not caused by abnormal expression of SdhA.
The genome region of LPE509 coding for ravZ, the gene involved in inhibition of autophagy is identical to that of Philadelphia 1 (13), and the introduction of a plasmid expressing His6-RavZ did not rescue the cell death induced by LPE509. Similarly, addition of the autophagy inhibitor 3-methyladenine did not detectably lead to gain of bacterial intracellular growth (see Fig. S3 in the supplemental material). These results suggest that the cell death induced by LPE509 is not caused by the inability to inhibit autophagy by this strain.
In order to determine whether the subset of Dot/Icm substrates whose translocation requires the IcmS/W chaperone (58) are responsible for the induction of cells death, we constructed strain LPE509ΔicmW and examined the relevant phenotypes. This mutant failed to induce cytotoxicity in A/J macrophages and was unable to replicate in this host (see Fig. S4 in the supplemental material), suggesting that the protein(s) responsible for the induction of cell death in LPE509 is translocated in an IcmS/W-dependent manner.
We also examined several host pathways potentially responsible for the cell death. First, necrostatin, the effective inhibitor for necrosis (59) did not protect macrophages infected by LPE509 from cell death (see Fig. S5 in the supplemental material), suggesting that necrosis play only neglectable roles, if any, in the cell death caused by LPE509. Similarly, neither macrophage from mice deficient in caspase 3 nor the addition of the pan-caspase inhibitor z-VAD prevent the cell death upon challenge with strain LPE509 (see Fig. S6 in the supplemental material), indicating that the classical apoptotic pathway is not responsible for the cell death induced by LPE509.
The cytotoxicity induced by LPE509 is independent of caspase 1 and caspase 11 and can be inhibited by glycine.
Two pathways are known to be important for the activation of pyroptosis in response to infection of murine macrophages by L. pneumophila with a functional Dot/Icm system. The NLRC4/NAIP5 inflammasome pathway that senses flagellin by the leucine-rich repeat-containing protein (NLR) family, apoptosis inhibitory protein 5 (NAIP5) (24, 25), and the caspase 11-dependent pathway that engages as-yet-unidentified ligand(s) (60, 61). To determine whether any of these two pathways is involved in the cell death induced by strain LPE509, we examined the intracellular growth and cytotoxicity of this strain in macrophages from mice deficient in both caspase 1 and caspase 11 (Casp1/11−/−). Consistent with earlier results, macrophages from C57BL/6 or Casp1/11−/− supported robust intracellular growth the flaA mutant of JR32 (22, 56), but no productive intracellular growth of LPE509ΔflaA was detected (Fig. 8A and B). Further, mutants without a functional Dot/Icm transporter were unable to cause cytotoxicity in C57BL/6 macrophages (Fig. 8C). Importantly, whereas the flaA mutant of JR32 almost completely failed to cause cytotoxicity on C57BL/6 macrophages, the flagellin-deficient mutant of LPE509 is still able to induce LDH release from these cells at levels similar to its wild-type parent strain (Fig. 8C).
Fig 8.

Intracellular growth and cytotoxicity of strain LPE509 in macrophages from mouse deficient in caspase 1 and caspase 11. (A and B) Bone marrow-derived macrophages from C57BL/6 mice (A) and capase1/11-deficient mice (Casp1/11−/−) (B) were infected with the indicated bacterial strains at an MOI of 0.05, and the intracellular replication of the bacteria was determined by obtaining total CFU at a 24-h interval. (C and D) The cytotoxicity of the relevant bacterial strains to these cells was examined by measuring the release of lactate dehydrogenase (LDH) upon being challenged by these bacterial at an MOI of 1 for 4 h. All assays were performed in triplicate, and similar results were obtained in four independent experiments.
When macrophages from Casp1/11−/− mice were tested, we found that strain LPE509ΔflaA still strongly induced cytotoxicity (Fig. 8D). In contrast, the flaA-deficient strain of JR32 failed to induce membrane damage and LDH release (Fig. 8D). Taken together, these results indicate that the rapid cell death induced by strain LPE509 occurs in a way that requires the Dot/Icm system but not caspase 1 or caspase 11. Notably, we consistently observed that strain JR32 still caused considerable LDH release from Casp1/11−/− macrophages (Fig. 8D). Although JR32 and Lp02 are derivatives of strain Philadelphia 1, the latter strain was not able to induce cytotoxicity on macrophages from Casp1/11−/− mice (60, 61). Such differences may result from the genetic variations between these strains. For example, the type IVB secretion system Lvh found in JR32 (62) is absent in the genome of Lp02 (63, 64).
Pathogen-induced pyroptosis can be prevented by exogenous glycine, which blocks the formation of nonspecific plasma membrane leaks for small ions such as sodium (65, 66). We thus determined the nature of cell death induced by infections with our bacterial strains in macrophages from caspase1/11−/− mice. Macrophages infected by strains LPE509, JR32, and LPE509ΔflaA all exhibited extensive cytotoxicity (Fig. 8D). On the other hand, similar to results in previously experiments, JR32ΔflaA failed to induce LDH release in these cells (Fig. 8D). Importantly, glycine treatment significantly blocked cytotoxicity induced by these bacterial strains, including LPE509ΔflaA (Fig. 8D). Interestingly, glycine cannot rescue the defect in intracellular growth of LPE509 in A/J macrophages (see Fig. S7 in the supplemental material). This result is consistent with the fact that exogenous glycine functions to prevent nonspecific plasma membrane damage, which is downstream of the cell death (65, 66). Taken together, these results suggest that cytotoxicity induced by strain LPE509ΔflaA is caused by a form of cell death that may differ from classical pyroptosis involved in caspase 1.
DISCUSSION
L. pneumophila is an aquatic pathogen and environmental bacteria are the primary sources of infection. Although primary mouse macrophages have been extensively used to analyze the virulence of L. pneumophila, how these cells respond to environmental strains of this pathogen is largely unknown. Here, we present evidence that the environmental L. pneumophila strain LPE509 causes cell death in primary mouse macrophages permissive to derivatives of the Philadelphia 1 strain, thus restricting its replication.
Several lines of evidence indicate that the restriction of LPE509 replication in macrophages from A/J mice is due to host cell death but not the targeting of the bacteria into the lysosomal network. First, LPE509 robustly replicates in D. discoideum, a protozoan host known to be more restrictive for L. pneumophila replication (64). Similarly, it replicates well in cultured human macrophages such as U937 cells. Further, intracellular growth in both hosts requires the Dot/Icm system. Second, vacuoles containing LPE509 did not acquire lysosomal markers such as LAMP-1 and was able to efficiently recruit Rab1, the small GTPase important for intracellular life cycle of L. pneumophila (67). Third, a portion of the LPE509 vacuoles developed into large phagosomes containing multiple bacteria, this is reminiscent to the infection of strain Lp02 in the restrictive macrophages from C57BL/6 (68). Finally, LPE509 causes severe damage to plasma membranes of A/J macrophages at low MOIs (i.e., and MOI of 1), which is in contrast to the high MOI (>500) required for strain Lp02 to nonspecifically form pores on the same cell type (69). Thus, the cytotoxicity of LPE509 almost certainly is not caused by the pore formation activity of its Dot/Icm system.
At lower MOIs, derivatives of strain Philadelphia 1 cause inflammatory cell death in murine macrophages by two distinct mechanisms. First, in macrophages from mice coding for a nonpermissive NAIP5 allele, flagellin introduced into the cytosol by the Dot/Icm transporter is sensed by NAIP5, leading to the activation of the NLRC4 inflammasome and subsequent caspase 1 activation and pyroptosis (24, 25). In macrophages primed by ligands of TLRs, a parallel pyroptotic pathway involved in caspase 11 is also induced by L. pneumophila with a competent Dot/Icm transporter (60, 61). Apparently, the induction of cytotoxicity by strain LPE509 is independent of the flagellin/NAIP5/NLRC4 axis or the caspase 11-dependent pathway. Furthermore, the cell death was not caused by abnormal expression of SdhA, because either overexpression of SdhA or deletion of plaA, the phospholipase involved in maintaining the membrane integrity of the bacterial vacuole, failed to gain productive intracellular bacterial replication (see Fig. S2 in the supplemental material). Finally, autophagy, necrosis, or the classical apoptosis pathway appeared not responsible for the cell death (see Fig. S3, S5, and S6 in the supplemental material). Thus, we postulate that murine macrophages code for receptor(s) that can detect one or more factors specifically coded for by LPE509 and that the engagement of the receptor with the ligand may lead to the activation of enzymes such as caspases capable of causing membrane permeabilization and subsequent cell death. That the cytotoxicity induced by LPE509 or its flaA mutant can be blocked by glycine suggests the cell death induced by these bacterial strains is pyroptosis.
The primary function of the L. pneumophila Dot/Icm system is to deliver effectors into host cells to construct the vacuoles permissive for bacterial growth in phagocytes. Recent evidence suggests that these effectors assume a modular structure to accommodate the expansion of the host range in the environment (64). Comparing to strain Philadelphia 1, the genome of LPE509 is predicted to encode 496 unique proteins (45). Thus, it is possible that LPE509 codes for more effectors, and some of these extra effectors serve as ligands for the putative receptor in murine macrophages. Our observation that the icmW mutant no longer induced cell death in A/J macrophages (see Fig. S4 in the supplemental material) suggests that this chaperone complex is important for the translocation of the protein(s) responsible for the phenotypes. However, because laboratory strains such Lp01 lacking the chaperone complex is severely defective in intracellular replication in A/J macrophages (47), suggesting that the number of Dot/Icm substrates whose translocation requires IcmS/W is large. Thus, the usefulness of using the IcmS/W-dependent phenotype to identify such substrates may be limited. Alternatively, some PAMP molecules associated with LPE509 may be able to prime murine macrophages, rendering them more sensitive to factors shared between itself and Philadelphia 1 that are capable of activating a novel cell death pathway. The fact that human cells such as U937 did not respond similarly to LPE509 challenge suggests the absence of such receptor in these cells or that the allele in human bears mutations that render it no longer sensitive to the ligand.
It has now been recognized that specialized protein transporters such as the Dot/Icm of L. pneumophila play essential roles not only in their virulence but also in functioning as messengers that trigger host immune responses (70, 71). The latter activity has been exploited as a highly effective tool to dissect the mechanisms used by which host to distinguish pathogenic microorganisms from nonharmful ones (70). The robust response with a readily detectable cell death phenotype to LPE509 from macrophages has provided an excellent model for future identification of the bacterial and host factors responsible for the signaling pathway that leads to restriction of intracellular bacterial growth, which may reveal novel receptors and their cognate ligands for immune surveillance in mammalian systems. It will also be interesting to determine whether the factors from LPE509 responsible for triggering this cell death are present in other L. pneumophila strains, either from environmental or from clinical settings.
Supplementary Material
ACKNOWLEDGMENTS
We thank Russell E. Vance (University of California at Berkeley, Berkeley, CA) for mice and for helpful discussions. We also thank Ralph Isberg (Tufts University Medical School, Boston, MA), Michele Swanson (University of Michigan, School of Medicine, Ann Arbor, MI), and Craig Roy (Yale University School of Medicine, New Haven, CT) for bacterial strains, plasmids, or antibodies.
This study was supported by NIH-NIAID grants K02AI085403 and R21AI092043 (Z.-Q.L.) from the National Institutes of Health and Shanghai Leading Talent Projects (no. 036, 2010) (J.-M.Q.).
Footnotes
Published ahead of print 10 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00252-13.
REFERENCES
- 1.Fields BS, Benson RF, Besser RE. 2002. Legionella and Legionnaires' disease: 25 years of investigation. Clin. Microbiol. Rev. 15:506–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diederen BM. 2008. Legionella spp. and Legionnaires' disease. J. Infect. 56:1–12 [DOI] [PubMed] [Google Scholar]
- 3.Isberg RR, O'Connor TJ, Heidtman M. 2009. The Legionella pneumophila replication vacuole: making a cozy niche inside host cells. Nat. Rev. Microbiol. 7:13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagai H, Kubori T. 2011. Type IVB secretion systems of Legionella and other gram-negative bacteria. Front. Microbiol. 2:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schroeder GN, Petty NK, Mousnier A, Harding CR, Vogrin AJ, Wee B, Fry NK, Harrison TG, Newton HJ, Thomson NR, Beatson SA, Dougan G, Hartland EL, Frankel G. 2010. Legionella pneumophila strain 130b possesses a unique combination of type IV secretion systems and novel Dot/Icm secretion system effector proteins. J. Bacteriol. 192:6001–6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, Luo ZQ. 2011. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6:e17638. 10.1371/journal.pone.0017638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gomez-Valero L, Rusniok C, Cazalet C, Buchrieser C. 2011. Comparative and functional genomics of Legionella identified eukaryotic like proteins as key players in host-pathogen interactions. Front. Microbiol. 2:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hubber A, Roy CR. 2010. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26:261–283 [DOI] [PubMed] [Google Scholar]
- 9.Xu L, Luo ZQ. 2013. Cell biology of infection by Legionella pneumophila. Microbes Infect. 15:157–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardiman CA, McDonough JA, Newton HJ, Roy CR. 2013. The role of Rab GTPases in the transport of vacuoles containing Legionella pneumophila and Coxiella burnetii. Biochem. Soc. Trans. 40:1353–1359 [DOI] [PubMed] [Google Scholar]
- 11.Luo ZQ. 2011. Striking a balance: modulation of host cell death pathways by Legionella pneumophila. Front. Microbiol. 2:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu F, Zhu W, Brennan L, Tao L, Luo ZQ, Mao Y. 2012. Structural basis for substrate recognition by a unique Legionella phosphoinositide phosphatase. Proc. Natl. Acad. Sci. U. S. A. 109:13567–13572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choy A, Dancourt J, Mugo B, O'Connor TJ, Isberg RR, Melia TJ, Roy CR. 2012. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338:1072–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muller MP, Peters H, Blumer J, Blankenfeldt W, Goody RS, Itzen A. 2010. The Legionella effector protein DrrA AMPylates the membrane traffic regulator Rab1b. Science 329:946–949 [DOI] [PubMed] [Google Scholar]
- 15.Neunuebel MR, Chen Y, Gaspar AH, Backlund PS, Jr, Yergey A, Machner MP. 2011. De-AMPylation of the small GTPase Rab1 by the pathogen Legionella pneumophila. Science 333:453–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan Y, Luo ZQ. 2011. Legionella pneumophila SidD is a deAMPylase that modifies Rab1. Nature 475:506–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukherjee S, Liu X, Arasaki K, McDonough J, Galan JE, Roy CR. 2011. Modulation of Rab GTPase function by a protein phosphocholine transferase. Nature 477:103–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan Y, Arnold RJ, Luo ZQ. 2011. Legionella pneumophila regulates the small GTPase Rab1 activity by reversible phosphorylcholination. Proc. Natl. Acad. Sci. U. S. A. 108:21212–21217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oesterlin LK, Goody RS, Itzen A. 2012. Posttranslational modifications of Rab proteins cause effective displacement of GDP dissociation inhibitor. Proc. Natl. Acad. Sci. U. S. A. 109:5621–5626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boshuizen HC, Neppelenbroek SE, van Vliet H, Schellekens JF, den Boer JW, Peeters MF, Conyn-van Spaendonck MA. 2001. Subclinical Legionella infection in workers near the source of a large outbreak of Legionnaires disease. J. Infect. Dis. 184:515–518 [DOI] [PubMed] [Google Scholar]
- 21.Neild AL, Roy CR. 2004. Immunity to vacuolar pathogens: what can we learn from Legionella? Cell Microbiol. 6:1011–1018 [DOI] [PubMed] [Google Scholar]
- 22.Ren T, Zamboni DS, Roy CR, Dietrich WF, Vance RE. 2006. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2:e18. 10.1371/journal.ppat.0020018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, Henry T, Sun YH, Cado D, Dietrich WF, Monack DM, Tsolis RM, Vance RE. 2008. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 9:1171–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kofoed EM, Vance RE. 2011. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477:592–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600 [DOI] [PubMed] [Google Scholar]
- 26.von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. 2013. Recognition of bacteria by inflammasomes. Annu. Rev. Immunol. 31:73–106 [DOI] [PubMed] [Google Scholar]
- 27.Abu-Zant A, Jones S, Asare R, Suttles J, Price C, Graham J, Kwaik YA. 2007. Anti-apoptotic signaling by the Dot/Icm secretion system of Legionella pneumophila. Cell Microbiol. 9:246–264 [DOI] [PubMed] [Google Scholar]
- 28.Losick VP, Isberg RR. 2006. NF-κB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J. Exp. Med. 203:2177–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shin S, Case CL, Archer KA, Nogueira CV, Kobayashi KS, Flavell RA, Roy CR, Zamboni DS. 2008. Type IV secretion-dependent activation of host MAP kinases induces an increased proinflammatory cytokine response to Legionella pneumophila. PLoS Pathog. 4:e1000220. 10.1371/journal.ppat.1000220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fontana MF, Banga S, Barry KC, Shen X, Tan Y, Luo ZQ, Vance RE. 2011. Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog. 7:e1001289. 10.1371/journal.ppat.1001289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fontana MF, Shin S, Vance RE. 2012. Activation of host mitogen-activated protein kinases by secreted Legionella pneumophila effectors that inhibit host protein translation. Infect. Immun. 80:3570–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monroe KM, McWhirter SM, Vance RE. 2009. Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog. 5:e1000665. 10.1371/journal.ppat.1000665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vance RE, Isberg RR, Portnoy DA. 2009. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6:10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Molmeret M, Jarraud S, Mori JP, Pernin P, Forey F, Reyrolle M, Vandenesch F, Etienne J, Farge P. 2001. Different growth rates in amoeba of genotypically related environmental and clinical Legionella pneumophila strains isolated from a thermal spa. Epidemiol. Infect. 126:231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin T, Yan G, Ren H, Zhou H, Wang H, Xu Y, Zhao M, Guan H, Li M, Shao Z. 2013. High prevalence, genetic diversity and intracellular growth ability of legionella in hot spring environments. PLoS One 8:e59018. 10.1371/journal.pone.0059018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Izu K, Yoshida S, Miyamoto H, Chang B, Ogawa M, Yamamoto H, Goto Y, Taniguchi H. 1999. Grouping of 20 reference strains of Legionella species by the growth ability within mouse and guinea pig macrophages. FEMS Immunol. Med. Microbiol. 26:61–68 [DOI] [PubMed] [Google Scholar]
- 37.Messi P, Bargellini A, Anacarso I, Marchesi I, de Niederhausern S, Bondi M. 2013. Protozoa and human macrophages infection by Legionella pneumophila environmental strains belonging to different serogroups. Arch. Microbiol. 195:89–96 [DOI] [PubMed] [Google Scholar]
- 38.Tao LL, Hu BJ, Yu LL, Zhou ZY, Xie HM, Zhou CM. 2011. Genetic diversity of Legionella pneumophila serogroup 1 in hospital water distribution systems in Shanghai. Zhonghua Jie He He Hu Xi Za Zhi 34:100–103. (In Chinese.) [PubMed] [Google Scholar]
- 39.Sadosky AB, Wiater LA, Shuman HA. 1993. Identification of Legionella pneumophila genes required for growth within and killing of human macrophages. Infect. Immun. 61:5361–5373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feeley JC, Gibson RJ, Gorman GW, Langford NC, Rasheed JK, Mackel DC, Baine WB. 1979. Charcoal-yeast extract agar: primary isolation medium for Legionella pneumophila. J. Clin. Microbiol. 10:437–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conover GM, Derre I, Vogel JP, Isberg RR. 2003. The Legionella pneumophila LidA protein: a translocated substrate of the Dot/Icm system associated with maintenance of bacterial integrity. Mol. Microbiol. 48:305–321 [DOI] [PubMed] [Google Scholar]
- 42.Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo ZQ. 2010. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog. 6:e1000822. 10.1371/journal.ppat.1000822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dumenil G, Isberg RR. 2001. The Legionella pneumophila IcmR protein exhibits chaperone activity for IcmQ by preventing its participation in high-molecular-weight complexes. Mol. Microbiol. 40:1113–1127 [DOI] [PubMed] [Google Scholar]
- 44.Rankin S, Li Z, Isberg RR. 2002. Macrophage-induced genes of Legionella pneumophila: protection from reactive intermediates and solute imbalance during intracellular growth. Infect. Immun. 70:3637–3648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma J, He Y, Hu BJ, Luo Z-Q. 2013. Genome sequence of an environmental isolate of the bacterial pathogen Legionella pneumophila. Genome Announc. 1(3):e00320-13. 10.1128/genomeA.00320-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y, Gao P, Banga S, Luo ZQ. 2008. An in vivo gene deletion system for determining temporal requirement of bacterial virulence factors. Proc. Natl. Acad. Sci. U. S. A. 105:9385–9390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ninio S, Zuckman-Cholon DM, Cambronne ED, Roy CR. 2005. The Legionella IcmS-IcmW protein complex is important for Dot/Icm-mediated protein translocation. Mol. Microbiol. 55:912–926 [DOI] [PubMed] [Google Scholar]
- 48.Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. 2010. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host. Microbe 8:471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swanson MS, Isberg RR. 1995. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect. Immun. 63:3609–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tilney LG, Harbour OS, Connelly PS, Robinson CG, Roy CR. 2001. How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J. Cell Sci. 114:4637–4650 [DOI] [PubMed] [Google Scholar]
- 51.Solomon JM, Rupper A, Cardelli JA, Isberg RR. 2000. Intracellular growth of Legionella pneumophila in Dictyostelium discoideum, a system for genetic analysis of host-pathogen interactions. Infect. Immun. 68:2939–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Segal G, Shuman HA. 1999. Legionella pneumophila utilizes the same genes to multiply within Acanthamoeba castellanii and human macrophages. Infect. Immun. 67:2117–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edgeworth JD, Spencer J, Phalipon A, Griffin GE, Sansonetti PJ. 2002. Cytotoxicity and interleukin-1β processing following Shigella flexneri infection of human monocyte-derived dendritic cells. Eur. J. Immunol. 32:1464–1471 [DOI] [PubMed] [Google Scholar]
- 54.McFarland AJ, Anoopkumar-Dukie S, Perkins AV, Davey AK, Grant GD. 2011. Inhibition of autophagy by 3-methyladenine protects 1321N1 astrocytoma cells against pyocyanin- and 1-hydroxyphenazine-induced toxicity. Arch. Toxicol. 86:275–284 [DOI] [PubMed] [Google Scholar]
- 55.Creasey EA, Isberg RR. 2012. The protein SdhA maintains the integrity of the Legionella-containing vacuole. Proc. Natl. Acad. Sci. U. S. A. 109:3481–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Molofsky AB, Byrne BG, Whitfield NN, Madigan CA, Fuse ET, Tateda K, Swanson MS. 2006. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J. Exp. Med. 203:1093–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laguna RK, Creasey EA, Li Z, Valtz N, Isberg RR. 2006. A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. U. S. A. 103:18745–18750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cambronne ED, Roy CR. 2007. The Legionella pneumophila IcmSW complex interacts with multiple Dot/Icm effectors to facilitate type IV translocation. PLoS Pathog. 3:e188. 10.1371/journal.ppat.0030188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. 2008. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4:313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, Zamboni DS, Roy CR. 2013. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc. Natl. Acad. Sci. U. S. A. 110:1851–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, Miao EA. 2013. Caspase-11 protects against bacteria that escape the vacuole. Science 339:975–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Segal G, Russo JJ, Shuman HA. 1999. Relationships between a new type IV secretion system and the icm/dot virulence system of Legionella pneumophila. Mol. Microbiol. 34:799–809 [DOI] [PubMed] [Google Scholar]
- 63.Ensminger AW, Yassin Y, Miron A, Isberg RR. 2012. Experimental evolution of Legionella pneumophila in mouse macrophages leads to strains with altered determinants of environmental survival. PLoS Pathog. 8:e1002731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O'Connor TJ, Adepoju Y, Boyd D, Isberg RR. 2011. Minimization of the Legionella pneumophila genome reveals chromosomal regions involved in host range expansion. Proc. Natl. Acad. Sci. U. S. A. 108:14733–14740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brennan MA, Cookson BT. 2000. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 38:31–40 [DOI] [PubMed] [Google Scholar]
- 66.Frank A, Rauen U, de Groot H. 2000. Protection by glycine against hypoxic injury of rat hepatocytes: inhibition of ion fluxes through nonspecific leaks. J. Hepatol. 32:58–66 [DOI] [PubMed] [Google Scholar]
- 67.Kagan JC, Roy CR. 2002. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 4:945–954 [DOI] [PubMed] [Google Scholar]
- 68.Derre I, Isberg RR. 2004. Macrophages from mice with the restrictive Lgn1 allele exhibit multifactorial resistance to Legionella pneumophila. Infect. Immun. 72:6221–6229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kirby JE, Vogel JP, Andrews HL, Isberg RR. 1998. Evidence for pore-forming ability by Legionella pneumophila. Mol. Microbiol. 27:323–336 [DOI] [PubMed] [Google Scholar]
- 70.Vance RE. 2010. Immunology taught by bacteria. J. Clin. Immunol. 30:507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luo ZQ. 2012. Legionella secreted effectors and innate immune responses. Cell Microbiol. 14:19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Z, Solomon JM, Isberg RR. 2005. Dictyostelium discoideum strains lacking the RtoA protein are defective for maturation of the Legionella pneumophila replication vacuole. Cell Microbiol. 7:431–442 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.