

Abstract
Lactate, once considered a waste product of glycolysis, has emerged as a critical regulator of cancer development, maintenance, and metastasis. Indeed, tumor lactate levels correlate with increased metastasis, tumor recurrence, and poor outcome. Lactate mediates cancer cell intrinsic effects on metabolism and has additional non–tumor cell autonomous effects that drive tumorigenesis. Tumor cells can metabolize lactate as an energy source and shuttle lactate to neighboring cancer cells, adjacent stroma, and vascular endothelial cells, which induces metabolic reprogramming. Lactate also plays roles in promoting tumor inflammation and in functioning as a signaling molecule that stimulates tumor angiogenesis. Here we review the mechanisms of lactate production and transport and highlight emerging evidence indicating that targeting lactate metabolism is a promising approach for cancer therapeutics.
Lactate homeostasis in cancer cells
Cancer cells are programmed to rely on aerobic glycolysis to support their proliferation and anabolic growth, an observation known as the Warburg effect (1, 2). Aerobic glycolysis rapidly generates ATP and diverts carbon from glucose into precursors for the synthesis of nucleotides, proteins, and lipids. As a consequence of this switch, glucose is preferentially catabolized to lactate, rather than fully metabolized to carbon dioxide via mitochondrial oxidative phosphorylation (OXPHOS). Glutamine catabolism by cancer cells also supports anabolic growth, sustains TCA cycle intermediates, and regulates redox homeostasis (3–5). These activities augment lactate production, albeit to a lesser extent than the lactate derived from glucose catabolism. Specifically, glutaminase (GLS) directs the conversion of glutamine to glutamate, which is then converted to α-ketoglutarate (αKG) by glutamate dehydrogenase that enters the TCA cycle. Malate that is then generated from αKG can exit the TCA cycle and be converted to pyruvate by malic enzyme (ME), which contributes to redox homeostasis via NADPH production. An alternate use of glutamine in pancreatic ductal carcinomas involves the transamination of glutamate and oxaloacetate (OAA) to αKG and aspartate (5). Aspartate exits the mitochondria and is transaminated back to OAA and glutamate; OAA is then converted to malate and subsequently to pyruvate. Finally, pyruvate is converted to lactate by the enzyme lactate dehydrogenase A (LDHA, Figure 1).
Figure 1. Aerobic glycolysis and glutaminolysis in cancer cells.

Oncoproteins drive the expression of genes involved in glycolysis and glutaminolysis, which results in production of excess amounts of lactate. Aberrant PI3K/AKT signaling and the transcriptional oncoproteins HIF-1α and MYC regulate the transcription of GLUT, HK2, TPI, ENO, and LDHA. HIF-1α induces the transcription of PFKFB3, which favors the production of F2,6BP, an allosteric activator of PFK1. The tumor suppressor protein p53 induces the expression of TIGAR, which dephosphorylates F2,6BP, blocking activation of PFK1 and inhibiting glycolysis. HIF-1α and MYC regulate the expression and splicing of the PKM2 isoform. MYC also regulates the expression of the glutamine transporter ASCT2 and GLS. Monocarboxylic acid transporters (MCTs) export lactate and protons and are regulated by HIF-1α and MYC. AcCoA, acetyl-CoA; ASP, aspartate; ASCT2, glutamine transporter; G, glucose; G6P, glucose-6-phosphate, F6P, fructose-6-phosphate; DHAP, dihydroxyacetone phosphate; GA3P, glyceraldehyde-3-phosphate; 1,3BPG, 1,3-bisphosphoglycerate; 2PG, 2-phosphoglycerate; 3PG, 3-phosphoglycerate; PEP, phosphoenolpyruvate; MDH, malate dehydrogenase; GOT, glutamic-oxaloacetic transaminase; GLUD1, glutamate dehydrogenase.
Oncogenic lesions in cancer drive the switch to aerobic glycolysis and lactate production by inducing the expression and activation of several glycolytic enzymes (Figure 1). First, aberrant PI3K/AKT signaling induces the expression and cell surface expression of high-affinity glucose transporters (i.e., GLUT1 and GLUT4) and the activation of hexokinase 2 (HK2) and 6-phosphofructokinase 1 (PFK1) (6–8). Second, the transcriptional oncoproteins MYC and HIF-1α induce the expression of several glycolytic enzymes, including HK2, glucose-6-phosphate isomerase (GPI), PFK1, aldolase (ALDO), fructose bisphosphate (FBP), triose phosphate isomerase (TPI), GAPDH, phosphoglycerate kinase (PGK1), enolase 1 (ENO1), pyruvate kinase, muscle (PKM), and LDHA (9–12). In addition, MYC augments glutamine catabolism by inducing the transcription of the glutamine transporter ASC-like Na+-dependent neutral amino acid transporter (ASCT2; also known as SLC1a5) and by repressing microRNA 23a/b (miR-23a/b), which normally blocks GLS translation (13, 14). Higher GLS expression results in increased glutamine uptake and catabolism, again augmenting lactate production. Third, feed-forward pathways manifest in cancer cells accelerate glycolytic flux: (a) LDHA generates NAD+ that is used by GAPDH; (b) loss-of-function mutations in the p53 tumor suppressor lead to reductions in TIGAR (TP53-induced glycolysis and apoptosis regulator, a fructose-2,6-bisphosphatase), which lead to increases in fructose-2,6-bisphosphate (F2,6BP) (Figure 1), which allosterically activates PFK1 (15, 16); and (c) HIF-1α induces the bifunctional enzyme 6-phosphofructo-2-kinase/F2,6BP (PFKFB3) to augment F2,6BP levels and activate PFK1 (17, 18). Fourth, MYC induces the transcription of select splicing factors to promote production of PKM2, a normally embryonic isoform of pyruvate kinase that is catalytically inefficient and favors aerobic glycolysis (19, 20). Finally, coupling of glycolysis to OXPHOS is disabled by MYC- or HIF-1α–directed induction of pyruvate dehydrogenase (PDH) kinase 1 (PDK1), which phosphorylates and inactivates PDH (Figure 1 and refs. 21, 22).
Lactate homeostasis in both normal cells and cancer cells requires its transport by four members of the solute carrier 16a family of 12-membrane pass, facilitative and proton-linked monocarboxylic acid symporters: MCT1 (also known as SLC16a1), MCT2 (also known as SLC16a7), MCT3 (also known as SLC16a8), and MCT4 (also known as SLC16a3) (reviewed in ref. 23). These transporters direct both the influx and efflux of lactate across the plasma membrane, and the excessive levels of lactate that are produced by cancer cells are removed by MCTs. Transport depends on the pH, the intracellular versus extracellular concentration of lactate, and the levels of other substrates of MCTs, including acetate, pyruvate, butyrate, and ketone bodies (24, 25). The cotransport of protons with lactate prevents the toxic buildup of lactate and acidification of the intracellular milieu. Accordingly, lactate transport by MCTs represents a therapeutic vulnerability for cancer cells.
LDH and its roles in tumorigenesis
LDH mediates bidirectional conversion of pyruvate and lactate and is an emerging anticancer target (Figure 2). LDH is a tetramer composed of two different subunits, LDHA and LDHB, which can assemble into five different combinations (LDH1 is comprised of four LDHB subunits; LDH2 contains three LDHB and one LDHA subunit; LDH3 contains two LDHB and two LDHA subunits; LDH4 contains one LDHB and three LDHA subunits; and LDH5 contains four LDHA subunits) (Figure 2 and ref. 26). LDHB is ubiquitously expressed, although it is the predominant isoform found in heart muscle. LDHA is the predominant isoform found in skeletal muscle and other highly glycolytic tissues. Notably, LDHA has a higher affinity for pyruvate and a higher Vmax for pyruvate reduction than LDHB. Given this, LDHA, and in particular the LDH5 tetramer, catabolizes pyruvate to lactate and produces NAD+, which is essential for other metabolic enzymes such as GAPDH (Figure 1 and ref. 27). By contrast, LDHB converts lactate to pyruvate, which allows cells to use lactate as a nutrient source for oxidative metabolism (e.g., heart tissue and neurons), and/or for gluconeogenesis (e.g., in the liver and kidney) (27).
Figure 2. Lactate dehydrogenase activity and tetramers.
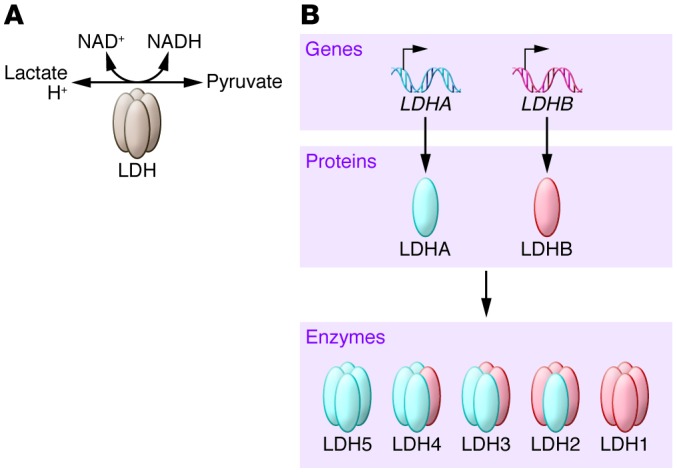
(A) LDH mediates the redox-coupled conversion between lactate and NAD+ with pyruvate and NADH. (B) The functional LDH enzyme is a tetramer containing differing ratios of the LDHA and LDHB subunits. The composition of the five LDH tetramers is shown.
Elevated levels of LDHA are a hallmark of many tumors, the majority of which are highly glycolytic, and high LDHA connotes poor prognosis in several human malignancies (28–30). The association of LDHB with cancer is more complex; LDHB is silenced by promoter methylation in several cancer types but is overexpressed or amplified in others, for example in human lung adenocarcinoma with KRAS mutation and in testicular germ cell tumors (31–35). Thus, the efficacy of anticancer agents that target LDHA, LDHB, or both will likely differ by cancer type and metabolic phenotype.
Genetic studies have provided proof-of-principle that targeting LDHA is an attractive strategy for cancer therapeutics. First, selective knockdown of LDHA inhibits anchorage-independent growth of several transformed cell lines (10) and the in vivo growth of transplanted breast tumors (36). Further, FX11, a small-molecule inhibitor of LDHA, impairs the growth of human pancreatic cancer and lymphoma xenografts. Interestingly, inactivation or inhibition of LDHA leads to the production of ROS, which is exacerbated by hypoxia, which drives glycolysis (36–38). While increases in ROS could be due to redirecting pyruvate toward oxidative metabolism, mechanistic studies of MCT1 inhibitors suggest that loss of LDHA activity may trigger elevated ROS through negative feedback effects on glycolysis and on the synthesis of the major antioxidant of the cell, glutathione (39).
Rather than using lactate as a nutrient, cancer cells generally export lactate, which acidifies the tumor environment (40). Interestingly, lactate efflux provokes a local inflammatory response that attracts immune cells such as macrophages, which secrete cytokines and growth factors that drive tumor cell growth and metastasis (41, 42). Indeed, the inflammatory response is often necessary for tumor progression, and elevated numbers of inflammatory cells, such as tumor-associated macrophages, connote poor prognosis (41). Furthermore, lactate in the tumor cell milieu impairs the adaptive immune response, disabling immune surveillance (43–46). Thus, lactate also appears to promote tumorigenesis via non–tumor cell–mediated effects on the inflammatory and immune responses.
Unexpectedly, a recent report of a mouse model of B cell lymphoma (λ-Myc transgenic mice) suggested that LDHA is dispensable for the initiation and progression of lymphoma (47). This finding was particularly surprising given the apparent importance of lactate in maintaining the malignant state and its propensity to provoke inflammation that promotes tumor progression. The authors found that a germline mutation of Ldha, which severely diminishes LDHA activity, does not affect lymphomagenesis in this model of human Burkitt lymphoma (47). Thus, high LDHA activity may not be required for all cancers, or tumor cell metabolism may adapt to other means of gaining energy, for example a shift to OXPHOS, in the face of strong oncogenic mutations such as MYC. These findings also suggest that the efficacy of LDHA inhibitors would be augmented if used in combination with other agents that force a reliance on glycolysis.
LDHA inhibitors as cancer therapeutics
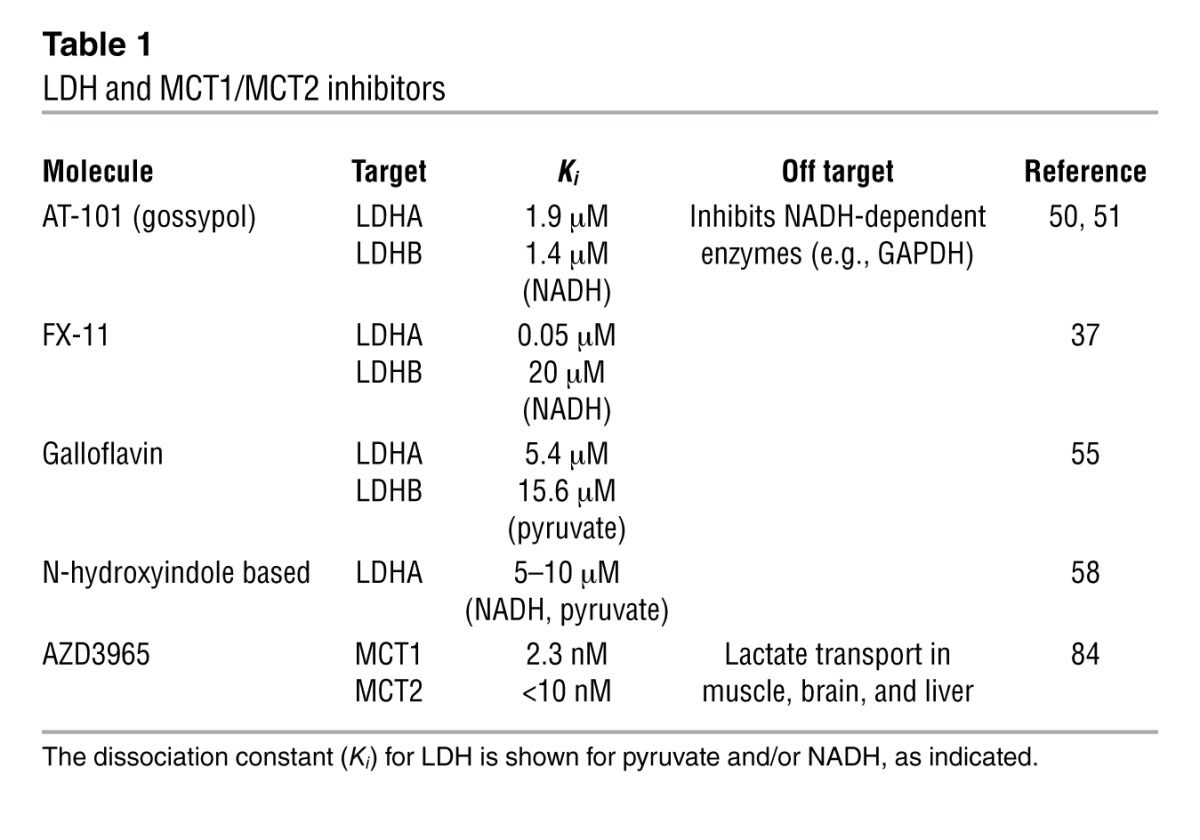
LDHA is generally considered a safe therapeutic target in humans because an inherited 20-bp deletion in the LDHA gene, which results in the loss of LDHA protein, has only relatively mild symptoms of exertional myopathy (48, 49). Accordingly, several LDHA inhibitors are being tested for their anticancer activity (Table 1). Gossypol (also known as AT-101), a natural product found in cottonseed, is a nonselective inhibitor of LDH that blocks the binding of NADH, with a Ki for LDHA and LDHB of 1.9 and 1.4 μM, respectively (50, 51). This compound was initially developed as an antimalarial therapeutic, but is now being used in Phase I and Phase II clinical oncology trials (52–54). However, gossypol also inhibits GAPDH, which is an NAD+-dependent enzyme, and thus its antitumor activity may also include the inhibition of GAPDH. In addition, FX-11, a gossypol derivative that shows selectivity for LDHA over LDHB (Ki of 0.05 vs. 20 μM, respectively) (55), has antitumor activity in xenograft models and has the potential to be developed into a cancer therapeutic (37, 56). Another compound, galloflavin, was identified from a structure-based screen using the Open Chemical Collection of the NCI. Galloflavin inhibits both LDH isoforms, induces apoptosis of hepatocellular carcinoma cell lines, and impairs breast cancer cell proliferation at rather high (250-μM) concentrations (55, 57). Finally, N-hydroxyindole–based compounds have been developed that inhibit LDHA more potently than LDHB, and these agents impair the proliferation of cancer cell lines and primary cancer cultures with micromolar IC50 activity (58). Collectively, these agents are promising starting points for LDHA inhibitors, although there is a clear need to develop much more potent and selective compounds.
Table 1.
LDH and MCT1/MCT2 inhibitors
Targeting the MCT lactate transporters as cancer therapeutics
Highly glycolytic cells utilize MCT transporters to export lactate produced by LDH as well as protons; thus, MCTs are critical regulators of intracellular lactate and pH. However, MCT functions are also necessary for lactate import into cells that use lactate as an oxidative metabolite (e.g., heart and skeletal muscle) or cells that use lactate as a substrate for gluconeogenesis (e.g., liver and kidney) (reviewed in ref. 59). MCT1 is expressed in most tissues at low levels, whereas MCT4 is expressed at high levels in white skeletal muscle fibers and at lower levels in other tissues, such as testis, lung, and placenta, and in some cell types such as chondrocytes, leukocytes, and astrocytes. MCT2 and MCT3 have a more restricted expression pattern; MCT2 is primarily expressed in liver, kidney, and neurons in the brain, while MCT3 expression is restricted to basolateral retinal pigment epithelium and the choroid plexus.
A notable, and exploitable, feature of MCT transporters is that cochaperone immunoglobulin-family single-membrane pass proteins are required for their transport to the surface of the cell membrane. Specifically, MCT1, MCT3, and MCT4 bind to CD147 (also known as basigin, EMMRIN, and OX-47), which is required for their expression on the cell surface, whereas MCT2 binds and requires embigin for cell surface expression (60, 61). Notably, the expression of CD147 on the cell surface also depends on co-expression with an MCT protein, and this interaction stabilizes both binding partners (62).
Marked increases in the levels of MCT1 and/or MCT4 are a hallmark of several human malignancies, and high levels of these transporters connote poor outcome. For example, elevated levels of MCT1 have been detected in breast, colorectal, gastric, and cervical cancer as well as in neuroblastoma and glioma (63–68). In contrast, MCT4 expression is highly elevated in renal cell carcinoma as well as in cervical and prostate cancer (65, 69, 70). Importantly, the elevated expression of MCT1 and MCT4 in cancer provides a therapeutic window for disabling these transporters with small-molecule inhibitors and/or by targeting their cochaperone CD147, for example using CD147-specific antibody therapeutics.
While essentially nothing is known regarding the control of MCT2 and MCT3 expression, several studies have shed light on the regulation of MCT1 and MCT4 expression. MCT4 is a direct transcription target of HIF-1α and is markedly upregulated in response to hypoxia (71, 72). MCT4 is also expressed at basal levels in several tissues and cell types (e.g., astrocytes), though the mechanism(s) underlying the regulation of basal expression are not clear. MCT1 expression is normally silenced in pancreatic β cells by both histone methylation (H3K27me3) at the promoter and by miR targeting of the transcript (73, 74). Further, MCT1 promoter methylation of CpG islands and transcriptional repression have been documented in the breast cancer cell line MDA-MB-231 (75). Other cues also appear to regulate MCT1 expression. In muscle cells, increases in extracellular lactate that occur following high-intensity exercise induce MCT1 expression, apparently via the generation of ROS (76), and this response may be relevant to MCT1 control in highly glycolytic, oxidative tumor cells and in adjacent stromal cells that are bathed in lactate. Furthermore, p53 represses MCT1 transcription and decreases the stability of MCT1 transcripts during hypoxia; thus, loss of p53 during tumorigenesis or the acquisition of therapeutic resistance can also augment MCT1 expression (77). Stage IV human neuroblastomas that contain MYCN amplification express high levels of MCT1 transcripts (64), suggesting that MYC regulates MCT1 transcription. Increased levels of MCT1 in tumors expressing high levels of MYC have also been noted in expression arrays (78, 79), and genome-wide chromatin immunoprecipitation analyses of MYC binding indicate that MYC binds to the MCT1 promoter of both normal and cancer cell lines (80). Collectively, these findings suggest that MCT1 may be a transcriptional target of MYC and that targeting MCT1 may be an attractive strategy to disable MYC-driven malignancies.
Genetic knockdown studies and the development of several small-molecule inhibitors have established the relevance of targeting lactate transporters in cancer therapeutics. Knockdown of MCT1 (and in some cases, MCT2) in glioblastoma (GBM) cells or inhibition of MCTs with the small molecule a-cyano-4-hydroxy-cinnamate (CHC) impairs GBM cell proliferation, migration, and survival (39, 63, 81). Furthermore, CHC impairs tumorigenic potential of GBM in intracranial xenografts (82). More convincingly, inhibition of MCT1 with highly selective and potent MCT1/MCT2 inhibitors that were originally developed by AstraZeneca to block the proliferation of activated T cells (e.g., AR-C155858; refs. 83, 84) impairs the growth and tumorigenicity of Ras-transformed fibroblasts (85). Furthermore, a second-generation MCT1/MCT2 inhibitor (AZD3965) is currently in Phase I clinical trials for advanced solid tumors and for diffuse large B cell lymphoma (86). However, these drugs fail to clear hypoxic regions of otherwise-MCT1-expressing tumors, due to HIF-1α–directed induction of MCT4 (85). In addition, these agents do not bind to MCT4 and are thus ineffective against MCT4-expressing tumors and transformed cells lines. MCT4 also plays key roles in tumorigenesis, and knockdown experiments have shown that MCT4 is required for migration and invasion of MCT4-expressing tumor cells (62, 87, 88) and for the interplay of cancer cells with stromal cells in the tumor microenvironment (see below). Thus, there is a clear need to develop potent small molecules that function as pan-MCT inhibitors, or that selectively disable MCT4.
Targeting MCT chaperones for cancer therapeutics
Given its requirement for stabilizing and localizing MCTs to the cell membrane (60), targeting CD147 is also an attractive antitumor strategy. One approach involves the generation of humanized anti-CD147 antibodies that can induce antibody-dependent cell-mediated cytotoxicity (ADCC). Such antibodies might also be utilized to deliver covalently attached drug payloads to CD147/MCT-expressing tumor cells, which will kill cancer cells irrespective of their MCT expression status. Furthermore, the organomercurial reagent p-chloromercuribenzene sulfonate (pCMBS) inhibits the activity of MCT1 and MCT4 by disrupting their association with CD147 (89). Mechanistically, this disruption occurs via pCMBS-directed reduction of the disulfide bond in the Ig-like domain of CD147. However, this agent is likely promiscuous and may target other immunoglobulin family proteins. Proof-of-principle that targeting CD147 is an attractive approach has been established by knockdown studies and by using anti-CD147 antibodies, which showed that, as expected, loss of CD147 function markedly reduces the levels of MCT1 and MCT4 proteins and impairs the growth of tumor xenografts (85, 90, 91). However, on a cautionary note, CD147 is ubiquitously expressed and interacts with other proteins at the cell surface, most notably matrix metalloproteinases, integrin-β1, and cyclophilins (92). Thus, approaches that selectively target CD147-MCT interactions are likely needed to establish a therapeutic window.
The lactate shuttle
MCTs also facilitate the shuttling of lactate between cell populations under normal conditions, for example between astrocytes, which produce lactate and express MCT1 and MCT4, and neurons, which express MCT2 (93). This shuttling of lactate, as opposed to a unidirectional export of the acid into the extracellular milieu, appears to play important roles in the development, growth, and metastasis of tumors. Indeed, three models of lactate shuttling have been described in cancer: the “reverse Warburg effect,” metabolic symbiosis, and vascular endothelial cell shuttling. Notably, inhibiting MCT function in any of these situations contributes to the anticancer activity of MCT inhibitors in vivo.
The reverse Warburg effect proposes that tumor cells can provoke neighboring stromal cells to become glycolytic and export lactate (and ketones and perhaps pyruvate), which is then taken up by cancer cells and used for oxidative metabolism (94). In this model, cancer cells are thought to create a “pseudo-hypoxic” environment for stromal fibroblasts by secreting hydrogen peroxide (95), which activates HIF-1α, glycolysis, and MCT4 expression in stromal cells. The newly generated lactate is then exported from stromal cells via MCT4 and imported into cancer cells via MCT1 (Figure 3). In support of this model, analyses of breast cancer xenografts have shown that PKM2 and MCT4 are indeed expressed in stromal fibroblasts, while markers of mitochondrial mass (TOMM20), OXPHOS (COX), and MCT1 can be detected in breast cancer cells (96–98). The induction of glycolysis in fibroblasts and lactate oxidation in cancer cells can be recapitulated in co-culture experiments, and the tumor cell–intrinsic roles of MCT1 in this shuttling have been demonstrated by MCT1 knockdown and by inhibition with CHC (99, 100). However, the reverse Warburg effect is certainly not a universal occurrence in tumors, because many tumor types are highly glycolytic and/or express MCT4. Nonetheless, it is easy to envision how this interplay could be manifest during tumor development, for example in scenarios where ATP production is more important than anabolic growth.
Figure 3. Three models of lactate shuttling in cancer.
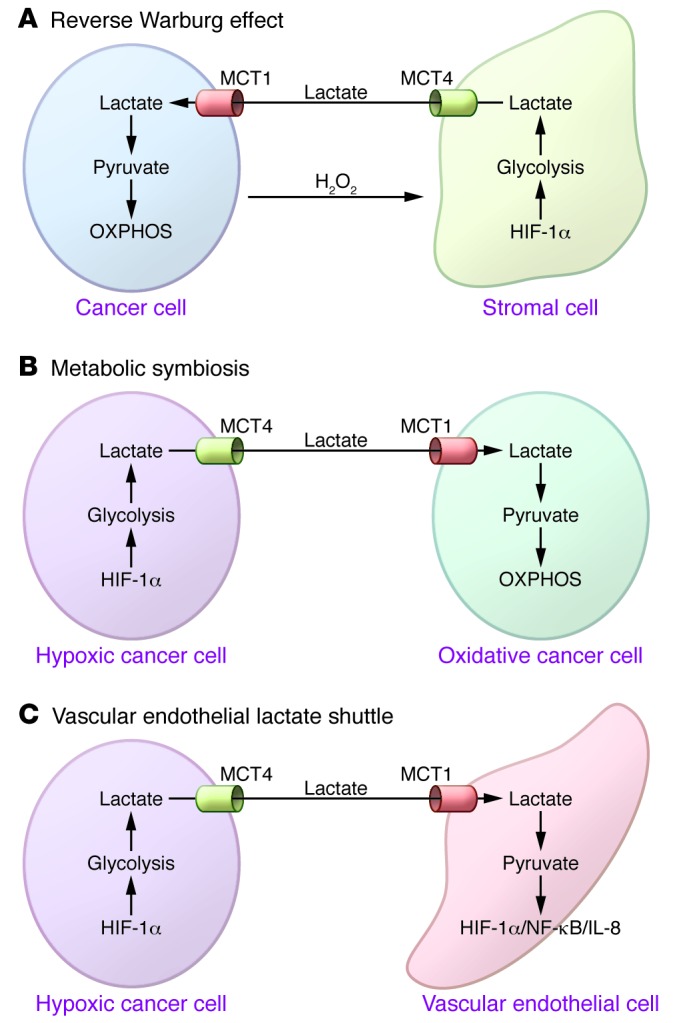
(A) The reverse Warburg effect occurs when cancer cells secrete hydrogen peroxide, which is thought to generate a pseudo-hypoxic environment in the stroma. In turn, this induces HIF-1α, MCT4 expression, and glycolysis in stromal fibroblasts, which then efflux excess lactate via MCT4. Stromal-derived lactate is then imported by tumor cells via MCT1 and used as an oxidative metabolite. (B) In metabolic symbiosis, tumor cells in hypoxic regions of the tumor efflux lactate through MCT4, which is then imported by tumor cells in less hypoxic regions via MCT1 and used as an oxidative metabolite. This shuttling facilitates delivery of glucose to the hypoxic regions of the tumor. (C) In the vascular endothelial lactate shuttle, tumor cells efflux lactate via MCT4, which is imported by vascular endothelial cells by MCT1. Lactate is then converted to pyruvate, which activates HIF-1α and NF-κB/IL-8 signaling.
Lactate shuttling has also been suggested between cancer cells within a tumor, a form of metabolic symbiosis (101). In this model, highly glycolytic, hypoxic tumor cells produce and efflux lactate, which is then imported and metabolized by more oxidative tumor cells. This shuttling is thought to facilitate glucose delivery from the vasculature into the tumor to supply hypoxic tumor cells (Figure 3). Sonveaux and colleagues have demonstrated that MCT1 expression and hypoxic regions of xenograft tumors are mutually exclusive (101). Furthermore, the growth of xenograft tumors can be impaired by CHC (101). These findings suggest that MCT1 can direct lactate uptake into tumor cells that rely on OXPHOS, and that MCT functions are critical for tumor growth. Moreover, CHC appears to effectively starve the more hypoxic regions of the tumor by forcing oxidative tumor cells to rely on glucose rather than lactate. In support of this model, CHC treatment can provoke a temporary redistribution of glucose uptake from strictly hypoxic to both hypoxic and nonhypoxic regions in tumor xenografts (102).
Finally, lactate shuttling is implicated in the interplay of tumor cells with vascular endothelial cells. Here, lactate functions as a signaling molecule rather than a metabolic intermediate, where, following import via MCT1, lactate induces endothelial cell migration, tube formation, and tumor angiogenesis. Lactate-induced signaling in endothelial cells may stabilize HIF-1α and activate an autocrine NFB/IL-8 pathway (103, 104) (Figure 3). Mechanistically, HIF-1α stabilization is thought to occur via the conversion of lactate to pyruvate, which competes with αKG for binding to prolylhydroxylases (PHDs), which normally hydroxylate proline residues in HIF-1α and mark them for ubiquitylation and destruction by the proteasome (105, 106). In support of this model, knockdown or inhibition of MCT1 prevents lactate-mediated stabilization of HIF-1α ex vivo, and CHC inhibits angiogenesis in tumor xenografts (103, 104). On the other hand, MCT1 was not detected on the surface of tumor-associated blood vessels for a large collection (over 500) of human tumors (107). However, the ability of lactate to induce HIF-1α is not restricted to vascular endothelial cells and occurs in some tumor cell lines (105, 108).
The promise of targeting lactate metabolism and transport
The new-found roles of lactate on so many components of tumor cell biology — from a tumor cell–intrinsic role as a key metabolic regulator to the interactions of cancer cells with neighboring cancer cells, stroma, and the vasculature — as well as the potent anticancer activity of LDHA and MCT inhibitors, validate this circuit as a high-priority target for next-generation cancer metabolism therapeutics. Top goals include the development of more potent and selective LDHA inhibitors, the identification and optimization of MCT4 inhibitors or pan-MCT inhibitors, and agents that target CD147 (e.g., small molecules and humanized anti-CD147 therapeutic antibodies). Of course, concerns regarding an appropriate therapeutic window will arise in the course of developing such agents given, for example, the high levels of MCTs that are expressed in muscle tissue. On the other hand, such agents appear safe, and top MCT1/MCT2 and LDHA inhibitors are in clinical oncology trials (Table 1). Therefore, there is real hope that agents that target lactate metabolism and transport will add to our anticancer armament. Finally, to optimize the efficacy of such agents, a better understanding of lactate shuttling in human malignancies is clearly needed, including studies that address tumor type and the stages of tumor development at which shuttling occurs.
Acknowledgments
Research on cancer cell metabolism in the Cleveland lab is supported by grants from the NCI (R01CA100603 and R01CA154739) and by monies from the State of Florida to Scripps Florida.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(9):3685–3692. doi:10.1172/JCI69741.
References
- 1.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeBerardinis RJ, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104(49):19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29(3):313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Son J, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271(49):31372–31378. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- 7.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15(11):1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem. 1997;272(28):17269–17275. doi: 10.1074/jbc.272.28.17269. [DOI] [PubMed] [Google Scholar]
- 9.Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000;88(4):1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- 10.Shim H, et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997;94(13):6658–6663. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JW, et al. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol. 2004;24(13):5923–5936. doi: 10.1128/MCB.24.13.5923-5936.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osthus RC, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275(29):21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- 13.Gao P, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wise DR, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105(48):18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Jogl G. Structural and biochemical studies of TIGAR (TP53-induced glycolysis and apoptosis regulator). J Biol Chem. 2009;284(3):1748–1754. doi: 10.1074/jbc.M807821200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bensaad K, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 17.Minchenko O, Opentanova I, Caro J. Hypoxic regulation of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene family (PFKFB-1-4) expression in vivo. FEBS Lett. 2003;554(3):264–270. doi: 10.1016/S0014-5793(03)01179-7. [DOI] [PubMed] [Google Scholar]
- 18.Minchenko A, et al. Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J Biol Chem. 2002;277(8):6183–6187. doi: 10.1074/jbc.M110978200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christofk HR, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 20.David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463(7279):364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27(21):7381–7393. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halestrap AP. The monocarboxylate transporter family —Structure and functional characterization. IUBMB Life. 2012;64(1):1–9. doi: 10.1002/iub.573. [DOI] [PubMed] [Google Scholar]
- 24.Bröer S, Schneider HP, Bröer A, Rahman B, Hamprecht B, Deitmer JW. Characterization of the monocarboxylate transporter 1 expressed in Xenopus laevis oocytes by changes in cytosolic pH. Biochem J. 1998;333(pt 1):167–174. doi: 10.1042/bj3330167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dimmer KS, Friedrich B, Lang F, Deitmer JW, Bröer S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem J. 2000;350(pt 1):219–227. doi: 10.1042/0264-6021:3500219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markert CL, Shaklee JB, Whitt GS. Evolution of a gene. Multiple genes for LDH isozymes provide a model of the evolution of gene structure, function and regulation. Science. 1975;189(4197):102–114. doi: 10.1126/science.1138367. [DOI] [PubMed] [Google Scholar]
- 27.Dawson DM, Goodfriend TL, Kaplan NO. Lactic dehydrogenases: functions of the two types rates of synthesis of the two major forms can be correlated with metabolic differentiation. Science. 1964;143(3609):929–933. doi: 10.1126/science.143.3609.929. [DOI] [PubMed] [Google Scholar]
- 28.Koukourakis MI, Giatromanolaki A, Winter S, Leek R, Sivridis E, Harris AL. Lactate dehydrogenase 5 expression in squamous cell head and neck cancer relates to prognosis following radical or postoperative radiotherapy. Oncology. 2009;77(5):285–292. doi: 10.1159/000259260. [DOI] [PubMed] [Google Scholar]
- 29.Koukourakis MI, Giatromanolaki A, Simopoulos C, Polychronidis A, Sivridis E. Lactate dehydrogenase 5 (LDH5) relates to up-regulated hypoxia inducible factor pathway and metastasis in colorectal cancer. Clin Exp Metastasis. 2005;22(1):25–30. doi: 10.1007/s10585-005-2343-7. [DOI] [PubMed] [Google Scholar]
- 30.Koukourakis MI, et al. Lactate dehydrogenase-5 (LDH-5) overexpression in non-small-cell lung cancer tissues is linked to tumour hypoxia, angiogenic factor production and poor prognosis. Br J Cancer. 2003;89(5):877–885. doi: 10.1038/sj.bjc.6601205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCleland ML, et al. Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin Cancer Res. 2013;19(4):773–784. doi: 10.1158/1078-0432.CCR-12-2638. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez S, et al. Expression profile of genes from 12p in testicular germ cell tumors of adolescents and adults associated with i(12p) and amplification at 12p11.2-p12.1. Oncogene. 2003;22(12):1880–1891. doi: 10.1038/sj.onc.1206302. [DOI] [PubMed] [Google Scholar]
- 33.Leiblich A, et al. Lactate dehydrogenase-B is silenced by promoter hypermethylation in human prostate cancer. Oncogene. 2006;25(20):2953–2960. doi: 10.1038/sj.onc.1209262. [DOI] [PubMed] [Google Scholar]
- 34.Thangaraju M, Carswell KN, Prasad PD, Ganapathy V. Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem J. 2009;417(1):379–389. doi: 10.1042/BJ20081132. [DOI] [PubMed] [Google Scholar]
- 35.Maekawa M, Taniguchi T, Ishikawa J, Sugimura H, Sugano K, Kanno T. Promoter hypermethylation in cancer silences LDHB, eliminating lactate dehydrogenase isoenzymes 1–4. Clin Chem. 2003;49(9):1518–1520. doi: 10.1373/49.9.1518. [DOI] [PubMed] [Google Scholar]
- 36.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9(6):425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 37.Le A, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107(5):2037–2042. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie H, et al. LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther. 2009;8(3):626–635. doi: 10.1158/1535-7163.MCT-08-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colen CB, Seraji-Bozorgzad N, Marples B, Galloway MP, Sloan AE, Mathupala SP. Metabolic remodeling of malignant gliomas for enhanced sensitization during radiotherapy: an in vitro study. Neurosurgery. 2006;59(6):1313–1323. doi: 10.1227/01.NEU.0000249218.65332.BF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wike-Hooley JL, Haveman J, Reinhold HS. The relevance of tumour pH to the treatment of malignant disease. Radiother Oncol. 1984;2(4):343–366. doi: 10.1016/S0167-8140(84)80077-8. [DOI] [PubMed] [Google Scholar]
- 41.Yabu M, et al. IL-23-dependent and -independent enhancement pathways of IL-17A production by lactic acid. Int Immunol. 2011;23(1):29–41. doi: 10.1093/intimm/dxq455. [DOI] [PubMed] [Google Scholar]
- 42.Shime H, et al. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J Immunol. 2008;180(11):7175–7183. doi: 10.4049/jimmunol.180.11.7175. [DOI] [PubMed] [Google Scholar]
- 43.Dietl K, et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol. 2010;184(3):1200–1209. doi: 10.4049/jimmunol.0902584. [DOI] [PubMed] [Google Scholar]
- 44.Fischer K, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109(9):3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 45.Gottfried E, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. 2006;107(5):2013–2021. doi: 10.1182/blood-2005-05-1795. [DOI] [PubMed] [Google Scholar]
- 46.Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. Int J Cancer. 2012;131(3):633–640. doi: 10.1002/ijc.26410. [DOI] [PubMed] [Google Scholar]
- 47.Nilsson LM, et al. Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet. 2012;8(3):e1002573. doi: 10.1371/journal.pgen.1002573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maekawa M, Sudo K, Kanno T, Li SS. Molecular characterization of genetic mutation in human lactate dehydrogenase-A (M) deficiency. Biochem Biophys Res Commun. 1990;168(2):677–682. doi: 10.1016/0006-291X(90)92374-9. [DOI] [PubMed] [Google Scholar]
- 49.Kanno T, Sudo K, Maekawa M, Nishimura Y, Ukita M, Fukutake K. Lactate dehydrogenase M-subunit deficiency: a new type of hereditary exertional myopathy. Clin Chim Acta. 1988;173(1):89–98. doi: 10.1016/0009-8981(88)90359-2. [DOI] [PubMed] [Google Scholar]
- 50.Yu Y, et al. Selective active site inhibitors of human lactate dehydrogenases A4, B4, and C4. Biochem Pharmacol. 2001;62(1):81–89. doi: 10.1016/S0006-2952(01)00636-0. [DOI] [PubMed] [Google Scholar]
- 51.Vander Jagt DL, Deck LM, Royer RE. Gossypol: prototype of inhibitors targeted to dinucleotide folds. Curr Med Chem. 2000;7(4):479–498. doi: 10.2174/0929867003375119. [DOI] [PubMed] [Google Scholar]
- 52.Liu G, Kelly WK, Wilding G, Leopold L, Brill K, Somer B. An open-label, multicenter, phase I/II study of single-agent AT-101 in men with castrate-resistant prostate cancer. Clin Cancer Res. 2009;15(9):3172–3176. doi: 10.1158/1078-0432.CCR-08-2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heist RS, et al. Phase I/II study of AT-101 with topotecan in relapsed and refractory small cell lung cancer. J Thorac Oncol. 2010;5(10):1637–1643. doi: 10.1097/JTO.0b013e3181e8f4dc. [DOI] [PubMed] [Google Scholar]
- 54.Baggstrom MQ, et al. A phase II study of AT-101 (Gossypol) in chemotherapy-sensitive recurrent extensive-stage small cell lung cancer. J Thorac Oncol. 2011;6(10):1757–1760. doi: 10.1097/JTO.0b013e31822e2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manerba M, et al. Galloflavin (CAS 568-80-9): a novel inhibitor of lactate dehydrogenase. ChemMedChem. 2012;7(2):311–317. doi: 10.1002/cmdc.201100471. [DOI] [PubMed] [Google Scholar]
- 56.Deck LM, et al. Selective inhibitors of human lactate dehydrogenases lactate dehydrogenase from the malarial parasite Plasmodium falciparum. J Med Chem. 1998;41(20):3879–3887. doi: 10.1021/jm980334n. [DOI] [PubMed] [Google Scholar]
- 57.Farabegoli F, Vettraino M, Manerba M, Fiume L, Roberti M, Di Stefano G. Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur J Pharm Sci. 2012;47(4):729–738. doi: 10.1016/j.ejps.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 58.Granchi C, et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J Med Chem. 2011;54(6):1599–1612. doi: 10.1021/jm101007q. [DOI] [PubMed] [Google Scholar]
- 59.Halestrap AP, Wilson MC. The monocarboxylate transporter family — role and regulation. IUBMB Life. 2012;64(2):109–119. doi: 10.1002/iub.572. [DOI] [PubMed] [Google Scholar]
- 60.Kirk P, Wilson MC, Heddle C, Brown MH, Barclay AN, Halestrap AP. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J. 2000;19(15):3896–3904. doi: 10.1093/emboj/19.15.3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ovens MJ, Manoharan C, Wilson MC, Murray CM, Halestrap AP. The inhibition of monocarboxylate transporter 2 (MCT2) by AR-C155858 is modulated by the associated ancillary protein. Biochem J. 2010;431(2):217–225. doi: 10.1042/BJ20100890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gallagher SM, Castorino JJ, Wang D, Philp NJ. Monocarboxylate transporter 4 regulates maturation and trafficking of CD147 to the plasma membrane in the metastatic breast cancer cell line MDA-MB-231. Cancer Res. 2007;67(9):4182–4189. doi: 10.1158/0008-5472.CAN-06-3184. [DOI] [PubMed] [Google Scholar]
- 63.Miranda-Goncalves V, et al. Monocarboxylate transporters (MCTs) in gliomas: expression and exploitation as therapeutic targets. Neuro Oncol. 2013;15(2):172–188. doi: 10.1093/neuonc/nos298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fang J, et al. The H+-linked monocarboxylate transporter (MCT1/SLC16A1): a potential therapeutic target for high-risk neuroblastoma. Mol Pharmacol. 2006;70(6):2108–2115. doi: 10.1124/mol.106.026245. [DOI] [PubMed] [Google Scholar]
- 65.Pinheiro C, et al. Increasing expression of monocarboxylate transporters 1 and 4 along progression to invasive cervical carcinoma. Int J Gynecol Pathol. 2008;27(4):568–574. doi: 10.1097/PGP.0b013e31817b5b40. [DOI] [PubMed] [Google Scholar]
- 66.de Oliveira AT, et al. Co-expression of monocarboxylate transporter 1 (MCT1) and its chaperone (CD147) is associated with low survival in patients with gastrointestinal stromal tumors (GISTs). J Bioenerg Biomembr. 2012;44(1):171–178. doi: 10.1007/s10863-012-9408-5. [DOI] [PubMed] [Google Scholar]
- 67.Pinheiro C, et al. Increased expression of monocarboxylate transporters 1, 2, and 4 in colorectal carcinomas. Virchows Arch. 2008;452(2):139–146. doi: 10.1007/s00428-007-0558-5. [DOI] [PubMed] [Google Scholar]
- 68.Pinheiro C, et al. Monocarboxylate transporter 1 is up-regulated in basal-like breast carcinoma. Histopathology. 2010;56(7):860–867. doi: 10.1111/j.1365-2559.2010.03560.x. [DOI] [PubMed] [Google Scholar]
- 69.Pértega-Gomes N, et al. Monocarboxylate transporter 4 (MCT4) and CD147 overexpression is associated with poor prognosis in prostate cancer. BMC Cancer. 2011;11:312. doi: 10.1186/1471-2407-11-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gerlinger M, et al. Genome-wide RNA interference analysis of renal carcinoma survival regulators identifies MCT4 as a Warburg effect metabolic target. J Pathol. 2012;227(2):146–156. doi: 10.1002/path.4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pérez de Heredia F, Wood IS, Trayhurn P. Hypoxia stimulates lactate release and modulates monocarboxylate transporter (MCT1, MCT2, and MCT4) expression in human adipocytes. Pflugers Arch. 2010;459(3):509–518. doi: 10.1007/s00424-009-0750-3. [DOI] [PubMed] [Google Scholar]
- 72.Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem. 2006;281(14):9030–9037. doi: 10.1074/jbc.M511397200. [DOI] [PubMed] [Google Scholar]
- 73.van Arensbergen J, et al. Derepression of Polycomb targets during pancreatic organogenesis allows insulin-producing beta-cells to adopt a neural gene activity program. Genome Res. 2010;20(6):722–732. doi: 10.1101/gr.101709.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pullen TJ, da Silva Xavier G, Kelsey G, Rutter GA. miR-29a and miR-29b contribute to pancreatic β-cell-specific silencing of monocarboxylate transporter 1 (Mct1). Mol Cell Biol. 2011;31(15):3182–3194. doi: 10.1128/MCB.01433-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Asada K, et al. Reduced expression of GNA11 and silencing of MCT1 in human breast cancers. Oncology. 2003;64(4):380–388. doi: 10.1159/000070297. [DOI] [PubMed] [Google Scholar]
- 76.Hashimoto T, Hussien R, Oommen S, Gohil K, Brooks GA. Lactate sensitive transcription factor network in L6 cells: activation of MCT1 and mitochondrial biogenesis. FASEB J. 2007;21(10):2602–2612. doi: 10.1096/fj.07-8174com. [DOI] [PubMed] [Google Scholar]
- 77.Boidot R, et al. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res. 2012;72(4):939–948. doi: 10.1158/0008-5472.CAN-11-2474. [DOI] [PubMed] [Google Scholar]
- 78.Coller HA, et al. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci U S A. 2000;97(7):3260–3265. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schuhmacher M, et al. The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res. 2001;29(2):397–406. doi: 10.1093/nar/29.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Encyclopedia of DNA Elements. University of California Santa Cruz Web site. http://genome.ucsc.edu/ENCODE/ . Accessed July 3, 2013.
- 81.Mathupala SP, Parajuli P, Sloan AE. Silencing of monocarboxylate transporters via small interfering ribonucleic acid inhibits glycolysis and induces cell death in malignant glioma: an in vitro study. Neurosurgery. 2004;55(6):1410–1419. doi: 10.1227/01.neu.0000143034.62913.59. [DOI] [PubMed] [Google Scholar]
- 82.Colen CB, et al. Metabolic targeting of lactate efflux by malignant glioma inhibits invasiveness and induces necrosis: an in vivo study. Neoplasia. 2011;13(7):620–632. doi: 10.1593/neo.11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Murray CM, et al. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat Chem Biol. 2005;1(7):371–376. doi: 10.1038/nchembio744. [DOI] [PubMed] [Google Scholar]
- 84.Ovens MJ, Davies AJ, Wilson MC, Murray CM, Halestrap AP. AR-C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7-10. Biochem J. 2010;425(3):523–530. doi: 10.1042/BJ20091515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Le Floch R, et al. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc Natl Acad Sci U S A. 2011;108(40):16663–16668. doi: 10.1073/pnas.1106123108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cancer Research UK. A Phase I Trial of AZD3965 in Patients With Advanced Cancer. NIH Web site. http://www.clinicaltrials.gov/ct2/show/NCT01791595 . Updated June 24, 2013. Accessed July 3, 2013.
- 87.Kobayashi M, Otsuka Y, Itagaki S, Hirano T, Iseki K. Inhibitory effects of statins on human monocarboxylate transporter 4. Int J Pharm. 2006;317(1):19–25. doi: 10.1016/j.ijpharm.2006.02.043. [DOI] [PubMed] [Google Scholar]
- 88.Izumi H, et al. Monocarboxylate transporters 1 and 4 are involved in the invasion activity of human lung cancer cells. Cancer Sci. 2011;102(5):1007–1013. doi: 10.1111/j.1349-7006.2011.01908.x. [DOI] [PubMed] [Google Scholar]
- 89.Wilson MC, Meredith D, Fox JE, Manoharan C, Davies AJ, Halestrap AP. Basigin (CD147) is the target for organomercurial inhibition of monocarboxylate transporter isoforms 1 and 4: the ancillary protein for the insensitive MCT2 is EMBIGIN (gp70). J Biol Chem. 2005;280(29):27213–27221. doi: 10.1074/jbc.M411950200. [DOI] [PubMed] [Google Scholar]
- 90.Baba M, Inoue M, Itoh K, Nishizawa Y. Blocking CD147 induces cell death in cancer cells through impairment of glycolytic energy metabolism. Biochem Biophys Res Commun. 2008;374(1):111–116. doi: 10.1016/j.bbrc.2008.06.122. [DOI] [PubMed] [Google Scholar]
- 91.Schneiderhan W, et al. CD147 silencing inhibits lactate transport and reduces malignant potential of pancreatic cancer cells in in vivo and in vitro models. Gut. 2009;58(10):1391–1398. doi: 10.1136/gut.2009.181412. [DOI] [PubMed] [Google Scholar]
- 92.Iacono KT, Brown AL, Greene MI, Saouaf SJ. CD147 immunoglobulin superfamily receptor function and role in pathology. Exp Mol Pathol. 2007;83(3):283–295. doi: 10.1016/j.yexmp.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pellerin L, et al. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55(12):1251–1262. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- 94.Lisanti MP, et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor micro-environment. Cancer Biol Ther. 2010;10(6):537–542. doi: 10.4161/cbt.10.6.13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Martinez-Outschoorn UE, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9(16):3256–3276. doi: 10.4161/cc.9.16.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sotgia F, et al. Mitochondrial metabolism in cancer metastasis: visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle. 2012;11(7):1445–1454. doi: 10.4161/cc.19841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pavlides S, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8(23):3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 98.Bonuccelli G, et al. The reverse Warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle. 2010;9(10):1960–1971. doi: 10.4161/cc.9.10.11601. [DOI] [PubMed] [Google Scholar]
- 99.Whitaker-Menezes D, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10(11):1772–1783. doi: 10.4161/cc.10.11.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fiaschi T, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012;72(19):5130–5140. doi: 10.1158/0008-5472.CAN-12-1949. [DOI] [PubMed] [Google Scholar]
- 101.Sonveaux P, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118(12):3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Busk M, et al. Inhibition of tumor lactate oxidation: consequences for the tumor microenvironment. Radiother Oncol. 2011;99(3):404–411. doi: 10.1016/j.radonc.2011.05.053. [DOI] [PubMed] [Google Scholar]
- 103.Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71(7):2550–2560. doi: 10.1158/0008-5472.CAN-10-2828. [DOI] [PubMed] [Google Scholar]
- 104.Sonveaux P, et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One. 2012;7(3):e33418. doi: 10.1371/journal.pone.0033418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem. 2002;277(26):23111–23115. doi: 10.1074/jbc.M202487200. [DOI] [PubMed] [Google Scholar]
- 106.Lu H, Dalgard CL, Mohyeldin A, McFate T, Tait AS, Verma A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J Biol Chem. 2005;280(51):41928–41939. doi: 10.1074/jbc.M508718200. [DOI] [PubMed] [Google Scholar]
- 107.Pinheiro C, Longatto-Filho A, Nogueira R, Schmitt F, Baltazar F. Lactate-induced IL-8 pathway in endothelial cells — letter. Cancer Res. 2012;72(7):1901–1902. doi: 10.1158/0008-5472.CAN-11-1540. [DOI] [PubMed] [Google Scholar]
- 108.De Saedeleer CJ, Copetti T, Porporato PE, Verrax J, Feron O, Sonveaux P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS One. 2012;7(10):e46571. doi: 10.1371/journal.pone.0046571. [DOI] [PMC free article] [PubMed] [Google Scholar]