

Abstract
Salmonella enterica subsp. enterica serovar Typhimurium is a leading cause of food-borne salmonellosis in the United States. The number of antibiotic-resistant isolates identified in humans is steadily increasing, suggesting that the spread of antibiotic-resistant strains is a major threat to public health. S. Typhimurium is commonly identified in a wide range of animal hosts, food sources, and environments, but little is known about the factors mediating the spread of antibiotic resistance in this ecologically complex serovar. Previously, we developed a subtyping method, CRISPR–multi-virulence-locus sequence typing (MVLST), which discriminates among strains of several common S. enterica serovars. Here, CRISPR-MVLST identified 22 sequence types within a collection of 76 S. Typhimurium isolates from a variety of animal sources throughout central Pennsylvania. Six of the sequence types were identified in more than one isolate, and we observed statistically significant differences in resistance among these sequence types to 7 antibiotics commonly used in veterinary and human medicine, such as ceftiofur and ampicillin (P < 0.05). Importantly, five of these sequence types were subsequently identified in human clinical isolates, and a subset of these isolates had identical antibiotic resistance patterns, suggesting that these subpopulations are being transmitted through the food system. Therefore, CRISPR-MVLST is a promising subtyping method for monitoring the farm-to-fork spread of antibiotic resistance in S. Typhimurium.
INTRODUCTION
Salmonella is the leading cause of bacterial food-borne illness in the United States. It is responsible for an estimated 1 million illnesses, 20,000 hospitalizations, and 400 deaths annually at an economic cost of $3.3 to 4.4 billion (1–3), and its substantial burden to public health has persisted over the last several decades even as the overall incidence of bacterial food-borne illness has been in steady decline. Salmonella enterica subsp. enterica serovar Typhimurium is one of the most common serovars associated with clinically reported salmonellosis in humans, accounting for at least 15% of infections (1). S. Typhimurium infections have exhibited a gradual decline in susceptibility to traditional antibiotics, a trend which is concerning in light of this pathogen's broad host range and its potential to spread antibiotic resistance determinants to other bacteria (4). Now more than ever, it is imperative to effectively monitor the clonal transmission of S. Typhimurium throughout the food system in order to implement effective control measures.
Ecological characterization of S. Typhimurium requires effective identification and discrimination of clonal populations that are often widely disseminated across space and time. Molecular subtyping methods such as pulsed-field gel electrophoresis (PFGE), multilocus sequence typing (MLST), and multiple-locus variable-number tandem-repeat analysis (MLVA) have been used extensively to this end and have revealed important insights about the population structure and distribution of S. Typhimurium (see reference 5 for a review). These methods differ greatly in their resolution and are often combined in order to unravel phylogenetic and epidemiologically relevant relationships among strains. For instance, a recent study by Soyer et al. used a combination of PFGE and MLST to identify overlapping populations of S. Typhimurium from bovine and human sources (6). Several MLST subtypes were found repeatedly on the same farms over time, but PFGE patterns from these isolates showed diversification, suggesting that PFGE might not be an appropriate method for monitoring the persistence of clonal populations. Another study by Hernandez et al. (7) used a combination of PFGE and MLVA to trace the transmission of several S. Typhimurium strains from wild birds to humans. Interestingly, the authors identified strains which appear only in wild birds, but it is unclear if these strains are closely related to more broadly transmissible strains (7).
Extensive efforts have also been devoted to characterizing the emergence and spread of antibiotic resistance in S. Typhimurium. For these studies, molecular subtyping methods are often used in conjunction with phenotypic subtyping methods, such as phage typing or antimicrobial susceptibility testing (8). For instance, a recent study by Molla et al. used a combination of antimicrobial susceptibility testing and PFGE to identify overlapping populations of multidrug-resistant S. enterica in feed and pig fecal samples, but the clonal relationship between the populations was not determined (9). A stable, high-resolution subtyping marker would be needed in this case to evaluate the success of intervention strategies in limiting the transmission of S. enterica throughout the pig production system. Certain phage types, like the epidemic strain DT104, have distinct multidrug resistance patterns (see reference 10 for a review). However, molecular subtyping techniques have shown that DT104 is not clonal (11, 12), and there are several known permutations of its characteristic five-drug resistance pattern (11). Moreover, identical phage sensitivity patterns (i.e., phage types) can manifest in unrelated isolates, so phage typing does not necessarily identify populations with a single clonal origin (13). The limitations of each available subtyping technique can be overcome to some extent by complementing the weakness of one technique with the strength of another, but there is still no reliable, high-throughput method for monitoring the clonal spread of antibiotic resistance in S. Typhimurium over time.
We recently developed a subtyping method based on sequence analysis of clustered regularly interspaced short palindromic repeats (CRISPRs) and the virulence genes fimH and sseL (14). Mechanistically, CRISPRs are an integral part of a system which provides defense against bacteriophage attack in bacteria and archaea (15). Spacer sequences are acquired after exposure to foreign DNA and incorporated between palindromic direct repeat sequences to provide immunity against future attack through sequence homology in a manner similar to that of RNA interference (see references 16 to 21 for reviews). Newly acquired spacers are added at the 5′ end of CRISPR loci proximal to an AT-rich region called the leader. Two CRISPR loci have been identified in the S. enterica genome, and each locus contains a distinct array of spacers, the composition of which is referred to as a CRISPR allele. Direct repeat sequences are typically highly conserved within alleles, although sequence variants have been observed (14, 22–24). Spacer acquisition in response to challenge with a plasmid or phage has yet to be reported in wild-type S. enterica. Nonetheless, several studies have shown considerable variation in the composition and architecture of spacers among CRISPR alleles from related strains of S. enterica, and there is growing evidence that CRISPRs in S. enterica and other species are evolving at a rate which makes them useful subtyping markers (14, 24–27). We have shown that a subtyping method based on CRISPRs and multi-virulence-locus sequence typing (MVLST) of fimH and sseL, designated CRISPR-MVLST, provides a high discriminatory power and strong epidemiological relevance for outbreak investigations (14, 24). We also demonstrated the utility of CRISPR-MVLST for identifying common strains of S. Enteritidis in eggs, the environment, and humans over a period of several years (22). Together, these studies suggest that CRISPR-MVLST may be a robust subtyping method for monitoring the transmission of S. enterica throughout the food system.
Here, we used CRISPR-MVLST to investigate the diversity and distribution of S. Typhimurium isolates from different animal sources in central Pennsylvania over a period of 3 years. We compared the spacer content of CRISPR alleles from these isolates to those of previously published CRISPR alleles from an international collection of S. Typhimurium isolates and found a high degree of spacer conservation across a wide range of animal, environmental, clinical, and geographical sources. Six CRISPR-MVLST sequence types (STs) were predominant in our isolate collection, five of which were also identified in a collection of human clinical isolates (N. Shariat, C. H. Sandt, M. J. DiMarzio, R. Barrangou, and E. G. Dudley, submitted for publication). We observed significant differences in the frequencies and patterns of antibiotic resistance among these six sequence types which were consistent in both animal and human clinical isolates. Our study provides the first evidence that CRISPR-MVLST is an effective tool for monitoring the spread of antibiotic-resistant subpopulations of S. Typhimurium.
MATERIALS AND METHODS
Bacterial isolates and DNA extraction.
A total of 76 S. Typhimurium isolates were obtained from the Animal Diagnostic Laboratory (ADL) at the Pennsylvania State University (see Table S1 in the supplemental material). The isolates represent all of the S. Typhimurium isolates collected by the ADL from 45 different farms and wildlife sampling locations located throughout central Pennsylvania over a period from July 2008 to December 2011. All isolates were harvested from pathology samples of sick or dead animals, with the exception of one isolate from an on-farm water source. Immediately after collection, isolates were stored either at −80°C in tryptic soy broth (Remel, Lenexa, KS) with 20% glycerol or at room temperature on tryptic soy agar slants (Remel). Prior to genomic DNA (gDNA) extraction, all isolates were colony purified on lysogeny broth (LB) agar plates. Single colonies were grown overnight with shaking agitation at 37°C in LB, and a portion of the overnight culture was stored for future work at −80°C in 20% glycerol. The remaining 1 ml of overnight culture was used to prepare a gDNA extraction using the Wizard genomic DNA purification kit (Promega) according to the manufacturer's protocol. Purified gDNA was stored at −20°C until use.
PCR amplification.
Primers used for PCR amplification of all four CRISPR-MVLST loci have been reported previously (25). Each reaction mixture contained a 25-μl mixture with 2 μl of purified gDNA template, 2.5 μl of standard 10× Taq reaction buffer (New England BioLabs), 0.25 μl 10 mM deoxynucleoside triphosphates (dNTPs), 1.0 μl of each 10 μM primer solution, 0.5 μl Taq polymerase (2.5 units; New England BioLabs), and 17.75 μl sterile distilled water (dH2O). Reaction conditions included a single cycle at 95°C for 10 min, followed by 35 cycles at 95°C for 1 min, annealing temperature for 1 min, and 72°C for 1 min. A final extension followed at 72°C for 10 min. The annealing temperatures for PCR amplification of sseL, fimH, CRISPR1, and CRISPR2 were 60°C, 56.7°C, 55°C, and 55°C, respectively. PCR amplicons were visualized on a 1.2% agarose gel with a 1-kb DNA ladder (New England BioLabs).
DNA sequencing.
PCR amplicons were treated with 10 units of exonuclease I (New England BioLabs), and 1 unit of Antarctic phosphatase (New England BioLabs) at 37°C for 45 min to remove unincorporated primers and dNTPs and then at 85°C for 15 min to inactivate the enzymes. Purified PCR amplicons were sequenced at the Huck Institute's Nucleic Acid Facility at the Pennsylvania State University using 3′ BigDye-labeled dideoxynucleotide triphosphates (v3.1 dye terminators; Life Technologies) and run on an ABI 3730XL DNA analyzer with the ABI Data Collection Program (v2.0). Data were analyzed with ABI Sequencing Analysis software (version 5.1.1). As needed, additional primers were designed to complete double-stranded sequencing (see Table S2 in the supplemental material).
Sequence analysis and ST assignment.
Individual sequences were assembled, and contigs were compared to previously identified alleles for all four CRISPR-MVLST loci (i.e., CRISPR1, CRISPR2, fimH, and sseL) using the DNAStar Lasergene 9 software suite (DNAStar Inc.). Contig sizes were corroborated with the sizes of PCR amplicons as determined by gel electrophoresis. Complete contigs that did not share 100% identity to previously identified alleles were assigned a new allele number. S. Typhimurium CRISPR-MVLST sequence types (TSTs) were assigned based on the combined allelic profile of all four loci and are listed in Table 1. Spacers were identified using the web-based CRISPR-Finder program and visualized as described previously (28, 29). CRISPR alleles identified in the ADL S. Typhimurium collection were compared to alleles in the Fabre et al. popset in the National Center for Biotechnology Information database using a BLASTn search targeted to GenBank accession numbers JF724159 to JF725640 (24, 30).
Table 1.
Sources of isolates in the ADL S. Typhimurium collection and allelic composition of TSTs
| CRISPR-MVLST TST | Total no. of isolates | Source(s) of isolation (no. of isolates) | Allele |
|||
|---|---|---|---|---|---|---|
| fimH | sseL | CRISPR1 | CRISPR2 | |||
| TST 9 | 2 | Cattle (2) | 6 | 15 | 129 | 159 |
| TST 10 | 13 | Cattle (8), poultry (2), swine (2), French guinea fowl (1) | 8 | 15 | 11 | 160 |
| TST 11 | 1 | Cattle (1) | 6 | 15 | 10 | 163 |
| TST 12 | 2 | Cattle (2) | 6 | 15 | 10 | 164 |
| TST 13 | 6 | Cattle (3), poultry (2), deer (1) | 6 | 15 | 129 | 162 |
| TST 17 | 5 | Cattle (5) | 6 | 15 | 10 | 167 |
| TST 19 | 12 | Cattle (11), turkey vulture (1) | 6 | 62 | 10 | 164 |
| TST 42 | 13 | Cattle (11), poultry (1), water source (1) | 6 | 15 | 10 | 181 |
| TST 43a | 1 | Cattle (1) | 6 | 15 | 10 | 182 |
| TST 44a | 8 | Perching birds (7), red-tailed hawk (1) | 7 | 15 | 144 | 14 |
| TST 45a | 2 | Poultry (2) | 6 | 15 | 145 | 183 |
| TST 46a | 1 | Commercial turkey (1) | 6 | 15 | 10 | 184 |
| TST 47a | 1 | Cattle (1) | 6 | 62 | 10 | 185 |
| TST 48a | 1 | Horse (1) | 6 | 15 | 146 | 164 |
| TST 49a | 1 | Cattle (1) | 6 | 15 | 147 | 164 |
| TST 50a | 1 | Cattle (1) | 6 | 20 | 148 | 166 |
| TST 51a | 1 | Cattle (1) | 6 | 20 | 148 | 166 |
| TST 52a | 1 | Poultry (1) | 6 | 65 | 129 | 162 |
| TST 53a | 1 | Cattle (1) | 6 | 15 | 11 | 160 |
| TST 54a | 1 | Deer (1) | 8 | 15 | 11 | 186 |
| TST 55a | 1 | Cattle (1) | 6 | 15 | 129 | 187 |
| TST 61a | 1 | Pigeon (1) | 6 | 66 | 165 | 206 |
This TST has not previously been identified in clinical isolates.
Antibiotic susceptibility testing.
Antimicrobial susceptibility testing was performed with an automated system (Sensititre; Trek Diagnostic Systems) using bovine/porcine (with tulathromycin) MIC format (BOPO6F) in accordance with the manufacturer's instructions and Clinical and Laboratory Standards Institute (CLSI) guidelines (31). The MIC results were interpreted according to the breakpoints of the CLSI guidelines (31).
Statistics.
Associations between resistance to each antimicrobial agent and CRISPR-MVLST sequence types were determined using Fisher's exact test (SAS, v9.3) with an alpha set at 0.05.
Nucleotide accession numbers.
The CRISPR1, CRISPR2, fimH, and sseL sequences for each isolate were deposited in GenBank under accession numbers KC854555 to KC854706 and KC916785 to KC916936.
RESULTS
CRISPR allelic diversity.
Previously, we demonstrated that CRISPR sequences in S. Typhimurium are sufficiently variable for use as molecular subtyping markers (14). Here, we explored the potential relationship between CRISPR sequence variability and ecological distribution within a diverse veterinary collection of S. Typhimurium isolates. The adaptive nature of CRISPR-Cas systems led us to hypothesize that the spacer content of the CRISPR1 and CRISPR2 loci in S. Typhimurium would identify source-associated clonal populations due to the unique pool of foreign DNA elements in each source. In order to test our hypothesis, we assembled a collection of 76 S. Typhimurium isolates from a variety of animal sources throughout central Pennsylvania over a period from 2006 to 2011 (see Table S1 in the supplemental material). Isolates from cattle, swine, poultry, commercial turkey, and French guinea fowl were obtained from farm animals, while isolates from deer, pigeon, perching birds, a red-tailed hawk, and a turkey vulture were obtained from necropsies of wild animals. A single isolate was obtained from a water source on a farm.
The CRISPR1 and CRISPR2 loci of each isolate were sequenced, and 9 CRISPR1 alleles and 16 CRISPR2 alleles were identified (Fig. 1). CRISPR1 alleles range in size from 6 to 32 spacers, and CRISPR2 alleles range in size from 4 to 40 spacers. The wide range of allele sizes reflects deletions or duplications of a conserved order of 32 unique CRISPR1 and 36 unique CRISPR2 spacers. For instance, CRISPR2 allele 184 differs from allele 164 by a deletion of the spacer at position 4 in allele 184, and CRISPR2 alleles 181 and 182 have a five-spacer duplication at positions 7 to 11 and 14 to 18. The highest degree of spacer conservation is apparent at both the leader-proximal and leader-distal ends of both CRISPR1 and CRISPR2 alleles. All nine CRISPR1 alleles have a conserved spacer at position 31, and only allele 145 lacks the conserved spacer at position 1. Similarly, only CRISPR2 allele 166 lacks the conserved spacer at position 44, and all 16 CRISPR2 alleles have a conserved spacer at position 1. CRISPR1 allele 148 is a notable exception, as it has a unique spacer at its leader-proximal end (i.e., spacer position 32).
Fig 1.

Graphic representation of spacer content for all CRISPR1 (A) and CRISPR2 (B) alleles identified in the ADL S. Typhimurium collection. Alleles marked in bold were also identified in the Fabre et al. isolate collection. A uniquely colored box and symbol combination designates each spacer sequence, and the shape of the symbol inside the box designates the length of the spacer. The ° symbol represents a truncated repeat that runs into an adjacent spacer, each of which is indistinguishable from the other. The box designated “IS” represents the position of IS10. Spacers have been aligned in order to facilitate comparison among alleles.
Generally, spacers are 32 bp in length. Exceptions include four CRISPR1 alleles with a 33-bp spacer at position 26, seven CRISPR1 alleles with a 33-bp spacer at position 12, and 11 CRISPR2 alleles with a 33-bp spacer at position 9 (Fig. 1). Additionally, six CRISPR1 alleles have a 42-bp unit comprised of a spacer fused with a truncated direct repeat at position 10. Interestingly, CRISPR2 allele 182 has IS10 in place of the consensus spacer at position 34. The insertion sequence is 1,308 bp in length, and it is integrated exactly between two perfect direct repeats.
The CRISPR alleles that we identified are comprised of a conserved pool of 31 CRISPR1 and 38 CRISPR2 spacers, and the most common CRISPR alleles were identified in isolates from several animal sources. For instance, CRISPR1 allele 10 was identified in isolates from cattle, poultry, a turkey vulture, and a water source, and CRISPR2 allele 162 was identified in isolates from cattle, poultry, and deer (Table 1). Overall, our analysis does not support the hypothesis that the CRISPR-Cas system of S. Typhimurium is acquiring new spacers in a manner that would be useful for identifying source-adapted clonal populations.
Geographical comparison of CRISPR alleles.
The recent publication of a large collection of CRISPR sequences from 156 European, African, and Asian S. Typhimurium isolates enabled us to compare the distribution of CRISPR alleles in a geographically diverse isolate collection to that of CRISPR alleles in our collection of isolates, which are closely linked geographically (24). We observed that CRISPR1 alleles 10, 11, 129, 148, and 165 and CRISPR2 alleles 14, 160, 162, 166, and 181 are common to the two isolate collections (Fig. 1). These were also some of the most frequently observed alleles within the ADL collection. For example, CRISPR1 allele 10 was identified in 35 isolates in the ADL collection and 7 isolates in the Fabre et al. collection, or 44% and 5% of isolates, respectively, and CRISPR1 allele 11 was identified in 15 isolates in the ADL collection and 32 isolates in the Fabre et al. collection, or 19% and 21% of isolates, respectively. Similarly, CRISPR2 allele 160 was identified in 14 isolates in the ADL collection and 31 isolates in the Fabre et al. collection, or 18% and 21% of isolates, respectively, and CRISPR2 allele 181 was identified in 12 isolates in the ADL collection and 5 isolates in the Fabre et al. collection, or 15% and 3% of isolates, respectively. Conversely, CRISPR1 alleles 145, 146, and 147 and CRISPR2 alleles 182, 183, 184, 185, and 187 are all unique to the ADL collection and were identified in only one or two isolates. CRISPR1 allele 144 is a notable exception to this observation. It was identified exclusively in the ADL collection in eight isolates from an avian wildlife outbreak that lasted only 2 months (see Table S1 in the supplemental material).
The identification of multiple CRISPR alleles common to the two isolate collections suggests that strains with these alleles are globally disseminated. However, it remains to be determined if CRISPR alleles that are unique to the ADL collection are indicative of geographically limited strains of S. Typhimurium.
CRISPR-MVLST.
In a previous publication, we described a CRISPR-based subtyping method, CRISPR-MVLST, that combines CRISPR allele sequencing with single nucleotide polymorphism (SNP) analysis of the virulence genes fimH and sseL (14). We showed that CRISPR-MVLST has a higher discriminatory power than does a subtyping method based on CRISPR sequences alone for separating strains of S. Typhimurium. CRISPR-MVLST has also identified differences in the distribution of S. Enteritidis among sources within the egg production system (22). Here, we hypothesized that CRISPR-MVLST would identify similar differences in the distribution of S. Typhimurium within the ADL collection.
In total, 3 fimH alleles and 5 sseL alleles were identified in addition to the abovementioned 9 CRISPR1 and 16 CRISPR2 alleles (Table 1). By combining the four CRISPR-MVLST markers to generate a unique allelic profile for each isolate, 22 distinct CRISPR-MVLST sequence types, or TSTs, were identified in the ADL collection. Sixteen TSTs were identified in only one or two isolates, while TSTs 10, 13, 17, 19, 42, and 44 were identified in more than two isolates. Generally, sources of isolation varied within the six most frequently identified TSTs. For instance, TST 10 was identified in 13 isolates with sources of isolation including cattle, poultry, swine, and French guinea fowl. However, TSTs 17 and 44 are notable exceptions to this observation, as they were identified exclusively in cattle and avian wildlife isolates, respectively.
Association between CRISPR-MVLST sequence types and antibiotic resistance.
Previous publications have shown that CRISPR sequences may be useful for determining the relatedness of Salmonella isolates (14, 24, 32) and that CRISPR-MVLST provides insights into the ecology of S. Enteritidis (22). In light of these observations, we hypothesized that CRISPR-MVLST would correlate with important phenotypes such as antibiotic resistance, which has been shown to vary extensively within serovars (33). Therefore, we tested each isolate in the ADL collection for resistance to a panel of 18 common antibiotics in order to determine if there are differences in antibiotic resistance among TSTs. Data are not shown for TSTs identified in fewer than two isolates in the ADL collection. Six epidemiologically unrelated human clinical isolates from each of the five most frequently identified TSTs were also tested to see if any differences in antibiotic resistance among sequence types persist as strains are transmitted from animals to humans. Only two clinical isolates were available with TST 42, and there were no available clinical isolates with TST 44.
We found a statistically significant association between TST and frequency of resistance to 7 of the 18 antibiotics included in our bovine antibiotic resistance panel (Fig. 2; see also Table S3 in the supplemental material). Isolates within TSTs 10 and 42 were more frequently resistant to these seven antibiotics than were isolates within TSTs 13, 17, 19, and 44. Antibiotics for which resistance is not significantly associated with TST include clindamycin, danofloxacin, enrofloxacin, florfenicol, penicillin, spectinomycin, sulfadimethoxazine, tiamulin, tilmicosin, tulathromycin, and tylosin (see Table S1). The association between resistance and TST is generally dependent on the frequency of resistance to a particular antibiotic within the total ADL collection. For example, there is no significant association between TST and resistance to penicillin due to the ubiquity of penicillin resistance within the ADL collection.
Fig 2.
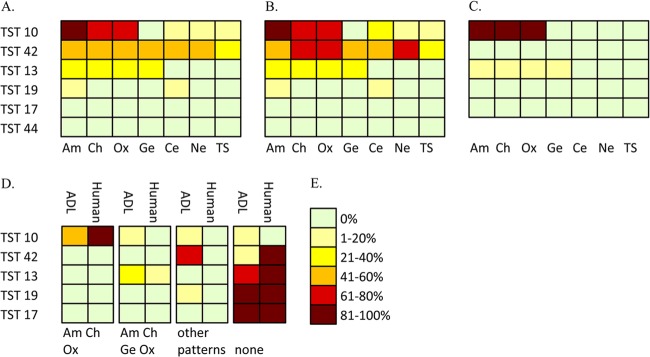
(A to C) Heat map showing the frequency of resistance to individual antibiotics by sequence type for ADL isolates and human clinical isolates combined (A), ADL isolates only (B), and clinical isolates only (C). (D) The frequencies of the most common antibiotic resistance patterns for ADL and human clinical isolates compared by sequence type. (E) Heat map key. Antibiotics are represented by the following abbreviations: Am, ampicillin; Ce, ceftiofur; Ch, chlortetracycline; Ge, gentamicin; Ne, neomycin; Ox, oxytetracycline; TS, trimethoprim-sulfamethoxazole.
Isolates within each TST were further separated based on their pattern of resistance, revealing several subpopulations within each sequence type (Fig. 2). Interestingly, while clinical isolates exhibited a lower diversity of resistance patterns, the predominant resistance pattern within each TST was generally the same for clinical and ADL isolates. An ampicillin, chlortetracycline, and oxytetracycline resistance pattern was the most frequent in human clinical and ADL isolates within TST 10, and a subpopulation resistant to ampicillin, chlortetracycline, gentamicin, and oxytetracycline was observed in both ADL and clinical isolates within TST 13. However, isolates within TST 42 were an exception, as ADL isolates within TST 42 have four unique resistance patterns, but no human clinical isolates within TST 42 were resistant to the subset of seven antibiotics in our panel. Admittedly, our small sample size precludes a more formal analysis of these subpopulations, but our data provide evidence that resistant subpopulations within TSTs 10 and 13 are being transmitted from animals to humans. The identification of multiple resistance patterns within sequence types also suggests that CRISPR sequences do not evolve as rapidly as does antibiotic resistance in S. Typhimurium.
DISCUSSION
Despite extensive surveillance, tracking, and control efforts, S. Typhimurium stubbornly persists as one of the leading causes of food-borne illness throughout the world. Its extensive host range and its ability to survive a wide range of processing conditions are thought to contribute to its successful transmission throughout the food system (see reference 46 for a recent review). Currently available molecular subtyping methods lack an adequate mix of resolution and stability to effectively monitor the acquisition of antimicrobial resistance and the transmission of strains within clonal serovars of S. enterica over time (33). Recently published work from our lab has shown that CRISPR-MVLST can discriminate between outbreak-associated and non-outbreak-associated strains within several S. enterica serovars and that it can identify differences in the distribution of S. Enteritidis across different ecological niches (14, 25). Here, we have shown that CRISPR-MVLST can also separate strains of S. Typhimurium with differences in distribution, and we provide the first evidence that CRISPR-MVLST sequence types are different with respect to antibiotic resistance. This study complements previous work demonstrating a correlation between CRISPR type and streptomycin resistance in Erwinia amylovora (26) and correlation of CRISPR loss with the acquisition of antibiotic resistance in Enterococcus faecalis (34).
The majority of S. enterica infections are self-limiting and do not require treatment, but systemic infections in immunologically compromised individuals can be fatal and require treatment with antibiotics (35). It is now recommended that these infections be treated with expanded-spectrum cephalosporins or fluoroquinolones as a consequence of increasingly common resistance to traditional antibiotics such as ampicillin, chloramphenicol, and trimethoprim-sulfamethoxazole (35, 36). Our data support this trend, as nearly 40% of isolates in the ADL collection were resistant to ampicillin and 15% of isolates were resistant to trimethoprim-sulfamethoxazole (see Table S1 in the supplemental material). None of the isolates in the ADL collection were resistant to either fluoroquinolone tested in our panel, but it is concerning that over 20% of isolates in the ADL collection were resistant to ceftiofur, an expanded-spectrum cephalosporin of which a close analog, ceftriaxone, is used in human medicine. Resistance to ceftiofur was observed in 15 of 55 cattle isolates, only two isolates from other sources, and none of the human isolates that we tested. The prevalence of ceftiofur-resistant S. Typhimurium isolates in the ADL collection is similar to that reported nationally in isolates from cattle over the same period and further suggests that cattle are a major reservoir for ceftiofur resistance in S. Typhimurium (33, 37).
The potential for transmission of ceftiofur-resistant strains of S. Typhimurium from cattle to humans through the food system is an important public health concern. Ceftiofur resistance has been linked to the presence of blaCMY-2, a gene which encodes an AmpC-like β-lactamase and has been identified on several different Salmonella plasmids (38–40). The horizontal transfer of blaCMY-2 across multiple distinct evolutionary lineages and subtypes in Salmonella was demonstrated in a study by Alcaine et al. using a collection of isolates from dairy farms (41). The same study also provided evidence for the clonal spread of ceftiofur resistance within a local geographic area—a finding that supports the notion that effective control measures for limiting the spread of antibiotic resistance should focus on limiting the transmission of individual strains and thus preventing opportunities for horizontal gene transfer between strains (41, 42). Therefore, our finding that TSTs correspond with ceftiofur resistance suggests that CRISPR-MVLST is a promising tool for monitoring the transmission of ceftiofur-resistant S. Typhimurium from cattle to humans.
It has been previously suggested that certain strains of S. Typhimurium are host adapted (43). In support of this, two recent publications used molecular subtyping techniques to provide evidence for songbird-adapted strains of S. Typhimurium, and in our study, we identified TST 44 exclusively in wild birds (7, 44). Interestingly, none of the isolates within TST 44 were susceptible to the subset of seven antibiotics in our resistance panel, which may reflect a decreased selective pressure for antibiotic resistance in a wild-host-adapted strain. Additionally 13 sequence types were identified only once within the ADL collection, and only one of these sequence types was subsequently identified in humans. Further study is needed to determine whether CRISPR-MVLST is a useful tool for studying the ecology of S. Typhimurium.
It has not escaped our attention that the virulence genes fimH and sseL are contributing only minimally to the discriminatory power of CRISPR-MVLST. In fact, the six most commonly identified TSTs in our collection could all be separated using only their CRISPR1 and CRISPR2 allelic profile, although they do exhibit some variation in their fimH and sseL alleles. These markers were originally included in our subtyping scheme because of their ability to distinguish between unrelated serovars of S. enterica, while CRISPRs provided additional discriminatory power for separating strains within serovars of S. enterica (14). At the time, we suggested that environmental isolates of S. Enteritidis have distinct CRISPR alleles compared to isolates identified in broilers, eggs, and humans. However, it was later shown that these isolates had been assigned to the incorrect serotype. As we have accumulated sequencing data for a larger number of isolates and compared our data to similar data sets, it is becoming increasingly clear that CRISPR alleles strongly correlate with serovar and might be reliable serotyping markers (14, 24, 25). There is also some evidence to suggest that CRISPR alleles reflect phylogenetic relationships among strains (23, 32). Future work must clarify the significance of microvariations underlying CRISPR allelic diversity and their implications for predicting phenotypic differences within S. Typhimurium, other serovars of S. enterica, and even similar pathogens such as Shiga toxin-producing Escherichia coli and Yersinia pestis.
The best-characterized eubacterial CRISPR-Cas systems have been shown to function as an immune system which derives spacers from foreign DNA (15, 45). However, it is becoming increasingly clear that the CRISPR-Cas system of S. enterica is not evolving as quickly as are similar CRISPR-Cas systems found in Streptococcus thermophilus or Pseudomonas aeruginosa. If spacers in S. Typhimurium were derived from pools of foreign DNA, then we would hypothesize that the unique environmental history of an isolate would be reflected in its spacer composition, particularly at the leader-proximal end of the CRISPR allele. However, the CRISPR alleles that we have identified here and previously in a diverse assemblage of isolates from a multitude of animal, food, environmental, clinical, and geographically distinct sources are composed of a common pool of spacer sequences and show a high degree of spacer conservation at their leader-proximal ends (14, 22, 25). Variations among these alleles stem from deletions and duplications of a conserved order of spacers and are consistent with S. enterica CRISPR allelic diversity reported by others (24). The identification of common CRISPR alleles in isolates from a wide range of sources provides further evidence that CRISPR alleles are stable subtyping markers for the identification of widely disseminated strains of S. Typhimurium. Moreover, it suggests that CRISPRs in S. Typhimurium are evolving vertically and therefore are valuable markers for tracking the clonal dissemination of individual strains. Consequently, we hypothesize that the association between antibiotic resistance and sequence type is not mediated directly by CRISPRs but instead reflects unexplored biological differences among strains. Future study is needed to reveal the nature of these differences and may provide valuable insight into the complex range of factors which influence the dissemination of S. Typhimurium.
Ultimately, we demonstrated the ability of CRISPR-MVLST to identify and distinguish subpopulations of S. Typhimurium with differences in antibiotic resistance. We identified common strains from both human and animal sources, highlighting the need for monitoring the transmission of antibiotic resistance determinants throughout the greater population of S. Typhimurium isolates in order to minimize this important pathogen's risk to human health. It has become increasingly clear that CRISPRs in S. enterica are stable and highly discriminatory subtyping markers for tracking its transmission throughout the global food system and monitoring for the emergence of new strains.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a United States Army Research Office grant to E.G.D. (W911NF-11-1-0442) and a U.S. Department of Agriculture Special Milk Safety grant to the Pennsylvania State University (contract 2009-34163-20132).
Sequencing support for this work was provided by the Huck Institute's Nucleic Acid Facility at Penn State University.
Footnotes
Published ahead of print 24 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00913-13.
REFERENCES
- 1.Scallan E. 2011. Foodborne illness acquired in the United States—major pathogens. Emerg. Infect. Dis. 17:7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scharff RL. 2012. Economic burden from health losses due to foodborne illness in the United States. J. Food Prot. 75:123–131 [DOI] [PubMed] [Google Scholar]
- 3.Hoffmann S, Batz MB, Morris JG. 2012. Annual cost of illness and quality-adjusted life year losses in the United States due to 14 foodborne pathogens. J. Food Prot. 75:1292–1302 [DOI] [PubMed] [Google Scholar]
- 4.Blake DP, Hillman K, Fenlon DR, Low JC. 2003. Transfer of antibiotic resistance between commensal and pathogenic members of the Enterobacteriaceae under ileal conditions. J. Appl. Microbiol. 95:428–436 [DOI] [PubMed] [Google Scholar]
- 5.Wattiau P, Boland C, Bertrand S. 2011. Methodologies for Salmonella enterica subsp. enterica subtyping: gold standards and alternatives. Appl. Environ. Microbiol. 77:7877–7885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soyer Y, Alcaine SD, Schoonmaker-Bopp DJ, Root TP, Warnick LD, McDonough PL, Dumas NB, Gröhn YT, Wiedmann M. 2010. Pulsed-field gel electrophoresis diversity of human and bovine clinical Salmonella isolates. Foodborne Pathog. Dis. 7:707–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hernandez SM, Keel K, Sanchez S, Trees E, Gerner-Smidt P, Adams JK, Cheng Y, Ray A, Martin G, Presotto A, Ruder MG, Brown J, Blehert DS, Cottrell W, Maurer JJ. 2012. Epidemiology of a Salmonella enterica subsp. enterica serovar Typhimurium strain associated with a songbird outbreak. Appl. Environ. Microbiol. 78:7290–7298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foley SL, White DG, McDermott PF, Walker RD, Rhodes B, Fedorka-Cray PJ, Simjee S, Zhao S. 2006. Comparison of subtyping methods for differentiating Salmonella enterica serovar Typhimurium isolates obtained from food animal sources. J. Clin. Microbiol. 44:3569–3577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molla B, Sterman A, Mathews J, Artuso-Ponte V, Abley M, Farmer W, Rajala-Schultz P, Morrow WEM, Gebreyes WA. 2010. Salmonella enterica in commercial swine feed and subsequent isolation of phenotypically and genotypically related strains from fecal samples. Appl. Environ. Microbiol. 76:7188–7193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Threlfall EJ, Ward LR, Frost JA, Willshaw GA. 2000. The emergence and spread of antibiotic resistance in food-borne bacteria. Int. J. Food Microbiol. 62:1–5 [DOI] [PubMed] [Google Scholar]
- 11.Markogiannakis A, Tassios PT, Lambiri M, Ward LR, Kourea-Kremastinou J, Legakis NJ, Vatopoulos AC. 2000. Multiple clones within multidrug-resistant Salmonella enterica serotype Typhimurium phage type DT104. The Greek Nontyphoidal Salmonella Study Group. J. Clin. Microbiol. 38:1269–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gebreyes WA, Altier C, Thakur S. 2006. Molecular epidemiology and diversity of Salmonella serovar Typhimurium in pigs using phenotypic and genotypic approaches. Epidemiol. Infect. 134:187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lan R, Reeves PR, Octavia S. 2009. Population structure, origins and evolution of major Salmonella enterica clones. Infect. Genet. Evol. 9:996–1005 [DOI] [PubMed] [Google Scholar]
- 14.Liu F, Barrangou R, Gerner-Smidt P, Ribot EM, Knabel SJ, Dudley EG. 2011. Novel virulence gene and clustered regularly interspaced short palindromic repeat (CRISPR) multilocus sequence typing scheme for subtyping of the major serovars of Salmonella enterica subsp. enterica. Appl. Environ. Microbiol. 77:1946–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712 [DOI] [PubMed] [Google Scholar]
- 16.Deveau H, Garneau JE, Moineau S. 2010. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 64:475–493 [DOI] [PubMed] [Google Scholar]
- 17.Bhaya D, Davison M, Barrangou R. 2011. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 45:273–297 [DOI] [PubMed] [Google Scholar]
- 18.Barrangou R, Horvath P. 2012. CRISPR: new horizons in phage resistance and strain identification. Annu. Rev. Food Sci. Technol. 3:143–162 [DOI] [PubMed] [Google Scholar]
- 19.Westra ER, Swarts DC, Staals RHJ, Jore MM, Brouns SJJ, Van der Oost J. 2012. The CRISPRs, they are a-changin': how prokaryotes generate adaptive immunity. Annu. Rev. Genet. 46:311–339 [DOI] [PubMed] [Google Scholar]
- 20.Wiedenheft B, Sternberg SH, Doudna JA. 2012. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482:331–338 [DOI] [PubMed] [Google Scholar]
- 21.Barrangou R. 2013. CRISPR-Cas systems and RNA-guided interference. Wiley Interdiscip. Rev. RNA 4:267–278 [DOI] [PubMed] [Google Scholar]
- 22.Liu F, Kariyawasam S, Jayarao BM, Barrangou R, Gerner-Smidt P, Ribot EM, Knabel SJ, Dudley EG. 2011. Subtyping Salmonella enterica serovar enteritidis isolates from different sources by using sequence typing based on virulence genes and clustered regularly interspaced short palindromic repeats (CRISPRs). Appl. Environ. Microbiol. 77:4520–4526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Touchon M, Rocha EPC. 2010. The small, slow and specialized CRISPR and anti-CRISPR of Escherichia and Salmonella. PLoS One 5:e11126. 10.1371/journal.pone.0011126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fabre L, Zhang J, Guigon G, Le Hello S, Guibert V, Accou-Demartin M, De Romans S, Lim C, Roux C, Passet V, Diancourt L, Guibourdenche M, Issenhuth-Jeanjean S, Achtman M, Brisse S, Sola C, Weill F-X. 2012. CRISPR typing and subtyping for improved laboratory surveillance of Salmonella infections. PLoS One 7:e36995. 10.1371/journal.pone.0036995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shariat N, DiMarzio MJ, Yin S, Dettinger L, Sandt CH, Lute JR, Barrangou R, Dudley EG. 2013. The combination of CRISPR-MVLST and PFGE provides increased discriminatory power for differentiating human clinical isolates of Salmonella enterica subsp. enterica serovar Enteritidis. Food Microbiol. 34:164–173 [DOI] [PubMed] [Google Scholar]
- 26.McGhee GC, Sundin GW. 2012. Erwinia amylovora CRISPR elements provide new tools for evaluating strain diversity and for microbial source tracking. PLoS One 7:e41706. 10.1371/journal.pone.0041706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pourcel C, Salvignol G, Vergnaud G. 2005. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151:653–663 [DOI] [PubMed] [Google Scholar]
- 28.Horvath P, Romero DA, Coûté-Monvoisin A-C, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, Barrangou R. 2008. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 190:1401–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35:W52–W57. 10.1093/nar/gkm360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 31.Watts JL, Shryock TR, Apley M, Bade D, Brown S, Grey JT, Heine H, Hunter RP, Mevius DJ, Papich MG, Silley P, Zurenko GE. 2008. Performance standards for antimicrobial disk and dilution susceptibility tests for bacteria isolated from animals; approved standard, 3rd ed. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 32.Fricke WF, Mammel MK, McDermott PF, Tartera C, White DG, Leclerc JE, Ravel J, Cebula TA. 2011. Comparative genomics of 28 Salmonella enterica isolates: evidence for CRISPR-mediated adaptive sublineage evolution. J. Bacteriol. 193:3556–3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoelzer K, Soyer Y, Rodriguez-Rivera LD, Cummings KJ, McDonough PL, Schoonmaker-Bopp DJ, Root TP, Dumas NB, Warnick LD, Gröhn YT, Wiedmann M, Baker KNK, Besser TE, Hancock DD, Davis MA. 2010. The prevalence of multidrug resistance is higher among bovine than human Salmonella enterica serotype Newport, Typhimurium, and 4,5,12:i:− isolates in the United States but differs by serotype and geographic region. Appl. Environ. Microbiol. 76:5947–5959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palmer KL, Gilmore MS. 2010. Multidrug-resistant enterococci lack CRISPR-cas. mBio 1(4):e00227–10. 10.1128/mBio.00227-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hohmann EL. 2001. Nontyphoidal salmonellosis. Clin. Infect. Dis. 32:263–269 [DOI] [PubMed] [Google Scholar]
- 36.Crump JA, Medalla FM, Joyce KW, Krueger AL, Hoekstra RM, Whichard JM, Barzilay EJ. 2011. Antimicrobial resistance among invasive nontyphoidal Salmonella enterica isolates in the United States: National Antimicrobial Resistance Monitoring System, 1996 to 2007. Antimicrob. Agents Chemother. 55:1148–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.US Department of Agriculture 2008. National Antimicrobial Resistance Monitoring System—Enteric Bacteria, Animal Arm (NARMS): 2006 NARMS animal arm annual report. Agricultural Research Service, US Department of Agriculture, Athens, GA [Google Scholar]
- 38.Gaillot O, Clément C, Simonet M, Philippon A. 1997. Novel transferable beta-lactam resistance with cephalosporinase characteristics in Salmonella enteritidis. J. Antimicrob. Chemother. 39:85–87 [DOI] [PubMed] [Google Scholar]
- 39.Winokur PL, Brueggemann A, DeSalvo DL, Hoffmann L, Apley MD, Uhlenhopp EK, Pfaller MA, Doern GV. 2000. Animal and human multidrug-resistant, cephalosporin-resistant Salmonella isolates expressing a plasmid-mediated CMY-2 AmpC beta-lactamase. Antimicrob. Agents Chemother. 44:2777–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winokur PL, Vonstein DL, Hoffman LJ, Uhlenhopp EK. 2001. Evidence for transfer of CMY-2 AmpC beta-lactamase plasmids between Escherichia coli and Salmonella isolates from food animals and humans. Antimicrob. Agents Chemother. 45:2716–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alcaine SD, Sukhnanand SS, Warnick LD, Su W, McGann P, McDonough P, Wiedmann M. 2005. Ceftiofur-resistant Salmonella strains isolated from dairy farms represent multiple widely distributed subtypes that evolved by independent horizontal gene transfer. Antimicrob. Agents Chemother. 49:4061–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis MA, Hancock DD, Besser TE. 2002. Multiresistant clones of Salmonella enterica: the importance of dissemination. J. Lab. Clin. Med. 140:135–141 [DOI] [PubMed] [Google Scholar]
- 43.Rabsch W. 2002. Salmonella enterica serotype Typhimurium and its host-adapted variants. Infect. Immun. 70:2249–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawson B, Hughes LA, Peters T, De Pinna E, John SK, Macgregor SK, Cunningham AA. 2011. Pulsed-field gel electrophoresis supports the presence of host-adapted Salmonella enterica subsp. enterica serovar Typhimurium strains in the British garden bird population. Appl. Environ. Microbiol. 77:8139–8144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cady KC, Bondy-Denomy J, Heussler GE, Davidson AR, O'Toole GA. 2012. The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J. Bacteriol. 194:5728–5738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waldner L, MacKenzie K, Köster W, White A. 2012. From exit to entry: long-term survival and transmission of Salmonella. Pathogens 1:128–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.