

Abstract
Spx, a member of the ArsC protein family, is a regulatory factor that interacts with RNA polymerase (RNAP). It is highly conserved in Gram-positive bacteria and controls transcription on a genome-wide scale in response to oxidative stress. The structural requirements for RNAP interaction and promoter DNA recognition by Spx were examined through mutational analysis. Residues near the CxxC redox disulfide center of Spx functioned in RNAP α subunit interaction and in promoter DNA binding. R60E and C10A mutants were shown previously to confer defects in transcriptional activation, but both were able to interact with RNAP. R92, which is conserved in ArsC-family proteins, is likely involved in redox control of Spx, as the C10A mutation, which blocks disulfide formation, was epistatic to the R92A mutation. The R91A mutation reduced transcriptional activation and repression, suggesting a defect in RNAP interaction, which was confirmed by interaction assays using an epitope-tagged mutant protein. Protein-DNA cross-linking detected contact between RNAP-bound Spx and the AGCA element at −44 that is conserved in Spx-controlled genes. This interaction caused repositioning of the RNAP σA subunit from a −35-like element upstream of the trxB (thioredoxin reductase) promoter to positions −36 and −11 of the core promoter. The study shows that RNAP-bound Spx contacts a conserved upstream promoter sequence element when bound to RNAP.
INTRODUCTION
Bacterial responses to environmental and metabolic changes often involve gene regulation at the level of transcription initiation. The essential enzyme catalyzing this first step of gene expression, RNA polymerase (RNAP), is targeted by a variety of regulatory factors that direct RNAP activity to specific transcription units (1). The core RNAP, bearing the catalytic component, is composed of large subunits, β and β′, the α subunit dimer, and the ω subunit. Interaction of core RNAP with the σ subunit gives rise to the holoenzyme endowed with gene promoter specificity (2, 3). Regulatory factors that are controlled by signal sensing/transducing systems that mediate stress responses can target one or more of the RNAP subunits to modulate activity and promoter specificity. Some regulatory factors exert positive control by engaging promoter DNA and recruiting RNAP through direct protein-RNAP subunit interaction (1). There is a growing list of regulatory proteins that prerecruit or appropriate RNAP by first contacting holoenzyme before mediating DNA target recognition (4). One of them, AsiA of phage T4, serves to mediate contact between RNAP σ subunit and a phage-encoded DNA-bound factor, MotA, which triggers prereplicative phage gene transcription (5). The DksA/Gre family proteins target the RNAP active site to affect transcription initiation and elongation at specific promoters (6). SoxS and related proteins engage in prerecruitment by interacting with holoenzyme before contacting specific cis-acting promoter elements (7). Other factors, such as Crl and 6S RNA, stabilize or inhibit specific holoenzyme forms bearing distinct σ subunits (8, 9).
In low-GC Gram-positive bacteria, the Spx family of proteins controls transcription initiation in response to thiol-reactive agents (10). A variety of genes/operons are activated by Spx, including those whose products function in thiol homeostasis, in low-molecular-weight redox buffer biosynthesis, and in cysteine biosynthesis/uptake (11–14). Spx is often encoded by paralogous genes within certain bacterial species (15), and in some cases the genes are essential and/or required for bacterial virulence in pathogenic species (16–18). Spx is a monomer in solution, and a single monomer engages RNAP by contacting the α dimer (19). Spx has higher affinity for holoenzyme bearing the σA subunit than for core RNAP, although there is little evidence that Spx directly contacts sigma (19, 20). Free Spx protein of B. subtilis does not bind to DNA, although when it interacts with holoenzyme it directs RNAP to promoter regions that bear a conserved upstream sequence motif, which is usually AGCA, centered approximately at position −44 with respect to the transcription start site (21, 22). This sequence assignment was supported by recent chromatin immunoprecipitation (ChIP)-chip analysis, which also showed that Spx/RNAP complex targeted 144 operons in the B. subtilis genome (14).
The expression of spx in B. subtilis is under multilevel control. Transcription is catalyzed by four different RNAP holoenzyme forms and is under negative control by PerR and YodB, which are sensitive to peroxide- and thiol-reactive agents, respectively (23–27). The activity of Spx is controlled by a redox switch involving an N-terminal CXXC thiol/disulfide center. Spx mediates RNAP-promoter interaction when in the oxidized, disulfide form, as Spx/RNAP contact with promoter DNA is abolished in the presence of reductant (21). Lastly, Spx is under proteolytic control mediated by the ATP-dependent protease, ClpXP, and a substrate recognition factor, YjbH (28, 29). Proteolytic control is responsive to oxidative stress, as Spx protein accumulates to elevated concentrations in cells that encounter toxic, thiol-reactive oxidants (30, 31).
Many questions remain to be answered with respect to the mechanism of Spx-activated transcription. It is not known how Spx maintains higher affinity for holoenzyme than for RNAP lacking σA, or if Spx contacts promoter DNA. It is not known why the oxidized form of Spx promotes RNAP-promoter DNA contact, and it is not clear why only one Spx monomer contacts the α dimer. Previous structural studies showed that Spx contacts the C-terminal domain of the α subunit (αCTD), and that an α helix in Spx, α4 (Fig. 1), becomes unfolded upon reduction of the Spx disulfide center (32). An amino acid substitution, R60E, in this region of the protein affects Spx-dependent activation and prevents a complex of Spx and αCTD from binding to promoter DNA.
Fig 1.
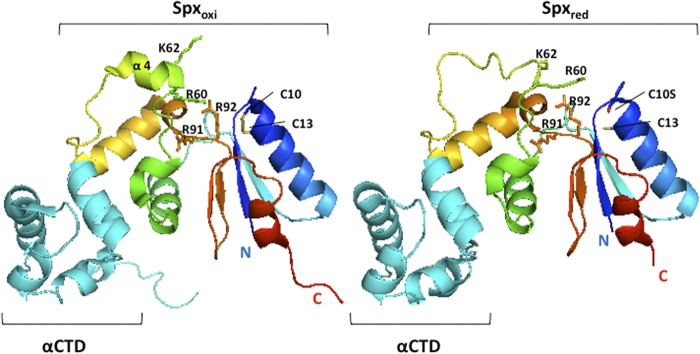
Structures of oxidized Spx (Spxoxi) and reduced C10S Spx (Spxred) in complex with αCTD. The crystal structures of Bacillus subtilis-oxidized Spx (Protein Data Bank [PDB] accession number 3GFK) (44) and reduced C10S Spx (PDB accession number 3IHQ) (32)/αCTD complexes were visualized by using the software PyMOL. αCTD and Spx are shown as cyan- and rainbow-colored ribbons, respectively. The side chains of C10, C13, R60, K62, R91, and R92 are shown and labeled.
The structural analysis of the Spx protein uncovered two domains: a redox domain consisting of the N- and C-terminal regions and containing the redox disulfide center (C10TSC13), and the central domain that contains the αCTD binding surface (defined by the spx codon substitution G52R [20, 33]) and the α4 helix (residue R60 to V69 [32]). The two domains are separated by a linker of two coils bearing residues which, in ArsC, function in catalysis (34). This report presents a study in which residue substitutions in the linker and α4 helix of Spx were generated that affect RNAP and α subunit interaction. Together, the data suggest the presence of an additional site on Spx required for α subunit contact. Data are also presented that demonstrate Spx direct contact with the conserved upstream AGCA element, and that defects in DNA contact are conferred by Spx residue substitutions that affect Spx-activated transcription. DNA-protein cross-linking data show that Spx interaction with RNAP and the AGCA upstream element results in a repositioning of the σA subunit, resulting in its contact with the core promoter.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
All bacterial strains and plasmids are listed in Table S1 in the supplemental material. The B. subtilis strains used in this study are derivatives of JH642 and were grown at 37°C in 2× yeast extract-tryptone (2× YT) or Difco sporulation medium (DSM) (35). Escherichia coli DH5α was used for plasmid construction. Strain ER2566 (New England BioLabs) was used for protein production by an intein-chitin system. Plasmid-bearing E. coli strains were grown at 37°C in 2× YT liquid or on lysogeny broth (LB) solid medium containing 1.2% agar (Difco). For overproduction and purification of Spx proteins in E. coli strain ER2566, cells were grown at 37°C in LB liquid medium. Antibiotic concentrations used were as previously reported (35, 36). For trpC2 pheA1 phenotype confirmation, JH642 derivatives were streaked on a TSS minimal medium agar plate with or without tryptophan and phenylalanine supplements.
Construction of Spx amino acid substitution mutants.
The effect of Spx amino acid substitution was examined using spx constructs bearing amino acid codon substitutions (AN to DD) at the carboxyl-terminal coding end, which gives rise to the spxDD allele. The product of spxDD is resistant to ClpXP proteolysis (13). The previously constructed plasmid pSN56 (13) is a pDR111 derivative that was used to express spxDD and mutant versions. pDR111 is an amyE integration vector, and the cloned spx alleles were expressed from the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible Pspankhy promoter of the plasmid (37). The codon substitutions were generated by two-step PCR mutagenesis with pairs of complementary mutagenic oligonucleotides (listed in Table S2 in the supplemental material) as described previously (32). Plasmids were used to transform ORB4566 (spx::neo thrC::trxB-lacZ) for assays of trxB-directed β-galactosidase activity.
To construct the expression plasmids containing Spx mutant proteins used for in vitro transcription and in vitro affinity interaction assay, two-step PCR-based mutagenesis was performed using plasmids pMMN470 (38) and pAL46 (19) as the templates to generate the desired amino acid substitution. The pMMN470 and pAL46 plasmids are pTYB4 derivatives carrying wild-type spx and spxΔCHA, respectively. DNA fragments were digested with NcoI and SmaI and then inserted into pTYB4, which is an E. coli expression vector used in the Impact kit (New England BioLabs). The recombinant plasmids were introduced by transformation into E. coli strain ER2566 for protein expression.
β-Galactosidase assays.
Strains were grown in liquid DS medium. Strains bearing a trxB-lacZ fusion (see Table S1 in the supplemental material) were grown at 37°C overnight on DS medium agar plates supplemented with the appropriate antibiotics. The overnight cultures were used to inoculate the same liquid medium at a starting optical density at 600 nm (OD600) of 0.02. When the OD600 of the cultures reached 0.4, the cultures were divided into two flasks and 1 mM IPTG was added to one of the flasks. Samples were collected every 30 min, and β-galactosidase activity was assayed as previously described (39); data are presented as Miller units (40).
Western blot analysis.
Samples were collected at the same time when growing the strains for β-galactosidase assays, and the cells were lysed in lysis buffer (30 mM Tris-HCl, pH 8.0, 1 mM EDTA) by the bead-beating method. The cell pellet was suspended in lysis buffer, mixed with 0.1-mm disruption glass beads (RPI Corp.) by vortexing for 5 min, and transferred onto ice for 5 min. Two more vortexing cycles were repeated, followed by centrifugation. The supernatant of each sample was collected, and the protein concentration was measured with Coomassie protein assay reagent (Pierce). Ten μg of the total protein for each sample was loaded onto an SDS-PAGE gel. The Western blot was undertaken as previously described using an anti-Spx antibody (35).
Protein purification.
His-tagged σA-depleted RNAP (SAd-RNAP) was purified from the σA subunit mutant B. subtilis strain ORB5853 (rpoC-His10, sigA-L366A), in which the Leu366 substitution in σA weakens the interaction with the RNAP core enzyme (41). As described previously (19), B. subtilis cells were grown in 2× YT liquid containing chloramphenicol and neomycin at 37°C until the OD600 of the culture reached 0.8 to 0.9. The cells were harvested and lysed by passage through a French press. The protein was purified stepwise with three columns, a HisPur nickel-nitrilotriacetic acid (Ni-NTA; Thermo Scientific) affinity column, a heparin column, and a Bio-Rad High Q column, as previously described (22, 32), and then stored at −20°C in buffer containing 10 mM Tris-HCl, pH 7.8, 100 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 50% glycerol.
The genes specifying σA subunit and Spx variants were cloned in plasmid pTYB4 (terminus Impact CN system; New England BioLabs). The products of the recombinant plasmids bear self-cleavable intein- and chitin-binding domains positioned at the C termini. σA was overproduced from plasmid pSN64 (42) in E. coli ER2566 and purified by using chitin resins (New England BioLabs) followed by a Bio-Rad High Q column. Purified σA was dialyzed and stored at −80°C in buffer containing 25 mM Tris-HCl, pH 8.0, 100 mM KCl, 0.1 mM EDTA, 1 mM MgCl2, and 10% glycerol. Spx variants were expressed from pTYB4 derivatives listed in Table S1 in the supplemental material. As previously described, Spx proteins were purified by using a chitin column followed by a Bio-Rad High S column (35). Spx proteins were stored at −80°C in buffer containing 10 mM Tris-HCl, pH 8.0, 100 mM KCl, 5% glycerol.
In vitro transcription.
The trxB promoter DNA (−200 to +30) fragment was generated by PCR amplification with oligonucleotides oDW7 and oDY8 and inserted into the TOPO vector with a Zero Blunt TOPO PCR cloning kit (Invitrogen, Life Technologies) to generate pDW4. The trxB promoter DNA fragment was further cleaved out of pDW4 with restriction endonucleases EcoRI and HindIII and inserted into plasmid pRLG770 (43) to generate plasmid pDW10. pRLG770 was designed as a supercoiled DNA template for in vitro transcription, and it contains a cloning site for inserting promoter DNA fragments of interest, two terminators, and an internal control RNA-1 transcription unit (43).
For the transcription reaction, a 3-fold molar excess of σA subunit was preincubated with SAd-RNAP at 37°C in buffer containing 10 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 30 mM KCl, and 50% glycerol for 1 h to assemble the RNAP holoenzyme (holo-RNAP). In each reaction, 100 ng of pDW10 template was incubated with 10 nM Spx, 10 nM holo-RNAP, and 5 μCi of [α-32P]UTP (3,000 Ci/mmol) in 50 μl of 10 mM Tris-HCl, pH 8.0, 30 mM KCl, 0.5 mM MgCl2, and 1 mg/ml bovine serum albumin (BSA) at 37°C. After 10 min, the nucleotide mixture (0.2 mM rATP/rGTP/rCTP and 10 nM UTP, final concentration) was then added to initiate the reaction. Ten μl of sample was taken and mixed with an equal volume of sequencing stop solution (95% formamide, 25 mM EDTA, 0.05% bromophenol blue) to stop the reaction after 2, 5, 10, and 20 min of incubation. The samples were heated at 90°C for 2 min and applied to a 6% polyacrylamide-urea gel. The gel was dried, and gels were scanned on a Typhoon Trio+ variable imager (GE Healthcare).
In vitro affinity interaction assay.
To examine RNAP affinity to HA-tagged Spx variants in vitro, the anti-hemagglutinin (HA) affinity matrix was used for the affinity binding assay as described previously (19). Briefly, 0.25 μM His-tagged SAd-RNAP and 2.5 μM SpxΔCHA were incubated with or without 0.25 μM σA in 150 μl of reaction buffer (RB; 10 mM Tris-HCl, pH 8.0, 50 mM NaCl, 5 mM MgCl2) at room temperature for 20 min, and the protein mixture then was applied to the anti-HA affinity column, followed by washing with washing buffer (0.05% Tween 20 in RB). The protein complex was eluted from the column with 100 mM triethylamine, pH 11.5, and neutralized with 1/10 volume of 1 M Tris-HCl, pH 6.8. The composition of protein complex was analyzed on the SDS-polyacrylamide gel, followed by Coomassie blue G250 staining.
For individual RNAP subunit binding assays, equal concentrations of α, αCTD, and/or σA proteins were incubated with 2.5 μM Spx in RB for 20 min. The pulldown procedure detailed above then was conducted.
Quantification of band intensities was performed by using ImageJ software on multiple gel images, and the ratio of each RNAP subunit binding to the mutant Spx to that of wild-type Spx was calculated and is presented in histograms.
Promoter DNA-protein cross-linking.
Cross-linking probes were synthesized as described previously (22), with some modifications. Briefly, biotinylated trxB promoter DNA fragment was generated by PCR using forward 5′-biotinyl-oligonucleotide oDYR06-02 and reverse oligonucleotide oDYR06-03 specifically amplifying trxB promoter DNA from −98 to +22. This DNA fragment was purified using low-melting-point agarose gel extraction and then incubated with streptavidin-conjugated magnetic beads (Dynabeads M280 streptavidin; Invitrogen). Biotinylated trxB DNA bound to the beads was denatured with 0.1 M NaOH and 1 M NaCl, and streptavidin-bound single-stranded trxB promoter was collected by centrifugation and washed with 10 mM Tris-HCl, 1 mM EDTA (TE; pH 7.5) buffer containing 0.1% BSA. Oligonucleotides containing a single 3′ phosphorothioate substitution at the desired nucleotide positions on the trxB promoter (see Table S2 in the supplemental material) were then annealed to the streptavidin-bound single-stranded DNA template and radiolabeled with 120 mCi [α-32P]dATP (MP Biomedicals) using Klenow fragment (3′→5′ exonuclease) enzyme (New England BioLabs) at an adjacent position. After washing off excess free [α-32P]dATP, the full-length extension was performed by adding Klenow fragment (3′→5′ exonuclease) and deoxynucleoside triphosphate (dNTP) mix. The extension reaction was stopped by washing off Klenow enzyme and free dNTP by centrifugation and resuspension in NEBuffer 2 (New England BioLabs). The radiolabeled DNA was released from the streptavidin beads by cleavage with HaeIII (New England BioLabs). The released, radiolabeled DNA was extracted with phenol-chloroform-isoamyl alcohol (PCI) solution and precipitated with ethanol. The promoter DNA was derivatized with 100 mM p-azidophenacylbromide (APB) in 100 mM triethylammonium bicarbonate buffer (pH 8.0) in the dark at room temperature overnight. The derivatized products were then PCI extracted, ethanol precipitated, and dissolved in distilled water.
For the cross-linking experiments, 0.25 μM RNAP with 2.5 μM Spx or Spx variants were incubated with radiolabeled probes (10,000 cpm) at 37°C for 30 min in the dark in buffer containing 20 mM Tris-HCl (pH 8.0), 50 mM KCl, 5 mM MgCl2, 0.1 mg/ml BSA, and 5% glycerol. Cross-linking was carried out by UV irradiation (UV Stratalinker 1800; Stratagene) for 10 s, and then samples were immediately transferred to ice. Samples were treated with 80 U DNase I at 37°C for 1 h, and the cross-linked products were resolved on an 18% SDS-PAGE gel. The dried gels were scanned on a Typhoon Trio+ variable imager (GE Healthcare).
RESULTS
Mutational analysis identified residues near the redox switch and within the helix α4 that are important for trxB transcriptional activation.
Optimal Spx-stimulated transcription requires the formation of a disulfide bond between C10 and C13 in Spx. The structures of the oxidized Spx and reduced C10S Spx in complex with RNAP αCTD have been resolved (20, 32, 44). A significant conformational change was found between the oxidized and reduced forms of Spx (Fig. 1), whereby the helix α4 in mutant C10S Spx was unfolded when the disulfide bond could not be formed. It is likely that the formation of a disulfide bond leads to the structural change in helix α4 of Spx and further enables Spx to activate transcription by facilitating target promoter or RNAP contact. To investigate whether the residues within helix α4 are necessary for the function of Spx and whether residues in the vicinity of the redox center conserved in ArsC are involved in transcriptional activation, single-amino-acid substitutions were introduced within the helix α4 region and near the redox disulfide center in the linker region separating the redox and central domains of the Spx protein to examine their effects on Spx activity. To prevent the degradation of Spx by ClpXP protease under nonstress conditions, we expressed the ClpXP-resistant forms (SpxDD) of the wild-type and mutant proteins from an IPTG-inducible promoter and evaluated the effect of Spx amino acid substitutions on Spx-dependent trxB-lacZ transcription in vivo in the presence of IPTG. The relative activity of the mutant to the wild-type Spx product and production of each Spx protein, as observed by Western blot analysis, are shown in Fig. 2. Previous mutational analyses showed that the residue C10 in the redox disulfide center and R60 and K62 residues in the helix α4 region are important for the transcriptional activation of trxB (21, 32). The Spx(R60E) mutant markedly reduced transcriptional activation in vivo and in vitro, and the Spx(R60E)/αCTD complex could not interact with DNA (32). As shown in a previous study, Spx(C10A) and Spx(R60E) mutants significantly reduced the trxB transcription, and the K62E mutant was also moderately defective in transcription activation (Fig. 2A). This confirmed that the R60 residue plays an important role in the interaction between Spx/RNAP and promoter DNA. The F64A mutant showed an effect similar to that of K62E. When F64 was replaced with tryptophan, another amino acid bearing an aromatic side chain, the activity of Spx was restored, which implied that the aromatic ring at this position in α4 was involved in forming a proper structure for Spx function. The other three mutants, Q65A, N68A, and N70A, in the helix α4 region showed no effect on transcription activation. We also investigated the effect of residues around the redox disulfide center required for trxB transcriptional activation. The Y5 residue in the N-terminal sheet β1 near the redox center was replaced with alanine, but the production of mutant protein was not detected by Western blot analysis (Fig. 2B); hence, the decreased transcriptional activity is due to low levels of Spx (Fig. 2A). However, the Y5F mutant protein is stable, and its activity is similar to that of wild-type Spx, indicating that an aromatic side chain is required at this position. The function of Y5 is structural apparently and is not directly involved in Spx-dependent transcriptional activation.
Fig 2.

Effect of amino acid substitutions near the redox switch and within helix α4 of Spx on trxB-lacZ transcription. (A) The IPTG-inducible alleles encoding a proteolysis-resistant form of SpxDD or SpxDD with other amino acid substitutions was introduced into the amyE locus of the Spx null mutant strain bearing a trxB-lacZ fusion. Strains were grown in DS medium, and when the OD600 was 0.4, 1 mM IPTG was added to induce the SpxDD proteins. Samples were taken at time intervals, and β-galactosidase activities were measured. The highest activities of Spx mutants were selected and used to calculate the ratio of mutant activity to SpxDD activity, where 1 represents the activity observed in the strain expressing the parent SpxDD. (B) Cells of each strain were harvested 1.5 h following IPTG addition and lysed with the bead-beating method to extract total cellular protein. Spx expression levels in these strains were determined by Western blotting (WB) using anti-Spx antibody.
The R92 residue is conserved in arsenate reductase and is thought to heighten the C10 thiolate reactivity (45). The side chain of R92, which is in the vicinity of C10 and the helix α4 region in Spx, turns away from the disulfide upon Spx oxidation (Fig. 1). Next to R92 is another conserved arginine, R91, which in some Spx orthologs is replaced with a lysine. Both Spx mutations, R91A and R92A, reduced the trxB-lacZ expression up to 30 to 40% (Fig. 2). However, the R91A R92A double mutant showed synergistic effects on the trxB transcription and almost abolished transcription (Fig. 3A). This result implies that the two arginine residues play different roles in Spx-dependent transcription regulation. To determine whether the effect of these two arginine substitutions is specific to Spx transcription activation, the involvement of the two substitutions in the negative control was examined by measuring the expression of an srfA-lacZ construct. As reported previously, ComA-dependent transcriptional activation of srfA is repressed by Spx in vivo and in vitro (31). The R92A mutant form of Spx produced in the presence of IPTG could still repress srfA transcription as well as the wild-type Spx, but the R91A mutant lost the ability to repress srfA transcription (Fig. 3B). This result indicated that Spx R92A mutation affected transcription activation but was able to engage RNAP. In contrast, R91A mutant Spx is defective in RNAP interaction, since neither Spx-dependent positive nor negative control was fully operational. For the R92 residue, the crystal structure of Spx in the sulfate-free condition showed movement of the R92 residue when the disulfide formed, but a conformational change in the helix α4 region was not detected (Fig. 1, left) (44). To examine whether the structure of the side chain of R92 is important for the transcriptional activation, the R92 residue was replaced with a glutamine residue, which has a similar side chain geometry but is uncharged. However, LacZ assay results showed that the Spx(R92Q) mutant also had 30% reduction of trxB transcription, which was similar to the defect conferred by the R92A mutation (see Fig. S1 in the supplemental material). To test if the positive charge of the Arg side chain is required for optimal activity, R92 was replaced with a lysine, but this did not restore the level of Spx activity in vivo to that of the wild-type parent protein (see Fig. S1). This suggests that the role of R92 is not to contribute a positive charge for the electrostatic interaction of Spx with DNA, which its interaction with a sulfate ion within the crystal structure was thought to imply (20, 44).
Fig 3.
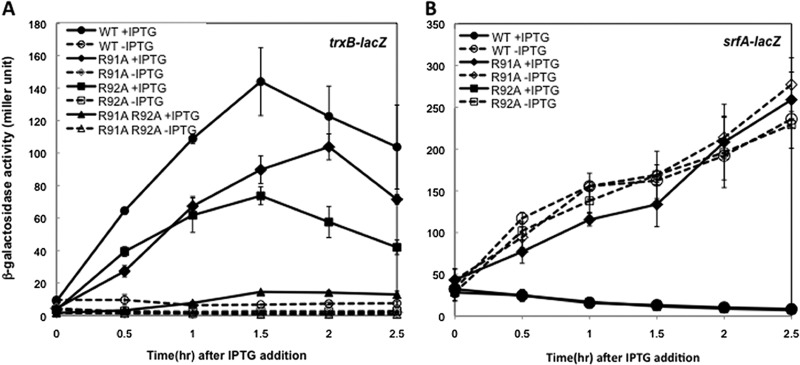
Effect of Spx R91A and R92A mutants on the transcription of trxB-lacZ and srfA-lacZ. Strains bearing trxB-lacZ (A) or srfA-lacZ (B) were grown in DS medium as described in the legend to Fig. 2. When the OD600 reached 0.4, each culture was divided into two flasks and 1 mM IPTG was added to one flask to induce SpxDD expression. Samples were taken at time intervals, and β-galactosidase activities were measured. Symbols: circles, SpxDD; diamonds, Spx(R91A)DD; squares, Spx(R92A)DD; triangles, Spx(R91A, R92A)DD. Open symbols with broken lines represent cells cultured without IPTG, and closed symbols with solid lines represent cell culture with IPTG.
The phenotype of the double mutants suggests a role of R92 in redox control of Spx.
Several additional residue substitutions were introduced in the spx(R92A) mutant to create a series of double mutants, including Spx(C10A, R92A), Spx(R60E, R92A), and Spx(K62E, R92A), and trxB-lacZ activity was examined in these mutant strains. Western blot analysis indicated that mutant Spx proteins were within a 2-fold range of intracellular concentrations (Fig. 4B). The results indicate that the C10A mutation is epistatic to R92A in the Spx(C10A, R92A)-expressing strain, as the same level of activity as that of the Spx(C10A) mutant was observed (Fig. 4A and B). In contrast, R60E or K62E, together with R92A, showed synergistic negative effects on trxB transcription, because Spx(R60E, R92A) nearly abolished trxB-lacZ expression (Fig. 4A), and Spx(K62E, R92A) reduced transcription to the same extent as the Spx(C10A) mutant (Fig. 4B). These results indicate that the R92A phenotype cannot be observed in the C10A background and is observed only if the C10-C13 disulfide bond is able to form. It suggests that the effect of the R92A residue on transcription is connected to disulfide formation and redox control of Spx. In contrast, combining the R92A mutation with R60E results in an additive effect, suggesting that the two residues perform separate functions that contribute to Spx-activated transcription.
Fig 4.

Effect of Spx R92A mutant on the amino acid substitutions in the redox switch or in the helix α4 region on trxB-lacZ transcription. The IPTG-inducible alleles encoding Spx(R92A)DD with amino acid substitutions in either the redox switch (C10) or helix α4 region R60 (A) or K62 (B) were introduced into the amyE locus of the Spx null mutant strain bearing trxB-lacZ. The β-galactosidase assay was performed as described in the legend to Fig. 3, and the highest activities of Spx mutants were taken and calculated as a ratio to that of SpxDD (as explained in the legend to Fig. 2). Spx levels in these strains were determined by Western blotting using anti-Spx antibody, and results are shown in the bottom panel.
Residue substitutions at R91 and R92 reduce Spx activity in vitro.
To verify the effect of Spx(R91A) and Spx(R92A) mutant proteins in vitro, each protein, purified as shown in Materials and Methods, was applied to a transcription reaction with purified RNA polymerase and the supercoiled template (43) containing trxB promoter DNA. A time course transcription was performed (Fig. 5), and the effect of Spx mutants was compared to that of wild-type Spx at each time point. The intensity of the transcript was quantified and normalized by using an internal control transcript encoded by plasmid-borne rna1. The effect of Spx mutants was presented as percent normalized trxB transcription relative to that observed in reaction mixtures containing wild-type Spx (Fig. 5, bottom). Both R91A and R92A mutations conferred reduced transcription-stimulating activity, with levels of in vitro-generated transcripts below those synthesized by wild-type Spx/RNAP.
Fig 5.

In vitro transcription from the plasmid carrying the trxB promoter in the absence and presence of the wild-type Spx or the Spx(R91A) or Spx(R92A) mutant. The plasmid DNA containing the trxB promoter region (10 nM) was incubated with 10 nM reconstituted B. subtilis RNAP (SAd-RNAP, σA = 1:3) and 10 nM Spx, Spx(R91A), or Spx(R92A), and the reaction was performed in a time course manner. Band intensity of each trxB transcript was quantified and normalized to that of the RNA-1 transcript. The trxB transcription level activated by each Spx mutant was calculated and is presented as a percentage compared to that activated by wild-type Spx.
Spx(R91A) mutant is defective in RNAP binding.
To examine whether the R91 and R92 residue substitutions affect Spx-RNAP interaction, an epitope affinity chromatography system designed to capture Spx bound to RNAP was performed as described previously (19). Briefly, epitope-tagged versions of the Spx mutants (SpxΔCHA) were created in which the last 12 C-terminal residues were replaced with an HA (influenza hemagglutinin) tag. The SpxΔCHA protein is functional in vivo (19). The tagged Spx mutant proteins were produced and purified by using a chitin-binding affinity chromatography system, followed by two more column purification steps. His-tagged σA-depleted RNAP (SAd-RNAP) was purified from the sigA(L366A) B. subtilis strain as previously described (19), and recombinant σA protein was purified with a chitin-intein affinity column system (19). The binding of SAd-RNAP or SAd-RNAP with σA subunit (holo-RNAP) to Spx(R91AΔCHA) and Spx(R92AΔCHA) was examined by the anti-HA Affi-Gel pulldown reaction. After RNAP and HA-tagged Spx were incubated and applied to an anti-HA affinity column, protein complexes were eluted with high pH buffer and analyzed by SDS-PAGE (Fig. 6A and C). The association of each RNAP subunit of the complex with the mutant Spx was quantified and is presented as a ratio to the values derived from reaction mixtures containing wild-type Spx (Fig. 6B and D). Both of the arginine substitutions significantly affected RNAP binding. Spx(R91AΔCHA) exhibited severely reduced binding to SAd-RNAP (Fig. 6A and B), but the binding was improved in the presence of σA subunit (Fig. 6C and D). Spx(R92AΔCHA) showed higher affinity to SAd-RNAP than the R91A derivative, but this level was still lower than that of wild-type Spx. Addition of σA subunit improved Spx(R92AΔCHA)-RNAP interaction (Fig. 6B and D).
Fig 6.

Effect of Spx mutants on RNAP interaction in vitro. The σA-depleted RNAP (SAd-RNAP) or RNAP holoenzyme (Holo-RNAP) was incubated with SpxΔCHA, Spx(R91A)ΔCHA, or Spx(R92A)ΔCHA in binding buffer (10 mM Tris-HCl, pH 8.0, 100 mM KCl, 5 mM MgCl2). By pulldown assay with anti-HA affinity chromatography, the interaction between Spx mutants and SAd-RNAP (A) or Holo-RNAP (C) was analyzed by SDS-PAGE. The intensity of each subunit of RNAP holoenzyme (B) or SAd-RNAP (D) on the gel was quantified and normalized, and results are presented as a ratio to the intensity of SpxΔCHA. Abbreviations: I, input; FT, flowthrough; E, eluate. The band labeled “milk” is protein from the blocking agent (dissolved powdered milk).
To examine whether the mutants affect binding of individual RNAP subunits, the purified α C-terminal domain (αCTD) and σA subunit were applied to the anti-HA Affi-Gel pulldown assay. First, the binding of αCTD and σA subunit to the SpxΔCHA mutant was tested. As expected, Spx did not interact with the σA subunit; however, surprisingly, the αCTD showed little affinity for SpxΔCHA (Fig. 7A), although interaction was detected with a yeast two-hybrid system (31) and the crystal structure of the Spx/αCTD complex was solved (Fig. 1) (19, 20, 44). This indicates that the interaction between Spx and αCTD is weak and could be disrupted under the conditions of our pulldown experiment. Hence, we purified intact α subunit to examine whether α subunit-Spx interaction requires the α dimer. The purified α subunit was confirmed as a dimer by gel filtration chromatography (data not shown). The result of the affinity interaction assay showed that Spx interacts with intact α but not αCTD (Fig. 7B). Evidence that intact α subunit bears an Spx-binding surface was obtained by far-Western blotting using preincubation of gel-resolved and immobilized RNAP proteins with Spx, followed by reaction with anti-Spx antiserum (data not shown). These experiments showed that only intact α subunit, but not αCTD or σA, could interact with Spx. This suggests that contact surfaces, apart from αCTD, are required for optimal α-Spx interaction.
Fig 7.
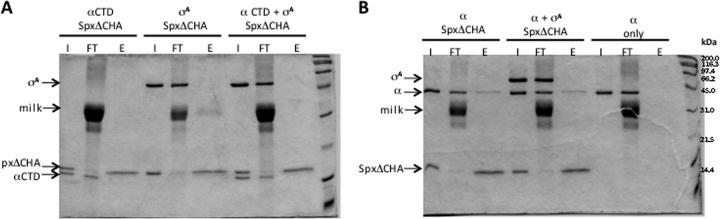
In vitro interaction of RNAP subunits with Spx. Spx or Spx mutant protein was incubated with RNAP subunit, and an in vitro affinity interaction assay was performed with anti-HA affinity chromatography. The result was analyzed by SDS-PAGE. (A) Equal molar ratio of αCTD and/or σA subunit was incubated with SpxΔCHA. (B) Equal molar ratio of intact α subunit was incubated with SpxΔCHA or together with σA subunit. The α subunit was also applied to the chromatography to confirm that α subunit itself did not bind to anti-HA resin.
To examine the effect of Spx mutant proteins on α/Spx interaction, the affinity of SpxΔCHA and mutant derivatives for the α subunit was examined with an anti-HA Affi-Gel interaction assay (19). The Spx R60E and R92A mutants did not show a significant defect in α binding (Fig. 8A, lanes 5 and 6, and B), and the C10A mutant only slightly reduced affinity to the α subunit (Fig. 8A, lane 4, and B). It has been known that the G52 residue constitutes part of the αCTD-Spx interaction interface (20, 21), and as previous results showed, α binding to the Spx(G52R)ΔCHA mutant was significantly reduced to 40% compared to that of SpxΔCHA (Fig. 8A, lane 2, and B). The Spx(R91A)ΔCHA protein, which is defective in binding to SAd-RNAP (Fig. 6A and C), showed a more severe effect on α binding than the G52R mutant (Fig. 8A, lane 3, and B). The result suggests that the defective SAd-RNAP binding to the Spx(R91A) mutant was attributed to the weakened α-Spx interaction and indicates that the R91 residue is important for α interaction, and it further suggests that Spx establishes multiple contacts with the α subunit dimer.
Fig 8.
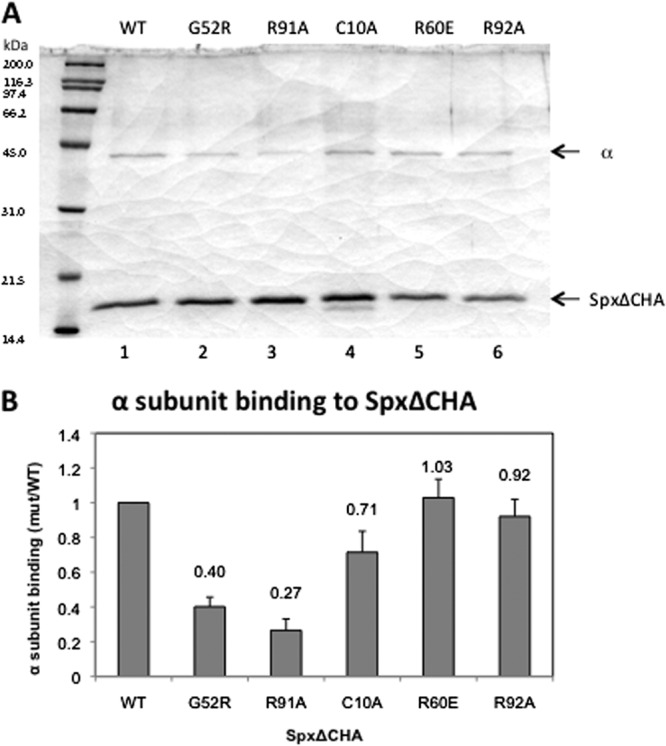
Effect of Spx mutants on interaction with α subunit in vitro. SpxΔCHA or SpxΔCHA variants with single-amino-acid substitutions were incubated with α subunit, and an in vitro affinity interaction assay was performed with anti-HA affinity chromatography. The result was analyzed by SDS-PAGE (A), and the band intensity of α with different Spx mutants on the gel was quantified, normalized, and presented as a ratio to the α subunit binding of SpxΔCHA.
We reasoned that R91 could be another contact point between Spx and the α dimer, or that the R91A mutation alters indirectly the previously identified α-binding surface of Spx, defined by the G52R substitution. Previous studies showed that the G52R substitution resulted in a 75% reduction in Spx-RNAP interaction according to affinity interaction assay results (19). The R91A G52R double mutant was constructed to determine if the phenotype resembled that of the G52R mutant, suggesting that R91 participates in supporting the structure of the αCTD-binding surface of Spx. The Spx(G52R, R91A) mutant protein was produced in B. subtilis in the stable DD form, and the effect of its expression was tested by measuring the activity of the trxB-lacZ fusion. Introduction of the R91A substitution into the Spx(G52R) mutant resulted in nearly complete elimination of activity (see Fig. S2 in the supplemental material). The affinity interaction assay using a version of Spx(G52R, R91A) that bears the C-terminal HA tag and using an anti-HA Affi-Gel column showed that interaction between R91 or G52R mutant protein and the α dimer is eliminated when the two substitutions are combined in the Spx monomer (see Fig. S2). This result, along with the observation of weak interaction of the Spx monomer with αCTD and stable interaction with the α dimer, suggests that the Spx protein has an additional α contact surface, defined by the R91 residue, within the linker region separating the central and redox domains of the Spx protein.
Nucleotide-specific DNA-protein cross-linking shows that Spx interacts with the conserved AGCA motif and repositions σA in the trxB promoter region.
Thus far, there is no evidence showing that Spx directly interacts with DNA. Our previous work identified two potential cis elements required for Spx-activated trxB transcription at positions −44 and −33 (Fig. 9A, boxes) (22, 32). A sequence in nfrA, trxA, and trxB promoter regions centered at −44 and bearing the sequence AGCA was found to be the location of mutations that reduced Spx-stimulated transcription. These findings were supported by the recently reported ChIP analysis (14). The A/TGCA/T sequence upstream of the −35 core promoter element is conserved in promoters that interact with Spx/RNAP in vivo. The element resides in the DNase I-protected region when trxA or trxB promoter DNA is bound to Spx/RNAP (14, 21, 22). These results suggest that the upstream −44 cis element is a site where Spx contacts Spx-activated promoter DNA when bound to RNAP.
Fig 9.

Effect of wild-type and mutant Spx on RNAP/trxB promoter DNA cross-linking. APB-derivatized and radiolabeled trxB promoter DNA was incubated with 0.25 μM RNAP in the absence or presence of 2.5 μM Spx or mutant proteins. After UV irradiation and DNase I treatment, the cross-linking result was analyzed by SDS-PAGE, and cross-linked proteins were detected by phosphorimaging. (A) Cross-linking result at positions −36, −40, and −44 on trxB promoter. (B and C) Summary of the results of cross-linking experiment at all positions investigated. The sequence of the trxB promoter is shown in the middle, and the APB (UV-activated cross-linker)-derivatized position is labeled gray. The binding intensity of each cross-linked protein in the absence (B) and presence (C) of Spx is indicated by an asterisk. Two or three asterisks represent more binding of the specified protein detected at the position examined on the trxB promoter.
The interaction of RNAP and Spx with the trxB promoter DNA was examined by nucleotide-specific protein-DNA cross-linking (22). This was conducted by incubating RNAP and Spx with DNA fragments that were APB (p-azidophenacyl bromide) derivatized and radioactively labeled at specific nucleotide positions in the trxB promoter, followed by photo-cross-linking and DNase I treatment. Radiolabeled, cross-linked proteins were then identified by SDS-PAGE and phosphorimaging. Radiolabeled promoter DNA fragments that were modified with phosphothioate and APB were active as the templates for in vitro transcription reaction mixtures containing Spx/RNAP (data not shown). The binding of Spx/RNAP complex to the eight positions, −11, −21, −36, −40, −44, −46, −49, and −51 (Fig. 9; also see Fig. S3 in the supplemental material), on the trxB promoter was examined. A previous report showed that in the presence of Spx, a strong σA cross-linking signal using a probe labeled at position −11 was generated (22), and no significant cross-linked Spx was detected at −11. Spx also caused elevated generation of a cross-linked product containing the large RNAP subunits and modified position −21 of the trxB promoter. This result was confirmed in the current study (see Fig. S3, lane 4). Previously reported results also showed that Spx(C10A) and Spx(G52R) mutations failed to induce σA contact with a −11 region cross-linker-modified probe (22). The result suggests that oxidized Spx promotes σA contact with the −10 region on the trxB promoter during transcription initiation. Among the eight positions tested, there were no significant changes in the interactions of each RNAP subunit to DNA at −46, −49, and −52 when Spx was present in the cross-linking reaction (see Fig. S3), although at position −46, stronger cross-linked σA signal was detected (see Fig. S3, lanes 10 and 12) than at the other two positions.
In the absence of Spx and in a reaction mixture containing only RNAP incubated with DNA, significant cross-linking of ββ′ with the −44 probe was detected (Fig. 9A, lane 3), especially compared to the level of cross-linked product at −36 (lane 1). Strong σA cross-linking to the −44 probe was also observed, which could be due to the −35-like element located in the −44 region of the trxB promoter (Fig. 9A, lane 3, and B). In the presence of Spx, a strong band representing Spx cross-linked to position −44 was observed, and less cross-linked product was formed in reaction mixtures containing DNA modified at positions −40 and −36 (Fig. 9A, lanes 4 to 6). The −44 position is within the AGCA motif believed to serve as the Spx-specific cis-acting control element. In the presence of Spx, enhanced cross-linked σA protein at position −36 was observed (Fig. 9A, compare lanes 4, 10, and 19 to lane 1). This result indicates that Spx interaction with RNAP repositions the σA subunit from the nonproductive position, −44 in the trxB promoter region, to −36, where σA protein normally interacts when RNAP contacts the core promoter elements. Higher concentrations of RNAP (to 0.5 μM) resulted in reduced cross-linking of Spx to position −44 and less binding of σA to −36 (data not shown). Part of the reason for this result is the heightened competition for the −44 sequence on the part of σA, where some σA is observed to contact in the overlapping −35-like element.
The effect of Spx residue substitutions on DNA binding was also examined. The R60E mutant, which, when combined with the αCTD, showed defective binding to trxB DNA in previous electrophoretic mobility shift assays (EMSAs) (32), showed significantly reduced binding at position −44 compared to that of wild-type Spx (Fig. 9A, lane 9). σA cross-linking at this position was also reduced. This indicates that the Spx(R60E) mutant is still capable of interacting with RNAP, preventing σA contact with DNA at −44, but fails to promote Spx/RNAP interaction with the trxB core promoter. The C10A mutant, conferring a defect in the redox center, almost abolished the binding ability of Spx to the DNA at −44 (Fig. 9A, lane 15), and, as with the R60E mutant, the nonproductive σA binding was reduced (Fig. 9A, lane 15 versus lane 3). This implies that the C10A mutant still retains some affinity to RNAP, as shown in previous affinity interaction assays; however, since it fails to facilitate a conformational change in the helix α4 region under oxidizing conditions, the DNA-binding ability is affected (21, 32), which prevents productive Spx/RNAP-promoter contact. This conclusion is supported by previous work showing reduced interaction of σA with position −11 when the SpxC10A mutant protein is complexed with RNAP (22). The residue substitutions G52R, R91A, and R92A all caused reductions in the level of contact between RNAP subunits and positions −36, −40, and −44 (Fig. 9A, lanes 15 to 18 and 22 to 27) compared to the pattern of RNAP cross-linking in reaction mixtures containing wild-type Spx. The two Spx mutants, G52R and R91A, which show defective binding to RNAP and α, also showed significant reductions in DNA cross-linking at −44 (Fig. 9A, lanes 18 and 24). The R92A mutant cross-linked to position 44 and showed a slight defect in σA binding to position −36 (Fig. 9A, lane 27), as the mutation had a modest, albeit reproducible, effect on transcriptional activation (Fig. 2 and 5).
The enhanced cross-linked σA protein observed at positions −11 and −36 in the presence of Spx indicates that Spx, when bound to RNAP and contacting the −44 promoter element, redirects σA subunit to properly interact with −10 and −35 consensus elements on the trxB promoter as part of its mode of action during transcriptional activation. Figure 9B and C present a summary of the cross-linking data involving interaction of RNAP subunits with the trxB promoter in the absence and presence of Spx.
DISCUSSION
Previous studies indicated that the region of Spx that includes the small α4 helix and residues within the linker region separating the redox and central domains (Fig. 1) undergoes conformational changes upon transition from reduced/thiol to oxidized/disulfide states (32). This transition affects interaction of Spx-αCTD with target promoter DNA. Previous work also showed that a single monomer of Spx engages the RNAP holoenzyme, suggesting that Spx targets not only the αCTD but also another subunit/domain within the holoenzyme complex (19). Evidence presented here shows that three arginine residues at the end of α4 and within the Spx linker region (R60, R91, and R92) exert effects on (i) Spx-RNAP interaction, (ii) contact of Spx with a conserved cis-acting element in the target promoter of trxB, and/or (iii) Spx-dependent positioning of the RNAP σA subunit with the core promoter element trxB.
Spx interaction with RNAP.
One of the questions regarding Spx function is how it interacts with RNAP. In an earlier work, Spx was shown to have higher affinity for RNAP holoenzyme than for RNAP depleted of σA subunit (19). However, several studies have not detected contact between Spx and σA (current study and K. J. Newberry and P. Z. R. G. Brennan, unpublished data). Affinity interaction assays and far-Western blot analysis using Spx and anti-Spx antibody, along with gel-resolved and filter-immobilized RNAP subunits (data not shown), showed interaction of Spx only with the α subunit and not the σA subunit or the αCTD. The data presented above indicate that Spx targets the α dimer and is shown to have higher affinity for intact α subunit than the αCTD, despite the fact that an Spx/αCTD complex has been obtained by coexpression in E. coli (44). Mutational analysis suggests that in addition to the αCTD-binding interface in the central domain of Spx, there is also an α subunit contact point in the linker defined by the R91 residue. The phenotype of R91 might relate to the observation that only one Spx monomer is required to productively interact with RNAP (19). The two α contact points defined by G52R and R91A might mediate formation of a bridge between the two α monomers, thereby occupying a position in the α dimer that can only accommodate a single Spx monomer. It is not clear why Spx prefers contact with RNAP holoenzyme over that with SAd-RNAP, but the presence of σA in the holoenzyme might promote a conformation of the α dimer that maximizes Spx contact. It is also possible that Spx contacts σA only when the two proteins are incorporated into the holoenyzme complex.
Redox control of Spx.
It is known that Spx can undergo oxidation to the disulfide form, a reaction that heightens Spx activity in terms of promoting RNAP interaction with the regulatory regions of some Spx-activated genes (21). The mechanism behind this process is currently unclear, but evidence provided here suggests that R92 participates in thiol/disulfide redox control. The C10A mutation eliminates the transcription-stimulating activity of Spx in vitro, while SpxDD(C10A) shows an 80% reduction in activity in vivo compared to SpxDD. Introduction of a second substitution at R92 does not change the level of mutant Spx activity in vivo, indicating that C10A is epistatic to R92A, and the mutant phenotype of R92A is observed only when a disulfide can be generated. Such epistasis experiments using multiple residue substitutions has been applied to uncover protein-protein contact surfaces on SoxS and its binding partner, σ70 of E. coli RNAP (46, 47). It is not clear how the R92 side chain transduces a redox signal from the disulfide center to the target-interacting surfaces of Spx and ultimately to RNAP. The reduced Spx structure of Lamour et al. shows a subtle rotation of the R92 side chain compared to the structure of oxidized Spx determined by Newberry et al. (20, 44). On the other hand, the R92 side chain in the structure of the SpxC10S mutant veers away from the C10 position and toward α4 helix. The possibility of other residues functioning in the transmission of redox control is currently being investigated.
Spx/RNAP interaction with promoter DNA.
The presumed cis-acting element within the regulatory region of Spx-activated genes is located in a sequence around position −44 with respect to the transcriptional start site; it is usually AGCA and is followed by an AT-rich sequence. Construction of hybrid promoters, site-directed mutagenesis, and alignment of sequences that interact with Spx/RNAP in vivo (14, 22, 32) all indicate that the conserved upstream A/TGCA/T sequence centered around position −44 is required for Spx/RNAP interaction with promoter DNA. This is supported by nucleotide-specific protein-DNA cross-linking, in which wild-type Spx, when complexed with RNAP, is shown to contact the derivatized −44 nucleotide position (Fig. 9). This is accompanied by strong cross-linking signals caused by contact between σA and the −10 and −35 regions of the trxB promoter. Indications from EMSA that Spx, in combination with the αCTD, could generate a DNA-binding complex were reported previously (32). As previously reported and presented here, Spx(R60E) can form a complex with αCTD and RNAP (19, 32) but does not promote transcriptional activation, most likely due to the failure to contact target DNA, which is necessary to induce σA binding to core promoter elements (Fig. 9). Codon substitutions (G52R and R91A) in spx that confer defects in RNAP binding also compromised cis-element contact, as shown in the cross-linking data.
In the trxB promoter region, there is a −35-like element that overlaps with the AGCA sequence, and cross-linking indicates that this element contacts the σA subunit of holoenzyme. In the absence of Spx, the −36 position is poorly contacted by σA, but contact between σA and −36 is evident in the presence of Spx (Fig. 9). Thus, it seems that Spx repositions σA when it is bound to RNAP. The data also suggest that some Spx-dependent remodeling of holoenzyme takes place prior to DNA interaction. Mutant Spx proteins [Spx(R60E), for example] also reduce interaction of σA with position −44 without significant Spx contact with the −44 cis element. While this suggests that remodeling of RNAP by Spx occurs at the pre-DNA-binding stage, σA-promoter interaction involves Spx contact with the −44 element as part of the Spx/RNAP/promoter complex.
Data presented here and reported previously (32) indicate that R60 is important for contact with the −44 element. Amino acid residue substitutions affecting RNAP binding (G52R and R91A) cause a reduction in contact between Spx at position −44 and σA at −36 (Fig. 9). The R92A mutation affects interaction of Spx at −44 and σA at −36 as well, which could account for its intermediate negative effect on in vivo and in vitro Spx activity. There is little, if any, specific contact between Spx/RNAP complex and DNA upstream at positions −49 and −52 (see Fig. S3 in the supplemental material).
A model of Spx/RNAP-trxB promoter contact is shown in Fig. 10. This model does not explicitly include the possible involvement of multiple α contact surfaces on Spx. The interaction of α subunits with DNA (Fig. 10) also is speculative at this point, but we are of the opinion that one of the αCTDs contacts the AT-rich region between the −44 element and the −35 region.
Fig 10.
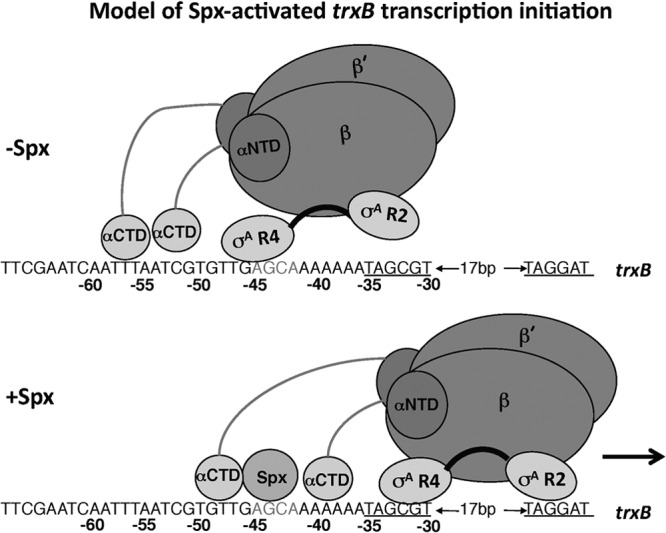
Diagram showing a model of RNAP interaction with the trxB promoter region in the presence and absence of Spx protein. Proposed contact between region 4 of the σA subunit at the −44 sequence, which overlaps a −35-like element when Spx has been shown to be absent. The interaction of RNAP-bound Spx with the −44 element causes σA to engage the core promoter −35 and −10 elements. αNTD, N-terminal domain of the α subunit.
The current study provides evidence for multiple RNAP-binding surfaces on Spx, the resulting interaction of which causes remodeling of RNAP so that σA contacts the −35 and −10 core promoter elements. This is further facilitated by interaction of RNAP-bound Spx protein with the −44 element that is conserved among the Spx-activated genes. The results reported here also suggest that redox control is transmitted in part through changes in positioning of the R92 side chain but will likely involve other conformational changes in the important linker region separating the redox and central domains of Spx.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. M. Nakano for critical reading of the manuscript.
Research reported here was supported by grant GM045898 to P.Z. from the National Institutes of Health.
Footnotes
Published ahead of print 28 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00645-13.
REFERENCES
- 1.Lee DJ, Minchin SD, Busby SJ. 2012. Activating transcription in bacteria. Annu. Rev. Microbiol. 66:125–152 [DOI] [PubMed] [Google Scholar]
- 2.Murakami KS, Darst SA. 2003. Bacterial RNA polymerases: the wholo story. Curr. Opin. Struct. Biol. 13:31–39 [DOI] [PubMed] [Google Scholar]
- 3.Murakami KS, Masuda S, Darst SA. 2003. Crystallographic analysis of Thermus aquaticus RNA polymerase holoenzyme and a holoenzyme/promoter DNA complex. Methods Enzymol. 370:42–53 [DOI] [PubMed] [Google Scholar]
- 4.Haugen SP, Ross W, Gourse RL. 2008. Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat. Rev. Microbiol. 6:507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pineda M, Gregory BD, Szczypinski B, Baxter KR, Hochschild A, Miller ES, Hinton DM. 2004. A family of anti-sigma70 proteins in T4-type phages and bacteria that are similar to AsiA, a transcription inhibitor and co-activator of bacteriophage T4. J. Mol. Biol. 344:1183–1197 [DOI] [PubMed] [Google Scholar]
- 6.Paul BJ, Barker MM, Ross W, Schneider DA, Webb C, Foster JW, Gourse RL. 2004. DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell 118:311–322 [DOI] [PubMed] [Google Scholar]
- 7.Griffith KL, Shah IM, Myers TE, O'Neill MC, Wolf RE., Jr 2002. Evidence for “pre-recruitment” as a new mechanism of transcription activation in Escherichia coli: the large excess of SoxS binding sites per cell relative to the number of SoxS molecules per cell. Biochem. Biophys. Res. Commun. 291:979–986 [DOI] [PubMed] [Google Scholar]
- 8.Barrick JE, Sudarsan N, Weinberg Z, Ruzzo WL, Breaker RR. 2005. 6S RNA is a widespread regulator of eubacterial RNA polymerase that resembles an open promoter. RNA 11:774–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bougdour A, Lelong C, Geiselmann J. 2004. Crl, a low temperature-induced protein in Escherichia coli that binds directly to the stationary phase sigma subunit of RNA polymerase. J. Biol. Chem. 279:19540–19550 [DOI] [PubMed] [Google Scholar]
- 10.Zuber P. 2004. Spx-RNA polymerase interaction and global transcriptional control during oxidative stress. J. Bacteriol. 186:1911–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chi BK, Gronau K, Maeder U, Hessling B, Becher D, Antelmann H. 2011. S-bacillithiolation protects against hypochlorite stress in Bacillus subtilis as revealed by transcriptomics and redox proteomics. Mol. Cell. Proteomics 10:M111.009506. 10.1074/mcp.M111.009506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaballa A, Newton GL, Antelmann H, Parsonage D, Upton H, Rawat M, Claiborne A, Fahey RC, Helmann JD. 2010. Biosynthesis and functions of bacillithiol, a major low-molecular-weight thiol in bacilli. Proc. Natl. Acad. Sci. U. S. A. 107:6482–6486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakano S, Küster-Schöck E, Grossman AD, Zuber P. 2003. Spx-dependent global transcriptional control is induced by thiol-specific oxidative stress in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 100:13603–13608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rochat T, Nicolas P, Delumeau O, Rabatinova A, Korelusova J, Leduc A, Bessieres P, Dervyn E, Krasny L, Noirot P. 2012. Genome-wide identification of genes directly regulated by the pleiotropic transcription factor Spx in Bacillus subtilis. Nucleic Acids Res. 40:9571–9583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turlan C, Prudhomme M, Fichant G, Martin B, Gutierrez C. 2009. SpxA1, a novel transcriptional regulator involved in X-state (competence) development in Streptococcus pneumoniae. Mol. Microbiol. 73:492–506 [DOI] [PubMed] [Google Scholar]
- 16.Chen L, Ge X, Wang X, Patel JR, Xu P. 2012. SpxA1 involved in hydrogen peroxide production, stress tolerance and endocarditis virulence in Streptococcus sanguinis. PLoS One 7:e40034. 10.1371/journal.pone.0040034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kajfasz JK, Mendoza JE, Gaca AO, Miller JH, Koselny KA, Giambiagi-Demarval M, Wellington M, Abranches J, Lemos JA. 2012. The Spx regulator modulates stress responses and virulence in Enterococcus faecalis. Infect. Immun. 80:2265–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kajfasz JK, Rivera-Ramos I, Abranches J, Martinez AR, Rosalen PL, Derr AM, Quivey RG, Lemos JA. 2010. Two Spx proteins modulate stress tolerance, survival, and virulence in Streptococcus mutans. J. Bacteriol. 192:2546–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin AA, Zuber P. 2012. Evidence that a single monomer of Spx can productively interact with RNA polymerase in Bacillus subtilis. J. Bacteriol. 194:1697–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newberry KJ, Nakano S, Zuber P, Brennan RG. 2005. Crystal structure of the Bacillus subtilis anti-alpha, global transcriptional regulator, Spx, in complex with the alpha C-terminal domain of RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 102:15839–15844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakano S, Erwin KN, Ralle M, Zuber P. 2005. Redox-sensitive transcriptional control by a thiol/disulphide switch in the global regulator, Spx. Mol. Microbiol. 55:498–510 [DOI] [PubMed] [Google Scholar]
- 22.Reyes DY, Zuber P. 2008. Activation of transcription initiation by Spx: formation of transcription complex and identification of a cis-acting element required for transcriptional activation. Mol. Microbiol. 69:765–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antelmann H, Scharf C, Hecker M. 2000. Phosphate starvation-inducible proteins of Bacillus subtilis: proteomics and transcriptional analysis. J. Bacteriol. 182:4478–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eiamphungporn W, Helmann JD. 2008. The Bacillus subtilis sigma(M) regulon and its contribution to cell envelope stress responses. Mol. Microbiol. 67:830–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarvis B. 1967. Resistance to nisin and production of nisin-inactivating enzymes by several Bacillus species. J. Gen. Microbiol. 47:33–48 [DOI] [PubMed] [Google Scholar]
- 26.Leelakriangsak M, Kobayashi K, Zuber P. 2007. Dual negative control of spx transcription initiation from the P3 promoter by repressors PerR and YodB in Bacillus subtilis. J. Bacteriol. 189:1736–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thackray PD, Moir A. 2003. SigM, an extracytoplasmic function sigma factor of Bacillus subtilis, is activated in response to cell wall antibiotics, ethanol, heat, acid, and superoxide stress. J. Bacteriol. 185:3491–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garg SK, Kommineni S, Henslee L, Zhang Y, Zuber P. 2009. The YjbH protein of Bacillus subtilis enhances ClpXP-catalyzed proteolysis of Spx. J. Bacteriol. 191:1268–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larsson JT, Rogstam A, von Wachenfeldt C. 2007. YjbH is a novel negative effector of the disulphide stress regulator, Spx, in Bacillus subtilis. Mol. Microbiol. 66:669–684 [DOI] [PubMed] [Google Scholar]
- 30.Engman J, Rogstam A, Frees D, Ingmer H, von Wachenfeldt C. 2012. The YjbH adaptor protein enhances proteolysis of the transcriptional regulator Spx in Staphylococcus aureus. J. Bacteriol. 194:1186–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakano S, Nakano MM, Zhang Y, Leelakriangsak M, Zuber P. 2003. A regulatory protein that interferes with activator-stimulated transcription in bacteria. Proc. Natl. Acad. Sci. U. S. A. 100:4233–4238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakano MM, Lin A, Zuber CS, Newberry KJ, Brennan RG, Zuber P. 2010. Promoter recognition by a complex of Spx and the C-terminal domain of the RNA polymerase. PLoS One 5:e8664. 10.1371/journal.pone.0008664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakano MM, Zhu Y, Liu J, Reyes DY, Yoshikawa H, Zuber P. 2000. Mutations conferring amino acid residue substitutions in the carboxy-terminal domain of RNA polymerase α can suppress clpX and clpP with respect to developmentally regulated transcription in Bacillus subtilis. Mol. Microbiol. 37:869–884 [DOI] [PubMed] [Google Scholar]
- 34.Martin P, DeMel S, Shi J, Gladysheva T, Gatti DL, Rosen BP, Edwards BF. 2001. Insights into the structure, solvation, and mechanism of ArsC arsenate reductase, a novel arsenic detoxification enzyme. Structure (Cambridge) 9:1071–1081 [DOI] [PubMed] [Google Scholar]
- 35.Nakano MM, Hajarizadeh F, Zhu Y, Zuber P. 2001. Loss-of-function mutations in yjbD result in ClpX- and ClpP-independent competence development of Bacillus subtilis. Mol. Microbiol. 42:383–394 [DOI] [PubMed] [Google Scholar]
- 36.Choi SY, Reyes D, Leelakriangsak M, Zuber P. 2006. The global regulator Spx functions in the control of organosulfur metabolism in Bacillus subtilis. J. Bacteriol. 188:5741–5751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Britton RA, Eichenberger P, Gonzalez-Pastor JE, Fawcett P, Monson R, Losick R, Grossman AD. 2002. Genome-wide analysis of the stationary-phase sigma factor (sigma-H) regulon of Bacillus subtilis. J. Bacteriol. 184:4881–4890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakano S, Zheng G, Nakano MM, Zuber P. 2002. Multiple pathways of Spx (YjbD) proteolysis in Bacillus subtilis. J. Bacteriol. 184:3664–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakano MM, Hoffmann T, Zhu Y, Jahn D. 1998. Nitrogen and oxygen regulation of Bacillus subtilis nasDEF encoding NADH-dependent nitrite reductase by TnrA and ResDE. J. Bacteriol. 180:5344–5350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 41.Zuber P, Chauhan S, Pilaka P, Nakano MM, Gurumoorthy S, Lin AA, Barendt SM, Chi BK, Antelmann H, Mader U. 2011. Phenotype enhancement screen of a regulatory spx mutant unveils a role for the ytpQ gene in the control of iron homeostasis. PLoS One 6:e25066. 10.1371/journal.pone.0025066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakano MM, Nakano S, Zuber P. 2002. Spx (YjbD), a negative effector of competence in Bacillus subtilis, enhances ClpC-MecA-ComK interaction. Mol. Microbiol. 44:1341–1349 [DOI] [PubMed] [Google Scholar]
- 43.Ross W, Thompson JF, Newlands JT, Gourse RL. 1990. E. coli Fis protein activates ribosomal RNA transcription in vitro and in vivo. EMBO J. 9:3733–3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lamour V, Westblade LF, Campbell EA, Darst SA. 2009. Crystal structure of the in vivo-assembled Bacillus subtilis Spx/RNA polymerase α subunit C-terminal domain complex. J. Struct. Biol. 168:352–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi J, Mukhopadhyay R, Rosen BP. 2003. Identification of a triad of arginine residues in the active site of the ArsC arsenate reductase of plasmid R773. FEMS Microbiol. Lett. 227:295–301 [DOI] [PubMed] [Google Scholar]
- 46.Zafar MA, Sanchez-Alberola N, Wolf RE., Jr 2011. Genetic evidence for a novel interaction between transcriptional activator SoxS and region 4 of the sigma(70) subunit of RNA polymerase at class II SoxS-dependent promoters in Escherichia coli. J. Mol. Biol. 407:333–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zafar MA, Shah IM, Wolf RE., Jr 2010. Protein-protein interactions between sigma(70) region 4 of RNA polymerase and Escherichia coli SoxS, a transcription activator that functions by the prerecruitment mechanism: evidence for “off-DNA” and “on-DNA” interactions. J. Mol. Biol. 401:13–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guerout-Fleury AM, Frandsen N, Stragier P. 1996. Plasmids for ectopic integration in Bacillus subtilis. Gene 180:57–61 [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Nakano S, Choi SY, Zuber P. 2006. Mutational analysis of the Bacillus subtilis RNA polymerase alpha C-terminal domain supports the interference model of Spx-dependent repression. J. Bacteriol. 188:4300–4311 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.