

Abstract
Coxiella burnetii, the etiological agent of acute and chronic Q fever in humans, is a naturally intracellular pathogen that directs the formation of an acidic Coxiella-containing vacuole (CCV) derived from the host lysosomal network. Central to its pathogenesis is a specialized type IVB secretion system (T4SS) that delivers effectors essential for intracellular replication and CCV formation. Using a bioinformatics-guided approach, 234 T4SS candidate substrates were identified. Expression of each candidate as a TEM-1 β-lactamase fusion protein led to the identification of 53 substrates that were translocated in a Dot/Icm-dependent manner. Ectopic expression in HeLa cells revealed that these substrates trafficked to distinct subcellular sites, including the endoplasmic reticulum, mitochondrion, and nucleus. Expression in Saccharomyces cerevisiae identified several substrates that were capable of interfering with yeast growth, suggesting that these substrates target crucial host processes. To determine if any of these T4SS substrates are necessary for intracellular replication, we isolated 20 clonal T4SS substrate mutants using the Himar1 transposon and transposase. Among these, 10 mutants exhibited defects in intracellular growth and CCV formation in HeLa and J774A.1 cells but displayed normal growth in bacteriological medium. Collectively, these results indicate that C. burnetii encodes a large repertoire of T4SS substrates that play integral roles in host cell subversion and CCV formation and suggest less redundancy in effector function than has been found in the comparative Legionella Dot/Icm model.
INTRODUCTION
Coxiella burnetii, a Gram-negative naturally obligate intracellular pathogen, is the etiological agent of Q fever, a zoonotic disease predominately associated with domestic livestock. This highly infectious organism is usually acquired via inhalation of contaminated aerosols, where it manifests as an acute, influenza-like illness that is typically self-limiting and readily resolves without antibiotic treatment. However, in rare but serious instances, chronic Q fever can develop and generally presents as endocarditis or hepatitis (1).
Actin-dependent endocytosis of C. burnetii by macrophages leads to internalization in an early endosome and trafficking through the default endocytic pathway to ultimately establish an acidic Coxiella-containing vacuole (CCV) derived from the host lysosomal network (2). While the nascent CCV readily acquires Rab5 and Rab7, markers of early and late endosomes, respectively, fusion with the lysosome is delayed compared to that seen with phagosomes harboring latex beads (3). During this initial stall, the Coxiella-containing phagosome undergoes repeated fusions with autophagosomes, an interaction that is believed to deliver nutrients to the organism (4). Following this delay, the phagosome fuses with the lysosome and the bacteria transition from a nonreplicative small-cell variant (SCV) to the replicative large-cell variant (LCV) (5). As replication ensues, the Coxiella-containing vacuole expands to ultimately occupy most of the host cell cytoplasm and undergoes repeated fusions with autophagosomes, endolysosomes, and, at later time points, vesicles from the early secretory pathway (4, 6). Establishment of this intracellular niche is dependent on active bacterial protein synthesis (7) and requires manipulation of numerous host cell processes. These include induction of autophagy (4), inhibition of apoptosis (8), modification of kinases and phosphatases (9, 10), modulation of the host transcriptome (11), and recruitment of secretory components (6).
Intracellular replication and CCV formation are dependent on a type IVB secretion system (T4SS) that is homologous to the Dot/Icm system of Legionella pneumophila (12, 13). In Legionella, this system is encoded by 26 dot/icm (defect in organelle trafficking/intracellular multiplication) genes (14, 15), 18 of which are homologous to components of the tra/trb-encoded DNA conjugative machinery (1). Together, these components form a pilus-like structure that presumably spans the inner and outer bacterial membranes and forms a translocation pore through the Legionella-containing vacuole (LCV), allowing delivery of bacterial effector proteins into the host cell cytosol (10). The Dot/Icm system of Coxiella consists of 23 of these Dot/Icm components, lacking homologs of IcmR, DotJ, and DotV (16).
These systems are functionally analogous, as demonstrated by cross-complementation of Legionella mutants with Coxiella dotB, icmS, icmW, and icmT (17). This similarity has allowed researchers to succeed in using the genetically tractable L. pneumophila as a surrogate host to identify C. burnetii T4SS substrates (8, 18–20). However, only a few of the effectors are conserved, which is consistent with the different properties of the vacuoles occupied by these two pathogens. Whereas C. burnetii thrives in an acidic, autophagosome-like vacuole, L. pneumophila resides in a vacuole with a neutral pH derived from the host endoplasmic reticulum (ER) (1). Additionally, substantial differences in host range exist, with Legionella predominately associated with protists and Coxiella associated with a variety of mammalian species. To date, approximately 50 T4SS substrates have been identified in C. burnetii using bacterial two-hybrid assays (18), genomic assays (8), and open reading frames (ORFs) with predicted eukaryote-like domains (18–21) as possessing the ability to serve as Dot/Icm-dependent secretion substrates.
The goal of the current study was to more comprehensively identify C. burnetii T4SS substrates using an enhanced bioinformatics-guided approach. The C. burnetii genome was screened for ORFs that contained a PmrA regulatory consensus sequence within the predicted promoter region (18, 22) or an E-block motif (23) or were homologs of known effectors. Using this approach, we identified 53 T4SS substrates, 46 of which have not been previously reported. Ectopic expression in mammalian cells and heterologous expression in Saccharomyces cervisiae revealed that these substrates traffic to distinct subcellular compartments and modulate crucial host processes. The use of the Himar1-mediated random mutagenesis method resulted in the generation of 20 clonal T4SS substrate mutants, 10 of which were defective in intracellular replication and CCV formation. Overall, the results of this study suggest that C. burnetii encodes a large repertoire of secretion substrates that play integral roles in host cell subversion, intracellular replication, and CCV formation and suggest less functional redundancy than is seen with the comparative Legionella model.
MATERIALS AND METHODS
Bacteria and host cell lines.
Bacteria used for this study are listed in Table S1 in the supplemental material. C. burnetii Nine Mile phase II (NMII), clone 4 (RSA439), was propagated in modified acidified citrate cysteine medium (ACCM-2) under microaerophilic conditions as previously described (24). When required, 350 μg/ml kanamycin or 5 μg/ml chloramphenicol was added. L. pneumophila strains Lp02 and Lp03 (a mutant lacking dotA) were maintained on charcoal yeast extract (CYE) agar or N-(2-acetamido)-2-aminoethanesulfonic acid (ACES) broth–yeast extract (AYE) broth, and 5 μg/ml chloramphenicol was added as needed.
HeLa, Hek293T, and J774A.1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS). THP-1 and U937 cells were propagated in RPMI medium with 10% FBS. All cell lines were maintained at 37°C with 5% CO2.
Bioinformatics analysis.
For bioinformatics prediction of C. burnetii T4SS candidate substrates, RSA493 and L. pneumophila Philadelphia 1 genomes were acquired from the J. Craig Venter Institute Comprehensive Microbial Resource (http://cmr.jcvi.org/cgi-bin/CMR/GenomePage.cgi?org=gcb). T4SS candidate substrates were selected based on the presence of one of the following three features: a PmrA consensus regulatory sequence in the promoter region, an E-block motif, or a homolog to a Legionella effector.
We recently reported the identification of 126 putative PmrA-regulated C. burnetii ORFs; however, only 35 of these candidate substrates were tested for Dot/Icm-dependent translocation (18). We included 83 of the untested putative PmrA-regulated genes in the current study.
We scanned the last 30 amino acids corresponding to all C. burnetii RSA493 genes encoding proteins for enrichment in glutamate, termed the E-block motif (23). Using this algorithm, we identified 328 ORFs that encoded similar motifs. Among these, 122 ORFs encode hypothetical proteins that were retained in the candidate pool.
Lastly, we scanned the C. burnetii genome for similarity to the newly identified 70 Legionella effector proteins (25). To accomplish this, we used all-against-all searches of C. burnetii RSA493 proteins using local BLAST-P. This approach resulted in the identification of 42 candidates, 29 of which were excluded based on their predicted function or conservation among Gram-negative bacteria.
Plasmid construction.
To examine Dot/Icm-dependent protein translocation in C. burnetii, we generated the kanamycin-resistant TEM vector, pCC108, by removing the chloramphenicol resistance marker (Cmr) and mCherry from pKM244 (Katja Mertens, James P. Berg, Robert E. Brennan, Mary M. Weber, Chen Chen, and James E. Samuel, unpublished data) using HindIII digestion and replaced it with a PCR-amplified pHSP10-EGFP-pCbu1910-Kanr selection marker cassette. A pCbu1910-Flag tags-TEM1-BamHI cassette was assembled using splicing by overlap extension (SOEing) PCR, and the resulting cassette was cloned into the SacI/SalI sites to generate pCC108. Identified T4SS substrates were cloned into the BamHI/SalI sites of pCC108 or pCBTEM to generate N-terminal TEM1 fusion proteins.
For ectopic expression and yeast toxicity experiments, all identified T4SS substrates were PCR amplified and cloned into BglII/SalI sites of pEGFPC1 (BD Clontech, Mountain View, CA) to generate C-terminal enhanced green fluorescent protein (EGFP) fusions or BamHI/SalI sites of pYesNTA2 (Invitrogen, Carlsbad, CA). For substrates possessing BamHI or SalI sites in the target gene, BglII or XhoI was used.
TEM translocation assays.
TEM translocation assays for L. pneumophila and C. burnetii in U937 and THP-1 cells, respectively, were conducted as previously described (18). L. pneumophila was transformed with pCBTEM and C. burnetii with pCC108 or pCBTEM. Individual L. pneumophila and C. burnetii colonies expressing the β-lactamase fusion protein were selected and propagated in AYE and ACCM-2 broth, respectively. Where needed, expression of fusion proteins was induced with 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) for 48 h. Host cells were seeded at 105/ml in 24-well plates and were inoculated at a multiplicity of infection (MOI) of 30 and incubated for 3 h for infection with L. pneumophila or at an MOI of 100 and incubated for 48 h for C. burnetii. The L. pneumophila- or C. burnetii-infected cells were loaded with 50 μl 6XCCF4/AM solution (LiveBLAzer-FRET B/G loading kit; Invitrogen) and incubated at room temperature for 2 h or 4 h, respectively. The percentage of blue cells was quantified using a Nikon A1 imaging system equipped with a Chroma β-lactamase filter set (Chroma no. 41031) (excitation, 405 nm; emission, 435 nm [long pass]). For substrates exhibiting translocation rates above 10%, approximately 1,000 cells were counted; however, for substrates exhibiting low (less than 10%) translocation rates, approximately 5,000 cells were counted. Results are presented as mean values determined from triplicate wells from at least two independent experiments.
Ectopic expression in HeLa cells.
To determine subcellular localization of C. burnetii T4SS substrates, HeLa cells were seeded in 24-well glass-bottom dishes at a density of 105/ml. Where indicated, cells were infected at an MOI of 100 with mCherry expressing C. burnetii-pKM244. At 72 h postinfection, infected or uninfected cells were transiently transfected with EGFP fusion constructs using Lipofectamine (Invitrogen) and following the guidelines outlined by the manufacturer. At 24 h posttransfection, cells were fixed with 4% formaldehyde and permeabilized with ice-cold methanol and nuclei were stained with 1× Hoechst stain. For colocalization analysis, primary antibodies were diluted in 1% bovine serum albumin (BSA) and used at the following concentrations: anti-CoxIV (GenScript, Piscataway, NJ) at 1:250 and anti-calnexin (GenScript) at 1:250. Primary antibodies were detected using Alexa Fluor 555 (Cell Signaling, Boston, MA) at 1:1,000 diluted in 1% BSA. To assess colocalization with autophagosomes, cells were cotransformed with EGFP-tagged substrates and pmRFP-LC3 (Addgene, Cambridge, MA). Fluorescence images were acquired with a Nikon-A1 microscope using the 60× oil immersion objective, and images were processed using NIS-Elements software. Similar results were obtained from at least three independent experiments performed with at least 100 transfected cells per experiment.
Yeast toxicity assay.
To assess the toxicity of C. burnetii T4SS substrates in yeast, each ORF was cloned into pYesNTA2 as BamHI/SalI PCR fragments, and the resulting constructs were transformed into Saccharomyces cerevisiae strain W303 using a standard transformation protocol (26). Transformants were plated on uracil dropout media, supplemented with glucose, and incubated for 72 h at 30°C. Five colonies from each transformation were expanded, and toxicity was assessed by serially diluting and spotting onto the appropriate dropout media (27). Toxicity results were determined from at least three independent experiments.
SEAP assay.
To identify T4SS substrates that disrupt the secretory pathway, Hek293T cells were cotransformed with pSEAP and pEGFP constructs expressing the T4SS substrates. Twenty-four hours posttransfection, cells were washed with 1× phosphate-buffered saline (PBS) and incubated for an additional 8 h. Following the allotted incubation period, the culture supernatant was collected to calculate the extracellular secreted embryonic alkaline phosphatase (SEAP) levels. For determination of intracellular SEAP levels, cells were lysed (28) and fractions measured for alkaline phosphatase activity using a Phospha-Light system (Invitrogen) as described by the manufacturer. Results were tabulated from at least 2 independent experiments with at least 3 replicates per experiment.
Generation of C. burnetii transposon mutants.
C. burnetii, axenically cultured in ACCM-2, was washed twice and resuspended in distilled water to an approximate concentration of 109 cells/ml. Bacterial cells were mixed with 1 μg of pKM225 plasmid DNA, and electroporation was performed as previously described (24). After 7 days, individual colonies were picked and expanded in ACCM-2. Genomic DNA was isolated for rescue cloning using a GenElute bacterial genomic DNA kit (Sigma, St. Louis, MO). Purified DNA was digested with HindIII (New England BioLabs, Ipswich, MA), ligated with T4 DNA ligase (New England BioLabs), and transformed into chemically competent Escherichia coli DH5α. To determine the genomic insertion site of the Himar1 transposon, three colonies per transformation were sequenced using ColE1-R (5′-CTTTCCTGCACTAGATCCCC-3′) and CatF (5′-GTACTGCGATGAGTGGCAG-3′).
Growth curve analysis of T4SS substrate mutants.
For growth curve comparisons of C. burnetii T4SS substrate mutants, the concentration of C. burnetii strains axenically cultured in ACCM-2 was determined using quantitative PCR (qPCR) with IS1111 gene-specific primers (29). To first verify that the mutants had comparable growth rates in axenic culture, determinations of curves in ACCM-2 were conducted by inoculating 20 ml ACCM-2 with 106 cells/ml of RSA439, RSA439 icmX::Tn, or RSA439 specific T4SS substrate mutants. At 1 day, 4 days, and 7 days postinoculation, 1 ml of culture was removed, pelleted, and resuspended in 200 μl of DNA lysis solution with 10 μl of 20 mg/ml proteinase K (30). For cell culture comparisons, J774A.1 or HeLa cells were seeded in 24-well plates to a density of 105 cells/ml. Cells were infected at an MOI of 50 with C. burnetii (RSA439), RSA439 icmX::Tn, or RSA439 specific T4SS substrate mutants in triplicate. At 4 h postinfection, cells were washed twice with 1× PBS to remove uninternalized bacteria, fresh medium was added to each well, and cultures were incubated at 37°C with 5% CO2. At 1 day, 4 days, and 7 days, cells were harvested using trypsinization (HeLa) or scraped (J774A.1) and resuspended in 200 μl of DNA lysis solution with 10 μl of 20 mg/ml proteinase K (30). DNA was isolated from the infected cells and ACCM-2 cultures using a High Pure PCR Template Prep kit (Roche Applied Sciences, Indianapolis, IN), and purified DNA was quantified using qPCR with TaqMan and primers specific for IS1111 (29). At least three independent growth curves for axenic and cell culture were determined with at least three samples per experiment.
Coinfection.
For coinfection experiments, HeLa cells were seeded at a density of 105 cells/ml onto glass-bottom 24-well plates. Cells were infected at an MOI of 100 with either wild-type RSA439 or specific T4SS substrate mutants or coinfected with wild-type and mutant strains at a 1:1 ratio. At 4 h postinfection, noninternalized bacteria were removed by two washes with 1× PBS and fresh culture medium was added. Cultures were propagated for an additional 7 days. Following infection, cells were washed, fixed with 4% formaldehyde, and DNA was stained with 1× Hoechst stain. Fluorescence images were acquired with a Nikon-A1 microscope using the 60× oil immersion objective, and images were processed using NIS-Elements software. Similar results were obtained from three independent experiments performed with at least 50 infected cells per experiment.
RESULTS
Identification of novel C. burnetii T4SS substrates by bioinformatics screening.
In silico analyses have succeeded in the prediction of putative secretion substrates in bacterial pathogens (18, 23, 31). The use of these computational tools allows one to reduce the number of candidates to an experimentally manageable level. In the current study, we expanded the number of C. burnetii type IV substrates using three parameters: a consensus promoter element in PmrA regulation (18, 22), an E-block motif (23), and homology to known effectors (25).
The two-component regulatory system PmrA-PmrB regulates several dot/icm genes and genes encoding T4SS substrates (22). A recent study revealed that 35 L. pneumophila and 68 C. burnetii ORFs contain the PmrA regulatory elements TTAA-N6-TTAA (22). In L. pneumophila, some of these ORFs code for bona fide Dot/Icm substrates, suggesting the usefulness of this prediction. Using 5 dot/icm genes (icmD, icmQ, icmV, icmW, and dotD) and several confirmed Coxiella secretion substrates (including Cbu1751, AnkB, AnkJ, and AnkM), we identified a consensus sequence (TTAATATTTTCTTAAGGTTTXTGXGXTAXAAT) (see Table S3 in the supplemental material) distinct from that reported by Zusman et al. (22). A genome-wide search with this element allowed us to obtain 126 candidates, 35 of which have been previously tested (18); the remaining 83 (see Table S3) were tested in the current study.
In the second method, we used the E-block feature found in a large number of Dot/Icm substrates of L. pneumophila (23). One of the three consensus sequences (EExxE, ExE, or EEx) was found in 49 Dot/Icm substrates of Coxiella, indicating the usefulness of this motif as a reliable predictor of substrate candidates. Using this feature, we identified 328 ORFs that possess similar motifs. Of these, 122 ORFs encode hypothetical proteins that were retained in the candidate pool (see Table S4 in the supplemental material).
Lastly, we scanned the C. burnetii genome for homologs of the recently identified L. pneumophila Dot/Icm substrates (25), leading to the identification of 42 candidates, 29 (see Table S5 in the supplemental material) of which were tested in the current study.
T4SS-dependent translocation of candidate substrates.
To determine Dot/Icm-dependent translocation of these candidates, each of the 234 genes was fused to the C-terminal end of β-lactamase on pCBTEM (18). Plasmids that direct the expression of the β-lactamase fusion proteins were transformed into Lp02 (14), a L. pneumophila strain with a functional Dot/Icm apparatus. The resulting transformants were used to infect U937 cells at an MOI of 30, and protein translocation was determined by β-lactamase-mediated cleavage of the fluorescence dye CCF4-AM. Fifty-three proteins were found to cause more than 1% of the U937 cells to display blue fluorescence. While the majority of these secretion substrates exhibited a low translocation rate of <10%, four substrates (Cbu0425, Cbu2007, Cbu1620, and CbuA0015) exhibited a high translocation rate of 90%. When expressed in the dotA mutant of L. pneumophila, none of these proteins was able to promote the delivery of the β-lactamase into host cytosol (Table 1 and Fig. 1). Collectively, these results support the designation of these 53 proteins as substrates of the Dot/Icm T4SS substrates.
Table 1.
Phenotypes associated with C. burnetii T4SS substrates
| Cbu no. | Screen and/or reference no.a | LP TEM (%)b | Ectopic localizationc | Yeast toxicityd | SEAPe | Transposon mutant/replication defectf |
|---|---|---|---|---|---|---|
| Cbu0012g | E-block | 1 | No pattern | No phenotype | No phenotype | Yes/slight defecth |
| Cbu0041 (cirA) | 18 | 70 | Punctate | Toxic | No phenotype | Yes/no intracellular replication |
| Cbu0069 | PmrA | 1 | No pattern | No phenotype | No phenotype | |
| Cbu0080 | E-block; 12 | 70 | No pattern | No phenotype | No phenotype | |
| Cbu0113 | E-block | 7 | No pattern | No phenotype | No phenotype | |
| Cbu0129 | 18 | 70 | Nucleic | No phenotype | No phenotype | |
| Cbu0175 | 18 | 20 | No pattern | No phenotype | No phenotype | |
| Cbu0183 | PmrA | 20 | No pattern | No phenotype | No phenotype | |
| Cbu0201g | PmrA | 1 | No pattern | No phenotype | No phenotype | Yes/no defect |
| Cbu0212 | E-block | 10 | No pattern | No phenotype | No phenotype | |
| Cbu0270g | E-block | 1 | No pattern | No phenotype | No phenotype | |
| Cbu0295g | E-block; 12 | 2 | No pattern | No phenotype | No phenotype | |
| Cbu0344 | E-block | 5 | No pattern | No phenotype | No phenotype | Yes/no defect |
| Cbu0372 | E-block | 20 | Endoplasmic reticulum | No phenotype | No phenotype | Yes/no defect |
| Cbu0375 | E-block | 35 | No pattern | No phenotype | No phenotype | |
| Cbu0376 | E-block | 7 | No pattern | No phenotype | No phenotype | |
| Cbu0388 | PmrA | 40 | Nucleic | No phenotype | No phenotype | Yes/no intracellular replicationh |
| Cbu0393g | PmrA | 1 | Nucleic | No phenotype | No phenotype | |
| Cbu0410 | 18 | 45 | ND | No phenotype | No phenotype | |
| Cbu0414 | 18 | 45 | No pattern | No phenotype | No phenotype | |
| Cbu0425 (cirB) | E-block; 12 | 90 | Cytoplasmic | No phenotype | No phenotype | Yes/no intracellular replication |
| Cbu0469g | E-block | 1 | No pattern | No phenotype | No phenotype | |
| Cbu0513 | E-block | 20 | Cytoplasmic | No phenotype | No phenotype | |
| Cbu0590 | E-block | 70 | No pattern | No phenotype | No phenotype | |
| Cbu0606 | E-block | 2 | Cytoplasmic | Toxic | No phenotype | |
| Cbu0637g | PmrA | 1 | No pattern | No phenotype | No phenotype | |
| Cbu0665 | PmrA | 3 | Cytoplasmic | No phenotype | No phenotype | |
| Cbu0773 | E-block | 5 | No pattern | No phenotype | No phenotype | |
| Cbu0794 | 18 | 80 | Nucleic | No phenotype | No phenotype | |
| Cbu0801 | 18 | 10 | No pattern | No phenotype | No phenotype | |
| Cbu0814 | 18 | 10 | No pattern | No phenotype | No phenotype | |
| Cbu0881 | 18 | 80 | ND | No phenotype | No phenotype | |
| Cbu0885 | E-block | 1 | Cytoplasmic | Toxic | No phenotype | |
| Cbu0937 (cirC) | 18 | 2 | Cytoplasmic | No phenotype | No phenotype | Yes/no intracellular replication |
| Cbu1045 | 18 | 40 | No pattern | No phenotype | No phenotype | |
| Cbu1079 | E-block | 5 | No pattern | No phenotype | No phenotype | |
| Cbu1102 | E-block | 70 | No pattern | No phenotype | No phenotype | |
| Cbu1110 | PmrA | 40 | No pattern | No phenotype | No phenotype | |
| Cbu1150g | E-block | 1 | No pattern | No phenotype | No phenotype | |
| Cbu1198 | PmrA | 75 | No pattern | No phenotype | No phenotype | Yes/slight defecth |
| Cbu1217 | 18 | 10 | No pattern | No phenotype | No phenotype | |
| Cbu1314 | 18 | 2 | Nucleic | No phenotype | No phenotype | |
| Cbu1379g | 18 | 1 | No pattern | No phenotype | No phenotype | |
| Cbu1406 | 18 | 40 | Cytoplasmic | No phenotype | No phenotype | |
| Cbu1425g | 18 | 1 | Mitochondrial | No phenotype | No phenotype | |
| Cbu1434g | E-block | 1 | Cytoplasmic | No phenotype | No phenotype | |
| Cbu1457 | 18 | 2 | ND | No phenotype | No phenotype | Yes/slight defecth |
| Cbu1460 | 18 | 40 | ND | No phenotype | No phenotype | |
| Cbu1461 | 18 | 2 | Cytoplasmic | No phenotype | No phenotype | |
| Cbu1524 | 12, 18 | 10 | Nucleic | Toxic | No phenotype | |
| Cbu1525 | E-block; 12 | Cytoplasmic | No phenotype | No phenotype | ||
| Cbu1543 | 18 | 25 | Punctate | No phenotype | No phenotype | |
| Cbu1556 | 18 | 50 | Cytoplasmic | No phenotype | 0.23 ± 0.01 | Yes/no defect |
| Cbu1566 | E-block | 5 | No pattern | No phenotype | No phenotype | |
| Cbu1569 | 18 | 90 | No pattern | No phenotype | No phenotype | Yes/no defect |
| Cbu1576 | PmrA | 2 | Endoplasmic reticulum | No phenotype | No phenotype | |
| Cbu1594 | Homolog | 5 | No pattern | No phenotype | No phenotype | |
| Cbu1599 | 18 | 10 | ND | No phenotype | No phenotype | |
| Cbu1607 | E-block | 5 | No pattern | No phenotype | No phenotype | |
| Cbu1620 | E-block | 90 | No pattern | No phenotype | No phenotype | |
| Cbu1636 | 18 | 1 | Punctate | No phenotype | No phenotype | Yes/no defect |
| Cbu1639 | PmrA | 4 | No pattern | No phenotype | No phenotype | Yes/no defect |
| Cbu1665g | E-block | 1 | No pattern | No phenotype | No phenotype | Yes/no defect |
| Cbu1677 | E-block | 5 | No pattern | No phenotype | No phenotype | |
| Cbu1685 | PmrA | 1 | No pattern | No phenotype | No phenotype | |
| Cbu1751 | 18 | 50 | No pattern | No phenotype | No phenotype | |
| Cbu1754 | E-block | 2 | No pattern | No phenotype | No phenotype | |
| Cbu1769 | 18 | 1 | No pattern | No phenotype | No phenotype | |
| Cbu1789g | E-block | 1 | No pattern | No phenotype | No phenotype | |
| Cbu1790g | E-block | 1 | Cytoplasmic | No phenotype | No phenotype | |
| Cbu1823 | 12, 18 | 50 | No pattern | No phenotype | No phenotype | |
| Cbu1825 | 12, 18 | 40 | Mitochondrial | No phenotype | 1.70 ± 0.01 | |
| Cbu2007 | E-block | 90 | Cytoplasmic | No phenotype | No phenotype | Yes/no defect |
| Cbu2013 | PmrA | 50 | Cytoplasmic | No phenotype | No phenotype | Yes/slight defecth |
| Cbu2016g | E-block | 1 | No pattern | Toxic | No phenotype | Yes/no defect |
| Cbu2028g | E-block | 1 | No pattern | No phenotype | No phenotype | |
| Cbu2052 (cirD) | 12, 18 | 90 | Cytoplasmic | Toxic | No phenotype | Yes/no intracellular replication |
| Cbu2059 (cirE) | E-block; 12 | 40 | Cytoplasmic | No phenotype | No phenotype | Yes/no intracellular replication |
| Cbu2064g | E-block; 12 | 1 | No pattern | No phenotype | No phenotype | |
| Cbu2076g | E-block | 1 | No pattern | No phenotype | No phenotype | |
| Cbu2078 | 12 | 10 | ND | No phenotype | No phenotype | |
| CbuA0014 | 12, 20 | 10 | No pattern | No phenotype | No phenotype | |
| CbuA0015 | E-block, 20 | 90 | No pattern | No phenotype | No phenotype | |
| CbuA0019g | E-block | 1 | Cytoplasmic | No phenotype | 1.36 ± 0.04 | |
| CbuA0020g | E-block | 1 | Mitochondrial | No phenotype | No phenotype |
Bioinformatic screen or reference used in identification of T4SS substrates.
Translocation efficiency of C. burnetii T4SS substrates by L. pneumophilia.
Subcellular localization of GFP-tagged substrates ectopically expressed in HeLa cells (Fig. 2). ND, not determined.
Heterologous expression in yeast diminishes replication (Fig. 3).
External/internal SEAP ratio (Fig. 4). Numbers in bold represent values representing significantly (P < 0.01) diminished growth compared to vector and wild-type results.
Transposon mutants were isolated using rescue cloning, and growth was monitored in J774 and HeLa cells (Fig. 5).
Putative T4SS substrate with low translocation in Legionella; translocation not confirmed in the natural C. burnetii host.
Growth defect in single mutant.
Fig 1.

Dot/Icm-dependent translocation of C. burnetii T4SS substrates by L. pneumophila. L. pneumophila wild-type (WT) strain Lp02 or a ΔdotA Lp03 mutant expressing β-lactamase-fused T4SS substrates was used to infect U937 cells at an MOI of 30. Infected cells were loaded with CCF4/AM, and substrate translocation was determined by counting the number of blue cells. For substrates exhibiting translocation rates above 10%, approximately 1,000 cells were counted; however, for substrates exhibiting low (less than 10%) translocation rates, approximately 5,000 cells were counted. Results are presented as mean values determined from triplicate wells from at least two independent experiments.
To determine whether the protein substrates identified with the L. pneumophila surrogate host can also be recognized by the Dot/Icm transporter of C. burnetii, C. burnetii RSA439 or icmX::Tn was transformed with β-lactamase TEM1 fusions listed in Table S2 in the supplemental material and the resulting transformants were used to infect THP-1 cells. At 48 h postinfection, cells were loaded with CCF4-AM and incubated for an additional 4 h prior to imaging. RSA439 expressing β-lactamase TEM-1 for each of the substrate fusions was positive for translocation as evident by the presence of at least 1% blue cells (see Fig. S1A in the supplemental material) for each tested substrate. Using the constitutive pCC108 and the inducible vector pCBTEM, we were able to demonstrate translocation of 11 substrates by C. burnetii; however, we did observe lower expression (data not shown) and lower translocation using the constitutive pCC108 vector, suggesting that this may be due to the presence of a promoter weaker than those in the comparative C. burnetii TEM vectors (8, 20). Notably, Cbu0069, Cbu0113, and Cbu0885 exhibited higher translocation when expressed in the natural C. burnetii host. No β-lactamase translocation was observed in host cells infected with the RSA439 icmX::Tn strain (see Fig. S1B), indicating that translocation of these C. burnetii T4SS substrates is dependent on a functional secretion system and suggesting that they are bona fide C. burnetii T4SS substrates.
Newly identified C. burnetii Dot/Icm substrates exhibit significant plasticity among bacterial isolates.
Different isolates of C. burnetii are associated with unique disease states characterized by distinct pathological features. For instance, the Dugway (D) rodent isolate is avirulent compared to NMI (32), whereas the Graves (G) and Kearns (K) isolates establish an infection characterized by less inflammation and dissemination (33). Comparison of these different isolates revealed that many effector proteins are intact in specific group isolates but disrupted in other isolates, suggesting that different effector proteins may be responsible for different host cell responses and consequently for distinct disease states (12, 21, 34).
To compare the heterogeneity characteristics of the newly described effectors, details of the genome information for NM, D, K, and G isolates were compared using PATRIC (35). Among the proteins identified in this study, only 12 (Cbu0270, Cbu0469, Cbu0637, Cbu0773, Cbu1079, Cbu1434, Cbu1566, Cbu1754, Cbu1789, Cbu2007, Cbu2016, and Cbu2076) are fully conserved among all isolates (see Table S6 in the supplemental material). Further comparison between the isolates showed that the highest conservation exists between NMI and Dugway isolates, with an additional 11 substrates fully conserved.
It is well established that the T4SS translocation signal resides in the last 100 residues of the C terminus of secretion substrates. Genome comparison of these isolates revealed that many of these substrates have C-terminal truncations, suggesting that these substrates may not be translocated by specific isolates and further supporting the idea of different effector pools for specific C. burnetii pathotypes. Many of the substrates have undergone frameshifts, resulting in the creation of several pseudogenes. For example, Cbu0388 has undergone several frameshifts, leading to the generation of three separate pseudogenes in K isolates and two pseudogenes in D isolates. Similarly, Cbu0375 and Cbu0376 have undergone several mutations resulting in two separate genes in NMI but in one gene in each of the other three isolates. Such variations may correlate with the requirement of different effector pools in different disease states during infection.
C. burnetii T4SS substrates target distinct subcellular compartments in mammalian cells.
Following translocation into a host cell, many effector proteins target specific subcellular compartments or decorate the CCV membrane where they interact with host proteins (8, 18, 20, 21, 36). To determine host subcellular compartments targeted by the C. burnetii T4SS substrates identified here, C. burnetii-infected or uninfected HeLa cells were transiently transfected with each substrate expressed as a fusion of EGFP. Imaging of 52 ectopically expressed substrates identified 17 substrates that displayed a pattern distinct from that seen with EGFP. Most of these EGFP fusions were diffusely localized throughout the cytosol and were excluded from the nuclei (Fig. 2A and Table 1). All of the tested substrates displayed similar localizations in infected and uninfected cells, and no protein was colocalized with the Coxiella-containing vacuole (data not shown).
Fig 2.

C. burnetii secretion substrates target distinct subcellular compartments when ectopically expressed in HeLa cells. Each T4SS substrate was expressed as a C-terminal fusion to EGFP and used for transfection of HeLa cells. (A) A diffuse cytoplasmic signal that excluded the nuclei (Cbu2059) or colocalization with the nucleus (Cbu0393 and Cbu0388) could be demonstrated. (B) Colocalization analysis identified two substrates (Cbu0372 and Cbu1576) that colocalize with the endoplasmic reticulum marker calnexin and one substrate (CbuA0020) that colocalizes with the mitochondrial marker CoxIV. Scale bars in merged images represent 10 μm. Arrows denote locations chosen for insets.
Interestingly, we identified two substrates, Cbu0393 and Cbu0388, that traffic to the nucleus, indicated by colocalization with the Hoechst marker (Fig. 2A). Bioinformatics analysis of these nuclear substrates identified a potential nuclear localization signal, PPTKRPRGL, in its N-terminal region beginning at residue 11 for Cbu0393 (37) and KKPSKKVKIKKSKPKKKK beginning at residue 922 for Cbu0388.
To better identify the subcellular organelles targeted by these EGFP-patterned substrate fusions, we conducted colocalization analysis with markers of relevant organelles. Two substrates, Cbu0372 and Cbu1576, localized to the endoplasmic reticulum (ER) as evident by colocalization with the ER marker calnexin (Fig. 2B). EGFP-CbuA0020 targeted the mitochondria as evident by colocalization with the mitochondrial marker CoxIV (Fig. 2B).
Overexpression of several substrates interferes with yeast growth.
Due to its genetic tractability and the high-level conservation of many cellular processes with mammalian cells, Saccharomyces cerevisiae has evolved as a useful model for studying bacterial effector proteins (38). A large number of bacterial effectors have been identified based on their ability to interfere with yeast growth (12, 27, 39) or by their ability to alter vesicle trafficking (27, 40). To determine whether any of the C. burnetii T4SS substrates are toxic to yeast, we inserted each of these genes, as well as previously reported substrates (18), into galactose-inducible vector pYesNTA2. S. cerevisiae W303, carrying C. burnetii substrates, was grown in uracil dropout media, serially diluted, and spotted onto appropriate dropout agar. Using this approach, we identified six substrates that severely impaired growth (Fig. 3).
Fig 3.
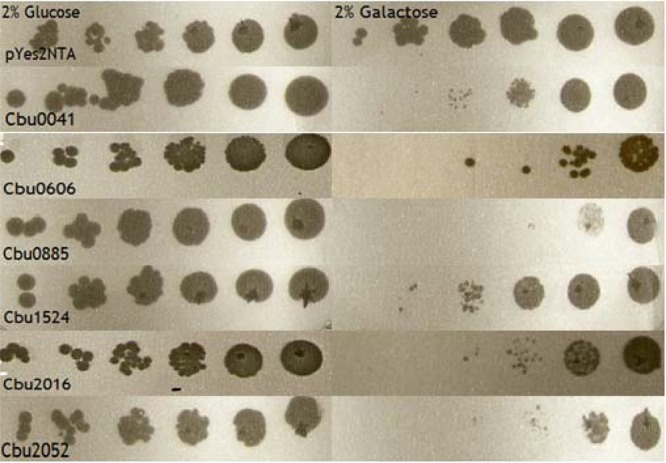
C. burnetii T4SS substrates interfere with yeast growth. S. cerevisiae W303 expressing C. burnetii T4SS substrates was serially diluted to an optical density at 600 nm (OD600) of 0.2 and spotted onto dropout media containing 2% glucose or 2% galactose. All strains exhibited similar growth characteristics under noninducing conditions, in the presence of glucose, but exhibited significantly diminished growth on galactose-containing media.
Overexpression of C. burnetii secretion substrates interferes with the host secretory pathway.
One C. burnetii T4SS effector has been shown to interfere with vesicle trafficking by disrupting membrane trafficking using the secreted embryonic alkaline phosphatase (SEAP) assay (12). To determine whether other Coxiella Dot/Icm substrates interfere with the secretory pathway of eukaryotic cells, Hek293T cells were transiently transfected to express both the SEAP protein and EGFP substrate fusions. The ratio of extracellular to intracellular SEAP activity was calculated (Table 1 and Fig. 4). Three substrates, Cbu1556, Cbu1825, and CbuA0019, exhibited extracellular/intracellular ratios of 0.23 ± 0.01 (P < 0.0001), 1.70 ± 0.01 (P = 0.0004), and 1.36 ± 0.04 (P = 0.0002), respectively, which were significantly reduced compared to the vector-alone ratio of 4.32 ± 0.16, indicating that these C. burnetii effectors interfere with the host secretory pathway.
Fig 4.

C. burnetii-secreted effectors interfere with host protein secretion. Hek293T cells were cotransfected with pSEAP and GFP-tagged T4SS substrates or a vector control. At 24 h posttransfection, external and internal SEAP levels were calculated. A significant decrease in SEAP activity was noted for Cbu1556 (P < 0.0001), CbuA0019 (P = 0.0002), and Cbu1825 (P = 0.0004) compared to the vector control results.
C. burnetii T4SS substrates are essential for intracellular replication and CCV formation.
The Legionella pneumophila Dot/Icm system is estimated to deliver over 250 proteins into the host cell cytosol. In the L. pneumophila model, loss of a single effector generally does not diminish bacterial intracellular replication, suggesting a degree of functional redundancy (41). To determine whether individual C. burnetii T4SS substrates are necessary for intracellular replication and CCV maturation, we generated a pool of mutants using the mariner-based Himar1 transposon for random mutagenesis. Rescue cloning and sequencing analysis led to the isolation of 20 unique T4SS substrate mutants (Table 1; see also Fig. S2 in the supplemental material).
To determine the effects of each of these mutations on bacterial intracellular replication or CCV formation, we infected J774A.1 and HeLa cells with these mutants and monitored bacterial growth. Whereas several of the substrates were dispensable for intracellular replication, six substrate mutants (Cbu0041, Cbu0388, Cbu0425, Cbu0937, Cbu2052, and Cbu2059) were unable to replicate in J774A.1 cells (Fig. 5A) or HeLa cells (data not shown). These mutants displayed a growth phenotype similar to that of the RSA439 icmX::Tn strain. An additional four substrate mutants (Cbu0012, Cbu1198, Cbu1457, and Cbu2013) were capable of replication in J774A.1; however, the bacterial yield was significantly reduced compared to that of the parental RSA439 (Fig. 5B). None of these mutants were capable of forming the characteristic large CCV and instead resided in a tight-fitting vacuole that did not expand even at late time points following infection (Fig. 5C). None of these mutants displayed a growth defect when cultured axenically in ACCM-2 (see Fig. S3A in the supplemental material), indicating that the growth defect is not caused by impairment in general bacterial physiology but likely by the inability of the mutants to properly interact with host cellular processes necessary for the establishment of the replicative niche.
Fig 5.

Individual C. burnetii effector proteins are essential for intracellular replication and CCV formation. NMII C. burnetii was transformed with the Himar1 transposon, and the resulting clonal substrate mutants were used to infect J774A.1 cells. (A) Tn insertion into Cbu0041 (cirA), Cbu0388, Cbu0425 (cirB), Cbu0937 (cirC), Cbu2052 (cirD), and Cbu2059 (cirE) resulted in an inability to replicate intracellularly, however. (B) Tn insertion into Cbu0012, Cbu1198, Cbu1457, and Cbu2013 resulted in reduced intracellular replication compared to RSA439 levels. (C) To determine if coinfection with parental NMII strain would rescue the growth defect of the Tn mutants, HeLa cells were infected with an MOI of 100 of each strain and incubated for 7 days. Images were captured using a Nikon A1 confocal microscope, and at least 100 infected cells were imaged per experiment. Coinfection with the wild type resulted in significant replication for both the wild-type and Tn substrate mutant strains, whereas cells infected only with the substrate mutant did not exhibit significant replication. Scale bars represent 6 μM.
Ten substrate mutants exhibited an inability to productively infect host cells. It was recently demonstrated that coinfection of an icmD::Tn strain with the isogenic NMII strain rescues this growth defect by supplying the T4SS functions in trans (13). If the inability of these effector mutants to replicate intracellularly is due to the loss of an individual effector function, supplying the effector function in trans should similarly rescue their growth defect. Confocal images of cells infected with individual substrate mutants resulted in the formation of tight-fitting vacuoles that do not undergo expansion at late time points of infection (Fig. 5C). However, coinfection of Cbu0041, Cbu0388, Cbu0937, Cbu2052, Cbu2059, Cbu0425, and Cbu1457 effector mutants with the isogenic NMII strain resulted in the formation of CCVs that consisted of roughly equivalent numbers of substrate mutant (red) and NMII (blue) cells (Fig. 5C), indicating that these transposon (Tn) effector mutants are capable of replicating in a vacuole coinhabited by the parental strain. No coinfected cells were observed for Cbu0012, Cbu1198, and Cbu2013. Therefore, at this time, we are unable to confirm that these individual substrates are necessary for intracellular replication and CCV formation.
To further verify that the affected substrate genes in these mutants are responsible for the observed phenotypes, we tested three additional Himar1 insertions each for Cbu0041, Cbu0425, Cbu0937, Cbu2052, and Cbu2059 that were inserted in the same positions (see Fig. S2B in the supplemental material) but were obtained from separate individual transformations. Growth curve analysis of each mutant in J774A.1 resulted in no intracellular replication (see Fig. S3), suggesting that the observed growth defect was due to the loss of that individual effector protein and was unlikely to have been the result of a random point mutation at a secondary site or due to polar effects. Given their crucial role in intracellular replication and CCV formation, we renamed Cbu0041, Cbu0425, Cbu0937, Cbu2052, and Cbu2059 cirA, cirB, cirC, cirD, and cirE (Coxiella effector for intracellular replication), respectively. Overall, these results indicate that the growth defect observed for cirA to -E is due to the loss of an individual effector protein and thus results in the inability of the mutant bacteria to form the proper CCV.
DISCUSSION
Using multiple bioinformatics predictions and analyses, we obtained 234 candidates for substrates of the Coxiella Dot/Icm T4SS. Translocation assays with L. pneumophila as the surrogate host resulted in the identification of 53 substrates that were secreted in a Dot/Icm-dependent manner, representing a 22.6% success rate for substrate identification. Seven substrates identified using this screen (Cbu0080, Cbu0295, Cbu0425, Cbu1525, Cbu2059, Cbu2064, and CbuA0015) were previously identified as Dot/Icm secretion substrates in two independent studies based on genomic (12) and plasmid QpH1 (20) screens. The identification of 46 novel substrates highlights the effectiveness of using computational approaches. However, the use of a PmrA consensus sequence and E-block motifs did not rediscover many of the previously reported substrates (12, 18–21). This suggests that the features governing the recognition of substrate secretion by the Dot/Icm complex are highly diverse and that their expression regulation characteristics differ significantly.
Cbu0201 (ankC) was previously reported as negative for secretion (19, 21); however, we observed a 1% translocation rate for this substrate using L. pneumophila as a surrogate host. The discrepancies between these studies can most likely be attributed to a difference in the secretion assays used. Here we used the β-lactamase TEM-1 assay and observed a 1% translocation rate, whereas the previous studies used a CyaA assay (19, 21). The differences between these studies in the results obtained highlight the importance of using multiple translocation approaches to more comprehensively define protein substrates of the Dot/Icm system. Although our results suggest that ankC is a T4SS substrate of Coxiella, further verification using independent secretion assays in C. burnetii is needed.
Following translocation into the host cell, many proteins traffic to specific subcellular compartments where they modulate the activity of specific host proteins. Subcellular localization has been used in numerous studies as an important starting point for elucidating effector function (12, 20, 39, 40). We transiently transfected infected and uninfected HeLa cells with EGFP-tagged T4SS substrates. Of the 52 substrates examined, 16 fusions displayed a pattern distinct from the EGFP pattern. Whereas the vast majority of these patterns were evenly distributed throughout the cytosol, distinct localizations of several substrates were observed.
The long-standing coexistence between bacterial pathogens and eukaryotic hosts has facilitated the evolution of novel methods for subverting the host innate immune response (42). In the past decade, several bacterial effectors have been identified that traffic to the nucleus, where they provoke histone modifications and facilitate chromatin remodeling (43–46), suggesting that targeting host gene expression is a conserved mechanism used by intracellular pathogens to combat the host defense. Given that many intracellular pathogens secrete nuclear effector proteins, we assessed colocalization of our T4SS substrates with the Hoechst nuclear marker. We identified two substrates, Cbu0393 and Cbu0388, which trafficked to the nucleus and encoded a predicted nuclear localization signal. Interestingly, several additional studies recently identified three other C. burnetii nuclear substrates, Cbu1524 (caeA), Cbu1314, and CbuK1976, suggesting that C. burnetii genes encode a large repertoire of nuclear effectors (12, 18, 47).
C. burnetii has been shown to overcome host defense and to inhibit host cell death by actively promoting host cell viability by inducing prosurvival pathways. Modulation of the interplay between Beclin 1 and Bcl-2 (48) and sustained activation of Akt and Erk1/2 (9) have been proposed to be mediated by T4SS effectors. This hypothesis is supported by the characterization of AnkG, a C. burnetii T4SS effector that was found to inhibit apoptosis by modulating p32 (8). An additional 2 effector proteins encoded by nucleus-localized caeA (Cbu1524) and mitochondrion-localized caeB (Cbu1532) were similarly found to prevent host cell apoptosis (47). In the current study, we identified another substrate, CbuA0020, that colocalized with the CoxIV mitochondrial marker. It will be interesting to determine whether this mitochondrion-localized protein is similarly capable of inhibiting host cell apoptosis.
Ectopic expression in yeast has been routinely used to study bacterial effector proteins. For example, 79 Legionella effectors are able to significantly impair yeast growth (49), a phenotype that has been exploited to analyze their biochemical function (50–52). The identification of Coxiella T4SS substrates capable of inhibiting yeast growth will provide additional tools in determining their role in infection. Similarly, specific targeting of these proteins to distinct host organelles will facilitate future functional analysis.
Recent advances in the field, including the ability to culture the bacteria axenically in ACCM-2 (24) and to genetically manipulate the pathogen using a Himar1 transposon (53) and, recently, the adaptation of a loop-in-loop-out-mediated site-directed mutagenesis method (54), have significantly advanced our understanding of C. burnetii pathogenesis. By applying these tools to our study, we were able to successfully generate and isolate a pool of transposon mutants which included 20 T4SS substrate mutants. Growth curve analysis of each mutant revealed that 10 of these substrate mutants were unable to replicate intracellularly or form a characteristic CCV. This intriguing finding suggests less functional redundancy than has been observed for the comparable L. pneumophila model in which only a few effectors, including SdhA (55) and AnkB (56), have been shown to be essential for intracellular replication in mammalian cells. Furthermore, a recent study by O'Connor et al. (41) demonstrated that deletions of large segments of the L. pneumophila genome result in no impairment in mammalian intracellular replication; however, these mutants were significantly impaired in replication in 3 amoeba species. The differences between these two related bacterial species in effector redundancy can mostly be attributed to the fact that L. pneumophila is an accidental human pathogen that has developed a large effector repertoire for growth in a diverse array of protozoa species, whereas C. burnetii is a highly adapted mammalian pathogen that is capable of replicating in numerous vertebrate species (41).
Each of the cirA-cirE::Tn mutants was observed to be incapable of efficient intracellular replication, except in cells coinfected with the isogenic NMII strain. Recent studies have shown that coinfection of the L. pneumophila ankB mutant with wild-type L. pneumophila was able to rescue the growth defect associated with this mutant by supplying the effector in trans, highlighting the importance of individual effector proteins in generating the appropriate intracellular niche. Similarly, it was demonstrated that the growth defect associated with a C. burnetii icmD::Tn mutant could also be rescued by coinfection with the wild type (13). The ability of the NMII strain to rescue the specific Tn mutants supports the hypothesis that the growth defect observed is the result of the inability of the mutants to form the appropriate CCV that results from the loss of an individual effector protein. This is an intriguing finding and highlights the importance of individual effector proteins in modulating interactions with the host to facilitate the appropriate intracellular niche.
Genome comparisons for cirA to -E revealed that only four substrates (cirA, cirB, cirC, and cirD) are fully conserved among the different C. burneti pathotypes (18), whereas significant heterogeneity exists for cirE. However, cirE has undergone several frameshifts, raising the issue of whether this substrate is secreted by other C. burnetii pathotypes. It will be interesting to determine if each of these substrates is translocated by each of the pathotypes and if the RSA439 mutants can be complemented by the corresponding K, G, or D DNA.
In conclusion, we have shown that C. burnetii genes encode a large, unique repertoire of T4SS substrates that traffic to distinct subcellular compartments and potentially interfere with crucial host cell processes. Through the use of a Himar1 transposon, we isolated 20 individual T4SS substrate mutants, 10 of which were found to be essential for efficient intracellular replication and CCV formation, indicating a lack of functional effector redundancy. Further study of these effectors, including mutant complementation, generation of site-directed mutants, and identification of host binding partners, is under way to better address the issue of how each effector facilitates bacterial pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Erin Van Schaik and Robert Faris for critical review of the manuscript and helpful discussions.
This work was supported by National Institutes of Health-National Institute of Allergy and Infectious Diseases grants AI088430 (J.E.S.), K02AI085403 (Z.-Q.L.), and AI090142 (J.E.S. and Z.-Q.L.).
Footnotes
Published ahead of print 28 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00071-13.
REFERENCES
- 1.Hubber A, Roy CR. 2010. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26:261–283 [DOI] [PubMed] [Google Scholar]
- 2.Omsland A, Heinzen RA. 2011. Life on the outside: the rescue of Coxiella burnetii from its host cell. Annu. Rev. Microbiol. 65:111–128 [DOI] [PubMed] [Google Scholar]
- 3.Howe D, Mallavia LP. 2000. Coxiella burnetii exhibits morphological change and delays phagolysosomal fusion after internalization by J774A. 1 cells. Infect. Immun. 68:3815–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romano PS, Gutierrez MG, Berón W, Rabinovitch M, Colombo MI. 2007. The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell. Microbiol. 9:891–909 [DOI] [PubMed] [Google Scholar]
- 5.Coleman SA, Fischer ER, Howe D, Mead J, Heinzen RA. 2004. Temporal analysis of Coxiella burnetii morphological differentiation temporal analysis of Coxiella burnetii morphological differentiation. J. Bacteriol. 186:7344–7352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campoy EM, Zoppino FCM, Colombo MI. 2011. The early secretory pathway contributes to the growth of the Coxiella-replicative niche. Infect. Immun. 79:402–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Howe D, Melnicáková J, Barák I, Heinzen RA. 2003. Maturation of the Coxiella burnetii parasitophorous vacuole requires bacterial protein synthesis but not replication. Cell. Microbiol. 5:469–480 [DOI] [PubMed] [Google Scholar]
- 8.Lührmann A, Nogueira CV, Carey KL, Roy CR. 2010. Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc. Natl. Acad. Sci. U. S. A. 107:18997–19001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voth DE, Heinzen RA. 2009. Sustained activation of Akt and Erk1/2 is required for Coxiella burnetii antiapoptotic activity. Infect. Immun. 77:205–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hussain SK, Broederdorf LJ, Sharma UM, Voth DE. 2010. Host kinase activity is required for Coxiella burnetii parasitophorous vacuole formation. Front. Microbiol. 1:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahapatra S, Ayoubi P, Shaw EI. 2010. Coxiella burnetii Nine Mile II proteins modulate gene expression of monocytic host cells during infection. BMC Microbiol. 10:244. 10.1186/1471-2180-10-244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carey KL, Newton HJ, Lührmann A, Roy CR. 2011. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog. 7:e1002056. 10.1371/journal.ppat.1002056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Coxrell DC, Howe D, Voth DE, Heinzen RA. 2011. Dot/Icm type IVB secretion system requirements for Coxiella. mBio 2:e00175–00111. 10.1128/mBio.00175-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger KH, Isberg RR. 1993. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 7:7–19 [DOI] [PubMed] [Google Scholar]
- 15.Marra A, Blander SJ, Horwitz MA, Shuman HA. 1992. Identification of a Legionella pneumophila locus required for intracellular multiplication in human macrophages. Proc. Natl. Acad. Sci. U. S. A. 89:9607–9611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seshadri R, Paulsen IT, Eisen JA, Read TD, Nelson KE, Ward NL, Tettelin H, Davidsen TM, Beanan MJ, Deboy RT, Daugherty SC, Brinkac LM, Madupu R, Dodson RJ, Khouri HM, Lee KH, Carty HA, Scanlan D, Heinzen RA, Thompson HA, Samuel JE, Fraser CM, Heidelberg JF. 2003. Complete genome sequence of the Q.-fever pathogen Coxiella burnetii. Proc. Natl. Acad. Sci. U. S. A. 100:5455–5460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zamboni DS, McGrath S, Rabinovitch M, Roy CR. 2003. Coxiella burnetii express type IV secretion system proteins that function similarly to components of the Legionella pneumophila Dot/Icm system. Mol. Microbiol. 49:965–976 [DOI] [PubMed] [Google Scholar]
- 18.Chen C, Banga S, Mertens K, Weber MM, Gorbaslieva I, Tan Y, Luo ZQ, Samuel JE. 2010. Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc. Natl. Acad. Sci. U. S. A. 107:21755–21760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan X, Lührmann A, Satoh A, Laskowski-Arce MA, Roy CR. 2008. Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science 320:1651–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Voth DE, Beare PA, Howe D, Sharma UM, Samoilis G, Cockrell DCA, Omsland Heinzen RA. 2011. The Coxiella burnetii cryptic plasmid is enriched in genes encoding type IV secretion system substrates. J. Bacteriol. 193:1493–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voth DE, Howe D, Beare PA, Vogel JP, Unsworth N, Samuel JE, Heinzen RA. 2009. The Coxiella burnetii ankyrin repeat domain-containing protein family is heterogeneous, with C-terminal truncations that influence Dot/Icm-mediated secretion. J. Bacteriol. 191:4232–4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zusman T, Aloni G, Halperin E, Kotzer H, Degtyar E, Feldman M, Segal G. 2007. The response regulator PmrA is a major regulator of the Icm/Dot type IV secretion system in Legionella pneumophila and Coxiella burnetii. Mol. Microbiol. 63:1508–1523 [DOI] [PubMed] [Google Scholar]
- 23.Huang L, Boyd D, Amyot WM, Hempstead AD, Luo ZQ, O'Connor TJ, Chen C, Machner M, Montminy T, Isberg RR. 2011. The E Block motif is associated with Legionella pneumophila translocated substrates. Cell. Microbiol. 13:227–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Omsland A, Beare PA, Hill J, Cockrell DC, Howe DD, Hansen B, Samuel JE, Heinzen RA. 2011. Isolation from animal tissue and genetic transformation of Coxiella burnetii are facilitated by an improved axenic growth medium. Appl. Environ. Microbiol. 77:3720–3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, Luo Z-Q. 2011. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6:e17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gietz D, St Jean A, Woods R, Schiestl RH. 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 20:1425. 10.1093/nar/20.6.1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sisko JL, Spaeth K, Kumar Y, Valdivia RH. 2006. Multifunctional analysis of Chlamydia-specific genes in a yeast expression system. Mol. Microbiol. 60:51–66 [DOI] [PubMed] [Google Scholar]
- 28.Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo ZQ. 2010. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog. 6:e1000822. 10.1371/journal.ppat.1000822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klee SR, Tyczka J, Ellerbrok H, Franz T, Linke S, Blajer G, Appel B. 2006. Highly sensitive real-time PCR for specific detection and quantification of Coxiella burnetii. BMC Microbiol. 6:2. 10.1186/1471-2180-6-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brennan RE, Samuel JE. 2003. Evaluation of Coxiella burnetii antibiotic susceptibilities by real-time PCR assay. J. Clin. Microbiol. 41:1869–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burstein D, Zusman T, Degtyar E, Viner R, Segal G, Pupko T. 2009. Genome-scale identification of Legionella pneumophila effectors using a machine learning approach. PLoS Pathog. 5:e1000508. 10.1371/journal.ppat.1000508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stoenner HG, Lackman DB. 1960. The biologic properties of Coxiella burnetii isolated from rodents collected in Utah. Am. J. Hyg. 71:45–51 [DOI] [PubMed] [Google Scholar]
- 33.Russell-Lodrigue KE, Andoh M, Poels MWJ, Shive HR, Weeks BR, Zhang GQ, Tersteeg C, Fukushi H, Hirai K, McMurray DN, Samuel JE. 2009. Coxiella burnetii isolates cause genogroup-specific virulence in mouse and guinea pig models of acute Q. fever. Infect. Immun. 77:5640–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beare PA, Unsworth N, Andoh M, Voth DE, Omsland AA, Gilk S, Williams KP, Sobral BW, Kupko JJ, Porcella SF, Samuel JE, Heinzen RA. 2009. Comparative genomics reveal extensive transposon-mediated genomic plasticity and diversity among potential effector proteins within the genus Coxiella. Infect. Immun. 77:642–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gillespie JJ, Wattam AR, Cammer SA, Gabbard JL, Shukla MP, Dalay O, Driscoll T, Hix D, Mane SP, Mao C, Nordberg EK, Scott M, Schulman JR, Snyder EE, Sullivan DE, Wang C, Warren A, Williams KP, Xue T, Yoo HS, Zhang C, Zhang Y, Will R, Kenyon RW, Sobral BW. 2011. PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect. Immun. 79:4286–4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo Z-Q, Isberg RR. 2004. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc. Natl. Acad. Sci. U. S. A. 101:841–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kosugi S, Hasebe M, Matsumura N, Takashima H, Miyamoto-Sato E, Tomita M, Yanagawa H. 2009. Six classes of nuclear localization signals specific to different binding grooves of importin alpha. J. Biol. Chem. 284:478–485 [DOI] [PubMed] [Google Scholar]
- 38.Valdivia RH. 2004. Modeling the function of bacterial virulence factors in Saccharomyces cerevisiae. Eukaryot. Cell 3:827–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Campodonico EM, Chesnel L, Roy CR. 2005. A yeast genetic system for the identification and characterization of substrate proteins transferred into host cells by the Legionella pneumophila Dot/Icm system. Mol. Microbiol. 56:918–933 [DOI] [PubMed] [Google Scholar]
- 40.de Felipe KS, Glover RT, Charpentier X, Anderson OR, Reyes M, Pericone CD, Shuman HA. 2008. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog. 4:e1000117. 10.1371/journal.ppat.1000117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Connor TJ, Adepoju Y, Boyd D, Isberg RR. 2011. Minimization of the Legionella pneumophila genome reveals chromosomal regions involved in host range expansion. Proc. Natl. Acad. Sci. U. S. A. 108:14733–14740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamon MA, Cossart P. 2008. Histone modifications and chromatin remodeling during bacterial infections. Cell Host Microbe 4:100–109 [DOI] [PubMed] [Google Scholar]
- 43.Garcia-Garcia JC, Rennoll-Bankert KE, Pelly S, Milstone AM, Dumler JS. 2009. Silencing of host cell CYBB gene expression by the nuclear effector AnkA of the intracellular pathogen Anaplasma phagocytophilum. Infect. Immun. 77:2385–2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hamon MA, Batsché E, Régnault B, Tham TN, Seveau SS, Muchardt C, Cossart P. 2007. Histone modifications induced by a family of bacterial toxins. Proc. Natl. Acad. Sci. U. S. A. 104:13467–13472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pennini ME, Perrinet S, Dautry-Varsat A, Subtil A. 2010. Histone methylation by NUE, a novel nuclear effector of the intracellular pathogen Chlamydia trachomatis. PLoS Pathog. 6:e1000995. 10.1371/journal.ppat.1000995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu B, Nethery KA, Kuriakose JA, Wakeel A, Zhang XX, McBride JW. 2009. Nuclear translocated Ehrlichia chaffeensis ankyrin protein interacts with a specific adenine-rich motif of host promoter and intronic Alu elements. Infect. Immun. 77:4243–4255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klingenbeck L, Eckart RA, Berens C, Lührmann A. 6 November 2012. The Coxiella burnetii type IV secretion system substrate CaeB inhibits intrinsic apoptosis at the mitochondrial level. Cell. Microbiol. [Epub ahead of print.] 10.1111/cmi.12066 [DOI] [PubMed] [Google Scholar]
- 48.Vázquez CL, Colombo MI. 2010. Coxiella burnetii modulates Beclin 1 and Bcl-2, preventing host cell apoptosis to generate a persistent bacterial infection. Cell Death Differ. 17:421–438 [DOI] [PubMed] [Google Scholar]
- 49.Heidtman M, Chen EJ, Moy M-Y, Isberg RR. 2009. Large-scale identification of Legionella pneumophila Dot/Icm substrates that modulate host cell vesicle trafficking pathways. Cell. Microbiol. 11:230–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan Y, Luo Z-Q. 2011. Legionella pneumophila SidD is a deAMPylase that modifies Rab1. Nature 475:506–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tan Y, Arnold RJ, Luo Z. 2011. Legionella pneumophila regulates the small GTPase Rab1 activity by reversible phosphorylcholination. Proc. Natl. Acad. Sci. U. S. A. 108:21212–21217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Viner R, Chetrit D, Ehrlich M, Segal G. 2012. Identification of two Legionella pneumophila effectors that manipulate host phospholipids biosynthesis. PLoS Pathog. 8:e1002988. 10.1371/journal.ppat.1002988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beare PA, Howe D, Cockrell DC, Omsland A, Hansen B, Heinzen RA. 2009. Characterization of a Coxiella burnetii ftsZ mutant generated by Himar1 transposon mutagenesis. J. Bacteriol. 191:1369–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beare PA, Larson CL, Gilk SD, Heinzen RA. 2012. Two systems for targeted gene deletion in Coxiella burnetii. Appl. Environ. Microbiol. 78:4580–4589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Laguna RK, Creasey EA, Li Z, Valtz N, Isberg RR. 2006. A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. U. S. A. 103:18745–18750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Al-Khodor S, Price CT, Habyarimana F, Kalia A, Abu Kwaik Y. 2008. A Dot/Icm-translocated ankyrin protein of Legionella pneumophila is required for intracellular proliferation within human macrophages and protozoa. Mol. Microbiol. 70:908–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.