

Abstract
Escherichia coli FOF1 ATP synthase, a rotary nanomachine, is composed of eight different subunits in a α3β3γδεab2c10 stoichiometry. Whereas FOF1 has been studied in detail with regard to its structure and function, much less is known about how this multisubunit enzyme complex is assembled. Single-subunit atp deletion mutants are known to be arrested in assembly, thus leading to formation of partially assembled subcomplexes. To determine whether those subcomplexes are preserved in a stable standby mode, a time-delayed in vivo assembly system was developed. To establish this approach, we targeted the time-delayed assembly of membrane-integrated subunit a into preformed FOF1 lacking subunit a (FOF1-a) which is known to form stable subcomplexes in vitro. Two expression systems (araBADp and T7p-laco) were adjusted to provide compatible, mutually independent, and sufficiently stringent induction and repression regimens. In detail, all structural atp genes except atpB (encoding subunit a) were expressed under the control of araBADp and induced by arabinose. Following synthesis of FOF1-a during growth, expression was repressed by glucose/d-fucose, and degradation of atp mRNA controlled by real-time reverse transcription-PCR. A time-delayed expression of atpB under T7p-laco control was subsequently induced in trans by addition of isopropyl-β-d-thiogalactopyranoside. Formation of fully assembled, and functional, FOF1 complexes was verified. This demonstrates that all subunits of FOF1-a remain in a stable preformed state capable to integrate subunit a as the last subunit. The results reveal that the approach presented here can be applied as a general method to study the assembly of heteromultimeric protein complexes in vivo.
INTRODUCTION
The FOF1 ATP synthase catalyzes the formation of ATP from ADP and inorganic phosphate by utilizing the energy stored in the ion motive force of the cytoplasmic membrane. In many bacteria, the reaction is reversible dependent on the physiological conditions and can generate a transmembrane ion gradient at the expense of ATP. In Escherichia coli ATP synthase, the membrane-embedded FO complex (ab2c10) couples proton translocation to ATP synthesis and hydrolysis within the peripherally associated catalytic F1 complex (α3β3γδε) via a rotary mechanism. The transmembrane flow of protons through two half-channels of subunit a drives the rotation of the rotor c10γε due to protonation and deprotonation of carboxyl groups of the subunit c ring. Subunit γ, which rotates inside a molecular bearing composed of the alternately arranged α3β3 hexamer, generates cyclic conformational changes due to its eccentric rotation within the three catalytic nucleotide binding sites, thereby allowing the synthesis of ATP. The α3β3 hexamer of F1, as well as subunit a of FO, is connected to the stator stalk composed of b2δ, thereby forming a stabilizing stator part counteracting the rotating c10γε unit (1, 2).
Whereas ample knowledge is available regarding the insertion of FO subunits into the membrane, only a few facts are at hand as to how a functional FOF1 complex is assembled in E. coli. The FOF1 complex consists of 22 polypeptide chains, which are expected to contain interaction areas with various affinities. Insertion of subunit c into the membrane involves YidC insertase and subunit b is dependent on the SecYEG translocon and the signal recognition particle pathway (Ffh), whereas subunit a requires all three systems for membrane insertion (3–5). In addition, subunits b and c are both essential for a stable incorporation of subunit a into the membrane in vivo (6, 7); otherwise, subunit a is rapidly degraded as a substrate of the membrane-integrated, ATP-dependent metalloprotease FtsH (8, 9). In contrast, subunits b and c insert into the membrane independently of other FO subunits (6) and the c10 ring is formed in a manner independent of the presence of other FOF1 subunits (10). For the Na+-pumping FO complexes of Propionigenium modestum and Acetobacterium woodii, a chaperone-like assistance of AtpI for the formation of the c ring has been shown to be essential (11–13). However, in E. coli, AtpI, a membrane-integrated protein of 14 kDa present in the membrane in substoichiometric amounts compared to FOF1 (14, 15), is not required for the assembly of a functional ATP synthase (16). However, studies of the H+-translocating ATP synthase of alkaliphilic Bacillus pseudofirmus OF4 revealed slightly reduced stability of the rotor as well as reduced ATPase activity (17). In the absence of subunit a, membrane-bound ATPase activities indicate an assembly of F1 to the subunit c ring (6), as has also been proposed for the assembly of the ATP synthase of yeast mitochondria (18, 19). The minimal catalytic unit stably present in the cytoplasm is composed of α3β3γ (20, 21), and complex formation of subunit α with other F1 subunits is a prerequisite for the binding of subunit δ to the N-terminal region of subunit α (22). In the case of both thermophilic Bacillus PS3 and human ρ0 cells, a stable FOF1 subcomplex lacking subunit a can be purified (23–25), clearly indicating that the F1 subunits are associated with the subunit c ring and the peripheral stalk prior to attachment of subunit a.
The analysis of partially assembled subcomplexes is a prerequisite for defining the sequence of events in the assembly process of the ATP synthase. In single-subunit knockout or deletion mutants, in which the synthesis of one of the subunits of the heteromultimeric complex is prevented, an accumulation of partially assembled subcomplexes can be expected until a status is reached at which the missing subunit has to be inserted into the preformed ensemble (6, 26, 27). To determine the stability of those preformed, partially assembled subcomplexes and, furthermore, to determine the preservation of their native molecular conformation in a standby mode ready for use, an in vivo assembly system was developed in which the missing subunit is synthesized in a time-delayed mode, thereby excluding de novo synthesis of FOF1. In addition, due to the formation of those subcomplexes under physiological conditions within the living cell, disintegration of unstable subcomplexes formed as intermediate states is minimized, since manipulations such as cell disruption by sonication or high shearing forces are avoided.
To establish this new approach, we targeted the time-delayed assembly of membrane-integrated subunit a into preformed FOF1 complexes lacking subunit a (FOF1-a) in vivo as an example. For FOF1 of thermophilic Bacillus PS3, it has been shown that a functional in vitro reconstitution of both components into liposomes was successful (23, 24). In detail, we established a system in which all structural atp genes except atpB (atpEFHAGDC) were expressed under the control of the tightly regulated araBAD promoter by induction with arabinose (28, 29). After synthesis of FOF1-a during growth, further expression of atpEFHAGDC was completely repressed by the catabolite repressor glucose and the anti-inducer d-fucose (28, 30). Complete degradation of the atp mRNA was controlled by real-time reverse transcription-PCR (RT-PCR), and the expression of atpB encoding subunit a was subsequently started in trans via induction of a isopropyl-β-d-thiogalactopyranoside (IPTG)-controlled T7-laco promoter (31). The time delay of the IPTG-controlled expression is the centerpiece of the in vivo assembly system introduced, since a complete degradation of the atp mRNA of the araBADp-controlled expression prior to IPTG induction is essential to exclude the formation of a functional enzyme complex by de novo protein synthesis from still-existing mRNA. In addition, the stringent repression of the T7-laco promoter prior to induction was controlled. The formation of a functionally assembled FOF1 complex was verified by N,N′-dicyclohexylcarbodiimide (DCCD)-sensitive ATPase activity and ATP-driven proton translocation as well as ATP synthesis, clearly demonstrating that all subunits of FOF1-a remain in a preformed state with stability comparable to that of the wild-type (WT) enzyme and are ready to integrate subunit a as the last subunit into the enzyme complex.
MATERIALS AND METHODS
Mutagenesis.
In most cases, a two-step PCR method that utilizes two mutagenic primers (see Table S1 in the supplemental material) and two wild-type primers with the corresponding restriction sites was used to generate stop codons in atpB (aY11amber or aW231opal) or an atpB deletion, which comprises the atpB coding region as well as the intergenic region between atpI and atpB (Table 1; see also Table S2 in the supplemental material). In addition, a KpnI site was introduced 49 bp upstream of the stop codon of atpI, i.e., downstream of the weak, constitutive atp promoter P3 (32). The atp genes present in pKH4 derivatives (33, 34) carrying one of the different modifications of atpB were subsequently cloned into the multiple-cloning site of pBAD33 via KpnI within atpI and XbaI being present 320 bp downstream of the 3′ end of the atpC gene, thereby allowing its expression under the control of the tightly regulated promoter araBADp. The atpB gene carrying variations in the start codon was cloned into plasmid pET-22b as described here (Table 1; see also Table S2 in the supplemental material). The presence of each mutation was confirmed by DNA sequencing.
Table 1.
E. coli strain and plasmids used in this study
| Strain or plasmida | Genotype/descriptionb | Reference or source |
|---|---|---|
| E. coli strain | ||
| DK8 | hfrPO1 bglR thi-1 relA1 ilv::Tn10 (Tetr) ΔatpBEFHAGDC | 35 |
| Plasmids | ||
| pBAD33 | Cmr araC pACYC184 ori | 29 |
| pBAD33.atp | pBAD33::atpBEFHAGDC | This study |
| pBAD33.Δa | pBAD33::atpBEFHAGDC, aW231end | This study |
| pBAD33.Δa2 | pBAD33::atpBEFHAGDC, aY11end | This study |
| pBAD33.Δa3 | pBAD33::atpEFHAGDC | This study |
| pBWU13 | Apr pMB1 ori atpI′BEFHAGDC | 66, 67 |
| pET-22b | Apr lacI pMB1 ori | Novagen |
| pET22-atpB | pET-22b::atpB, start codon ATG | This study |
| pET22-atpB-GTG | pET-22b::atpB, start codon GTG | This study |
| pET22-atpB-TTG | pET-22b::atpB, start codon TTG | This study |
| pJGA1 | Apr pMB1 ori atpI′EFHAGDC | This study |
| pT7POL26 | Kanr T7 gene 1 lacI pSC101 ori | 51 |
Construction of plasmids is described in Table S2 in the supplemental material.
Ap, ampicillin; Cm, chloramphenicol; Kan, kanamycin; Tet, tetracycline; T7 gene 1, T7 RNA polymerase.
(The following generalizations were chosen to facilitate reading. The term FOF1 is used for E. coli DK8 cells bearing plasmids pBAD33.atp, pET-22b, and pT7POL26 as well as for the enzyme complex synthesized by these cells, whereas the term FOF1-a is used for DK8 cells bearing plasmids pBAD33.Δa3, pET22-atpB-GTG, and pT7POL26 as well as for the enzyme complex lacking subunit a. The term FOF1-a + a denotes enzyme complexes obtained after time-delayed incubation of cells containing FOF1-a with IPTG during growth. The term WT [wild type] is used for ATP synthase complexes synthesized by DK8/pBWU13.)
Bacterial strains and growth conditions.
The genotype of E. coli strain DK8 (35) is described in Table 1. DK8 transformed with three different plasmids [pBAD33.atp, pET-22b, and pT7POL26 (FOF1)] or its derivatives (Table 1) were grown in 700 ml Luria-Bertani (LB) medium (36) at 37°C with 100 μg/ml ampicillin (Ap), 30 μg/ml chloramphenicol (Cm), and 50 μg/ml kanamycin (Kan) as selection markers. In each case, prior to cell growth, the LB medium was incubated for 10 to 16 h with 10 U/ml β-galactosidase (β-Gal) from Kluyveromyces lactis (Sigma G-3665), thereby degrading the lactose present in various concentrations in the yeast extract used for preparing LB medium (37). Additionally, the medium was supplemented with 0.03% (wt/vol) arabinose for induction of araBADp-controlled atp genes prior to inoculation of the medium to an optical density (OD; measured at 578 nm) of 0.05 with overnight cultures also grown in the presence of 0.03% (wt/vol) arabinose. At OD = 0.3, the araBAD promoter was repressed by the simultaneous addition of 0.5% (wt/vol) glucose and 0.045% (wt/vol) d-fucose (28, 29). After natural degradation of the atp mRNA during an additional incubation time of 30 min, the expression of atpB, being under the control of the IPTG-inducible T7 promoter, and of T7 gene1 coding for T7 RNA polymerase, being under the control of an IPTG-inducible T5N25 promoter, was induced by addition of 0.1 mM IPTG for 1 h.
RNA extraction, cDNA synthesis, and real-time RT-PCR.
Cells (2.5 × 108) of E. coli strain DK8 transformed with three different plasmids [pBAD33.atp, pET-22b, and pT7POL26 (FOF1)] or its FOF1-a derivatives, e.g., pBAD33.Δa3, pET22-atpB-GTG, and pT7POL26, were mixed with 2 vol of RNAprotect bacteria reagent (Qiagen), incubated for 5 min at room temperature, harvested at 5,000 × g, and stored at −20°C. The amount of cell culture needed was calculated according to the determination of Neidhardt et al. (38) that 1 ml of cells contains 109 viable cells at OD = 1.0. The different steps necessary were performed essentially as described by Strahl and Greie (39) with slight modifications. Total RNA was isolated by use of an RNeasy minikit (Qiagen) following the instructions of the supplier. RNA was diluted with RNase-free water to obtain 10 μg/ml and subsequently treated with DNase I (New England BioLabs) as indicated by the supplier to remove residual DNA contaminations prior to reverse transcription. Total RNA (65 ng) was reverse transcribed by use of a RevertAid First Strand cDNA synthesis kit (Fermentas) using the random hexamer primers supplied. For detection of gene expression levels, real-time RT-PCR was performed on a Bio-Rad iCycler with Bio-Rad iQ-SYBR green Supermix, 1 μl of cDNA, and 0.2 nmol of each primer. The primer pairs atpE'F (1 and 2) and atpA (3 and 4) (see Table S1 in the supplemental material) generate PCR products of approximately 200 bp. PCR was performed using an initial denaturation at 96°C for 1 min followed by 40 cycles of denaturation at 96°C for 15 s, annealing at 59°C for 30 s, and extension at 72°C for 20 s. The calculated threshold cycle (CT) values were normalized against CT values of reactions using a primer pair annealing within the rpsL gene as the housekeeping gene (primers 5 and 6; see Table S1 in the supplemental material) and analyzed with the program iCycler from Bio-Rad. As negative controls, reaction mixtures containing H2O (no template) or DNase I-treated RNA (without reverse transcription) were included. The data represent average values obtained in three independent measurements.
Preparative procedures.
Inverted membrane vesicles were prepared according to Krebstakies et al. (40) using 50 mM Tris-HCl (pH 7.5)–10 mM MgCl2–10% (vol/vol) glycerol as a buffer system.
Analytical procedures.
Protein concentrations were determined with the bicinchoninic acid (BCA) assay as recommended by the supplier (Pierce). Proteins were dissolved in SDS-PAGE sample loading buffer (41) and separated by SDS-PAGE using 10% separating gels (42) with a PageRuler prestained protein ladder (Fermentas) as the standard. Immunoblotting was performed according to Birkenhäger et al. (43) using 10 mM NaHCO3–3 mM Na2CO3 (pH 9.9)–20% (vol/vol) methanol as the transfer buffer (44). Blot membranes were incubated with corresponding primary antibodies raised in mice or rabbits (see figure legends), with IRDye800DX-labeled goat-anti-mouse IgG (H+L) or IRDye700DX-labeled goat anti-rabbit IgG (H+L) (Rockland or LI-COR) as the secondary antibody, and were finally detected with a two-channel Odyssey infrared detection system (Li-COR). Both secondary antibodies are affinity purified for low crossreactivities with serum proteins of other species and, therefore, allowed the simultaneous detection of fluorescence (shown in red for IRDye700DX and in green for IRDye800DX) after immunodecoration of two proteins on one blot membrane.
DCCD-sensitive ATPase activities of membrane vesicles were determined as described previously (45). ATP-driven proton translocation via 9-amino-6-chloro-2-methoxyacridine (ACMA; Sigma A-5806) fluorescence quenching was performed according to Deckers-Hebestreit et al. (46) using only half of the volume specified and 400 μg membrane protein. In the case of NADH-driven proton translocation, 200 μg of inverted membrane vesicles was applied. As an uncoupler, 20 μl of a saturated ammonium sulfate solution was used. ATP synthesis activity was measured at 37°C with 30 μg of membrane vesicle protein in 500 μl of buffer (25 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 10% [vol/vol] glycerol, 5 mM ADP, 5 mM K2HPO4) (47). The reaction was initiated by addition of 2 mM NADH and terminated after 0, 1, 2, and 3 min with 50 μl of 0.5 M trichloroacetic acid. Aliquots (1 μl) of the reaction mixture were diluted 100-fold in 0.1 M Tris-acetate (pH 7.8)–2 mM EDTA. In 10-μl samples of this dilution, ATP was determined luminometrically using a Promega Enliten luciferin/firefly luciferase system. Due to contamination of ADP with ATP, ADP was purified via anion-exchange chromatography on a 5 ml Hi-Trap-DEAE FF column (GE Healthcare). ADP (50 mg) was dissolved in 20 mM Tris-acetate (pH 8.5), loaded onto the column, and eluted with a linear NaCl gradient (0 to 400 mM). The eluted ADP was directly used for buffer preparation. The data represent average values obtained in three independent measurements.
RESULTS
FOF1-a is known to form a stable subcomplex.
Consistent with previous work (6, 23, 48), we observed stable synthesis of all FOF1 subunits except subunit a in cells of single-subunit knockout (aW231end or aY11end; data not shown) or deletion (ΔatpB; Fig. 1A) mutants expressing the atp genes from the corresponding plasmids. Evidence for a stable subcomplex formation resulted from the observation that FOF1-a can be isolated via a His6 tag fused to the N terminus of subunit β from E. coli membranes (68). In our setup for the in vivo approach, the single-subunit deletion mutant ΔatpB was applied, since in the knockout aY11 end mutant the amount of FOF1 protein present in the cell is comparably low (data not shown), probably due to a polar effect on the expression of the atp genes located downstream of atpB. On the other hand, the aW231end mutant, well described in the literature (6, 49, 50), showed a truncated subunit a in immunoblots, when expression of the mutated atp operon was under the control of the araBAD promoter. In contrast, during constitutive expression under the control of the weak atp promoter P3, the truncated subunit a product could not be detected (data not shown), a discrepancy that has been previously discussed in detail (6).
Fig 1.

Characterization of the FOF1-a subcomplex. (A) Cells of E. coli DK8 transformed with pBWU13 (WT) or pJGA1 (ΔatpB) were grown in LB medium with ampicillin and harvested at OD = 0.8 to 1.0. After resuspension in sample loading buffer, cells were incubated for 5 min at 99°C. The amount of cell extract applied per lane (20 μg) was calculated according to the determination of Neidhardt et al. (38) that 160 μg of protein was present per ml cell culture at OD = 1.0. Immunolabeling was performed using mouse (anti-β, anti-δ, anti-ε) or rabbit (anti-α, anti-γ, anti-b) polyclonal antisera or mouse monoclonal antibodies (anti-a, anti-c) raised against the individual subunits of the ATP synthase. (B and C) ATP hydrolysis (B) and ATP-driven proton-pumping activity (C) of membrane vesicles of DK8/pBAD33.atp and DK8/pBAD33.Δa3, respectively, determined using the WT as a control. Cells were grown in LB medium with chloramphenicol in the presence of different arabinose concentrations for induction of the araBADp expression system or in the presence of glucose for repression as indicated. Cells were harvested at OD = 0.8 to 1.0, and inverted membrane vesicles were prepared. ATP-driven proton translocation was measured via ACMA fluorescence quenching. The relative magnitudes of quenching induced by addition of ATP are shown. ara, arabinose; glu, glucose.
In membrane vesicles of the atpB deletion mutant (DK8/pBAD.Δa3), ATPase activities comparable to those seen with the corresponding wild-type enzyme (DK8/pBAD33.atp) were observed (Fig. 1B), an activity that can be detected only when catalytically active F1 or at least an α3β3γ subcomplex has been bound to membrane-integrated FO subunits, indicating the assembly of a stable subcomplex. Similar results have been obtained by Hermolin and Fillingame (reference 6 and references therein), whereas comparable studies on Bacillus PS3 FOF1-ATP synthase revealed that no ATPase activity could be detected in the absence of subunit a (23). Nevertheless, as expected, no ATP-driven proton translocation measured with the pH-sensitive dye ACMA could be observed due to the absence of the two half-channels within subunit a essential to enable proton translocation across the cytoplasmic membrane (Fig. 1C). Therefore, ATP-driven proton translocation has been used as the measure to discriminate between FOF1-a and intact FOF1.
In vivo system for time-delayed assembly of membrane-integrated subunit a into preformed FOF1-a.
The system used is based on the combination of two tightly regulated expression systems that work almost independently of each other and can be induced as well as completely repressed. For genes atpEFHAGDC, expression was controlled by the araBAD promoter (araBADp) chosen due to its specificity for induction with arabinose and, more importantly, its tight repression by a combination of the catabolite repressor glucose and the anti-inducer d-fucose, a nonmetabolizable methylpentose (6-deoxygalactose) (28–30). For atpB, expression was controlled by the lac operator-controlled T7 promoter (T7p-laco). The T7 promoter is not recognized by E. coli RNA polymerases but needs a separate source coding for T7 RNA polymerase (31), which is provided by plasmid-encoded expression of T7 gene1 under the control of the lac operator-controlled T5N25 promoter (51). To guarantee complete repression of the IPTG-inducible promoters, lactose present in LB medium was removed. In addition, the start codon of the atpB gene present under the control of T7p-laco was exchanged from ATG to GTG in order to prevent the synthesis of subunit a under noninduced conditions (see below).
Figure 2 shows the configuration of the test system and its different states during cell growth. The atp deletion strain DK8 was transformed with three different plasmids bearing different resistance cassettes and origins to generate compatibility within one cell: (i) a pBAD33 derivative (p15A ori; Cmr) carrying the structural genes of the atp operon with a deletion of the atpB gene (atpEFHAGDC) under the control of araBADp, (ii) a pET-22b derivative (pMB1 ori; Apr) encoding the atpB gene under the control of the IPTG-inducible T7-laco promoter, and (iii) a pSC101 derivative (Kanr) containing gene1 of phage T7, which codes for the T7 promoter-specific RNA polymerase, present under the control of the IPTG-inducible promoter T5N25-laco. The cartoon illustrates the induction or repression states of the different promoters present. As shown in the left panel, after addition of arabinose to cells inoculated at an OD of 0.05, araBADp was induced and allowed expression of the atpEFHAGDC genes such that all FOF1 subunits except subunit a (FOF1-a) were synthesized. Due to the absence of IPTG and further precautions, the lac operator-controlled promoters were completely repressed. As shown in the middle panel, at OD = 0.3, araBADp was repressed by the simultaneous addition of glucose and d-fucose, thereby stopping the transcription of atpEFHAGDC; furthermore, after incubation for 30 min, the atp mRNA present within the cell was completely degraded as indicated by real-time RT-PCR results (see below). Due to this time delay, the de novo biosynthesis of FOF1 subunits and, therefore, the formation of new FOF1-a complexes as well were completely prevented. As shown in the right panel, the delayed induction of the lac operator-controlled T7 and T5N25 promoters with IPTG then enabled the individual synthesis of subunit a, which can be integrated only into preformed partially assembled FOF1-a complexes present within the cytoplasmic membrane (referred to as FOF1-a + a). To control the synthesis of FOF1 subunits, subunit b as a representative for FOF1-a and subunit a itself were detected by immunoblotting. The formation of a functionally assembled FOF1 complex is controlled by assaying DCCD-sensitive ATPase activity and ATP-driven proton translocation as well as ATP synthesis. To facilitate reading, DK8/pBWU13 (atpBEFHAGDC, the expression system usually applied for studies of E. coli ATP synthase [10, 40]) is always referred to here as WT, whereas DK8 transformed with pBAD33.atp, containing the structural wild-type atp genes, as well as plasmids pET-22b and pT7POL26, is termed FOF1. FOF1 contains plasmids pET-22b and pT7POL26 in addition, in order to facilitate resemblance of the corresponding subsequent expression of FOF1-a by use of pBAD33.Δa3, pET22-atpB(-GTG), and pT7POL26.
Fig 2.

In vivo system for time-delayed assembly of membrane-integrated subunit a into preformed FOF1-a. A detailed description is given in the text.
Tightly regulated expression of atpB.
DK8 transformed only with pBAD33.Δa3 was not capable of ATP-driven proton translocation (Fig. 1C). However, in FOF1-a, in which plasmids pET22-atpB and pT7POL26 are also present, a signal comparable to that seen with the WT was observed (Fig. 3A), indicating that the lac-controlled promoters T7 and T5N25 were not completely repressed. To gain complete repression, in a first step, the LB medium used for growth was preincubated with β-Gal to remove residual lactose molecules (Fig. 3A) known to be present in various amounts in yeast extracts used for preparation of LB medium (37). With increasing amounts of β-Gal (up to 10 U/ml LB medium), a significant reduction of levels of subunit a present in membrane vesicles as well as a reduction of ATP-driven proton translocation could already be observed. However, the fluorescence quenching of FOF1-a in the absence of IPTG (see Fig. 3A; see also Fig. S1 in the supplemental material) revealed that complete repression could not be obtained. In a second step, the ATG start codon of atpB was exchanged to the weaker start codon GTG or TTG to reduce in general the synthesis of subunit a. In both cases, a further reduction in synthesis of subunit a, as well as of the ATP-driven proton translocation, could be observed (Fig. 3B; see also Fig. 6). Therefore, the following experiments were performed with preincubation of LB medium with β-Gal and the start codon of atpB being changed to GTG. For induction of lac-controlled promoters, an IPTG concentration of 0.1 mM was determined, allowing a synthesis rate of subunit a comparable to that seen with the WT or FOF1 as revealed by immunoblotting with anti-a antibodies as well as by ACMA fluorescence quenching (Fig. 3B; see also Fig. S1 in the supplemental material).
Fig 3.
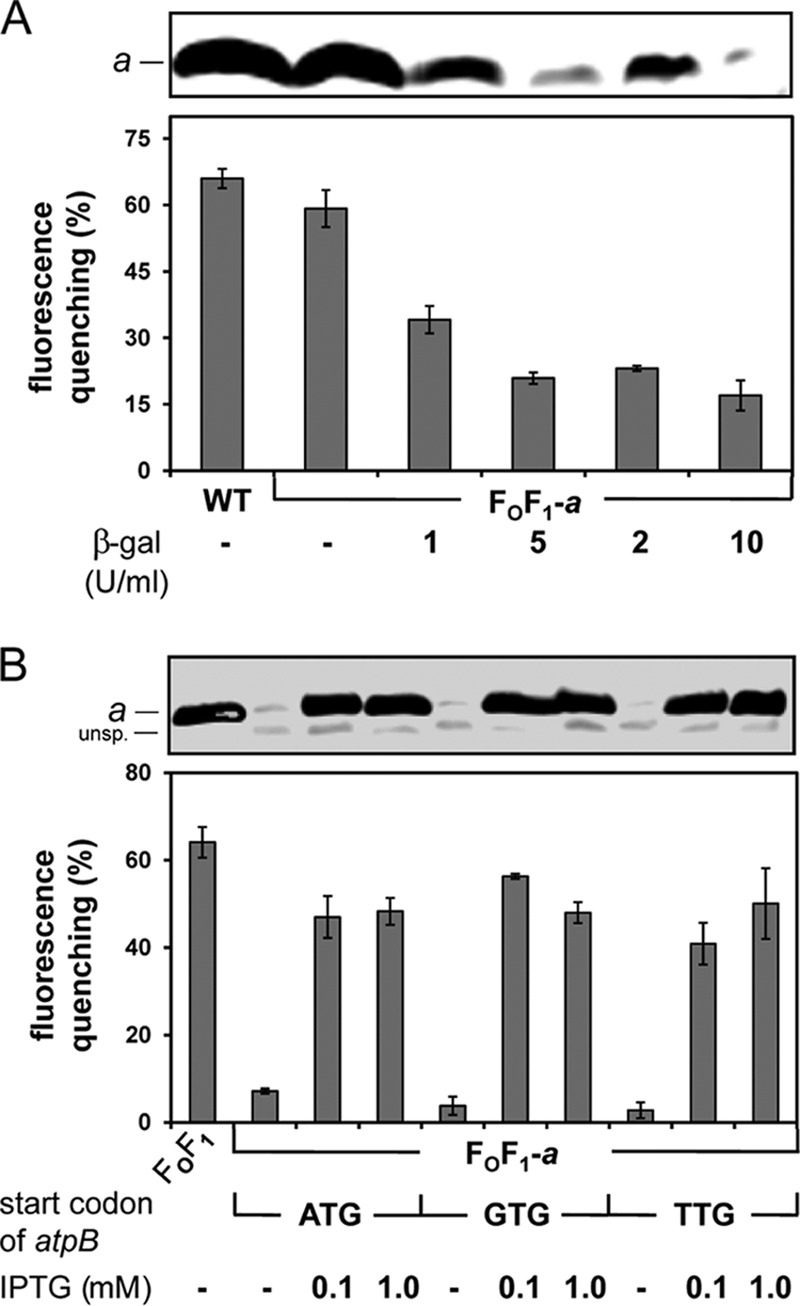
T7p-laco-driven expression of atpB. (A) β-Gal incubation for removal of lactose present in LB medium. DK8 transformed with pBWU13 (WT, control) or with pBAD33.Δa3, pET22-atpB, and pT7POL26 (FOF1-a) was grown in LB medium supplemented with antibiotics and, in the case of FOF1-a, with 0.03% arabinose for induction of araBADp-controlled atpEFHAGDC expression. Prior to cell growth, the LB medium was incubated overnight with β-Gal concentrations as indicated. (B) Variation of the IPTG concentration and the start codon of T7 promoter-controlled atpB. DK8 transformed with pBAD33.atp, pET-22b, pT7POL26 (FOF1), pBAD33.Δa3, pET22-atpB, pT7POL26 (FOF1-a; ATG), pBAD33.Δa3, pET22-atpB-GTG, pT7POL26 (FOF1-a; GTG), or pBAD33.Δa3, pET22-atpB-TTG, pT7POL26 (FOF1-a; TTG) was grown in β-Gal-preincubated LB medium supplemented with antibiotics, 0.03% arabinose, and IPTG as indicated. Cells were harvested at OD = 0.8 to 1.0 and membrane vesicles prepared. Upper panels, immunoblot analysis of membrane vesicles (20 μg/lane). Immunolabeling was performed using monoclonal mouse anti-a antibodies. Lower panels, ATP-driven proton translocation of membrane vesicles measured via ACMA fluorescence quenching. The relative magnitudes of quenching induced by the addition of ATP are shown. unsp., unspecific immunolabeling.
Fig 6.
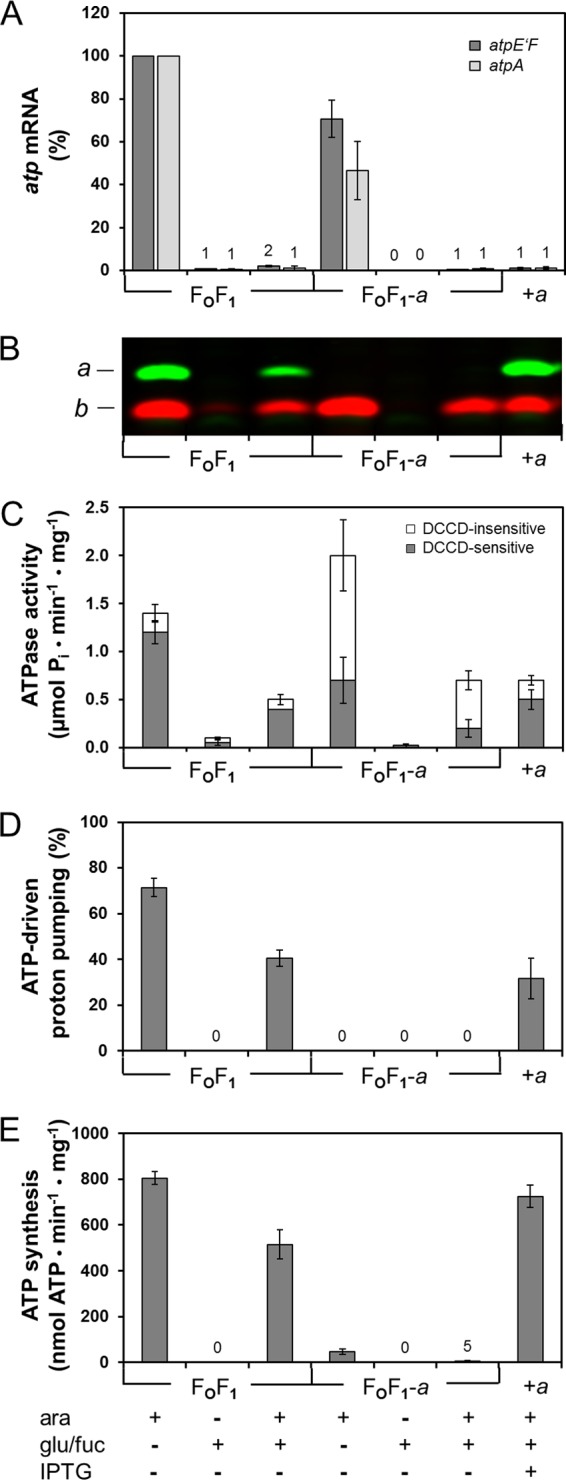
In vivo assembly of subunit a into preformed FOF1-a complexes. DK8 carrying plasmid pBAD33.atp, pET-22b, pT7POL26 (FOF1), pBAD33.Δa3, pET22-atpB-GTG, or pT7POL26 (FOF1-a) was grown in LB medium preincubated with β-Gal in the presence of antibiotics. Seven independent cell batches were prepared in parallel containing the additives as indicated. +ara or +glu/fuc, the addition of inducer or repressor, respectively, occurred directly after inoculation of the medium. +ara +glu/fuc, cells were grown in the presence of arabinose up to OD = 0.3 before addition of glucose/d-fucose. In the case of FOF1-a + a, 0.1 mM IPTG was added subsequently for 1 h as described in Materials and Methods. All cells were harvested at the same time, membrane vesicles were prepared, and a small volume of cell culture was used for isolation of total RNA. (A) Level of atp mRNA. The amount of atp mRNA was determined via real-time RT-PCR using primer pairs atpE'F (1 and 2) (dark gray) and atpA (3 and 4) (light gray). The amount of atp mRNA present in the samples grown with arabinose was set to 100% for FOF1. (B) Immunoblot analysis of membrane vesicles (20 μg protein/lane). Immunolabeling was performed using monoclonal mouse anti-a (green) or polyclonal rabbit anti-b (red) antibodies. (C) ATPase activity of membrane vesicles. The gray and white portions of the bars represent the DCCD-sensitive and DCCD-insensitive fractions of ATP hydrolysis, respectively. (D) ATP-driven proton translocation of membrane vesicles measured by ACMA fluorescence quenching. The relative magnitudes of quenching induced by the addition of ATP are shown. (E) ATP synthesis of membrane vesicles driven by NADH oxidation. The amount of ATP synthesized after addition of NADH was determined luminometrically with a luciferin/firefly luciferase system and corrected for nonspecific background activity. Experimental details are described in Materials and Methods. ara, arabinose; fuc, d-fucose; glu, glucose.
Tightly regulated expression of atpEFHAGDC.
During glucose repression in LB medium, neither target protein nor ACMA fluorescence quenching could be observed, whereas with increasing concentrations of arabinose (up to 0.03% [wt/vol]), the ATP-driven proton translocation as well as the presence of subunits a and b increased to values comparable to those seen with the WT (see Fig. S2 in the supplemental material).
The centerpiece of the in vivo assembly system is the time-delayed synthesis of subunit a to allow its integration into preformed FOF1-a subcomplexes. To prevent simultaneous de novo biosynthesis of FOF1, the complete degradation of the atpEFHAGDC mRNA prior to induction of atpB expression by IPTG is a prerequisite. The level of atp mRNA was measured by real-time RT-PCR using sequence-specific primer pairs atpE'F (1 and 2) and atpA (3 and 4) annealing in different genes of the atp operon. To verify the specific annealing of the primers, total RNA of the plasmid-free atp deletion strain DK8 was used as a control (data not shown).
For a complete repression of the araBADp expression system, FOF1-a was grown in the presence or absence of arabinose for induction (+ara) up to an OD of 0.3 before glucose or d-fucose or both (+glu or +fuc or +glu/fuc) were added for another 30 min. Cells were harvested in the late exponential phase, and total RNA was purified for analysis of atp mRNA by real-time RT-PCR. For a complete repression of the araBAD promoter, the addition of glucose as a catabolite repressor as well as of d-fucose as an anti-inducer was essential (Fig. 4). In the absence of arabinose, a background of atp mRNA within the range of 0% to 2% compared to the level seen with the system fully induced by arabinose could be observed in the presence of glucose or d-fucose or of both repressors (Fig. 4A). Nevertheless, under these conditions, ATP-driven proton translocation could not be observed (see Fig. S2 in the supplemental material), indicating that the amount of FOF1 subunits synthesized was extremely small. However, in the presence of arabinose, a comparable repression of the araBAD promoter could be obtained only by simultaneous addition of glucose and d-fucose (Fig. 4). Glucose was added at the concentration usually applied for growth of cells in minimal medium with glucose as the sole carbon and energy source (0.5% [wt/vol]) to maintain the repressed status also during the residual growth phase in the time-delayed in vivo assembly experiments (degradation of atp mRNA for 30 min and subsequent incubation with IPTG for 1 h), since glucose is metabolized in glycolysis. d-Fucose, a nonmetabolizable methylpentose (6-deoxygalactose), functions as a competitor to arabinose, resulting in a repression of araBADp when added in a slight molar excess over arabinose (52). In addition, IPTG is known to be an inhibitor of the araBADp expression system. Crosstalk between araBADp and lac operator-controlled promoters prevents them from being used simultaneously in the same cell (53). Under the time-delayed in vivo assembly conditions, this inhibitory effect of IPTG was advantageous, since the repression of the araBAD promoter was further strengthened.
Fig 4.
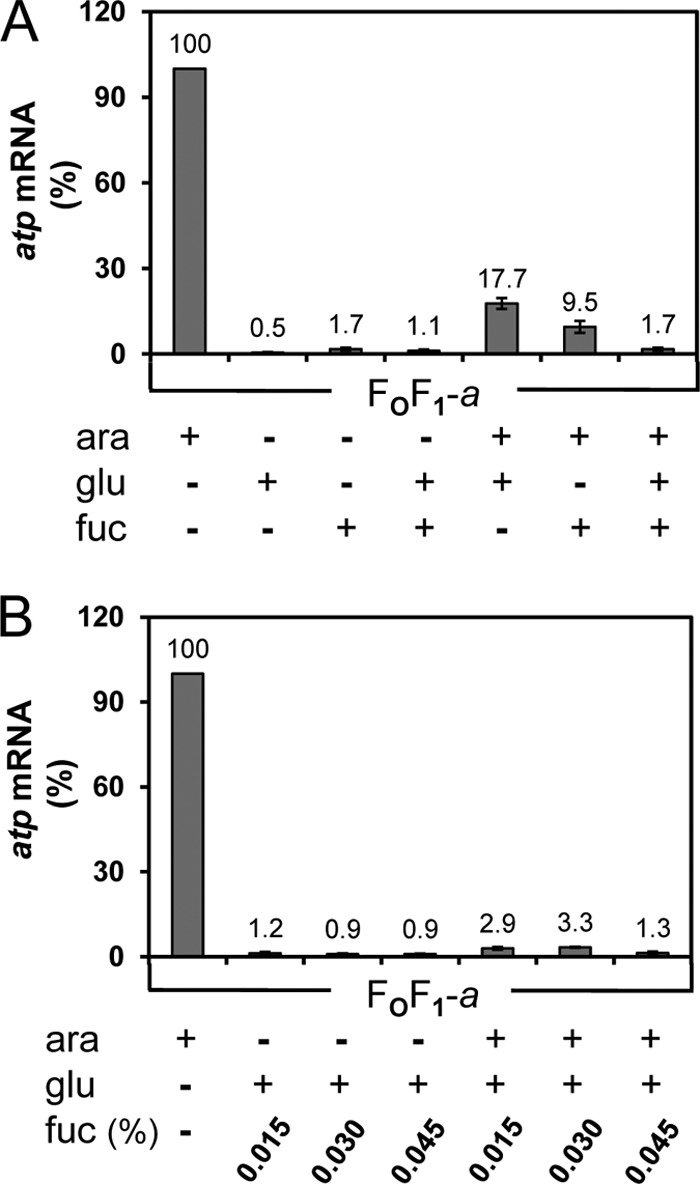
Repression of araBADp by glucose/d-fucose determined by real-time RT-PCR. Cells were grown in β-Gal-preincubated LB medium supplemented with antibiotics in the presence (+) or absence (−) of arabinose as an inducer up to OD = 0.3 before glucose/d-fucose was added as indicated for another 30 min. Total RNA was isolated from DK8 bearing pBAD33.Δa3, pET22-atpB-GTG, pT7POL26 (FOF1-a), and primer pair atpE'F (1 and 2) was applied for real-time RT-PCR. (A) Repression of the araBADp by glucose and d-fucose. (B) Determination of the d-fucose concentration necessary for repression. ara, arabinose; fuc, d-fucose; glu, glucose.
To determine the time delay necessary for a complete degradation of the atp mRNA, cells were grown in the presence of arabinose at up to OD = 0.3 before glucose/d-fucose was added as described above. Total RNA was purified from samples taken after several time intervals after the addition of glucose/d-fucose. In FOF1, the atp mRNA was rapidly degraded, revealing a half-life for its stability of approximately 3 min as shown by real-time RT-PCR with primer pairs atpE'F and atpA (Fig. 5A), which correlates well with the half-live of atpE mRNA determined to be 5 to 6 min with independent methods (54).
Fig 5.
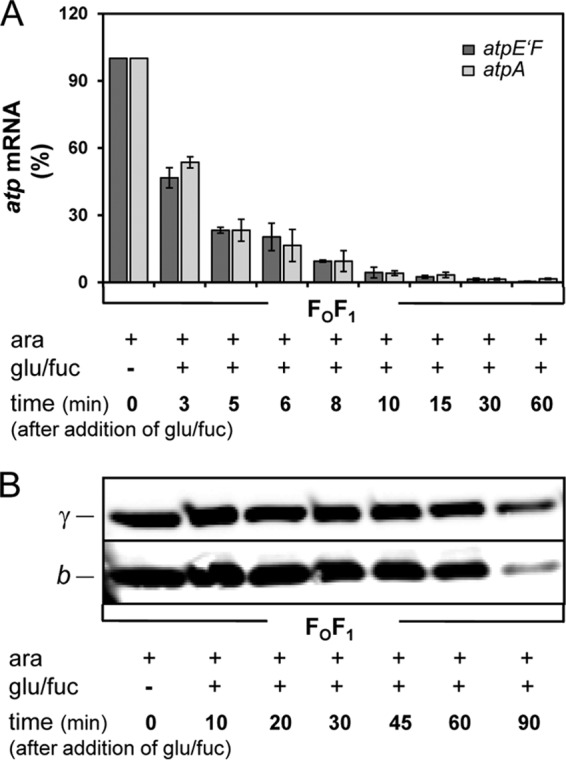
Degradation of atp mRNA (A) and stability of FOF1 (B) after repression of araBADp controlling expression of atpEFHAGDC. DK8 carrying plasmids pBAD33.atp, pET-22b, and pT7POL26 (FOF1) was grown as described in the Fig. 4 legend. At each time point indicated, cells were harvested for isolation of RNA (A) or immunoblot analysis (B). (A) Real-time RT-PCR was performed using primer pairs atpE'F (1 and 2) (dark gray) and atpA (3 and 4) (light gray). (B) Cell lysates were prepared and applied as described in the Fig. 1 legend. For immunolabeling, monoclonal mouse anti-b and polyclonal rabbit anti-γ antibodies were applied. ara, arabinose; fuc, d-fucose; glu, glucose.
A complete degradation of atp mRNA with a background level below 2% was always obtained within much less than 30 min (compare Fig. 5A to Fig. S3 in the supplemental material). In addition, the degrees of degradation of atp mRNA were comparable in FOF1 and FOF1-a as shown for primer pair atpE'F (Fig. S3), although, in general, the amount of atp mRNA present in FOF1-a under fully arabinose-induced conditions was always reduced compared to that present in FOF1 (compare Fig. S3 in the supplemental material to Fig. 6A).
Stability of FOF1 under araBADp-repressed conditions.
Due to the time delay necessary for removal of atp mRNA as well as the incubation time with IPTG for expression of the missing subunit, the stability of the ATP synthase complexes present in the membrane had to be guaranteed at least for the next 90 min in order to allow a subsequent analysis of the functional status of the enzyme complexes. To study the stability of FOF1, cells were grown in the presence of arabinose to an OD = 0.3 before addition of glucose/d-fucose. Subsequently, samples were taken after several time intervals and the cell lysates analyzed by immunoblotting. For immunolabeling, antibodies against subunit b and γ were applied. As shown in Fig. 5B, both subunits were highly stable for at least 60 min after the addition of glucose/d-fucose. After 90 min, a reduction of both subunits could be observed, whereupon subunit b appeared to be more degraded as subunit γ. While the b dimer was exposed at the periphery of the enzyme complex, subunit γ was embedded within its center, shielded by the α3β3 hexamer, subunit ε, and the c10 ring and, therefore, possibly more protected against proteolytic degradation. A comparison with Fig. 6B revealed that the degradation rate of subunit a, which was also located at the periphery of FOF1, was comparable to that of subunit b. Nevertheless, the amount of FOF1 still present in the membrane was sufficient to allow determination of its functional capabilities in ATP hydrolysis as well as ATP synthesis (compare Fig. 6C to E). A quantification using DCCD-sensitive ATPase activity revealed that approximately 30% of the enzyme complexes were still functional after 90 min of repression (compare Fig. 6C).
Time-delayed in vivo assembly of subunit a into preformed FOF1-a.
After determination of all parameters necessary, the following test procedure was applied. LB medium preincubated overnight with 10 U/ml of β-Gal, supplemented with antibiotics as well as 0.03% arabinose, was inoculated to OD = 0.05 with FOF1-a. At OD = 0.3, 0.5% glucose and 0.045% d-fucose were added and, after complete degradation of atpEFHAGDC mRNA within 30 min, 0.1 mM IPTG was added for 1 h to enable time-delayed synthesis of subunit a (compare Fig. 2). Subsequently, cells were harvested, usually at an OD of about 1, total RNA as well as inverted membrane vesicles was prepared, and the activity of the ATP synthase was analyzed. As a control, FOF1 samples were handled correspondingly except for the addition of IPTG. All in all, seven different cell batches were grown, each with the additives noted in Fig. 6 added at the time points indicated above.
Real-time RT-PCR revealed that under arabinose-induced conditions, atp mRNA was present within the cells, whereas in FOF1-a, the amount was slightly reduced compared to that seen with FOF1 (Fig. 6A). In samples containing glucose/fucose, the atp mRNA had been completely degraded, revealing that de novo synthesis of FOF1 subunits was no longer possible (Fig. 6A).
The immunoblot analysis showed a strong synthesis of subunit b under arabinose-induced conditions (Fig. 6B). After addition of glucose/d-fucose in the absence of arabinose, no subunit b could be detected in FOF1-a. Only in the case of FOF1, possibly due to the slightly increased stability of the atp mRNA, a small amount of subunit b could be detected, further promoted by the high avidity of the polyclonal antiserum with respect to its antigen. However, no subunit a could be observed in FOF1 under these conditions. The samples first induced with arabinose and then repressed by glucose/d-fucose showed significant bands for subunits a and b in the case of FOF1 and a comparable band for subunit b in the case of FOF1-a, thereby revealing that the enzyme (sub)complex was still present in membrane vesicles after 90 min. After addition of IPTG to FOF1-a, subunit a was slightly overproduced beyond its stoichiometric ratio. Nevertheless, all components necessary to allow the generation of a functional FOF1 complex, namely, FOF1-a + a, were present within the cytoplasmic membrane.
Bacterial ATP synthases can function either as ATP synthase or as ATP hydrolase in a manner dependent on the physiological status of the cells. Therefore, the functionality of FOF1-a + a was analyzed with respect to its DCCD-sensitive ATPase activity, ATP-driven proton translocation, and ATP synthesis (Fig. 6C, D, and E, respectively). As expected, both FOF1 and FOF1-a showed high levels of ATPase activities under arabinose-induced conditions. Under repressing conditions, only extremely low levels of ATPase activities could be observed in the case of FOF1, which correlates well with the amount of subunit b observed by immunoblotting. The ATPase activities observed in the samples, which were first induced with arabinose and then repressed with glucose/d-fucose, were also in good agreement with the immunoblot data and could, therefore, be used as a quantitative measure for protein stability. The results indicate that for FOF1 as well as for FOF1-a, about one-third of the enzyme complexes were still present in the membranes after repression for 90 min. ATPase activities of membrane vesicles were measured with or without pretreatment by DCCD. DCCD is known to label specifically the carboxyl group of cD61, thereby blocking the rotation of the subunit c ring and the proton translocation. Due to the inhibition of the rotation by DCCD, ATP hydrolysis cannot occur in coupled FOF1 complexes and, therefore, ATP hydrolysis is inhibited. Whereas the DCCD sensitivity of the ATPase activity was 80% to 85% in FOF1, a value usually observed in membrane vesicles (46, 55), FOF1-a showed comparably low sensitivity toward DCCD (Fig. 6C). In the sample synthesizing FOF1-a + a, the ATPase activity was comparable to FOF1-a activity, as expected; however, the DCCD sensitivity was increased from 29% to 71%. This indicates a functional incorporation of subunit a into FOF1.
In the next step, ATP-driven proton translocation was tested (Fig. 6D). Proton translocation into membrane vesicles was monitored by acidification of the lumen leading to fluorescence quenching of the pH-sensitive dye ACMA. Membranes containing FOF1 under arabinose-induced conditions generated a fluorescence quenching of 71%, whereas under repressed conditions no proton pumping could be observed. In the sample first induced by arabinose and then repressed by glucose/d-fucose, FOF1 produced a fluorescence quenching of 41%, with only one-third of FOF1 present in the membrane (compare Fig. 6C). In general, the quenching of ACMA fluorescence by proton gradients is known to be nonquantitative, so the ratios need not correspond exactly. In the case of ATP-driven proton pumping, results depend on the amount of FOF1 present in the membrane as well as on the proton permeability of the membrane itself (in this context, compare references 40 and 56). In the case of membranes containing FOF1-a, no ACMA fluorescence quenching could be detected in the absence of subunit a. To verify the functionality of inverted membrane vesicles, proton translocation through the respiratory chain energized by oxidation of NADH was monitored. NADH-driven proton translocation with and without pretreatment by DCCD was measured and revealed fluorescence quenching of 70% to 80% in both cases (data not shown). The protons pumped into the vesicles were retained in the lumen, thereby showing that FOF1-a does not increase proton leakage of the vesicle membrane. In contrast, membranes with FOF1-a + a showed ATP-driven proton translocation with a fluorescence quenching of 32%, again supporting the idea of functional assembly of subunit a into preformed FOF1-a complexes. Furthermore, FOF1-a + a restored proton translocation activity to the value observed for FOF1. For FOF1-a + a as well as FOF1, the proton-translocating activity was completely abolished after preincubation of the membranes with DCCD (data not shown).
For the synthesis of ATP, the proton motive force was generated by the respiratory chain upon oxidation of NADH (Fig. 6E). Under normal growth conditions, the ATP synthase is mainly used as an ATP-generating machine instead of hydrolyzing ATP to generate a proton motive force. Therefore, it is essential to verify that the assembly of subunit a into preformed FOF1-a complexes yielded an enzyme also functional in this aspect. Membrane vesicles containing FOF1-a + a exhibited ATP synthesis with a 90% yield compared to membranes with FOF1. This clearly demonstrates the presence of FOF1 complexes with tightly coupled proton translocation in FO and catalysis in F1. At first glance, the ATP synthesis rate obtained seems to be unusually high compared to the amount of functional FOF1 present in the membrane. However, the rate of ATP synthesis by the ATP synthase is generally limited by the rate of energy production via the respiratory chain in membrane vesicles, as has been shown by fractional inactivation of FOF1 with DCCD (57). Under these conditions, the rate of ATP synthesis per mol of FOF1 remaining active in the membrane is increased, using the ATP hydrolysis rates as the defining parameter for calculation of active ATP synthase complexes (57). In summary, all results obtained are consistent and demonstrate a functional time-delayed in vivo assembly of subunit a into preformed FOF1-a complexes.
DISCUSSION
Time-delayed in vivo assembly.
In the present work, a general approach to study the assembly of membrane-integrated as well as soluble heteromultimeric protein complexes in vivo has been established by the use of two independent expression systems as described in Results. Due to the time-delayed synthesis of at least one of the subunits, partially assembled complexes can be generated and studied with respect to stability and functionality and the missing subunit(s) subsequently synthesized and possibly integrated to obtain a functional enzyme complex. As an ideal tool to determine the essential parameters, the assembly of the membrane-integrated subunit a into preformed FOF1-a subcomplexes has been chosen due to the known stability of this subcomplex (23, 24, 68). Furthermore, the in vivo assembly system has already successfully been transferred to a FOF1-δ construct. A functional FOF1 complex could be assembled after time-delayed synthesis of missing subunit δ, thereby indicating that under these conditions also, the conformation of the preformed subcomplexes ab2 and c10α3β3γε, present in the membrane, remained in a ready-to-use state (68).
In vivo versus cell-free protein synthesis.
A major advantage of an in vivo system is the possibility to enable a sequential synthesis of proteins without any further manipulation. Recently, approaches involving cell-free protein synthesis have emerged as a promising tool for the study of membrane protein complexes, including the ATP synthase (24, 58, 59). However, in order to obtain sequential protein synthesis, a purification step of the partially assembled protein complex or at least of membrane vesicles has to be included, which is then followed by cell-free protein synthesis as described in detail by Kuruma et al. (24). In that approach, E. coli membrane vesicles or liposomes containing Bacillus PS3 FOF1-a subcomplexes were isolated or generated and, in a subsequent second step, applied to the cell-free PURE system in order to generate functional FOF1 by in vitro cell-free synthesis of subunit a (24), a procedure yielding results similar to those described in the text. However, under such conditions, the stability of the partially assembled subcomplex(es) is a prerequisite to allow for purification and subsequent reconstitution or for manipulation of cells by sonication, high pressure, and centrifugation, which is at hand in the case of FOF1-a, but not a general feature of partially assembled subcomplexes.
ATPase activity of partially assembled FOF1-a.
The presence of ATP hydrolysis in FOF1-a, at rates similar to those seen with wild-type FOF1, strengthens the view that comparable conformations at least of the catalytically active, membrane-bound core complex composed of c10α3β3γε are maintained in the two complexes. Whereas ATPase activities of E. coli FOF1-a have been determined in inverted membrane vesicles in a manner independent of their plasmid- or chromosome-based expression (compare reference 6), no ATPase activities could be observed for Bacillus PS3 FOF1-a (23). However, in the latter case, the FOF1-a complex had been solubilized from E. coli membranes, purified by affinity chromatography via His-tagged subunit β, and reconstituted into liposomes prior to ATPase activity measurements. Although a stable complex can be isolated under these conditions, the removal of the complex from its natural membrane environment might change the conformation or positioning of one or some of the partially assembled FOF1 subunits, thereby possibly inhibiting ATPase activity. Nevertheless, this process can be reversed by addition of subunit a (23). However, a putative function of the b dimer as an anchor rail to prevent the rotation of the c10 ring in the absence of subunit a, thereby inhibiting the ATPase activity of the partially assembled FOF1 complexes as proposed by Ono et al. (23), can be excluded for the ATP synthase of E. coli.
In FOF1, ATP hydrolysis in F1 is tightly coupled to proton translocation in FO and is, therefore, sensitive to DCCD inhibition. In the absence of subunit a, the two half-channels essential for proton translocation are missing and proton translocation across the cytoplasmic membrane is no longer possible. Therefore, with the common understanding of the rotary mechanism of the enzyme, the robust structure of FOF1-a as well as the observed ATPase activity implies that either the rotation of the central stalk (subunits γ and ε) is uncoupled from the subunit c oligomer or that the c10 ring remains coupled and corotates with subunits γε during ATP hydrolysis, thereby supporting a free rotation without proton translocation of the rotor part of the enzyme (c10γε) within the membrane. The latter interpretation could be favored, since free rotation without proton translocation has already been demonstrated for FOF1 purified from a mutant in which cD61 was exchanged to asparagine or glycine (60). Furthermore, the observed partial inhibition of the ATPase activity of FOF1-a by DCCD can be explained by sterical hindrance of the rotation due to the bulky structure of the hydrophobic carbodiimide but simultaneously indicates a tight coupling between F1 and the c10 ring in the membrane. In addition, it implies that the carboxyl groups of cD61 are in a protonated state at least in some of the c subunits, since binding of carbodiimides requires protonation as a prerequisite (61). Furthermore, the data support the view that no appreciable contact sites between the rotor and stator, namely, b2 and c10, are present in the FO part in the absence of subunit a.
Subunit a, the last subunit being assembled into FOF1?
All of these observations clearly demonstrate that a controlled synthesis of subunit a as well as a tightly regulated interaction with the other FOF1 subunits is essential for the viability of the cell. Subunit a is a protein of high hydrophobicity, and overexpression of the atpB gene causes growth inhibition of E. coli cells associated with cell elongation and an altered septum structure (62) and has been shown to dissipate the electrical potential of the membrane, probably due to an improper insertion of subunit a into the membrane (63). To avoid cell toxicity resulting from the presence of subunit a, in vivo overexpression is prevented by a rather unstable atpB gene transcript (54, 64) and a translational frameshifting at an additional ribosome binding site within the atpB reading frame (65) and, furthermore, improperly membrane-inserted subunit a is rapidly degraded by the membrane protease FtsH (8, 9).
The successful time-delayed assembly of subunit a into preformed FOF1-a complexes in vivo supports the view that subunit a is the last subunit being integrated into the enzyme complex (23–25), thereby finally generating a functional ATP synthase. From an energetic point of view, it appears favorable that the formation of the proton-conducting unit by interaction of subunit a with the c10 ring is the last step during assembly, ensuring that partially assembled subcomplexes do not result in proton leakage, which might be lethal for the cell. However, one has to keep in mind that the use of knockout or deletion mutants arrests the assembly pathway at a certain point to enable the investigation of intermediate states. Recent studies on FOF1 missing subunit δ revealed that also subunit δ can be integrated as the last subunit to assemble FOF1 from ab2 and c10α3β3γε using the same experimental design, a contradiction only at first glance, as discussed in detail elsewhere (68). Furthermore, for yeast mitochondria, a modular assembly of the ATP synthase has also been demonstrated which includes subcomplex formation between subunit a and the peripheral stalk prior to its binding to an FOF1 core complex consisting of F1 and the c ring (18). Nevertheless, also under these conditions, the formation of the proton translocating unit is the last step in assembly.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant of the Deutsche Forschungsgemeinschaft (DE482/1-1).
We thank Brigitte Herkenhoff-Hesselmann for expert technical assistance, Jörg-Christian Greie and Karlheinz Altendorf for critical reading of the manuscript, Julia Garrelfs and Judit Sauerland for generating plasmids pJGA1 and pJS1, and Stanley D. Dunn (University of Western Ontario, London, Canada) for providing antibodies against subunit γ.
Footnotes
Published ahead of print 8 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00468-13.
REFERENCES
- 1.von Ballmoos C, Cook GM, Dimroth P. 2008. Unique rotary ATP synthase and its biological diversity. Annu. Rev. Biophys. 37:43–64 [DOI] [PubMed] [Google Scholar]
- 2.von Ballmoos C, Wiedenmann A, Dimroth P. 2009. Essentials for ATP synthesis by F1FO ATP synthases. Annu. Rev. Biochem. 78:649–672 [DOI] [PubMed] [Google Scholar]
- 3.Kol S, Majczak W, Heerlien R, van der Berg JP, Nouwen N, Driessen AJM. 2009. Subunit a of the F1FO ATP synthase requires YidC and SecYEG for membrane insertion. J. Mol. Biol. 390:893–901 [DOI] [PubMed] [Google Scholar]
- 4.Yi L, Jiang F, Chen M, Cain BD, Bolhuis A, Dalbey RE. 2003. YidC is strictly required for membrane insertion of subunits a and c of the F1FO ATP synthase and SecE of the SecYEG translocase. Biochemistry 42:10537–10544 [DOI] [PubMed] [Google Scholar]
- 5.Yi L, Celebi N, Chen M, Dalbey RE. 2004. Sec/SRP requirements and energetics of membrane insertion of subunits a, b, and c of the Escherichia coli F1FO ATP synthase. J. Biol. Chem. 279:39260–39267 [DOI] [PubMed] [Google Scholar]
- 6.Hermolin J, Fillingame RH. 1995. Assembly of FO sector of Escherichia coli H+ ATP synthase. Interdependence of subunit insertion into the membrane. J. Biol. Chem. 270:2815–2817 [DOI] [PubMed] [Google Scholar]
- 7.Pierson HE, Uhlemann EME, Dmitriev OY. 2011. Interaction with monomeric subunit c drives insertion of ATP synthase subunit a into the membrane and primes a-c complex formation. J. Biol. Chem. 286:38583–38591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akiyama Y, Ito K. 2003. Reconstitution of membrane proteolysis by FtsH. J. Biol. Chem. 278:18146–18153 [DOI] [PubMed] [Google Scholar]
- 9.Akiyama Y, Kihara A, Ito K. 1996. Subunit a of proton ATPase FO sector is a substrate of the FtsH protease in Escherichia coli. FEBS Lett. 399:26–28 [DOI] [PubMed] [Google Scholar]
- 10.Ballhausen B, Altendorf K, Deckers-Hebestreit G. 2009. Constant c10 ring stoichiometry in the Escherichia coli ATP synthase analyzed by cross-linking. J. Bacteriol. 191:2400–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki T, Ozaki Y, Sone N, Feniouk BA, Yoshida M. 2007. The product of uncI gene in F1FO-ATP synthase operon plays a chaperone-like role to assist c-ring assembly. Proc. Natl. Acad. Sci. U. S. A. 104:20776–20781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozaki Y, Suzuki T, Kuruma Y, Ueda T, Yoshida M. 2008. UncI protein can mediate ring-assembly of c-subunits of FOF1-ATP synthase in vitro. Biochem. Biophys. Res. Commun. 367:663–666 [DOI] [PubMed] [Google Scholar]
- 13.Brandt K, Müller DB, Hoffmann J, Hübert C, Brutschy B, Deckers-Hebestreit G, Müller V. 2013. Functional production of the Na+ F1FO ATP synthase from Acetobacterium woodii in Escherichia coli requires the native AtpI. J. Bioenerg. Biomembr. 45:15–23 [DOI] [PubMed] [Google Scholar]
- 14.Schneppe B, Deckers-Hebestreit G, Altendorf K. 1990. Overproduction and purification of the uncI gene product of the ATP synthase of Escherichia coli. J. Biol. Chem. 265:389–395 [PubMed] [Google Scholar]
- 15.Schneppe B, Deckers-Hebestreit G, Altendorf K. 1991. Detection and localization of the i protein in Escherichia coli cells using antibodies. FEBS Lett. 292:145–147 [DOI] [PubMed] [Google Scholar]
- 16.Gay NJ. 1984. Construction and characterization of an Escherichia coli strain with a uncI mutation. J. Bacteriol. 158:820–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu J, Hicks DB, Krulwich TA. 2013. Roles of AtpI and two YidC-type proteins from alkaliphilic Bacillus pseudofirmus OF4 in ATP synthase assembly and nonfermentative growth. J. Bacteriol. 195:220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rak M, Gokova S, Tzagoloff A. 2011. Modular assembly of yeast mitochondrial ATP synthase. EMBO J. 30:920–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kucharczyk R, Zick M, Bietenhader M, Rak M, Couplan E, Blondel M, Caubet S-D, di Rago J-P. 2009. Mitochondrial ATP synthase disorders: molecular mechanisms and the quest for curative therapeutic approaches. Biochim. Biophys. Acta 1793:186–199 [DOI] [PubMed] [Google Scholar]
- 20.Koebmann BJ, Westerhoff HV, Snoep JL, Nilsson D, Jensen PR. 2002. The glycolytic flux in Escherichia coli is controlled by the demand for ATP. J. Bacteriol. 184:3909–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.al-Shawi MK, Parsonage D, Senior AE. 1990. Adenosine triphosphatase and nucleotide binding activity of isolated β-subunit preparations from Escherichia coli F1FO-ATP synthase. J. Biol. Chem. 265:5595–5601 [PubMed] [Google Scholar]
- 22.Senior AE, Muharemagić A, Wilke-Mounts S. 2006. Assembly of the stator in Escherichia coli ATP synthase. Complexation of α subunit with other F1 subunits is prerequisite for δ subunit binding to the N-terminal region of α. Biochemistry 45:15893–15902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ono S, Sone N, Yoshida M, Suzuki T. 2004. ATP synthase that lacks FOa-subunit. Isolation, properties, and indication of FOb2-subunits as an anchor rail of a rotating c-ring. J. Biol. Chem. 279:33409–33412 [DOI] [PubMed] [Google Scholar]
- 24.Kuruma Y, Suzuki T, Ono S, Yoshida M, Ueda T. 2012. Functional analysis of membranous FO-a subunit of F1FO-ATP synthase by in vitro protein synthesis. Biochem. J. 442:631–638 [DOI] [PubMed] [Google Scholar]
- 25.Wittig I, Meyer B, Heide H, Steger M, Bleier L, Wumaier Z, Karas M, Schägger H. 2010. Assembly and oligomerization of human ATP synthase lacking mitochondrial subunits a and A6L. Biochim. Biophys. Acta 1797:1004–1011 [DOI] [PubMed] [Google Scholar]
- 26.Ludlam A, Brunzelle J, Pribyl T, Xu X, Gatti DL, Ackerman SH. 2009. Chaperones of F1-ATPase. J. Biol. Chem. 284:17138–17146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Straffon AFL, Prescott M, Nagley P, Devenish RJ. 1998. The assembly of yeast mitochondrial ATP synthase: subunit depletion in vivo suggests ordered assembly of the stalk subunits b, OSCP and d. Biochim. Biophys. Acta 1371:157–162 [DOI] [PubMed] [Google Scholar]
- 28.Cagnon C, Valverde V, Masson J-M. 1991. A new family of sugar-inducible expression vectors for Escherichia coli. Protein Eng. 4:843–847 [DOI] [PubMed] [Google Scholar]
- 29.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beverin S, Sheppard DE, Park SS. 1971. D-Fucose as a gratuitous inducer of the L-arabinose operon in strains of Escherichia coli B/r mutant in gene araC. J. Bacteriol. 107:79–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185:60–89 [DOI] [PubMed] [Google Scholar]
- 32.Nielsen J, Hansen FG, Hoppe J, Friedl P, von Meyenburg K. 1981. The nucleotide sequence of the atp genes coding for the FO subunits a, b, c and the F1 subunit δ of the membrane-bound ATP synthase of Escherichia coli. Mol. Gen. Genet. 184:33–39 [DOI] [PubMed] [Google Scholar]
- 33.Kuo PH, Ketchum CJ, Nakamoto RK. 1998. Stability and functionality of cysteine-less FOF1 ATP synthase from Escherichia coli. FEBS Lett. 426:217–220 [DOI] [PubMed] [Google Scholar]
- 34.Noji H, Häsler K, Junge W, Kinosita K, Jr, Yoshida M, Engelbrecht S. 1999. Rotation of Escherichia coli F1-ATPase. Biochem. Biophys. Res. Commun. 260:597–599 [DOI] [PubMed] [Google Scholar]
- 35.Klionsky DJ, Brusilow WSA, Simoni RD. 1984. In vivo evidence for the role of the ε subunit as an inhibitor of the proton-translocating ATPase of Escherichia coli. J. Bacteriol. 160:1055–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 37.TaKaRa 2012. Single protein production system (SPP system). TaKaRa Bio Inc., Japan [Google Scholar]
- 38.Neidhardt FC, Ingraham JL, Schaechter M. 1990. Physiology of the bacterial cell. A molecular approach. Sinauer Associates, Sunderland, MA [Google Scholar]
- 39.Strahl H, Greie JC. 2008. The extremely halophilic archaeon Halobacterium salinarum R1 responds to potassium limitation by expression of the K+-transporting KdpFABC P-type ATPase and by a decrease in intracellular K+. Extremophiles 12:741–752 [DOI] [PubMed] [Google Scholar]
- 40.Krebstakies T, Aldag I, Altendorf K, Greie JC, Deckers-Hebestreit G. 2008. The stoichiometry of subunit c of Escherichia coli ATP synthase is independent of its rate of synthesis. Biochemistry 47:6907–6916 [DOI] [PubMed] [Google Scholar]
- 41.Douglas M, Finkelstein D, Butow RA. 1979. Analysis of products of mitochondrial protein synthesis in yeast: genetic and biochemical aspects. Methods Enzymol. 56:58–66 [DOI] [PubMed] [Google Scholar]
- 42.Schägger H, von Jagow G. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166:368–379 [DOI] [PubMed] [Google Scholar]
- 43.Birkenhäger R, Greie JC, Altendorf K, Deckers-Hebestreit G. 1999. FO complex of the Escherichia coli ATP synthase. Not all monomers of the subunit c oligomer are involved in F1 interaction. Eur. J. Biochem. 264:385–396 [DOI] [PubMed] [Google Scholar]
- 44.Dunn SD. 1986. Removal of the ε subunit from Escherichia coli F1-ATPase using monoclonal anti-ε antibody affinity chromatography. Anal. Biochem. 159:35–42 [DOI] [PubMed] [Google Scholar]
- 45.Stalz WD, Greie JC, Deckers-Hebestreit G, Altendorf K. 2003. Direct interaction of subunits a and b of the FO complex of Escherichia coli ATP synthase by forming an ab2 subcomplex. J. Biol. Chem. 278:27068–27071 [DOI] [PubMed] [Google Scholar]
- 46.Deckers-Hebestreit G, Simoni RD, Altendorf K. 1992. Influence of subunit-specific antibodies on the activity of the FO complex of the ATP synthase of Escherichia coli. I Effects of subunit b-specific polyclonal antibodies. J. Biol. Chem. 267:12364–12369 [PubMed] [Google Scholar]
- 47.Langemeyer L, Engelbrecht S. 2007. Essential arginine in subunit a and aspartate in subunit c of FOF1 ATP synthase: effect of repositioning within helix 4 of subunit a and helix 2 of subunit c. Biochim. Biophys. Acta 1767:998–1005 [DOI] [PubMed] [Google Scholar]
- 48.Vik SB, Simoni RD. 1987. F1FO-ATPase from Escherichia coli with mutant FO subunits. Partial purification and immunoprecipitation of F1FO complexes. J. Biol. Chem. 262:8340–8346 [PubMed] [Google Scholar]
- 49.Paule CR, Fillingame RH. 1989. Mutations in three of the putative transmembrane helices of subunit a of the Escherichia coli F1FO-ATPase disrupt ATP-driven proton translocation. Arch. Biochem. Biophys. 274:270–284 [DOI] [PubMed] [Google Scholar]
- 50.Eya S, Maeda M, Futai M. 1991. Role of the carboxyl terminal region of H+-ATPase (FOF1) a subunit from Escherichia coli. Arch. Biochem. Biophys. 284:71–77 [DOI] [PubMed] [Google Scholar]
- 51.Mertens N, Remaut E, Fiers W. 1995. Tight transcriptional control mechanism ensures stable high-level expression from T7 promoter-based expression plasmids. Biotechnology 13:175–179 [DOI] [PubMed] [Google Scholar]
- 52.Wilcox G. 1974. The interaction of L-arabinose and D-fucose with AraC protein. J. Biol. Chem. 249:6892–6894 [PubMed] [Google Scholar]
- 53.Lee SK, Chou HH, Pfleger BF, Newman JD, Yoshikuni Y, Keasling JD. 2007. Directed evolution of AraC for improved compatibility of arabinose- and lactose-inducible promoters. Appl. Environ. Microbiol. 73:5711–5715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schramm HC, Schneppe B, Birkenhäger R, McCarthy JEG. 1996. The promoter-proximal, unstable IB region of the atp mRNA of Escherichia coli: an independently degraded region that can act as a destabilizing element. Biochim. Biophys. Acta 1307:162–170 [DOI] [PubMed] [Google Scholar]
- 55.Deckers-Hebestreit G, Altendorf K. 1992. Influence of subunit-specific antibodies on the activity of the FO complex of the ATP synthase of Escherichia coli. II Effects of subunit c-specific polyclonal antibodies. J. Biol. Chem. 267:12370–12374 [PubMed] [Google Scholar]
- 56.Solomon KA, Brusilow WSA. 1988. Effect of an uncE ribosome-binding site mutation on the synthesis and assembly of the Escherichia coli proton-translocating ATPase. J. Biol. Chem. 263:5402–5407 [PubMed] [Google Scholar]
- 57.Etzold C, Deckers-Hebestreit G, Altendorf K. 1997. Turnover number of Escherichia coli FOF1 ATP synthase for ATP synthesis in membrane vesicles. Eur. J. Biochem. 243:336–343 [DOI] [PubMed] [Google Scholar]
- 58.Matthies D, Haberstock S, Joos F, Dötsch V, Vonck J, Bernhard F, Meier T. 2011. Cell-free expression and assembly of ATP synthase. J. Mol. Biol. 413:593–603 [DOI] [PubMed] [Google Scholar]
- 59.Uhlemann EME, Pierson HE, Fillingame RH, Dmitriev OY. 2012. Cell-free synthesis of membrane subunits of ATP synthase in phospholipid bicelles: NMR shows subunit a fold similar to the protein in the cell membrane. Protein Sci. 21:279–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gumbiowski K, Pänke O, Junge W, Engelbrecht S. 2002. Rotation of the c subunit oligomer in EFOEF1 mutant cD61N. J. Biol. Chem. 277:31287–31290 [DOI] [PubMed] [Google Scholar]
- 61.Kurzer F, Douraghi-Zadeh K. 1967. Advances in the chemistry of carbodiimides. Chem. Rev. 67:107–152 [DOI] [PubMed] [Google Scholar]
- 62.Eya S, Maeda M, Tomochika KI, Kanemasa Y, Futai M. 1989. Overproduction of truncated subunit a of H+-ATPase causes growth inhibition of Escherichia coli. J. Bacteriol. 171:6853–6858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.von Meyenburg K, Jørgensen BB, Michelsen O, Sørensen L, McCarthy JEG. 1985. Proton conduction by subunit a of the membrane-bound ATP synthase of Escherichia coli revealed after induced overproduction. EMBO J. 4:2357–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arechaga I, Miroux B, Runswick MJ, Walker JE. 2003. Over-expression of Escherichia coli F1FO-ATPase subunit a is inhibited by instability of the uncB gene transcript. FEBS Lett. 547:97–100 [DOI] [PubMed] [Google Scholar]
- 65.Matten SR, Schneider TD, Ringquist S, Brusilow WSA. 1998. Identification of an intragenic ribosome binding site that affects expression of the atpB gene of the Escherichia coli proton-translocating ATPase (unc) operon. J. Bacteriol. 180:3940–3945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iwamoto A, Omote H, Hanada H, Tomioka N, Itai A, Maeda M, Futai M. 1991. Mutations in Ser174 and the glycine-rich sequence (Gly149, Gly150, and Thr156) in the β subunit of Escherichia coli H+-ATPase. J. Biol. Chem. 266:16350–16355 [PubMed] [Google Scholar]
- 67.Moriyama Y, Iwamoto A, Hanada H, Maeda M, Futai M. 1991. One-step purification of Escherichia coli H+-ATPase (FOF1) and its reconstitution into liposomes with neurotransmitter transporters. J. Biol. Chem. 266:22141–22146 [PubMed] [Google Scholar]
- 68.Hilbers F, Eggers R, Pradela K, Friedrich K, Herkenhoff-Hesselmann B, Becker E, Deckers-Hebestreit G. Subunit Δ is the key player for assembly of the H+-translocating unit of Escherichia coli FOF1 ATP synthase. J. Biol. Chem., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.