

Abstract
Conventional methods for synthesizing protein/peptide–polymer conjugates, as a means to improve the pharmacological properties of therapeutic biomolecules, typically have drawbacks including low yield, non-trivial separation of conjugates from reactants, and lack of site-specificity, which results in heterogeneous products with significantly compromised bio activity. To address these limitations, the use of sortase A from Staphylococcus aureus is demonstrated to site-specifically attach an initiator solely at the C-terminus of green fluorescent protein (GFP), followed by in situ growth of a stealth polymer, poly(oligo(ethylene glycol) methyl ether methacrylate) by atom transfer radical polymerization (ATRP). Sortase-catalyzed initiator attachment proceeds with high specificity and near-complete (≈95%) product conversion. Subsequent in situ ATRP in aqueous buffer produces 1:1 stoichiometric conjugates with > 90% yield, low dispersity, and no denaturation of the protein. This approach introduces a simple and useful method for high yield synthesis of protein/peptide–polymer conjugates.
Keywords: atom transfer radical polymerization (ATRP), biological application of polymers, grafting from, protein–polymer conjugates, sortase
1. Introduction
Covalent conjugation with stealth polymers can improve the pharmacokinetics and biodistribution, and reduce the immunogenicity of therapeutic proteins and peptides. [1] Conjugation with “smart” polymers can also impart interesting and useful behaviors such as externally triggered activity switching of the biomolecule. [2] Conventional “grafting to” conjugation methods, which involve separate synthesis and attachment of a polymer to a biomolecule, often suffer from low yield due to steric hindrance and difficulty in product purification as a result of similar sizes and surface properties of the reactants and products. [3] The recent emergence of “grafting from” techniques, or growing polymers directly from biomolecular macroinitiators, effectively circumvents these issues. [3d, 4] However, most methods of initiator attachment still lack site specificity and general applicability. [3e, 4c-g, 5]
Aiming to develop a general method for a high yield “grafting from” synthesis of site-specific and stoichiometric bioconjugates, we selected sortase-mediated ligation (SML) for initiator attachment because its high specificity and mild reaction conditions are ideal for protein and peptide manipulation. [6] The versatility of SML has been demonstrated previously, [7] but it has not been exploited to install a polymerization initiator at a defined site on a protein. The sortase A (SrtA) transpeptidase from S taphylococcus aureus recognizes the penta-peptide sequence “LPXTG” (lysine-proline-X-threonineglycine, where “X” is any standard amino acid residue) embedded in or terminally attached to a protein or peptide, and its cysteine (C) nucleophilically attacks the amide bond between threonine and glycine within the recognition sequence, generating a relatively long-lived enzyme-thioacyl intermediate. [6a] To complete transpeptidation, a second (bio)molecule with an N-terminal nucleophilic group, typically an oligoglycine motif, attacks the intermediate, displacing SrtA and joining the two molecules via a native peptide bond. [6a]
Herein, SrtA was used to site-specifically attach an initiator solely at the C terminus of green fluorescent protein (GFP), followed by in situ growth of the stealth polymer, poly(oligo(ethylene glycol) methyl ether methacrylate) [poly(OEGMA)], from the protein by atom transfer radical polymerization (ATRP) to yield GFP–C–poly(OEGMA).
2. Experimental Section
2.1. Sortase-Catalyzed Initiator Attachment and Product Separation
All chemical reagents were purchased from Sigma Aldrich and used as received, unless otherwise specified. GFP–srt–ELP (ELP: elastin-like polypeptide) and SrtA were recombinantly expressed and purified as described in the Supporting Information. A reaction mixture consisting of GFP–srt–ELP, SrtA, and initiator N -(2-(2-(2-(2-aminoacetamido)acet-amido)acetamido) ethyl)-2-bromo-2-methylpropanamide (AEBMP, synthesis detail described in SI) at a 2:1:60 ratio in sortase buffer (50 m m tris-(hydroxymethyl)amino methane (Tris), 150 m m NaCl, 10 m m CaCl 2, pH adjusted to 7.5) was incubated at 37 °C for 5 h. Post reaction, a reverse histidine tag (His-tag) purification was used to isolate the GFP–C–Br macroinitiator, by exploiting the fact that the macroinitiator is the only species in the mixture without a hexahistidine tag (His 6 -tag). Equilibration and elution washes were done as described in the SI. The first two equilibration washes containing the eluted GFP–C–Br were collected and solvent exchanged by overnight dialysis against phosphate buffered saline (PBS, pH 7.4, EMD Millipore) in preparation for use. A control reaction was done by replacing AEBMP with triglycine (Gly 3), while keeping all other conditions the same. The resulting GFP–C–Gly 3 was used as a negative control in the subsequent in situ ATRP reaction.
2.2. In Situ ATRP from GFP–C–Br and Conjugate Purification
ATRP reactions were performed using conditions described previously with minor changes. [11] OEGMA (MW = 500) was eluted through a column packed with aluminum oxide to remove the polymerization inhibitors. Three sets of reaction conditions were attempted and the parameters are summarized in SI Table S1. Polymerization was typically carried out by first mixing specified amounts of CuCl, CuCl 2, and 1,1,2,7,10,10-hexamethyltriethylenetetramine (HMTETA) in 100 μ L of MilliQ water until all reagents were completely dissolved and then topped up with 400 μ L of PBS. A second solution was prepared by adding OEGMA to 2 mL of 100 μm GFP–C–Br in PBS. The two solutions were degassed by bubbling separately with argon for 30 min using a Schlenk line, after which the first solution was quickly transferred into the second solution by a cannula. Polymerization was allowed to proceed for a specified time at room temperature under argon and was quenched by bubbling with air. An initial separation of the conjugate from the low molecular weight (MW) reagents was carried out by gel filtration on disposable PD-10 columns (GE Life Science) before subsequent purification and characterization.
3. Results and Discussion
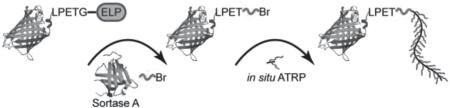
GFP was chosen as a model protein in this proof-of-concept study as its fluorescence allows easy tracking of the protein through the initiator installation and in situ polymerization, and it also serves as an indicator of the folding and activity of the protein. A ternary fusion protein, abbreviated as “GFP–srt–ELP”, was recombinantly expressed to serve as the sortase substrate (Scheme 1 a). Here, “srt” stands for the native SrtA recognition sequence “LPETG” [8] (“E”: glutamine) and ELP refers to an environmentally responsive elastin-like polypeptide that was included in the fusion to enable easy purification of the ternary fusion by inverse transition cycling (ITC), a non-chromatographic protein purification method that we previously developed. [9] The recognition sequence was deliberately located between the protein and the ELP, so that transpeptidation by SrtA not only attaches the initiator to GFP but also conveniently liberates the purification tag. As transpeptidation relies on the presence of the enzyme, cleavage does not begin until SrtA is added in vitro. Very little, if any, of the protein is thus expected to be lost in vivo before purification, hence increasing the overall product yield. This hypothesis was confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis of ITC purification of GFP–srt–ELP. As shown in SI Figure S2a, the only species that exhibited inverse transition behavior and thus was purified by ITC was GFP–srt–ELP. The lack of a free ELP band clearly demonstrates that no premature in vivo cleavage occurred. The fusion protein was obtained at high purity with an excellent yield of ≈300 mg L −1 from E. coli shaker flask culture.
Scheme 1.

Synthetic route of GFP–C–poly(OEGMA). a) Recombinant expression of ternary fusion protein GFP–srt–ELP and purification by inverse transition cycling (ITC). b) SrtA-catalyzed site-specific attachment of the ATRP initiator AEBMP to the C terminus of GFP. c) In situ ATRP of OEGMA yielding GFP–C–poly(OEGMA).
SrtA carrying an N-terminal His 6 -tag was also recombinantly expressed in E. coli with high yield (≈135 mg L −1) and obtained in high purity by immobilized metal affinity chromatography (IMAC) (Figure S2b, Supporting Information). The ATRP initiator AEBMP (Scheme 1) was chemically synthesized with an N-terminal Gly 3 motif serving as the nucleophile, as maximum reaction rates for SML have been reported when two or more glycines are incorporated. [7f]
Sortase-catalyzed initiator attachment (SCIA) was then carried out at a GFP–srt–ELP/SrtA/AEBMP ratio of 2:1:60 (Scheme 1 b). SDS-PAGE analysis of the reaction mixture showed near complete disappearance of the GFP–srt–ELP band close to 67 kDa, and the appearance of two bands around 39 and 28 kDa, corresponding to the cleaved ELP and the macroinitiator product, abbreviated as GFP–C–Br (Figure 1 a). A control reaction was done using Gly 3 as the nucleophile, to yield GFP–C–Gly 3 as a negative control for subsequent ATRP reaction. Quantification of band intensity in SDS-PAGE showed that initiator attachment efficiency was near quantitative (≈95% averaged across five samples, Table S2, Supporting Information).
Figure 1.

a) SDS-PAGE analysis of initiator attachment by SrtA. Lane 1: MW marker, lane 2: GFP–srt–ELP, lane 3: SrtA, lane 4: SCIA reaction mixture after 5 h of reaction, lane 5: purified GFP–C–Br macroinitiator. b) Isotopic distribution of GFP–C–Br C-terminal peptide [DHMVLLEFVTAAGITHGMDELYNVDGGGSLPET–“AEBMP”] 3+ detected by liquid chromatography tandem mass spectrometry (LC/MS-MS) after trypsin digestion.
To purify GFP–C–Br, a His 6 -tag was intentionally inserted between “srt” and ELP, such that upon trans-peptidation by SrtA, all species except GFP–C–Br carried a His 6 -tag. Consequently, elution through an IMAC column yielded pure macroinitiator in the eluent while leaving all other unwanted species bound to the resin. SDS-PAGE analysis (Figure S2c, Supporting Information) indicated that all of the GFP–C–Br was recovered by this method. The purified GFP–C–Br was then characterized by liquid chromatography/electrospray-ionization mass spectrometry (LC/ESI-MS) to confirm initiator attachment (Figure S3a, Supporting Information). A major peak was detected at 28 120.4 Da, which closely agrees with the theoretical mass of 28 123.8 Da for GFP–C–Br. To prove site specificity of initiator attachment, GFP–C–Br was subjected to trypsin digestion and the peptide fragments were analyzed by liquid chromatography/tandem mass spectrometry (LC-MS/MS). Only the C-terminal peptide fragment was detected as a brominated cation and its experimental isotope distribution (Figure 2 b) showed nearly perfect overlap with its theoretical distribution (Figure S3b). These results provided strong evidence that the brominated ATRP initiator was solely attached to the C-terminus of GFP by SrtA. Aside from the singly brominated C-terminal peptide, no other derivatives were detected.
Figure 2.

a) Size exclusion chromatography (SEC) traces of GFP-Br (dotted line), Rxn 1 (dashed dotted line), Rxn 2 (dashed line), and Rxn 3 (solid line). b) Fluorescence spectra of GFP before initiator attachment (solid triangle), after initiator attachment (empty square), and after in situ ATRP (solid circle, ATRP at Rxn 3 conditions: 263.1 kDa as measured by SEC-MALS); all samples at 20 μM.
Subsequently, in situ ATRP was performed to graft poly(OEGMA) from GFP–C–Br (Scheme 1 c). Three sets of polymerization conditions (Table S1, Supporting Information) were investigated to synthesize conjugates of increasing MWs, denoted herein as Rxn 1, Rxn 2, and Rxn 3. Size exclusion chromatography (SEC) was performed after ATRP to characterize the polymerization product. SEC of the product with UV/vis absorbance detection at 280 nm (Figure 2 a) showed a single peak at an elution time of 20.6 min, corresponding to GFP–C–Br prior to polymerization. This peak greatly diminished after polymerization, and was accompanied by the emergence of peaks at 17.9, 15.9, and 13.3 min, corresponding to GFP–C–poly(OEGMA) conjugates in each of the three reactions. The results from UV/vis detection were consistent with those from fluorescence detection (Figure S4, Supporting Information). Integration of peak areas showed that the conjugates constituted > 90% of the polymerization product on average (Table S3 and S4, Supporting Information), indicating that in situ ATRP from GFP–C–Br proceeds with extremely high efficiency.
SDS-PAGE analysis provided additional evidence for the successful growth of poly(OEGMA) from GFP–C–Br (Figure S2d, Supporting Information). After each reaction, the band corresponding to GFP–C–Br (≈28 kDa) decreased to a much lower intensity, accompanied by a new higher molecular weight band corresponding to the conjugate. In contrast, when the GFP–C–Gly 3 control was used in the polymerization, or when GFP–C–Br was physically mixed with pre-synthesized poly(OEGMA), only a single band was observed around 28 kDa, proving that poly(OEGMA) was only grown in situ from the C-terminal initiator attached by SCIA.
The conjugates were further characterized by light scattering (Table 1). First, size exclusion chromatography multi-angle light scattering (SEC-MALS) was performed to determine the weight-average molecular weight () and radius of gyration (Rg) of the conjugates. The of GFP–C–Br measured was 28.0 kDa and the polydispersity index (PDI) was 1.01, consistent with the theoretical value of 28 123.8 Da and the expected monodispersity of the macroinitiator. The s of the three conjugates measured by SEC-MALS were 61.2, 89.9, and 263.1 kDa, respectively, with corresponding PDIs of 1.23, 1.26, and 1.25. These results show that by tuning the ATRP conditions, conjugates can be synthesized from macroinitiators generated by SCIA with different molecular weights and fairly low polydispersity. The Rg s of GFP–C–Br and the Rxn 1 conjugate could not be accurately determined by SEC-MALS as they fell below the 10 nm lower limit of detection at a laser wavelength of 638 nm. Rg s of the products of Rxn 2 and 3 were 10.6 and 19.2 nm, respectively. Next, the hydrodynamic radius (Rh) of each species was measured by dynamic light scattering (DLS). The Rh of GFP–C–Br was determined to be 3.6 nm. In situ growth of poly(OEGMA) from the macroinitiator resulted in an increase of the Rh to 6.4, 10.0, and 18.3 nm, for the three polymerization reactions, respectively. With both Rg and Rh available for Rxn 2 and 3, their corresponding Rg/Rh ratios (ρ = form factor) were calculated, yielding values of 1.06 and 1.05, respectively. To put these values in perspective, ρ for globular proteins is ≈0.775, [10] while that of a monodisperse random coil polymer in theta solvent is 1.50. An increase in polymer polydispersity and the presence in a good solvent can increase ρ. [11] Thus, their ρ values suggest that the overall conformation of the GFP–C–poly(OEGMA) conjugates lies somewhere between that of their components. The conjugates could be easily and completely purified by preparative SEC. Fluorescence spectroscopy of unmodified GFP, GFP–C–Br, and purified GFP–C–poly(OEGMA) (Rxn 3 product, 263.1 kDa as measured by SEC-MALS) clearly shows that each step in the synthesis of the conjugate has minimal effect on the activity of the protein (Figure 2b).
Table 1.
Light scattering characterizations of the GFP–C–Br macroinitiator and GFP–C–poly(OEGMA) conjugates.
| Species |
a [kDa] |
PDIa |
Rga [nm] |
Rhb [nm] |
|---|---|---|---|---|
| GFP–C–Br | 28.0 | 1.01 | N/Ac | 3.6 |
| Rxn 1 | 61.2 | 1.23 | N/Ac | 6.4 |
| Rxn 2 | 89.9 | 1.26 | 10.6 | 10.0 |
| Rxn 3 | 263.1 | 1.25 | 19.2 | 18.3 |
Determined by SEC-MALS
Determined DLS
Below instrument lower limit of detection.
Comparison of these results with an alternative strategy for C-terminal growth of a polymer from biomolecules facilitated by intein-mediated initiator attachment (IMIA) is instructive. IMIA exploits cleavage of a mutated intein in a protein–intein fusion to generate a unique C-terminal reactive thioester on the protein, which can react with an N-terminal cysteine on an ATRP initiator to site-specifically attach the initiator solely at the C terminus of the protein. [12] Although useful, this method has a few limitations. First, its product yield is adversely affected by premature in vivo cleavage of the intein (≈30%) during expression, which results in loss of the appended purification tag from the protein, leading to appreciable product loss. [12] In contrast, the SCIA reaction occurs solely in vitro, and hence offers greater control over the reaction and product yield. Second, for IMIA, the protein of interest needs to be fused to a relatively large intein domain that can, in some instances, reduce the expression of the fusion protein. [7f] In addition, the presence of thiol groups at the ligation site in IMIA makes the method prone to generating small amounts of side-products through disulfide bonding, a problem that does not occur in SCIA. The one potential advantage of IMIA over SCIA is that SCIA requires the introduction of an extraneous LPET(G)n peptide tag at the C-terminus, which may have some consequence on the immunogenicity of the conjugate, although this remains to be clarified.
4. Conclusion
In conclusion, SrtA-catalyzed installation of the ATRP initiator on the C terminus of GFP proceeded to > 95% conversion with no side products. Subsequent in situ ATRP of poly(OEGMA) from the macroinitiator yielded site-specific and stoichiometric (1:1) GFP–C–poly(OEGMA) conjugates with well-controlled polydispersity and > 90% efficiency. The conjugates could be easily and completely purified and the overall yield of the conjugate normalized to purified protein was > 85%. We believe such a yield is notable, as it shows minimal loss during installation of the ATRP initiator, subsequent in situ ATRP or purification of conjugates, given that order of magnitude losses are considered routine in the synthesis of protein–polymer conjugates. This extraordinarily high yield is important for biomolecule–polymer conjugate synthesis, especially when the biomolecule of interest is a high value “biologic” peptide or protein drug. Enhanced pharmacokinetics, improved tissue biodistribution, and reduced immunogenicity of poly(OEGMA) conjugates of therapeutically relevant biomolecules are potential benefits of this polymer conjugation methodology, and will be investigated in future studies. We anticipate that this new method will provide a useful tool for the synthesis of protein (and peptide)–polymer conjugates for pharmaceutical applications.
Supplementary Material
Acknowledgements
The authors thank Dr. David Gooden at Duke Small Molecule Synthesis Facility for synthesis of AEBMP, Dr. George R. Dubay at Duke Chemistry Instrumentation Facility for help with LC/ESI-MS, and Dr. Erik J. Soderblom, Meredith E. Turner, Dr. J. Will Thompson, and Dr. M. Arthur Moseley at Duke Proteomics Facility for LC/MS-MS. This work was supported by National Institute of Health (R01-DK092665 to AC, and NIH R01-AI46611 to DGM). Y.Q. was supported by a fellowship from the Duke University Center of Biomolecular and Tissue Engineering.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Yizhi Qi, Department of Biomedical Engineering, Duke University, Durham, NC 27708, USA.
Dr. Miriam Amiram, Department of Biomedical Engineering, Duke University, Durham, NC 27708, USA.
Prof. Weiping Gao, Department of Biomedical Engineering, Duke University, Durham, NC 27708, USA; Center for Biologically Inspired Materials and Materials Systems, Duke University, Durham, NC 27708, USA.
Prof. Dewey G. McCafferty, Department of Chemistry, Duke University, Durham, NC 27708, USA
Prof. Ashutosh Chilkoti, Department of Biomedical Engineering, Duke University, Durham, NC 27708, USA; Center for Biologically Inspired Materials and Materials Systems, Duke University, Durham, NC 27708, USA.
References
- [1].a) Hamidi M, Azadi A, Rafiei P. Drug Deliv. 2006;13:399. doi: 10.1080/10717540600814402. [DOI] [PubMed] [Google Scholar]; b) Bansal R, Post E, Proost JH, de Jager-Krikken A, Poelstra K, Prakash J. J. Controlled Release. 2011;154:233. doi: 10.1016/j.jconrel.2011.05.027. [DOI] [PubMed] [Google Scholar]; c) Duncan R. Nat. Rev. Cancer. 2006;6:688. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]; d) Kim TH, Jiang HH, Lim SM, Youn YS, Choi KY, Lee S, Chen X, Byun Y, Lee KC. Bioconjugate Chem. 2012;23:2214. doi: 10.1021/bc300265n. [DOI] [PubMed] [Google Scholar]
- [2].Shimoboji T, Larenas E, Fowler T, Hoffman AS, Stayton PS. Bioconjugate Chem. 2003;14:517. doi: 10.1021/bc025615v. [DOI] [PubMed] [Google Scholar]
- [3].a) Broyer RM, Grover GN, Maynard HD. Chem. Commun. 2011;47:2212. doi: 10.1039/c0cc04062b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gaberc-Porekar V, Zore I, Podobnik B, Menart V. Curr. Opin. Drug Discov. Devel. 2008;11:242. [PubMed] [Google Scholar]; c) Le Droumaguet B, Nicolas J. Polym. Chem. 2010;1:563. [Google Scholar]; d) Sumerlin BS. ACS Macro Lett. 2012;1:141. doi: 10.1021/mz200176g. [DOI] [PubMed] [Google Scholar]; e) Gauthir MA, Klok H. Chem. Commun. 2008:2591. doi: 10.1039/b719689j. [DOI] [PubMed] [Google Scholar]
- [4].a) Averick S, Simakova A, Park S, Konkolewicz D, Magenau AJD, Mehl RA, Matyjaszewski K. ACS Macro. Lett. 2012;1:6. doi: 10.1021/mz200020c. [DOI] [PubMed] [Google Scholar]; b) Bontempo D, Maynard HD. J. Am. Chem. Soc. 2005:6508. doi: 10.1021/ja042230+. [DOI] [PubMed] [Google Scholar]; c) Heredia KL, Bontempo D, Ly T, Byers JT, Halstenber S, Maynard HD. J. Am. Chem. Soc. 2005;127:16955. doi: 10.1021/ja054482w. [DOI] [PubMed] [Google Scholar]; d) Nicolas J, San Miguel V, Mantovani G, Haddleton DM. Chem. Commun. 2006;45:4697. doi: 10.1039/b609935a. [DOI] [PubMed] [Google Scholar]; e) Liu J, Bulmus V, Herlambang DL, Barner-Kowollik C, Stenzel MH, Davis TP. Angew. Chem. Int. Ed. 2007;46:3099. doi: 10.1002/anie.200604922. [DOI] [PubMed] [Google Scholar]; f) Boyer C, Bulmus V, Liu J, Davis TP, Stenzel MH, Barner-Kowollik C. J. Am. Chem. Soc. 2007;129:7145. doi: 10.1021/ja070956a. [DOI] [PubMed] [Google Scholar]; g) De P, Li M, Gondi SR, Sumerlin BS. J. Am. Chem. Soc. 2008;130:11288. doi: 10.1021/ja804495v. [DOI] [PubMed] [Google Scholar]
- [5].a) Magnusson JP, Bersani S, Salmaso S, Alexander C, Caliceti P. Bioconjugate Chem. 2010;21:671. doi: 10.1021/bc900468v. [DOI] [PubMed] [Google Scholar]; b) Depp V, Alikhani A, Grammer V, Lele BS. Acta Biomater. 2009;5:560. doi: 10.1016/j.actbio.2008.08.010. [DOI] [PubMed] [Google Scholar]; c) Lele BS, Murata H, Matyjaszewski K, Russell AJ. Biomacromolecules. 2005;6:3380. doi: 10.1021/bm050428w. [DOI] [PubMed] [Google Scholar]
- [6].a) Popp MW, Antos JM, Ploegh HL. Curr. Protoc. Protein Sci. 2009;56:15.13.11–15.13.19. doi: 10.1002/0471140864.ps1503s56. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Leung KM, Hagemeyer CE, Johnston APR, Gonzales C, Kamphuis MMJ, Ardipradja K, Such GK, Peter K, Caruso F. Angew. Chem. Int. Ed. 2012;51:7132. doi: 10.1002/anie.201203612. [DOI] [PubMed] [Google Scholar]
- [7].a) Antos JM, Chew G, Guimaraes CP, Yoder NC, Grotenbreg GM, Popp MW, Ploegh HL. J. Am. Chem. Soc. 2009;131:10800. doi: 10.1021/ja902681k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Antos JM, Miller GM, Grotenbreg GM, Ploegh HL. J. Am. Chem. Soc. 2008;130:16338. doi: 10.1021/ja806779e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bellucci JJ, Amiram M, Bhattacharyya J, McCafferty D, Chilkoti A. Angew. Chem. Int. Ed. 2013;52:3703. doi: 10.1002/anie.201208292. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Popp MW, Dougan SK, Chuang T, Spooner E, Ploegh HL. Proc. Natl. Acad. Sci. USA. 2011;108:3169. doi: 10.1073/pnas.1016863108. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wu Z, Guo X, Wang Q, Swarts BM, Guo Z. J. Am. Chem. Soc. 2010;132:1567. doi: 10.1021/ja906611x. [DOI] [PubMed] [Google Scholar]; f) Mao H, Hart SA, Schink A, Pollok BA. J. Am. Chem. Soc. 2004;126:2670. doi: 10.1021/ja039915e. [DOI] [PubMed] [Google Scholar]
- [8].J Boekhorst, de Been MW, Kleerebezem M, Siezen RJ. J. Bacteriol. 2005;187:4928. doi: 10.1128/JB.187.14.4928-4934.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Meyer DE, Chilkoti A. Nat. Biotechnol. 1999;14:1112. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- [10].Burchard W, Schmidt M, Stockmayer WH. Macromolecules. 1980;13:1265. [Google Scholar]
- [11].W Burchard. In: Light Scattering: Principles and Development. Brown W, editor. Oxford University Press; New York: 1996. Ch. 13. [Google Scholar]
- [12].Gao W, Liu W, Christensen T, Zalutsky MR, Chilkoti A. Proc. Natl. Acad. Sci. USA. 2010;107:16432. doi: 10.1073/pnas.1006044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.