

Abstract
A facile hydroxyindole carboxylic acid-based focused amide library approach was designed to target both the PTP active site and a unique nearby pocket for enhanced affinity and selectivity. High throughput screening of the focused library let to the identification of a highly potent (Ki=50 nM) and selective (>100-fold against a large panel of PTPs) inhibitor 11a for mPTPB, an essential virulence factor for Mycobacterium tuberculosis. Importantly, 11a displayed highly efficacious cellular activity and was capable of reversing the altered immune responses induced by mPTPB in macrophages.
Keywords: Focused amide library, HydroxyIndole carboxylic acid, PTP, mPTPB inhibitor, Tuberculosis
Protein tyrosine phosphatases (PTPs) are key regulators of intracellular signaling pathways that control a broad range of physiological processes, including cell growth and differentiation, metabolism, apoptosis, and immune responses.[1] Not surprisingly, altered PTP activities result in aberrant tyrosine phosphorylation, which has been linked to the etiology of many human diseases, including cancer, diabetes, and immune dysfunctions.[2,3] The importance of PTPs in cellular physiology is further underscored by the fact that they are often exploited and subverted by pathogenic bacteria to cause infectious diseases. For example, Mycobacterium protein tyrosine phosphatase B (mPTPB) is a virulence factor for Mycobacterium tuberculosis and contributes to its survival within host macrophages and animal model.[4–6]
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis (TB), a decimating infectious disease causing two million deaths every year (World Health Organization, www.who.int). Despite the disease burden and the loss of life caused by TB, no new drugs have been developed for the treatment of the disease since the 1970s. Current treatment regimens with a combination of different antibiotics are too long, causing patient to stop taking the drugs too early, which leads to the emergence of drug-resistant strains of TB. Thus there is an urgent unmet need for improved therapeutic agents. As an intracellular pathogen, Mtb survives and replicates primarily in host macrophages. To evade the antimicrobial functions of the macrophage, Mtb encodes a number of virulence factors including mPTPB,[7] which is secreted by Mtb into the cytosol of the macrophage and is important for persistent mycobacterial infection.[4] Importantly, deletion of mPTPB blocks intracellular survival of Mtb in IFN-γ activated macrophages and severely reduces the bacterial load in a clinically-relevant guinea pig model of TB infection.[4] These findings suggest that specific inhibition of mPTPB activity may augment intrinsic host signaling pathways to eradicate TB infection. Consequently, there is considerable interest in developing mPTPB inhibitors.[8–17]
Although the PTPs have been implicated in a wide array of human diseases, they have proven to be exceptionally difficulty targets for the development of new medicine.[18] There are two main barriers to the acquisition of drugs targeting the PTPs. First, the PTPs are a large family of closely-related enzymes with a highly conserved active site, so it has been challenging to discover potent and selective inhibitors for individual members of the PTP family. Fortunately, it has been shown that pTyr alone is not sufficient for high-affinity binding and residues flanking pTyr also contribute to PTP substrate recognition.[19] Thus, an effective strategy to address the specificity issue is to link a nonhydrolyzable pTyr mimetic to an appropriately functionalized moiety to engage both the active site and a unique peripheral binding pocket.[20] Secondly, PTPs are difficult to drug because their pTyr binding pocket is highly positively charged so that high-throughput screening approaches commonly lead to highly polar series that are unable to cross cell membranes. Until recently, it was not thought possible to synthesize potent PTP inhibitors with drug-like properties.
To develop PTP inhibitory agents with more favorable pharmacological properties, we began to focus on natural products because they are evolved to interfere and interact with their biological targets in vivo. To this end, we discovered that salicylic acids could serve as non-phosphorus-containing pTyr mimetics.[21,22] More recently we showed that bicyclic hydroxybenzofuran and hydroxyindole carboxylic acid scaffolds are sufficiently polar to bind the PTP active site, yet remain capable of efficient cell penetration.[16,23,24] We hypothesized that hydroxybenzofuran or indole-based carboxylic acids may bind PTP active site in an analogous fashion as pTyr, but additional diversity elements attached to the benzofuran/indole core may interact with unique secondary pockets in the vicinity of the active site and therefore render the inhibitors PTP isozyme-selective. Click chemistry was initially employed to tether an alkyne-containing hydroxybenzofuran/indole carboxylic acid scaffold with a large number of azide-containing amines and hydrazines in order to target adjacent secondary binding pockets in the vicinity of the PTP active site. This led to the identification of several inhibitors for mPTPB, SHP2, and lymphoid-specific tyrosine phosphatase (Lyp).[16,23,24] However, despite the highly efficacious cellular activity, the potency and selectivity displayed by these inhibitors are relatively modest, and therefore are not adequate for chemical biological investigation and therapeutic development. The reasons for the less than spectacular results with the Click chemistry approach may be the following: 1) the hydroxybenzofuran/indole carboxylic acid cores were not optimal for the desired target PTPs; 2) the triazolidine linker formed as a result of the Click reaction was not conducive for ligand interactions with both the active site and nearby secondary pockets; and 3) the presence of trace amount of cuprous ion could compromise the quality of the hits identified from the libraries.
To overcome the potential problems associated with the Click chemistry approach and to obtain mPTPB inhibitors with improved potency and selectivity, we sought to first identify the most optimal hydroxyindole carboxylic acid core for mPTPB, which was then combined with a diverse collection of carboxylic acids through the well-established amide chemistry in order to target sub-pockets that border the active site (Figure 1).
Figure 1.

Strategy for the focused library design and combinatorial library approach.
Scheme 1 depicts a strategy for hydroxyindole carboxylic acid optimization. Compound 1 in methanol in the presence of concentrated sulfuric acid was refluxed overnight to provide ester 2 in 81.4% yields. Ester 2 was treated with paraformaldehyde and NaCNBH3 overnight to give product 3 with 78% yield. Iodination of compound 3 in ether and water yielded the desired isomer 4, which was then coupled with the corresponding commercially available alkynes by Sonogashira coupling to afford alkyne 5a-l with high yield. Electrophilic cyclizations of 5a-l by I2 furnished iodine 6a-l in 80–90% yield. After hydrolysis of 6a-l in 3.5 M NaOH for 2 days, 7a-l was purified by HPLC in 50–80% yield with over 95% purity.
Scheme 1.
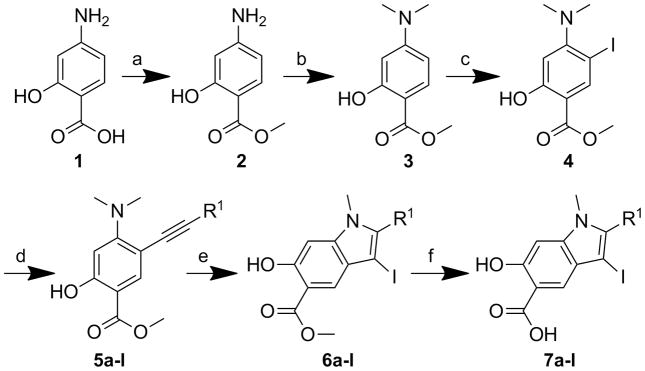
Synthesis of hydroxyindole scarboxylic acid cores 7a–l : a) Cat. Con. H2SO4, CH3OH, reflux, 24 h, 81.4%; b) (CHO)n, NaCNBH3, AcOH, rt, 24 h, 78%; c) I2, K2CO3, water, ether, rt, 4 h, 42%; d) corresponding Phenylacetylene, Pd(PPh3)2Cl2, CuI, Et3N, DMF, 80–90%; e) I2, CH2Cl2, rt, 80–90%; f) 3.5 M NaOH, THF/H2O, rt, 48 h, 50–80%.
The inhibitory activity of compounds 7a-l towards mPTPB was evaluated at pH 7 and 25 °C using p-nitrophenyl phosphate (pNPP) as a substrate (Table 1). It appears that compounds bearing electron-withdrawing group in the α-phenyl ring, such as 7a, 7b, 7e and 7f have enhanced inhibition compared to the parental compound 7g. Furthermore, ortho-trifluromethyl substituted inhibitor 7a exhibits better potency than its meta- (7b) and para- (7f) analogues. Compound 7c is 4 times more potent than the parent 7g, while the aliphatic substituted compound 7d displays a 3-fold increased affinity compared to compound 7g. Finally, fluoro-substitutions (7h, 7i, and 7j) in the α-phenyl ring have little influence on binding whereas hydroxyl (7k) or amino (7l) groups at the meta-position make a negative contribution to binding. Collectively, compound 7a surfaced as the most potent hydroxyindole carboxylic acid core (IC50 = 1.2 ± 0.1 μM) for mPTPB. Interestingly, compound 7a also displayed at least 47-fold selectivity for mPTPB over a large panel of PTPs, including mPTPA, YopH, PTP1B, TC-PTP, SHP1, SHP2, CD45, PTPε, PTPγ, PTPμ, PTPσ, Laforin, Cdc14A, VHR and VHX.
Table 1.
IC50 values (μM) of 7a-l for mPTPB
| Compound | R1 | IC50 (μM) |
|---|---|---|
| 7a | 2-CF3Ph | 1.2 ± 0.1 |
| 7b | 3-CF3Ph | 2.2 ± 0.2 |
| 7c | (1,1′-biphenyl)-4-yl | 2.4 ± 0.2 |
| 7d | Cyclohexyl | 3.6 ± 0.1 |
| 7e | 4-CF3OPh | 5.5 ± 0.5 |
| 7f | 4-CF3Ph | 7 ± 1 |
| 7g | Ph | 11 ± 1 |
| 7h | 3-FPh | 11 ± 1 |
| 7i | 4-FPh | 12 ± 2 |
| 7j | 2-FPh | 16 ± 2 |
| 7k | 3-OHPh | 18 ± 1 |
| 7l | 3-NH2Ph | 19 ± 1 |
To further increase the potency and selectivity of the optimized hydroxyindole carboxylic acid 7a for mPTPB, we employed a focused library approach to introduce molecular functionalities to its β-position in order to engage peripheral binding pockets adjacent to the active site (Figure 1). As discussed above, the rationale for this stems from our earlier observations that diversity elements appendaged to the β-position of the core enhance both inhibitor potency and selectivity,[16,23,24] likely through added interactions with secondary binding pockets adjacent to the PTP active site. However, the enhancement in binding affinity through this approach has been rather modest, likely due to the rigidity of the triazolidine linker formed as a result of the Click reaction. Indeed, no significant contact was observed between the triazolidine linker and the PTPs.[23,24] Here we chose amide chemistry because amide bond formation is one of the most efficient and reliable methods for library constructions; and it allows the use of the most common and commercially available amines and carboxylic acids as reactants. In addition, amide chemistry can be carried out in aqueous solution in the absence of deleterious reagents, thus allowing direct screening and identification of hits from the library. Finally, appropriately structured amide linker may impart flexibility necessary for optimal interaction with the enzyme. A short amine linker was attached to the β-position in compound 7a for tethering a structurally diverse set of carboxylic acids, aimed at capturing additional interactions with adjacent pockets surrounding the active site. In the interest of keeping the library to a reasonable size, we selected 102 commercially available carboxylic acids (Figure S1 in Supporting Information) that vary by molecular weight, charge, polarity, hydrophobicity, sterics, etc. and therefore provide a reasonable (albeit limited) structural diversity to increase the number and strength of noncovalent interactions between mPTPB and the inhibitor.
To construct the 102-member library, iodide 6a was coupled with tert-butyl prop-2-yn-1-ylcarbamate in the presence of Pd(PPh3)2Cl2 and CuI to afford alkyne 8 (Scheme 2). Alkyne 8 was hydrolyzed in 20% NaOH for 2 days to provide acid 9, which was treated with 20% TFA in DCM to produce Compound 10, as a result of exclusive conversion of alkyne to ketone under acid condition. Condensation of compound 10 with the 102 acids in the presence of HOBT, HBTU and TEA in DMF overnight furnished the amide library 11 in 96 well plates. The reactions were assessed by LC-MS, which indicated that ~70% of compound 10 were converted to target molecules.
Scheme 2.
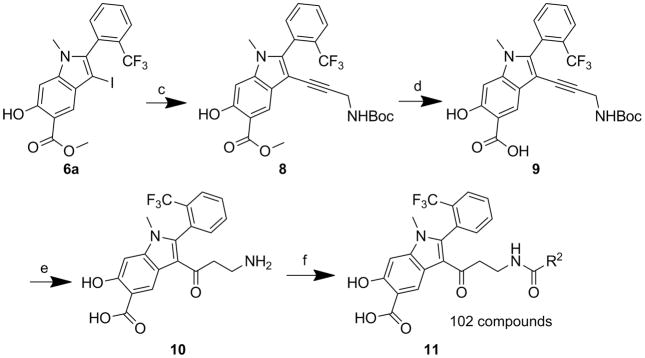
Synthesis of hydroxyindole carboxylic acid based library 11: a) corresponding phenylacetylene, Pd(PPh3)2Cl2, CuI, Et3N, DMF, 80 deg. 4–16 h, 80–90%; b) 20% NaOH, methanol, THF/H2O, rt, 48 h, 80–90%; c) tert-butyl prop-2-yn-1-ylcarbamate, Pd(PPh3)2Cl2, CuI, Et3N, DMF, 24 h, 68%; d) 20% NaOH, methanol, THF/H2O, rt, 48 h, 87%; e) 20% TFA in DCM, rt, 24 h, 91.5%; f) 102 acids, HOBT, HBTU, TEA, DMF, rt, 2h.
This library was directly screened at 1 μM concentration for inhibition of the mPTPB-catalyzed pNPP hydrolysis at pH 7 and 25 °C. Two of the library members, 11a and 11b, showed over 50% inhibition (81% for 11a and 52% for 11b) under these conditions (Table S1). Resynthesis of 11a and 11b confirmed that they were genuine inhibitors for mPTPB with IC50 values of 0.079 ± 0.010 μM and 0.68 ± 0.04 μM, respectively. As negative controls, we also resynthesized and HPLC purified compounds 11c, 11d and 11e, which showed less than 10% inhibition at 1 μM concentration (Table S1). As expected, the IC50 values for these three compounds were 3.3, 5.2, and >20 μM, much higher than those of 11a and 11b. The excellent agreement between the results from the primary screen and those from the resynthesized pure compounds indicated the success of the amide library approach. To determine the specificity of 11a, its inhibitory activity toward a large panel of PTPs, including the bacterial PTPs mPTPA and YopH, the mammalian cytosolic PTPs, SHP1, SHP2, PTP1B, TC-PTP, Lyp, HePTP and FAP1, the receptor-like PTPs, CD45, PTPε, PTPγ, PTPμ, and PTPσ, and the dual specificity phosphatases Laforin, VHR, VHX, VHZ, MKP3, and Cdc14A, were measured. As shown in Table 2, compound 11a exhibits at least 100-fold selectivity for mPTPB over all PTPs examined.
Table 2.
Selectivity of 11a against a panel of PTPs.
| PTP | IC50 (μM) | PTP | IC50 (μM) |
|---|---|---|---|
| mPTPB | 0.079 ± 0.01 | ||
| mPTPA | 7.8 ± 0.5 | SHP2 | 11.4 ± 0.4 |
| YopH | 16 ± 1 | Lyp | 12.2 ± 3.2 |
| CD45 | 21 ± 2 | TC-PTP | 16 ± 1 |
| PTPε | > 50 | HePTP | 10.7 ± 1.3 |
| PTPγ | 11 ± 2 | Laforin | 16 ± 3 |
| PTPμ | 30 ± 1 | VHR | 10.4 ± 0.7 |
| PTPσ | 56 ± 7 | VHX | 17 ± 1 |
| PTP1B | 12.8 ± 0.6 | VHZ | 16 ± 1 |
| FAP1 | 8.0 ± 0.7 | MKP3 | 14 ± 1 |
| SHP1 | 10.1 ± 0.3 | Cdc14A | > 50 |
To further characterize compound 11a as a mPTPB inhibitor, its IC50 value was determined under two different conditions: 1) 11a was pre-mixed with pNPP, and the reaction was initiated by the addition of mPTPB; and 2) 11a was pre-mixed with the enzyme for 30 minutes, and the reaction was initialized by the addition of pNPP. Irreversible, promiscuous nonspecific, or tight-binding inhibitors would be expected to exhibit significantly reduced IC50 values when they are pre-incubated with the enzyme. Similar IC50 values were obtained for compound 11a under these two conditions suggesting that 11a is a reversible mPTPB inhibitor. Further kinetic analyses indicated that 11a behaves as a noncompetitive inhibitor for mPTPB, with a Ki value of 50 ± 2 nM (Figure 2). Thus, compound 11a is the most potent and selective mPTPB inhibitor reported to date. Most importantly, compound 11a also displays excellent cellular activity as shown below.
Figure 2.

Lineweaver-Burk plot for Compound 11a mediated mPTPB inhibition. Compound 11a concentrations were 0 (●), 0.025 μM (○), 0.05 μM (▼), and 0.075 μM (▽) respectively
To facilitate the study of the cell biology of mPTPB without having to directly work with major human pathogens, we have established a murine macrophage Raw264.7 cell line ectopically expressing mPTPB. Using this system, we delineated two pathways by which mPTPB enables survival of the Mtb bacterium in the hostile environment of activated host macrophages.[16] Firstly, mPTPB can subvert innate immune responses by blocking ERK1/2-mediated IL-6 production. Secondly, macrophages expressing mPTPB are protected against programmed cell death when stimulated with IFN-γ and displayed a surge in AKT activity. As a result, the Raw264.7 cell line serves as a very convenient model system to evaluate the cellular efficacy of mPTPB inhibitors. We predicted that inhibition of mPTPB activity with compound 11a should reverse the altered immune responses induced by mPTPB by rescuing the ERK activity and decreasing the AKT activity in Raw264.7 cells.
As shown in Figure 3, Raw264.7 cells expressing mPTPB exhibited decreased IFN-γ stimulated ERK1/2 activation and increased AKT activity when compared to the vector control. Consistent with compound 11a being an mPTPB inhibitor, treatment of mPTPB expressing Raw264.7 macrophages with compound 11a restored the IFN-γ induced activation of ERK1/2 in a dose dependent manner (Figure 3). In addition, compound 11a normalized AKT activity in mPTPB cells to the same extend as the vector control cells (Figure 3). Moreover, the observed cellular activity by compound 11a also phenocopied those of several structurally unrelated small molecule mPTPB inhibitors.[16,17] Thus the ability of compound 11a to block the mPTPB-mediated cellular signaling is unlikely due to off-target effects. Taken together, the results demonstrate that compound 11a is highly efficacious in cell-based assays and capable of blocking mPTPB activity inside the cell.
Figure 3.
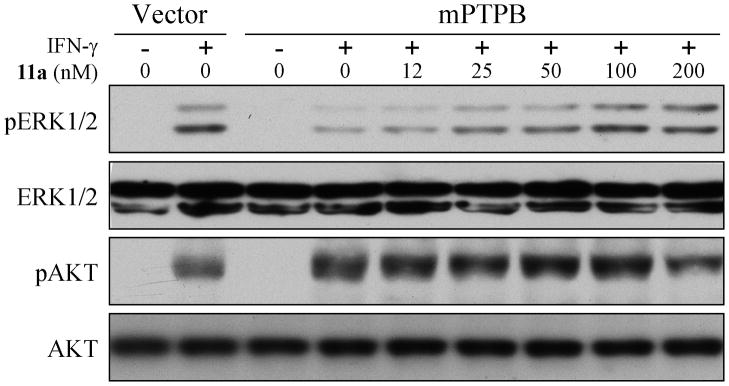
Cellular efficacy of mPTPB inhibitor 11a. Raw264.7 cells expressing mPTPB exhibited decreased IFN-γ stimulated ERK1/2 activation and increased AKT activity, and these can be reversed by treatment with mPTPB inhibitor 11a.
In summary, we describe a facile hydroxyindole carboxylic acid-based focused amide library approach designed to target both the PTP active site and a unique nearby pocket for enhanced affinity and selectivity. High throughput screening of the focused library let to the identification of a highly potent (Ki=50 nM) and selective (more than 2 orders of magnitude of selectivity against a large panel of PTPs) inhibitor 11a for mPTPB, an essential virulence factor for Mycobacterium tuberculosis. Importantly, compound 11a possesses highly efficacious cellular activity and is capable of reversing the altered immune responses induced by mPTPB in a murine macrophage Raw264.7 cell line. Therefore, 11a offers promise as an innovative therapeutic starting point for the development of potential anti-TB drugs. The results further support that the hydroxyindole-based carboxylic acid chemistry platform may provide a solution to overcome the inhibitor specificity and bioavailability issues that has plagued the PTP drug discovery field for many years. Given the ease and versatility of the amide chemistry based library approach described herein, we expect that the strategy can be used to identify cell permeable, potent and selective inhibitors for other members of the PTP superfamily.
Supplementary Material
Footnotes
This work was supported by in part the National Institutes of Health Grant CA152194.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.((Please delete if not appropriate))
Contributor Information
Dr. Li-Fan Zeng, Department of Biochemistry and Molecular Biology, Indiana, University School of Medicine, 635 Barnhill Drive, Indianapolis, Indiana 46202 USA
Dr. Jie Xu, Department of Biochemistry and Molecular Biology, Indiana, University School of Medicine, 635 Barnhill Drive, Indianapolis, Indiana 46202 USA
Dr. Yantao He, Department of Biochemistry and Molecular Biology, Indiana, University School of Medicine, 635 Barnhill Drive, Indianapolis, Indiana 46202 USA
Dr. Rongjun He, Department of Biochemistry and Molecular Biology, Indiana, University School of Medicine, 635 Barnhill Drive, Indianapolis, Indiana 46202 USA
Li Wu, Department of Biochemistry and Molecular Biology, Indiana, University School of Medicine, 635 Barnhill Drive, Indianapolis, Indiana 46202 USA. Chemical Genomics Core Facility, Indiana University, School of Medicine, 635 Barnhill Drive, Indianapolis, Indiana 46202 USA
Andrea M. Gunawan, Chemical Genomics Core Facility, Indiana University, School of Medicine, 635 Barnhill Drive, Indianapolis, Indiana 46202 USA
Prof. Dr Zhong-Yin Zhang, Email: zyzhang@iupui.edu, Department of Biochemistry and Molecular Biology, Indiana, University School of Medicine, 635 Barnhill Drive, Indianapolis, Indiana 46202 USA. Chemical Genomics Core Facility, Indiana University, School of Medicine, 635 Barnhill Drive, Indianapolis, Indiana 46202 USA.
References
- 1.Tonks NK. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 2.Zhang ZY. Curr Opin Chem Biol. 2001;5:416–423. doi: 10.1016/s1367-5931(00)00223-4. [DOI] [PubMed] [Google Scholar]
- 3.Ostman A, Hellberg C, Bohmer FD. Nat Rev Cancer. 2006;6:307–320. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 4.Singh R, Rao V, Shakila H, Gupta R, Khera A, Dhar N, Singh A, Koul A, Singh Y, Naseema M, Narayanan PR, Paramasivan CN, Ramanathan VD, Tyagi AK. Mol Microbiol. 2003;50:751–762. doi: 10.1046/j.1365-2958.2003.03712.x. [DOI] [PubMed] [Google Scholar]
- 5.Singh R, Singh A, Tyagi AK. Tuberculosis (Edinb) 2005;85:325–335. doi: 10.1016/j.tube.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 6.Koul A, Herget T, Klebl B, Ullrich A. Nat Rev Microbiol. 2004;2:189–202. doi: 10.1038/nrmicro840. [DOI] [PubMed] [Google Scholar]
- 7.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 8.Weide T, Arve L, Prinz H, Waldmann H, Kessler H. Bioorg Med Chem Lett. 2006;16:59–63. doi: 10.1016/j.bmcl.2005.09.051. [DOI] [PubMed] [Google Scholar]
- 9.Nören-Müller A, Reis-Correa I, Prinz H, Rosenbaum C, Saxena K, Schwalbe H, Vestweber D, Cagna G, Schunk S, Schwarz O, Schiewe H, Waldmann H. Proc Natl Acad Sci USA. 2006;103:10606–10611. doi: 10.1073/pnas.0601490103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reis-Correa I, Noren-Muller A, Ambrosi H, Jakupovic S, Saxena K, Schwalbe H, Kaiser M, Waldmann H. Chem Asian J. 2007;2:1109–1126. doi: 10.1002/asia.200700125. [DOI] [PubMed] [Google Scholar]
- 11.Soellner MB, Rawls KA, Grundner C, Alber T, Ellman JA. J Am Chem Soc. 2007;129:9613–9615. doi: 10.1021/ja0727520. [DOI] [PubMed] [Google Scholar]
- 12.Tan LP, Wu H, Yang PY, Kalesh K, Zhang X, Hu M, Srinivasan R, Yao S. Org Lett. 2009;11:5102–5105. doi: 10.1021/ol9023419. [DOI] [PubMed] [Google Scholar]
- 13.Nören-Müller A, Wilk W, Saxena K, Schwalbe H, Kaiser M, Waldmann H. Angew Chem Int Ed. 2008;47:5973–5977. doi: 10.1002/anie.200801566. [DOI] [PubMed] [Google Scholar]
- 14.Beresford NJ, Mulhearn D, Szczepankiewicz B, Liu G, Johnson ME, Fordham-Skelton A, Abad-Zapatero C, Cavet JS, Tabernero L. J Antimicrob Chemother. 2009;63:928–936. doi: 10.1093/jac/dkp031. [DOI] [PubMed] [Google Scholar]
- 15.Vintonyak VV, Warburg K, Kruse H, Grimme S, Hübel K, Rauh D, Waldmann H. Angew Chem Int Ed. 2010;49:5902–5905. doi: 10.1002/anie.201002138. [DOI] [PubMed] [Google Scholar]
- 16.Zhou B, He Y, Zhang X, Xu J, Luo Y, Wang Y, Franzblau SG, Yang Z, Chan RJ, Liu Y, Zheng J, Zhang ZY. Proc Natl Acad Sci USA. 2010;107:4573–4578. doi: 10.1073/pnas.0909133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Zhou B, Zhang S, Wu L, Wang Y, Franzblau SG, Zhang ZY. ACS Med Chem Lett. 2010;1:355–399. doi: 10.1021/ml1001135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barr AJ. Future Med Chem. 2010;2:1563–1576. doi: 10.4155/fmc.10.241. [DOI] [PubMed] [Google Scholar]
- 19.Zhang ZY. Annu Rev Pharmacol Toxicol. 2002;42:209–234. doi: 10.1146/annurev.pharmtox.42.083001.144616. [DOI] [PubMed] [Google Scholar]
- 20.Puius YA, Zhao Y, Sullivan M, Lawrence DS, Almo SC, Zhang ZY. Proc Natl Acad Sci USA. 1997;94:13420–13425. doi: 10.1073/pnas.94.25.13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarmiento M, Wu L, Keng YF, Song L, Luo Z, Huang Z, Wu GZ, Yuan AK, Zhang ZY. J Med Chem. 2000;43:146–155. doi: 10.1021/jm990329z. [DOI] [PubMed] [Google Scholar]
- 22.Liang F, Huang Z, Lee SY, Liang J, Ivanov MI, Alonso A, Bliska JB, Lawrence DS, Mustelin T, Zhang ZY. J Biol Chem. 2003;278:41734–41741. doi: 10.1074/jbc.M307152200. [DOI] [PubMed] [Google Scholar]
- 23.Yu X, Sun JP, He Y, Guo XL, Liu S, Zhou B, Hudmon A, Zhang ZY. Proc Natl Acad Sci USA. 2007;104:19767–19772. doi: 10.1073/pnas.0706233104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, He Y, Liu S, Yu Z, Jiang ZX, Yang Z, Dong Y, Nabinger SC, Wu L, Gunawan AM, Wang L, Chan RJ, Zhang ZY. J Med Chem. 2010;53:2482–2493. doi: 10.1021/jm901645u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.