

Background: Although universally conserved, ω is the only nonessential subunit of RNA polymerase, and its function is mostly undetermined.
Results: Dominant lethal mutants of ω were isolated and analyzed genetically and biochemically.
Conclusion: Mutations in ω that alter its structure interfere with RNA polymerase catalytic properties.
Significance: Results suggest a critical role for ω during transcription, bypassing the effects of compensatory proteins.
Keywords: Bacterial Genetics, Bacterial Transcription, Protein Conformation, Protein-Protein Interactions, RNA Polymerase, Dominant Lethal, Unstructured Protein, ω Subunit of RNA Polymerase
Abstract
The widely conserved ω subunit encoded by rpoZ is the smallest subunit of Escherichia coli RNA polymerase (RNAP) but is dispensable for bacterial growth. Function of ω is known to be substituted by GroEL in ω-null strain, which thus does not exhibit a discernable phenotype. In this work, we report isolation of ω variants whose expression in vivo leads to a dominant lethal phenotype. Studies show that in contrast to ω, which is largely unstructured, ω mutants display substantial acquisition of secondary structure. By detailed study with one of the mutants, ω6 bearing N60D substitution, the mechanism of lethality has been deciphered. Biochemical analysis reveals that ω6 binds to β′ subunit in vitro with greater affinity than that of ω. The reconstituted RNAP holoenzyme in the presence of ω6 in vitro is defective in transcription initiation. Formation of a faulty RNAP in the presence of mutant ω results in death of the cell. Furthermore, lethality of ω6 is relieved in cells expressing the rpoC2112 allele encoding β′2112, a variant β′ bearing Y457S substitution, immediately adjacent to the β′ catalytic center. Our results suggest that the enhanced ω6-β′ interaction may perturb the plasticity of the RNAP active center, implicating a role for ω and its flexible state.
Introduction
The bacterial RNAP2 holoenzyme is a multisubunit complex that performs the essential function of gene transcription (1, 2). In Escherichia coli, the core of RNAP contains two α subunits (αI and αII) and three distinct subunits designated as β, β′, and ω (3–6). The core RNAP associates with one of the various alternative σ subunits to form the holo-RNAP to initiate transcription from specific promoter sequences in the bacterial genome (7, 8).
Although extensive information is available on the contribution of various β, β′, and α subunits in the primary functioning of bacterial RNA polymerase (9, 10), the biological role of the ω subunit is less clear (11). The ω subunit encoded by the rpoZ gene is the smallest constituent of RNAP with a molecular mass of 10 kDa and is found in near stoichiometric amounts in preparations of both core and holo-RNAP from E. coli (3, 12). Although identified in the initial years of RNAP research, ω was not considered to be a bona fide subunit mainly because of two major observations. In vivo deletion of rpoZ is tolerated in bacterial cells (13) unlike deletion of the other subunits of RNAP. In addition, reconstitution of E. coli RNAP was achieved with purified α, β, and β′ subunits (14), thus pointing at the redundancy of ω.
However, two experiments in the last decade allowed us to contemplate for the role of ω. First, tethering experiment demonstrated that when ω is covalently linked to a DNA-binding protein it is able to activate transcription (15). On the other hand, the crystal structures of Thermus aquaticus RNAP (5) and that of E. coli RNAP published recently (16) confirm the presence of ω as a subunit of RNAP beyond ambiguity. Such contention was further supported by the analysis of the sequences of several bacterial genomes where the presence of rpoZ was noticed in addition to its similarity to a subunit of eukaryotic RNAP, Rpb6 (17), and archaeal RNAP, RpoK (18).
Previously we had observed that RNAP isolated from a ω-less strain of E. coli co-purifies with global chaperone GroEL, and attempts to remove GroEL ultimately destroy the enzyme (19). This indicates, albeit tenuously, that ω acts like a chaperone for RNAP. Interestingly, when the crystal structure was determined (5), it was mentioned that the arrangement of ω vis-à-vis β′ subunit was such that its role as a chaperone is conceivable. The ω subunit cross-links exclusively with β′ subunit in RNAP (20), and its association with the β′ subunit promotes the binding of ω-β′ with α2β subassembly (21). An analysis of the T. aquaticus RNAP core combined with functional studies has further defined the features of the ω/β′ interface in the RNAP core (17), leading to the proposal that ω may promote assembly and/or stability of β′ in RNAP by functioning as a clamp that latches the N-terminal half of β′ to its C terminus to conformationally constrain β′. The surface of ω subunit interacts extensively with the double ψ β-barrel (DPBB) domain of β′ subunit (22), which makes up the catalytic center of RNAP, suggesting an important role of ω subunit assembly for RNAP catalysis. Intriguingly, the E. coli RNAP crystal reveals an alternative conformation of the ω C-terminal tail that is very different from that of T. aquaticus RNAP (17), indicating a possible differential regulatory aspect of ω that has yet to be deciphered.
Other than its role in mediating effective assembly of RNAP, a physiological link between ω and the stringent response has been reported. This suggestion is based on the observation that in vitro a ω-less RNAP is rendered non-responsive to ppGpp, the effector of the stringent response, and that the presence of DksA, a coeffector (23), or externally added ω (24) rescues this defect. In addition, in strains lacking ω, expression of the relA promoter is impaired (25). Furthermore, recent crystal structures of the RNAP-ppGpp complex and a biochemical study showed that ppGpp binds at the interface between β′ and ω subunits, and the N terminus of ω is involved in transcription regulation mediated by ppGpp (26–28).
In this study, we used an alternative approach to delineate the function exclusive to ω. The genetic screen used acts to counter the influences of factors like GroEL and DksA known to elicit compensatory effects and complicate the elucidation of ω functionality. We have thus isolated a set of rpoZ variants whose conditional expression in vivo is associated with lethality. Apart from the lethality, all the mutant proteins exhibit a unique feature of structural reorganization into a more compact conformation. Studies with one variant, ω6, show its enhanced association with β′ without impeding the assembly of RNAP but resulting in inactivation of RNAP. As we probed into the various steps of transcription, it is evident that the enzyme with mutant ω is defective in a step of transcription initiation subsequent to the initial binding to the promoter. On a preformed elongation complex in the presence of RNA primer, the enzyme can catalyze transcription. Results from extragenic suppressor and in vitro reconstitution studies suggest that structural flexibility of ω is crucial for maintaining an active RNAP catalytic center. Changes that reorient ω to a more structured entity possibly affect RNAP plasticity by holding the DPBB catalytic domain of β′ in an unfavorable conformation.
MATERIALS AND METHODS
Bacterial Strains
All strain used in the study have been listed in Table 1. The bacterial strains DH5α, MG1655, and PD1 constructed by phage-based transduction (29) are derivatives of E. coli K12 and were used in genetic studies. All strains were propagated on Luria Bertani (LB) broth and agar. The rpoZ::kan allele was sourced from the strain JW3624 (30). The BL21(DE3) strain (31) was used for protein overproduction and purification. The antibiotics ampicillin and spectinomycin were used for selection of plasmid-borne antibiotic resistance phenotypes, namely AmpR and SpecR, respectively, at appropriate concentrations, and d-glucose and l-arabinose were used for repressing and inducing, respectively, the Para promoter (32) at the indicated concentrations.
TABLE 1.
List of strains and plasmids in this study
A brief description of each and their origin are given.
| Name | Description | Source/Ref. |
|---|---|---|
| Strains | ||
| DH5α | fhuA2 Δ(argF-lacZ) U169 phoA glnV44 Φ80 Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | |
| MG1655 | Wild-type K12 | |
| PD1 | MG1655 rpoZ::kan | This work |
| JW3624 | rpoZ::kan allele was sourced from the strain for construction of PD1 | |
| 30 | ||
| Bl21(DE3) | dcm ompT hsd S (rB− mB−) galλ (DE3) | 31 |
| RL916 | E. coli rpoC chromosomally His6-tagged | Gift from R. Landick |
| Plasmids | ||
| pBAD18 | Arabinose-inducible protein expression vector | 32 |
| pHYD3011 | Modified pBAD18 | Gift from J. Gowrishankar |
| pRPZ | rpoZ+ cloned in pHYD3011 | This work |
| pCL1920 | Low copy number vector bearing pSC101 replicon | 40 |
| pHYD535 | Plasmid pCL1920 with 10.2-kb HindIII DNA fragment bearing the rplL+ and the rpoB+C+ genes sourced from plasmid pDJJ12 | Gift from R. Harinarayan; 41 |
| pRPZM6 | rpoZ+ N60D cloned in pHYD3011 | This work |
| pRPZM2 | rpoZ+ D36Y,N60D cloned in pHYD3011 | This work |
| pRPZM15 | rpoZ+ A85T cloned in pHYD3011 | This work |
| pRPZM9 | Silent mutation in rpoZ+ cloned in pHYD3011 | This work |
| pRPC1 | rpoCY457S pHYD535 | This work |
| pET21b | Protein expression vector | Novagen |
| pRPC3 | rpoCY457S cloned in pET21b | This work |
| pRPC2 | rpoC cloned in pET21b | This work |
| pRW308 | For expression of RpoC | 38 |
| pETα | For expression of His6-RpoA | 21 |
| pGETB | For expression of RpoB | 43 |
| pARC8112 | For expression of RpoD | 44 |
| pAR1432 | T7A1 promoter fragment | 49 |
Isolation of Dominant Lethal rpoZ Variants
The open reading frame (ORF) encoding ω, rpoZ+ (wild type), was PCR-amplified from the chromosome of the strain MG1655 with a primer pair (5′-TGTGGAGCTTTTTACATATGGCACGAGTAACTTCAGCAATAG-3′ and 5′-ACAAGGGCGACCCGCTAAGCTTTTAACGACGACCTTCAGCAATAG-3′) and was cloned into the NdeI and HindIII sites of pHYD3011 (modified pBAD18 (32)) to generate plasmid pRPZ rendering the expression of rpoZ+ under the control of the Para promoter. With the genomic DNA of strain MG1655 as a template and the above mentioned primers, we generated pools of PCR-amplified rpoZ amplicons using error-prone PCR utilizing alterations in PCR conditions (33–36). The combined pool of rpoZ PCR amplicons was cloned into pHYD3011 (selectable marker AmpR), and the ligation mixture was transformed into DH5α. Transformants were isolated on LB agar plates supplemented with ampicillin and 0.2% d-glucose, and 2000 transformants were individually tested for their inability to grow on LB plates with ampicillin and 0.2% l-arabinose. Those that did not grow in l-arabinose medium but grew well on d-glucose medium were considered as bearing plasmids that expressed potential dominant lethal versions of rpoZ. Using plasmid DNA isolated from the counterpart colony on d-glucose LB plates, we tested by retransformation that the phenotype of dominant lethality was plasmid-borne. The rpoZ ORF from the chosen transformants was cloned into the vector pHYD3011 and sequenced after establishing the dominant lethal phenotype. The N60D substitution was recreated on the rpoZ+ ORF by site-directed mutagenesis using the megaprimer method (37). The sequences of mutagenic primer and flanking primer are 5′-GAAGGTCTGATCGACAACCACATC-3′ and 5′-AAGCTTTTAACGACG-3′, respectively. The mutagenized PCR amplicon was cloned in pHYD3011 identically to wild-type and mutagenized amplicon pools.
Total Cellular RNA Isolation
Primary cultures of DH5α cells bearing pRPZ and pRPZM6 were grown in the presence of 0.02% glucose for 5 h at 37 °C in LB. 1% of the primary culture was washed and subcultured in 0.02% l-arabinose in LB and grown for 3 h. A600 was normalized for the two cultures. Total cellular RNA was isolated using an RNeasy Mini kit (Qiagen). The RNA was checked by 1.2% formaldehyde agarose gel electrophoresis. For the uninduced sample, primary culture was subcultured in 0.02% d-glucose-containing LB.
ω-β′ and ω6-β′ Association Studies
Strain BL21(DE3) was used for protein preparation. Plasmids pRPZ and pRPZM6 were transformed in BL21(DE3) cells. Cultures of the transformed derivatives were grown to an A600 of 0.6, induced with 0.002% l-arabinose for 6–8 h at 37 °C, and purified using the protocol described previously (12). Plasmid pRW308 (38) was used for overproduction and purification of β′ using a previously published protocol (39). ω6-β′ complex formation was assessed by both immunoprecipitation and gel-based experiments. Purified proteins were mixed in a 1:1 molar ratio in denaturation buffer (6 m urea, 50 mm Tris HCl, pH 7.9, 200 mm KCl, 10 mm MgCl2, 10 mm ZnCl2, 10% glycerol, 1 mm EDTA, 10 mm DTT added fresh) and incubated for 1 h at 4 °C. The total protein concentration was kept at 0.5 mg ml−1. The mixture was then dialyzed against reconstitution buffer (50 mm Tris-HCI, pH 7.9, 300 mm KCl, 10 mm MgCl2, 10 μm ZnCl2, 0.1 mm EDTA, 1 mm DTT added fresh, 20% glycerol) at 4 °C to allow the formation of the complex. The dialyzed sample was spun at 10,000 × g at 4 °C for 15 min to settle debris, and the supernatant containing the complex was collected.
To detect protein-protein interaction, the supernatant containing ω6-β′ complex was electrophoresed on a 6% Tris borate-polyacrylamide gel. Western blot with anti-ω antibody identified the ω6-β′ complex species in a parallel gel run. A gel slice cut from the former gel identified to be ω6-β′ complex based upon migration of the immunodetected signal was minced, boiled with SDS loading buffer, and loaded onto an 8–20% gradient SDS-polyacrylamide gel. The protein species corresponding to ω6 and β′ were identified by silver staining as well as Western blot. The interaction was also confirmed by immunoprecipitation (data not shown).
Surface Plasmon Resonance (SPR) Studies
SPR studies to obtain quantitative estimates of ω-β′ and ω6-β′ associations were carried out in a Biacore 3000 instrument at 25 °C. Purified β′ was immobilized on a CM5 chip according to the manufacturer's recommendations. Solutions of both ω and ω6 were flowed over the chip separately in running buffer (10 mm HEPES, pH 7.5, 150 mm NaCl, 1 mm EDTA). Binding experiments used samples of at least four different concentrations of ω and ω6. After every injection of ω or ω6, the sensor chip surface was regenerated by rapid injection of 2–10 μl of 0.01–0.04% SDS. The data were analyzed globally by fitting both the association and dissociation phases simultaneously (BIAevaluation software, version 4.1) to a 1:1 (Langmuir) model. Uncertainties were calculated using three sets of curves for each of the two samples with each set comprising four curves of various concentrations of ω and ω6.
Isolation of the rpoC2112 Suppressor Allele
Plasmid pHYD535 is a derivative of the plasmid pCL1920 bearing the pSC101 replicon (40) and contains a 10.2-kb HindIII DNA fragment bearing the rplL+ and the rpoB+C+ genes sourced from plasmid pDJJ12 (41). pHYD535 was subjected to in vitro hydroxylamine mutagenesis (42), and the pool of mutagenized plasmid DNA was used to transform DH5α cells bearing pRPZM6. l-Arabinose-resistant (AraR) transformants able to survive the l-arabinose-induced lethality of rpoZM6 were selected on LB plates containing ampicillin, spectinomycin (for selection of the SpecR marker of pHYD535), and 0.02% l-arabinose.
One AraR transformant was isolated, and its AraR phenotype co-segregated with the SpecR marker on pHYD535 in DH5α cells bearing the plasmid pRPZM6. The plasmid so recovered was designated pRPC1 and is a derivative of pHYD535. DNA sequencing of the 10.2-kb insert in pRPC1 revealed a base alteration that would create Y457S substitution in rpoC+ (rpoC2112).
The Y457S amino acid substitution in β′ was created by site-directed mutagenesis of rpoC on plasmid pHYD535 using a primer pair (5′-CTGGTTTGTGCGGCATCTAACGCCGACTTCGATGGT-3′ and 5′-CCGGCAGCGGATTGTGCTAGCTCCGACGGGAGCAAATCCGTGAAAGATTTATTA-3′) with high fidelity Phusion (Thermo Scientific) DNA polymerase, and the amino acid substitution was verified by DNA sequencing. From pRPC1, rpoC2112 was amplified with a primer pair (5′-CCGGCAGCGGATTGTGCTAGCTCCGACGGGAGCAAATCCGTGAAAGATTTATTA-3′ and 5′-GTTTTTACGTTATTTGCGGATTAACTCGAGCTCGTTATCAGAACCGCCCAGACC-3′) and cloned into the BamHI-XhoI site of pET21b to generate plasmid pRPC3, which allows for overproduction of β′2112-C-His6 polypeptide. The same primer pair was used to clone β′ ORF identically in pET21b, generating pRPC2 to allow for overproduction of β′-C-His6. All PCRs described in this section were done using high fidelity Phusion (Thermo Scientific) DNA polymerase.
Reconstitution of RNAP
Individual subunits of RNAP α-C-His6 (21), β (43), and σ (44) were overproduced in BL21(DE3) using plasmids pETα, pGETB, and pARC8112, respectively, following previously published protocols. The β′-C-His6 and β′2112-C-His6 subunits were overproduced from plasmids pRPC2 and pRPC3, respectively (21). His6-tagged RNAP isolated from strain RL916 was purified using a procedure described previously (45). Reconstitution of core as well as RNA polymerase holoenzyme was essentially done as described previously (46). The reconstituted polymerase preparations were further clarified by passing through a heparin-Sepharose column equilibrated in TGED (10 mm Tris-HCl, pH 7.9 at 4 °C, 0.1 mm EDTA, 0.1 mm DTT, 5% glycerol) and 150 mm NaCl. The RNAP was eluted with TGED and 800 mm NaCl.
Promoter DNA Binding
An electrophoretic mobility shift assay for DNA binding was performed using an 85-bp 5′-GAATTCAATTTAAAAGAGTATTGACTTAAAGTCTAACCTATAGGATACTTACAGCCAGAGAGAGGGAGAAGGGAATCGGGGATCC-3′ double-stranded DNA fragment bearing T7A1 promoter. Promoter DNA was end-labeled using γ-32P-labeled ATP by polynucleotide kinase. Polymerase binding to detect the stable complex with promoter was carried out in a reaction mixture containing 45 mm Tris borate, 5 mm magnesium acetate, 0.1 mm DTT, 50 mm KCl, 50 μg ml−1 BSA, 5% glycerol, 2 pmol of labeled DNA, and polymerase in a 10-μl reaction for 15 min at 30 °C. Heparin was added to the mixture at a final concentration of 50 μg ml−1 to remove any nonspecific DNA-polymerase complex. The reaction products were separated in a 4% non-denaturing polyacrylamide gel.
In Vitro Gel-based Single Round Transcription Assay
The abortive transcription assay to detect first nucleotide bond formation was done as described (47). The single round in vitro gel-based transcription assay was performed as reported previously (48). Plasmid pAR1432 was used for the preparation of T7A1 promoter DNA template (49).
Reconstitution of Elongation Complex and Determining Its Transcriptional Activity
For in vitro reconstitution of elongation complex, the 30-nucleotide-long template strand and 9-mer RNA scaffold were designed as described previously (50). The reconstitution of elongation complex and transcription assay were carried out as described before (50).
RESULTS
Isolation of Dominant Lethal ω Variants and Rationale of the Genetic Screen
This work originated from our observation that ω-less RNAP has some inherent weakness that is not apparent in vivo as it secures protection from GroEL. Consequently, any phenotypic peculiarity that appears to be associated with mutational variation in ω gets masked by the global chaperone. We reasoned that if ω played a role in one or more steps of the RNAP transcription cycle, including those mediating promoter binding, opening, escape, RNA chain initiation, and elongation, then a mutation in ω that interfered with any of such essential steps might produce a lethal phenotype that would occur even in the presence of the wild-type rpoZ (encoding ω) allele.
To isolate such dominant lethal variants of ω, we generated libraries of rpoZ products obtained after in vitro PCR mutagenesis. The PCR products were cloned under the transcriptional control of a Para promoter-bearing plasmid, rendering expression of cloned rpoZ variants conditional. This screen allows selection of ω variants where the wild-type protein gets titrated by the faulty counterpart in vivo and GroEL is prevented from eliciting its rescue function on RNAP. The expression of dominant lethal rpoZ variants would be repressed in the presence of d-glucose (leading to their isolation) and induced in the presence of l-arabinose (32). A population of transformants of the strain DH5α bearing a library of PCR-mutagenized rpoZ genes was isolated in the presence of d-glucose in LB medium. Each transformant was screened for its inability to grow in the presence of l-arabinose. Such mutants were found at a frequency of 0.75%. We sequenced the rpoZ genes of four plasmids bearing dominant lethal rpoZ mutants whose conditional expression by l-arabinose at concentrations ranging from 0.002 to 0.2% produced a marked lethality. Most of the mutations were found to be clustered in the C-terminal tail region of the protein. One plasmid bore a base alteration in rpoZ leading to an N60D amino acid substitution in ω (pRPZM6; encoding ω6), whereas ω produced from the other two plasmids contained an A85T (pRPZM15; encoding ω15) substitution and a double D36Y and N60D substitution (pRPZM2; encoding ω2) (Table 2). We also isolated a dominant lethal variant bearing a silent third base alteration (GCC→GCT) marking generation of a rare codon for the 82nd amino acid (pRPZM9; encoding ω9).
TABLE 2.
List of ω variants indicating the resident nucleotide change and corresponding amino acid change of each mutated rpoZ
The percentage of α- helicity of each protein calculated with K2D2 software from their CD profile has been tabulated.
| Mutants | Nucleotide change | Amino acid change | α-Helicity |
|---|---|---|---|
| % | |||
| Wild-type ω | 15 | ||
| ω6 | A→G | N60D | 82 |
| ω2 | G→T | D36Y | 80 |
| A→G | N60D | ||
| ω15 | C→T | A85T | 60 |
| ω9 | G→A | Silent mutation | 61 |
Properties of the ω6 Variant
Because the N60D substitution was isolated twice and independently, we studied the properties of ω bearing this alteration, designated as ω6, in greater detail. Although the expression of rpoZ+ in the presence of l-arabinose produced no discernable alterations in growth phenotypes in the genetic backgrounds of DH5α, MG1655, and PD1 (MG1655 rpoZ::kan), expression of rpoZM6 (encoding ω6) yielded a dominant lethal phenotype in the above mentioned genetic backgrounds (Fig. 1, A, B, and C). The N60D amino acid substitution was recreated by site-specific mutagenesis of rpoZ+ to yield a similar dominant lethal phenotype. In addition, upon expression from the Para promoter, the levels of ω and ω6 were comparable on an SDS-polyacrylamide gel (data not shown). The N60D alteration occurs in the CR3 conserved region of ω (17) (Fig. 1D).
FIGURE 1.

The dominant lethal phenotype of ω6 and the location of the N60D substitution of ω6 in the ω/β′ interface. Cultures of DH5α (A), MG1655 (B), and PD1 (MG1655 rpoZ::kan (C) bearing plasmids pHYD3011 (Vector), pRPZ (encoding ω), and pRPZM6 (encoding ω6) were spotted at the indicated dilutions on LB agar plates containing 0.02% d-glucose (Glu) and 0.02% l-arabinose (Ara) with the appropriate antibiotic selection. D, location of the N60D mutation in relation to the postulated conserved regions (CR1–3; boxed) of ω (adapted from Minakhin et al. (17)).
The growth profile of DH5α/pRMZ6 in the presence of l-arabinose shows a marked reduction in cell viability (Fig. 2A). In an attempt to understand the extreme weakness of the mutants, the total RNA was isolated from l-arabinose (0.02%)-induced DH5α/pRZM6 cells. The total cellular RNA was also extracted from DH5α bearing pRPZ grown in l-arabinose as control. The A600 values of the induced cultures were normalized to have equal amounts of cells prior to RNA isolation. The comparative RNA profile indicates a decrease of the mRNA pool upon induction (Fig. 2B). This result implicated a possible defect in the transcription machinery in the mutant strain that eventually leads to cell death.
FIGURE 2.

Phenotypic effects of ω6-mediated lethality. A, growth curve of DH5α/pRPZM6 in the presence and absence of 0.02% l-arabinose. B, comparison of total cellular RNA profile from DH5α cells with plasmid compliment indicated at the top of each lane. Lanes 1 and 2 show RNA isolated from culture grown with 0.02% glucose (uninduced); lanes 3 and 4 show RNA isolated from 0.02% l-arabinose-induced culture.
Acquisition of Extensive Secondary Structure by ω6
The mutant protein ω6 (Fig. 3A) bearing N60D alteration results in an increment in molecular mass of ω by 0.98–1 Da that we have tested and verified in an independent study involving mass spectrometry (MS) (51). MS analysis also ensured that the isolated ω6 was 100% mutant variety with no detectable contamination of the wild-type ω (51). ω6 was found to be stable compared with ω, which tends to precipitate upon storage at 4 °C. Far-UV CD spectra of ω and ω6 revealed that ω6 had acquired extensive secondary structure in comparison with ω. The estimated percentage of α-helicity using K2D2 (52) software was 82% for ω6 and 15% for ω (Fig. 3B and Table 2).
FIGURE 3.

A, purified ω6. Lane M, molecular weight marker. B, comparative far-UV CD profile of ω (solid line) and ω6 (dashed line). deg, degrees. C, E. coli ω structure. The N- and C-terminal domains of ω and amino acid residues Asn60 and Ala85 are indicated. D, the ω and β′ interaction. A surface representation of the E. coli RNAP is shown. The surface of ω is partially transparent to see the surface behind ω. The four separated interfaces of β′ for ω binding are indicated (DPBB domain, yellow; residues 614–615, light blue; residues 905–913, green; resides 1360–1361, red). The orientation of ω is the same as that in the right panel of C.
As mentioned earlier, ω associates with the β′ subunit of RNAP (Fig. 3D and supplemental Movie 1). Thus, the structural alteration of ω6 prompted us to check whether the ω6-β′ interaction remained unaltered. To this effect, purified β′ and ω6 protein were allowed to reconstitute, any complex formed was tested in a gel-based assay wherein a preformed ω6-β′ complex was subjected to electrophoresis in a non-denaturing 6% polyacrylamide gel (Fig. 4A), and a similar gel run in parallel was probed with anti-ω antibody (Fig. 4B). The protein band corresponding to the position of the Western signal was excised from the former gel, and its constituents were separated and detected on an 8–20% gradient SDS-polyacrylamide gel and identified appropriately by anti-ω and anti-β′ antibodies (Fig. 4, C and D). The ω6-β′ interaction was further validated by an immunoprecipitation experiment showing that mutant ω associates as successfully with β′ as its wild-type counterpart (data not shown).
FIGURE 4.
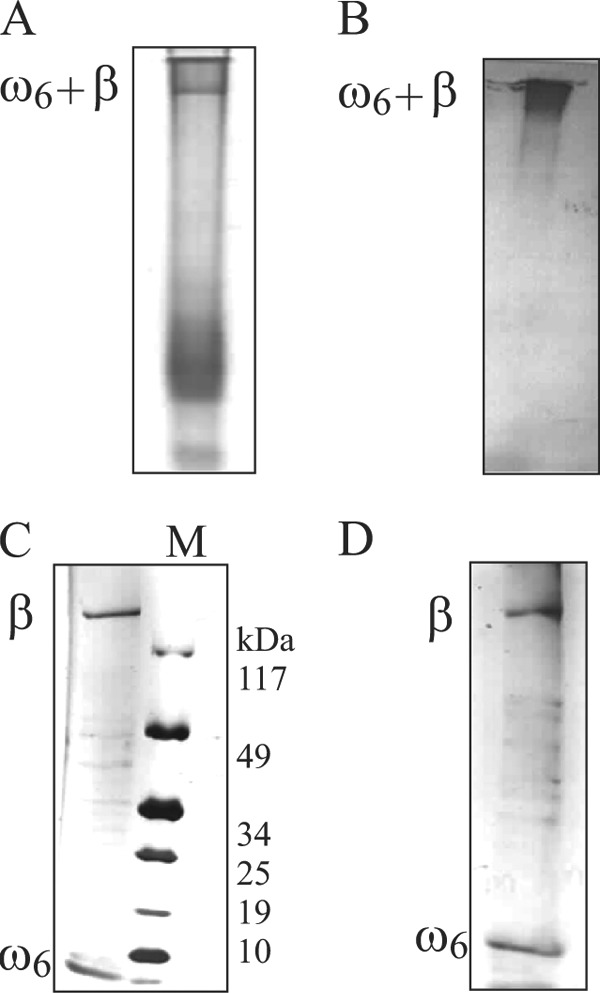
ω6-β′ association studies. Shown are 8–20% gradient SDS-PAGE of the ω6-β′ (A) and the position of ω6-β′ complex in A inferred by immunoblotting with anti-ω antibody (B). The gel slice bearing ω6-β′ complex in A was excised, and its contents were electrophoresed on an 8–20% gradient SDS-polyacrylamide gel (C), and the constituents of the preformed ω6-β′ complex were identified by immunoblotting with anti-ω and anti-β′ antibodies (D). M denotes the molecular weight marker.
The ω-β′ and ω6-β′ associations were further compared quantitatively by SPR studies wherein a range of concentrations of both proteins were flowed over immobilized β′. SPR analyses indicated that ω6 possessed ∼10-fold higher affinity for β′ as compared with ω with a significant increment in its rate of association (Table 3). It is worth mentioning that the SPR study shows a very small association rate constant (ka), which is indicative of a very large conformational change in the β′. This is expected because it is known that ω confers stability to β′ upon interaction.
TABLE 3.
Kinetic parameters of the ω-β′ and ω6-β′ interactions determined by surface plasmon resonance
χ2 values representing the corresponding “goodness of fit” are shown. S.D. is given.
| ka | kd | KD | χ2 value | |
|---|---|---|---|---|
| m−1 s−1 | s−1 | m | ||
| β′-ω | 89.0 ± 1.5 | 0.95 × 10−3 ± 0.02 | 1.07 × 10−5 ±0.2 | 0.045 |
| β′-ω6 | 341 ± 55 | 1.46 × 10−3 ± 0.15 | 4.28 × 10−6 ± 0.15 | 0.195 |
Reconstitution of RNAP with ω6
We reconstituted the holo-RNAP bearing ω and ω6 (ωRNAP and ω6RNAP, respectively) in vitro (see “Materials and Methods”) to monitor RNAP assembly and observed that both core and holoenzyme can be reconstituted in the presence of ω and ω6 (Fig. 5, A and B).
FIGURE 5.
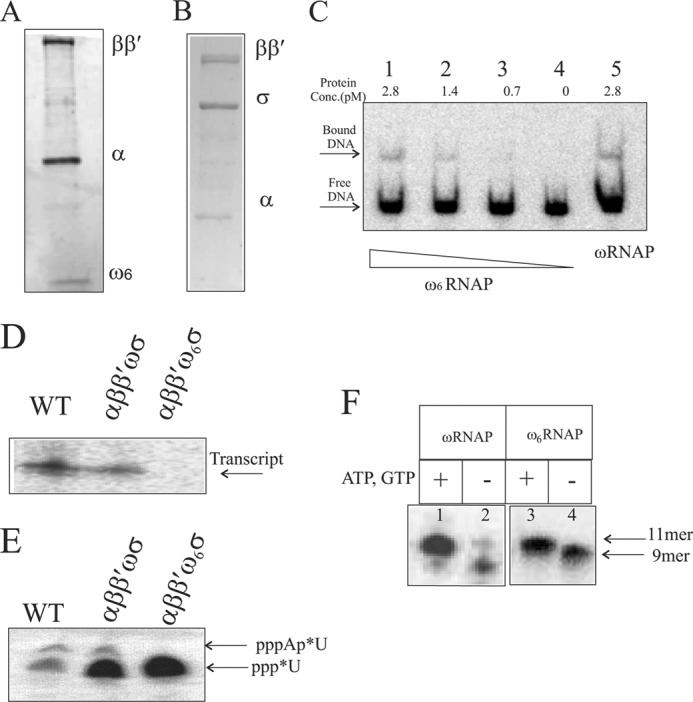
In vitro reconstitution and transcription activity of the ω6-bearing RNAP holoenzyme. Shown are the subunit compositions of reconstituted core RNAP with ω6 on 8–20% gradient SDS-PAGE (A) and reconstituted holo-RNAP with ω6 on10% SDS-PAGE (B). C, promoter DNA-polymerase complex formation. Lanes 1–4 show the indicated concentration of reconstituted ω6RNAP forming a heparin-resistant DNA-protein complex; lane 5 shows the complex formed by reconstituted wild-type polymerase. The free DNA and DNA-protein complex are marked with arrows. D, T7A1 promoter-specific in vitro single round transcription assay generating transcript with purified RNAP holoenzyme isolated from cells (WT) and holoenzyme versions of reconstituted ωRNAP and ω6RNAP. Enzyme and promoter concentrations used are 2 and 0.2 pmol, respectively. E, abortive transcription assay detecting first phosphodiester bond formation by 20% urea-PAGE. F, transcription activity of ωRNAP/ω6RNAP on preformed elongation complex with synthetic coding strand and 9-mer RNA primer along with bound enzyme. Lanes 1 and 3, with additional nucleotide added forming 11-mer transcript; lanes 2 and 4, no addition of nucleotide showing 9-mer RNA primer in elongation complex with template DNA strand and bound enzyme.
To ascertain whether the reconstituted ω6RNAP binds to promoter DNA, an electrophoretic mobility shift assay was performed. Radiolabeled T7A1 promoter DNA fragment (85 bp) was incubated with RNAP enzyme preparations, and complex formation was checked. ω6RNAP was found to form heparin-resistant stable complexes with labeled promoter fragment comparable with those formed by the reconstituted ωRNAP (Fig. 5C).
An RNAP Preparation Bearing ω6 Is Defective in Transcription Initiation
The transcriptional activity of the reconstituted RNAP preparations was tested in an in vitro heparin-resistant single round transcription assay. ω6RNAP was found to be inactive in mediating transcription at the T7A1 promoter (Fig. 5D). On the other hand, ωRNAP exhibited activity comparable with that displayed by an RNAP preparation isolated from the bacterial cell (Fig. 5D). ω6RNAP was found to be defective in the formation of the first phosphodiester bond in the transcription assay when only the first and the second (radiolabeled) nucleotides were provided (Fig. 5E), leading to the notion that ω6RNAP is defective for initiation of transcription. On a preformed elongation complex where the first few nucleotides were provided in the form of RNA primer, ω6RNAP was able to resume transcription (Fig. 5F).
Lethality of ω6 Is Suppressed in Cells Expressing the rpoC2112 Suppressor Mutation Encoding β′2112
To further the understanding of cellular lethality of ω6, we sought to obtain extragenic suppressors in gene(s) encoding RNAP subunits that may potentially suppress the cellular lethality imposed by the expression of rpoZM6. Our search for an extragenic suppressor was somewhat directed given the altered association of ω6 with β′. For this purpose, we subjected the rpoB+C+ genes cloned in pHYD535, a derivative of the low copy (pSC101 replicon) plasmid pCL1920, to hydroxylamine mutagenesis. In pHYD535, the expression of the rplL+ and rpoB+C+ genes that are located within a 10.2-kb HindIII fragment sourced in turn from the plasmid pDJJ12 (41) is expected to occur from the plasmid-borne Plac promoter. Nevertheless, the presence of plasmid pHYD535 rescued the thermosensitive growth phenotype of a strain bearing an rpoC(ts) mutation (data not shown). In this suppressor search, the suppressor mutation is expected to exert its phenotypic effect in a dominant manner to overcome the lethality of ω6 and in turn yield an l-arabinose-resistant (AraR) growth phenotype with the chromosomal rpoB+C+ genes behaving as neutral genetic entities. The presence of plasmid pHYD535 on its own does not yield an AraR phenotype (Fig. 6A). Following hydroxylamine mutagenesis of plasmid pHYD535 and the transformation of this pool of plasmids into DH5α bearing plasmid pRPZM6, the source of Para-expressing ω6, we selected for transformants that may survive the lethality imposed by the l-arabinose-induced production of ω6 under appropriate l-arabinose-inducing conditions and antibiotic selection on LB agar plates. We isolated a transformant exhibiting a weak AraR phenotype indicative of partial suppression. The l-arabinose-induced lethality conferred by rpoZM6 is seen with l-arabinose concentrations ranging from 0.002 to 0.2%, and at lower concentrations of l-arabinose (0.002–0.1%), the AraR phenotype of the said transformant was marked (Fig. 6A). Because the AraR phenotype in the co-transformant segregated with the antibiotic marker of plasmid pHYD535, we sequenced the entire rpoB+C+ insert DNA present on the pHYD535 derivative (pRPC1). We found that pRPC1 bore a base alteration in the rpoC gene leading to Y457S amino acid substitution in rpoC (Fig. 6, A and B) occurring adjacent to the 458NADFDGD464 active site motif of β′ that chelates the catalytic Mg2+ metals (53, 54). A possible reason for the partial suppression (AraR) phenotype conferred by pRPC1 is that the levels of RpoB and RpoC in plasmid pHYD535 (and pRPC1) may be limiting because the rpoBC genes are under heterologous expression control differing from that found in their chromosomal context. Recreation of the cognate base alteration in the rpoC gene present in pRPC1 led to a similar regeneration of the AraR phenotype conferred by pRPC1 (data not shown). The suppressor rpoC allele was further cloned into pET21b (Novagen) to introduce a His6 tag for ease of purification devoid of contamination from the chromosomal wild-type β′. The suppressor rpoC allele and its corresponding protein are referred to as rpoC2112 and β′2112, respectively.
FIGURE 6.
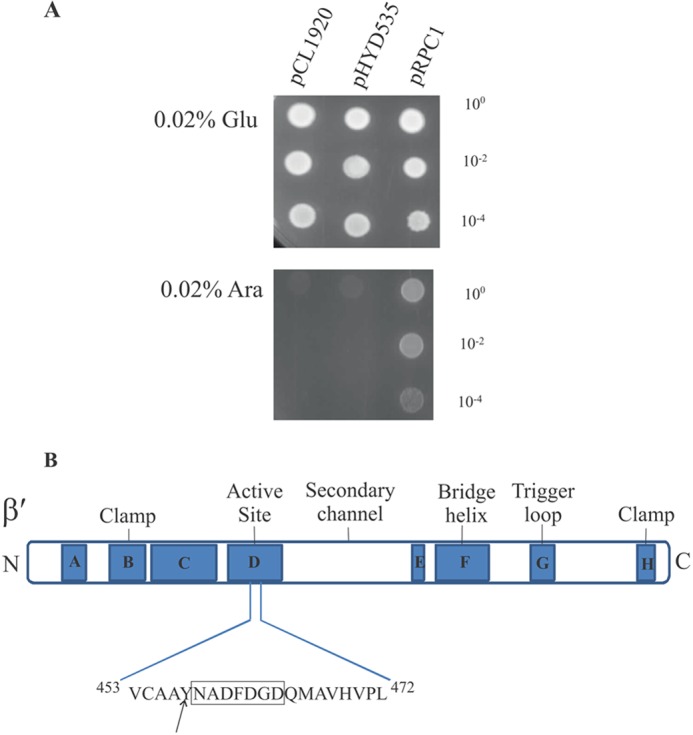
Suppression of the dominant lethal phenotype of rpoZM6 by the rpoC2112 allele. A, cultures of DH5α bearing pRPZM6 and either plasmid pCL1920 (Vector), pHYD535 (rpoB+rpoC+), or pRPC1 (rpoB+rpoC2112) were spotted at the indicated dilutions on LB agar plates containing 0.02% d-glucose (Glu) and 0.02% l-arabinose (Ara) with the appropriate antibiotic selection. B, sequence context of suppressor mutation in E. coli rpoC. The various domains and the active site sequence (boxed) are schematically represented (adapted from Landick and co-workers (68)).
Reconstituted ω6RNAP Bearing the β′2112 Subunit Is Transcriptionally Active in Vitro
Using purified β′ and β′2112 subunits in addition to α, wild-type ω, and σ70 subunits, we gauged the transcription capacities of reconstituted holo-RNAP preparations with in vitro single round gel-based transcription assays. ω6RNAP bearing the β′2112 subunit in contrast to ω6RNAP bearing the β′ subunit regained rifampicin-sensitive transcription activity at the T7A1 promoter template (Fig. 7A). In comparative transcriptional profiling (Fig. 7, B and C), ωRNAP bearing the β′2112 or β′ subunit displayed a transcriptional efficiency similar to that of the ω6-β′2112 RNAP preparation.
FIGURE 7.

Restoration of single round transcription by the ω6 RNAP holoenzyme bearing the β′2112 subunit. A, single round transcription assays at T7A1 promoter template were performed with reconstituted RNAP holoenzymes (enzyme and promoter concentrations are indicated) in the absence (−) or presence (+) of rifampicin (Rif). WT denotes purified RNAP holoenzyme isolated from cell extract. B, comparative transcription efficiencies of the WT RNAP holoenzyme and reconstituted RNAP holoenzymes. Enzyme and promoter concentrations used are 2 and 0.2 pmol, respectively. C, quantified relative transcriptional activity of the various RNAP preparations. Bar 1 shows transcription from isolated wild-type RNAP; bars 2–5 show transcription with reconstituted RNAP preparations. Transcription reactions contained 2 pmol of enzyme and 0.2 pmol of T7A1 promoter DNA. Error bars represent S.E.
Gain of Structure Is Common Feature of All Lethal ω Variants
We obtained far-UV CD spectra of other dominant lethal ω variants isolated in this study and found that in general all variants displayed a gain of helical content (Table 2). To our knowledge, such a result is unprecedented. This result indicates that not only are the WT residues at these positions somehow responsible for the unstructured state of the wild-type protein but also that each of these mutant residues favors folding (or assembly into multimers?) to impart a large structural change to the protein.
Correlation between Helical Content and Toxicity of Mutant ω Variants
Because all mutant ω variants that have been selected through the genetic screen show increment of helical content to different degrees, we tried to determine whether there is any correlation between the extent of toxicity and the percentage of helicity gained by the mutants. As the phenotype being scored in the genetic screen is lethality or absence of growth, establishing any direct quantitative correlation can be challenging. To this end, we performed an indirect genetic assessment. Plasmids bearing two of the mutants that show different helical content, ω6 (82%) and ω15 (60%), were introduced in wild-type DH5α. The cells were grown in the presence of l-arabinose (0.02%) until the A600 reached 0.2, allowing expression of the toxic protein. The cultures were then washed twice with LB, equal amount of cells were used to inoculate fresh LB with 0.2% glucose, and recovery of the cells as a function of time was monitored. We found that cells expressing more helical ω (ω6) recovered at a slower rate (Fig. 8). From the figure, it can be seen that the change in rate of growth between the two mutants is small; however, they are consistent over several experiments. This indicates that the more helical ω is comparatively more toxic than the less helical variant.
FIGURE 8.
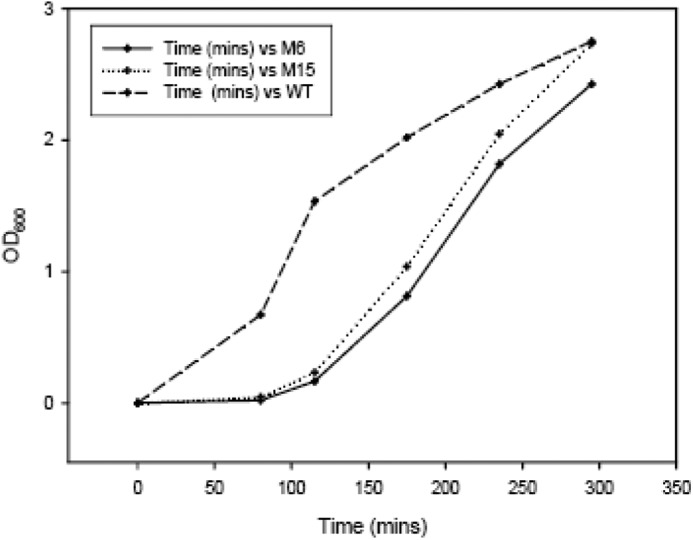
Recovery profile of DH5α cells with various ω-bearing plasmids. DH5α cells were transformed with pRPZM15 (M15), pRPZM6 (M6), and pRPZ (WT) and allowed to grow in the presence of 0.02% l-arabinose until A600 for each culture reached 0.2. Then the cultures were washed twice with LB and following A correction allowed to grow in the presence of glucose. The recovery has been plotted as A600 versus time.
DISCUSSION
Decades of extensive research have given us a clearer picture of how RNAP machinery functions, but the role of its smallest constituent subunit has remained controversial (11). Declared redundant from the very beginning, ω has resurfaced now and again with intriguing observations, proclaiming the need for renewed investigation.
The Unusual Phenotype of a Dominant Lethal rpoZ Mutation
The phenotype resulting from the conditional expression of rpoZM6 (encoding ω6) is one that is counterintuitive because a deficiency of ω does not overtly affect bacterial growth (Fig. 1, A, B, and C). The physiological perturbation caused by ω6, therefore, must occur at a step(s) that is essential for bacterial viability, in this case gene transcription. In general, dominant lethal mutations can occur both in proteins that are essential and in those that participate in an essential cellular process in an apparently redundant manner. However, they all possess one of two properties: (a) either the mutant proteins in some manner interfere with the functioning of their wild-type counterparts or (b) they exert their effects directly on the essential process. For example, aspartate to alanine replacements in the conserved NADFDGD active site residues of the β′ subunit produce a dominant lethal phenotype (53, 54) thought to occur because of increased persistence of open complex formation by the mutant RNAP that impedes the action of wild-type RNAP. On the other hand, the E96D substitution in RecA (RecA*), a nonessential protein, produces a dominant lethal phenotype (55) that is postulated to occur because the impaired RecA* DNA dissociation impedes essential DNA transactions. Such mutations have also been referred to as toxic mutations (55), and the ω6 mutant isolated in this study falls in the RecA* type category.
The toxic mutant isolation strategy used here where plasmid-based overexpression of the protein is used to detect phenotypic alteration can be argued as an artificial situation inside the cell. Because chromosomal deletion of rpoZ does not elicit any phenotype, this system appears to be promising and one of the few options to study the exclusive role of ω. The lack of phenotype in the rpoZ-null strain also prohibited us to move the mutations in the chromosome. The reduction in the total RNA profile, which is marked by the reduction of mRNA synthesis (Fig. 2B), specifically highlights that in vivo global transcription of the cell has been affected due to the ω mutation.
ω6 Binds to β′ with Greater Affinity than ω
The route to obtaining an answer to the general question “why is ω6 toxic to the cell?” probably begins with our observation that the dissociation constant between ω6 and β′ is significantly lower than that between ω and β′, suggesting that the structural transition in ω6 is causal to this effect (Table 3). The similarity between this observation and the two examples of dominant lethality cited (53, 55) is striking. In all three cases, the genesis of the lethal event occurs via shifting the equilibrium of a naturally occurring transient interaction toward a more stable association by the mutant protein. Nevertheless, these studies suggest that, in addition to promoting the assembly of β′ (19), ω may play a second role(s) once it is lodged in the confines of the RNAP core.
ω6-mediated Catalytic Inactivation of RNAP
As one of the major functions of ω is to assemble α2β subcomplex with properly folded β′ subunit (19), we were undoubtedly surprised to note that ω6 did not impair the assembly of RNAP holoenzyme (Fig. 5A). We detected the association of σ70 in ω6RNAP, suggesting that the lethal effect of ω6 is not due to a loss of σ70-ω6RNAP core interactions (Fig. 5B). As we dissected each step in the transcription initiation by ω6RNAP, it became clear that the enzyme retains its ability to form promoter complex (Fig. 5C) but is unable to initiate transcription (Fig. 5E) and thus rendered defective (Figs. 5D and 7C). When the initiation step is bypassed, the protein is able regain its transcriptional activity. This explanation for the lethality is further strengthened by the isolation of the rpoC2112 (encoding β′2112) extragenic suppressor whose presence provides a substantial bypass of both ω6 lethality in vivo (Fig. 6A) and ω6-mediated blockade of transcription in vitro (Fig. 7A). Approximately 60% of the transcriptional activity was recovered compared with reconstituted wild-type RNAP (Fig. 7C). Interestingly, β′2112 bears the Y457S substitution located immediately adjacent to the conserved 458NADFDGD464 active site residues located on β′ that coordinates the catalytic Mg2+ metals (Fig. 6B and supplemental Movie 1).
A Structural Transition in ω6 Is Critical to Its Lethality
Perhaps the most striking correlate of the pathology of ω6 is the remarkable structural transition (Fig. 3B) it bears caused by N60D substitution. This result suggests that biological RNAP activity becomes in some manner sensitive to a large scale alteration in the intrinsic protein disorder present in ω. Analyses of domain structure of ω by limited proteolysis (21) have shown that ω possesses a structured N-terminal region corresponding to the first 40–50 amino acids of ω comprising the CR1 and CR2 domains (17) (Fig. 1D). Using current nomenclature that includes proteins of partial structural disorder (56–58), ω can be bracketed in the category of an intrinsically disordered protein.
The CD profiles of all dominant lethal ω variants show acquisition of substantial secondary structure (Table 2). The nature of the variants is quite remarkable considering that all mutants have a single mutation (except ω2) and show extensive structural alteration. Intriguingly, we also isolated a silent mutation in rpoZ (GCC-GCT at the 82nd codon of rpoZ) whose expression caused a dominant lethal phenotype, and remarkably, its cognate protein also showed increment in structural content compared with ω (Table 2). This seemingly perplexing observation can be rationalized as an example of a synonymous single nucleotide polymorphism, which marks substitution of an amino acid codon with its rare counterpart. As has been reported (59), this change may alter the conformation due to slowdown of the protein synthesis rate, hence influencing its catalytic property. This observation strongly suggests a general mechanism whereby ordering of the unstructured ω tail results in lethality. It is plausible that the ordered proteins may function through inactivation of RNAP beyond promoter binding as established for ω6. The observation also highlights the importance of the unstructured nature of ω. The intrinsic disorder in proteins and their functional significance have been a subject of considerable interest (60–62). Chaperone proteins in particular are known to possess interspersed unstructured elements (63). It has been suggested that flexibility provided by these elements is key to their role (63). The chaperone activity provided by ω to the largest subunit serves as an example of this phenomenon.
Implications for the Physiological Role of ω
Detailed structure analysis of the ω/β′ interface in E. coli RNAP (17) has revealed that a globular N-terminal domain of ω (residues 2–59) makes contacts with β′ subunit in four regions, namely regions D and G (residues 905–913), residues 614–615, and a region proximal to the terminus (residues 1360–1361), whereas the C-terminal extended α-helices of ω (from residue 61 to the C terminus) does not interact with β′ subunit (Fig. 3, C and D, and supplemental Movie 1). The Asn60 residue of ω is positioned at a junction between these two domains and within a cavity of the ω N-terminal domain, suggesting that the N60D mutation changes amino acid interactions of the N-terminal domain for acquisition of secondary structure of ω.
Region D of β′ forms the DPBB domain, which contains the NADFDGD active site residues to enable chelation of Mg2+ in the RNAP active center (22, 54). The DPBB domain also makes extensive contacts with ω, but its binding surface is located at the side opposite the RNAP active site. It is worth mentioning that the interface between ω and β′ regulates RNAP transcription both positively and negatively depending on promoters and σ factors (64). In addition, the DPBB domain is a hot spot of compensatory mutations for reducing the fitness cost associated with rifampicin-resistant mutations of Mycobacterial tuberculosis RNAP (65).
The catalytic site architectural specificities β-D, β-H, β′-D, and β′-G form distinct domains that surround the central motif (Fig. 6B). Mutational studies supported by cross-linking experiments suggest that distortion of the catalytic site results in varying degrees of inactivation of the enzyme (43). It has been well established that β′-G along with region F constitutes the flexible/mobile ensemble of the RNAP catalytic center mediating the placement of cognate NTPs and unidirectional RNA extension at the RNAP active center (66–68). The inactivated polymerase that is reconstituted in the presence of ω6 retains its ability to bind promoter segment, but subsequent steps prior to elongation are impaired (Fig. 5, C, D, E, and F). Thus, we postulate that the large structural alteration in ω6 may affect the flexibility of the β′ mobile elements in the RNAP catalytic center needed for transcription initiation and act as a transcriptional inhibitor. It appears that the C-terminal end of ω subunit is important for regulation. If ω subunit is acting as a receiver of signals triggered by binding of a small molecule like ppGpp (26–28) or structural change as in this case, then the difference between the stringent response elicited by T. aquaticus enzyme vis-à-vis that of E. coli may be explained. We further speculate that ω might act in a modular fashion and transmit the molecular signal via the DPBB region to the active site. On the other hand, it is possible that ω6 may directly interfere with the functioning of the RNAP catalytic center or of another RNAP subunit. In addition, a previous finding that overproduction of ω mediates suppression (17) of the thermosensitive growth phenotype of a certain (but not all) ts allele of rpoC (rpoCtsX) that encodes an extremely unstable β′ subunit appears to be consistent with the present observation that ω-β′ maintains active site plasticity. In this regard, the survival of a strain lacking ω may be explained on the basis that in its absence the associated GroEL (19) prevents a total structural collapse possibly of β′ and that new stability acquired by β′ leads to reduced transcriptional activity because of compromised plasticity of the RNAP catalytic center.
Acknowledgments
We thank Robert Landick, J. Gowrishankar, and R. Harinarayanan for gifts of strains and plasmids. We also acknowledge the anonymous reviewers for many helpful suggestions.
This work was supported in part by the Department of Biotechnology, Government of India (to D. C.), Centre of Excellence, Department of Biotechnology, Government of India (to A. A. S.), National Institutes of Health Grant GM087350-A1 (to K. M. S.), and a research fellowship from Council of Scientific and Industrial Research, Government of India (to P. S.).

This article contains supplemental Movie 1.
- RNAP
- RNA polymerase
- DPBB
- double ψ β-barrel
- SPR
- surface plasmon resonance.
REFERENCES
- 1. Chamberlin M., Berg P. (1962) Deoxyribonucleic acid-directed synthesis of ribonucleic acid by an enzyme from Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 48, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chamberlin M. (1977) in RNA Polymerase (Losick R., Chamberlin M., eds) pp. 17–67, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 3. Burgess R. R. (1969) Separation and characterization of the subunits of ribonucleic acid polymerase. J. Biol. Chem. 244, 6168–6176 [PubMed] [Google Scholar]
- 4. Travers A. A., Burgess R. R. (1969) Cyclic re-use of the RNA polymerase sigma factor. Nature 222, 537–540 [DOI] [PubMed] [Google Scholar]
- 5. Zhang G., Campbell E. A., Minakhin L., Richter C., Severinov K., Darst S. A. (1999) Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 Å resolution. Cell 98, 811–824 [DOI] [PubMed] [Google Scholar]
- 6. Vassylyev D. G., Sekine S., Laptenko O., Lee J., Vassylyeva M. N., Borukhov S., Yokoyama S. (2002) Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 Å resolution. Nature 417, 712–719 [DOI] [PubMed] [Google Scholar]
- 7. Gross C. A., Lonetto M., Losick R. (1992) in Transcriptional Regulation (McKnight S. L., Yamamoto K. R., eds) Vol. 1, pp. 129–176, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 8. Gruber T. M., Gross C. A. (2003) Multiple σ subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol. 57, 441–466 [DOI] [PubMed] [Google Scholar]
- 9. Darst S. A. (2001) Bacterial RNA polymerase. Curr. Opin. Struct. Biol. 11, 155–162 [DOI] [PubMed] [Google Scholar]
- 10. Borukhov S., Nudler E. (2008) RNA polymerase: the vehicle of transcription. Trends Microbiol. 16, 126–134 [DOI] [PubMed] [Google Scholar]
- 11. Mathew R., Chatterji D. (2006) The evolving story of the ω subunit of bacterial RNA polymerase. Trends Microbiol. 14, 450–455 [DOI] [PubMed] [Google Scholar]
- 12. Gentry D. R., Burgess R. R., (1990) Overproduction and purification of the ω subunit of E. coli RNA polymerase. Protein Expr. Purif. 1, 81–86 [DOI] [PubMed] [Google Scholar]
- 13. Gentry D., Xiao H., Burgess R., Cashel M. (1991) The ω subunit of Escherichia coli K-12 RNA polymerase is not required for stringent RNA control in vivo. J. Bacteriol. 173, 3901–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heil A., Zillig W. (1970) Reconstitution of bacterial DNA-dependent RNA-polymerase from isolated subunits as a tool for the elucidation of the role of the subunits in transcription. FEBS Lett. 11, 165–168 [DOI] [PubMed] [Google Scholar]
- 15. Dove S. L., Hochschild A. (1998) Conversion of the ω subunit of Escherichia coli RNA polymerase into a transcriptional activator or an activation target. Genes Dev. 12, 745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murakami K. S. (2013) The x-ray crystal structure of Escherichia coli RNA polymerase σ70 holoenzyme. J. Biol. Chem. 288, 9126–9134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Minakhin L., Bhagat S., Brunning A., Campbell E. A., Darst S. A., Ebright R. H., Severinov K. (2001) Bacterial RNA polymerase subunit ω and eukaryotic RNA polymerase subunit RPB6 are sequence, structural, and functional homologs and promote RNA polymerase assembly. Proc. Natl. Acad. Sci. U.S.A. 98, 892–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hirata A., Klein B. J., Murakami K. S. (2008) The x-ray crystal structure of RNA polymerase from Archaea. Nature 451, 851–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mukherjee K., Nagai H., Shimamoto N., Chatterji D. (1999) GroEL is involved in activation of Escherichia coli RNA polymerase devoid of ω subunit in vivo. Eur. J. Biochem. 266, 228–235 [DOI] [PubMed] [Google Scholar]
- 20. Gentry D. R., Burgess R. R. (1993) Cross-linking of Escherichia coli RNA polymerase subunits: identification of β′ as the binding site of ω. Biochemistry 32, 11224–11227 [DOI] [PubMed] [Google Scholar]
- 21. Ghosh P., Ishihama A., Chatterji D. (2001) Escherichia coli RNA polymerase subunit ω and its N-terminal domain bind full-length β′ to facilitate incorporation into the α2β subassembly. Eur. J. Biochem. 268, 4621–4627 [DOI] [PubMed] [Google Scholar]
- 22. Iyer L. M., Koonin E. V., Aravind L. (2003) Evolutionary connection between the catalytic subunits of DNA-dependent RNA polymerases and eukaryotic RNA-dependent RNA polymerases and the origin of RNA polymerases. BMC Struct. Biol. 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vrentas C. E., Gaal T., Ross W., Ebright R. H., Gourse R. L. (2005) Response of RNA polymerase to ppGpp: requirement for the ω subunit and relief of this requirement by DksA. Genes Dev. 19, 2378–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Igarashi K., Fujita N., Ishihama A. (1989) Promoter selectivity of Escherichia coli RNA polymerase: ω factor is responsible for the ppGpp sensitivity. Nucleic Acids Res. 17, 8755–8765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chatterji D., Ogawa Y., Shimada T., Ishihama A. (2007) The role of the ω subunit of RNA polymerase in expression of the relA gene in Escherichia coli. FEMS Microbiol. Lett. 267, 51–55 [DOI] [PubMed] [Google Scholar]
- 26. Ross W., Vrentas C. E., Sanchez-Vazquez P., Gaal T., Gourse R. L. (2013) The magic spot: a ppGpp binding site on E. coli RNA polymerase responsible for regulation of transcription initiation. Mol. Cell 50, 420–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mechold U., Potrykus K., Murphy H., Murakami K. S., Cashel M. (2013) Differential regulation by ppGpp versus pppGpp in Escherichia coli. Nucleic Acids Res. 41, 6175–6189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zuo Y., Wang Y., Steitz T. A. (2013) The mechanism of E. coli RNA polymerase regulation by ppGpp is suggested by the structure of their complex. Mol. Cell 50, 430–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miller J. H. (1992) A Short Course in Bacterial Genetics: a Laboratory Manual and Handbook for Escherichia coli and Related Bacteria, pp. 263–278, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 30. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., Mori H. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Studier F. W., Moffatt B. A. (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130 [DOI] [PubMed] [Google Scholar]
- 32. Guzman L. M., Belin D., Carson M. J., Beckwith J. (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eckert K. A., Kunkel T. A. (1990) High fidelity DNA synthesis by the Thermus aquaticus DNA polymerase. Nucleic Acids Res. 18, 3739–3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou Y. H., Zhang X. P., Ebright R. H. (1991) Random mutagenesis of gene-sized DNA molecules by use of PCR with Taq DNA polymerase. Nucleic Acids Res. 19, 6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cadwell R. C., Joyce G. F. (1994) Mutagenic PCR. PCR Methods Appl. 3, S136–S140 [DOI] [PubMed] [Google Scholar]
- 36. Chen J. C., Minev M., Beckwith J. (2002) Analysis of ftsQ mutant alleles in Escherichia coli: complementation, septal localization, and recruitment of downstream cell division proteins. J Bacteriol. 184, 695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ke S. H., Madison E. L. (1997) Rapid and efficient site-directed mutagenesis by single-tube 'megaprimer' PCR method. Nucleic Acids Res. 16, 3371–3372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weilbaecher R., Hebron C., Feng G., Landick R. (1994) Termination-altering amino acid substitutions in the β′ subunit of Escherichia coli RNA polymerase identify regions involved in RNA chain elongation. Genes Dev. 8, 2913–2927 [DOI] [PubMed] [Google Scholar]
- 39. Borukhov S., Goldfarb A., (1993) Recombinant Escherichia coli RNA polymerase: purification of individually overexpressed subunits and in vitro assembly. Protein Expr. Purif. 4, 503–511 [DOI] [PubMed] [Google Scholar]
- 40. Lerner C. G., Inouye M. (1990) Low copy number plasmids for regulated low-level expression of cloned genes in Escherichia coli with blue/white insert screening capability. Nucleic Acids Res. 18, 4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jin D. J., Gross C. A. (1988) Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J. Mol. Biol. 202, 45–58 [DOI] [PubMed] [Google Scholar]
- 42. Ou X., Blount P., Hoffman R. J., Kung C. (1998) One face of a transmembrane helix is crucial in mechanosensitive channel gating. Proc. Natl. Acad. Sci. U.S.A. 95, 11471–11475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mustaev A., Kozlov M., Markovtsov V., Zaychikov E., Denissova L., Goldfarb A. (1997) Modular organization of the catalytic center of RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 94, 6641–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sharma U. K., Chatterji D. (2008) Differential mechanisms of binding of anti-σ factors Escherichia coli Rsd and bacteriophage T4 AsiA to E. coli RNA polymerase lead to diverse physiological consequences. J. Bacteriol. 190, 3434–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kashlev M., Martin E., Polyakov A., Severinov K., Nikiforov V., Goldfarb A. (1993) Histidine-tagged RNA polymerase: dissection of the transcription cycle using immobilized enzyme. Gene 130, 9–14 [DOI] [PubMed] [Google Scholar]
- 46. Tang H., Severinov K., Goldfarb A., Ebright R. H. (1995) Rapid RNA polymerase genetics: one-day, no-column preparation of reconstituted recombinant Escherichia coli RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 92, 4902–4906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Johnston D. E., McClure W. R. (1976) in RNA Polymerase (Losick R., Chamberlin M., eds) pp. 413–428, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 48. Chatterji D., Fujita N., Ishihama A. (1998) The mediator for stringent control, ppGpp, binds to the β-subunit of Escherichia coli RNA polymerase. Genes Cells 3, 279–287 [DOI] [PubMed] [Google Scholar]
- 49. Dunn J. J., Studier F. W. (1983) Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol. 166, 477–535 [DOI] [PubMed] [Google Scholar]
- 50. Sidorenkov I., Komissarova N., Kashlev M. (1998) Crucial role of the RNA:DNA hybrid in the processivity of transcription. Molecular Cell 2, 55–64 [DOI] [PubMed] [Google Scholar]
- 51. Sabareesh V., Sarkar P., Sardesai A. A., Chatterji D. (2010) Identifying N60D mutation in ω subunit of Escherichia coli RNA polymerase by bottom-up proteomic approach. Analyst 135, 2723–2729 [DOI] [PubMed] [Google Scholar]
- 52. Perez-Iratxeta C., Andrade-Navarro M. A. (2008) K2D2: estimation of protein secondary structure from circular dichroism spectra. BMC Struct. Biol. 8, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zaychikov E., Martin E., Denissova L., Kozlov M., Markovtsov V., Kashlev M., Heumann H., Nikiforov V., Goldfarb A., Mustaev A. (1996) Mapping of catalytic residues in the RNA polymerase active center. Science 273, 107–109 [DOI] [PubMed] [Google Scholar]
- 54. Sosunov V., Zorov S., Sosunova E., Nikolaev A., Zakeyeva I., Bass I., Goldfarb A., Nikiforov V., Severinov K., Mustaev A. (2005) The involvement of the aspartate triad of the active center in all catalytic activities of multisubunit RNA polymerase. Nucleic Acids Res. 33, 4202–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Campbell M. J., Davis R. W. (1999) Toxic mutations in the recA Gene of E. coli prevent proper chromosome segregation. J. Mol. Biol. 286, 417–435 [DOI] [PubMed] [Google Scholar]
- 56. Paliy O., Gargac S. M., Cheng Y., Uversky V. N., Dunker A. K. (2008) Protein disorder is positively correlated with gene expression in Escherichia coli. J. Proteome Res. 7, 2234–2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dyson H. J., Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 58. Uversky V. N., Dunker A. K., (2010) Understanding protein non-folding. Biochim. Biophys. Acta 1804, 1231–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kimchi-Sarfaty C., Oh J. M., Kim I. W., Sauna Z. E., Calcagno A. M., Ambudkar S. V., Gottesman M. M. (2007) A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315, 525–528 [DOI] [PubMed] [Google Scholar]
- 60. Babu M. M., van der Lee R., de Groot N. S., Gsponer J. (2011) Intrinsically disordered proteins: regulation and disease. Curr. Opin. Struct. Biol. 21, 432–440 [DOI] [PubMed] [Google Scholar]
- 61. Dunker A. K., Brown C. J., Lawson J. D., Iakoucheva L. M., Obradović Z. (2002) Intrinsic disorder and protein function. Biochemistry 41, 6573–6582 [DOI] [PubMed] [Google Scholar]
- 62. Tompa P. (2005) The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 579, 3346–3354 [DOI] [PubMed] [Google Scholar]
- 63. Tompa P., Csermely P. (2004) The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 18, 1169–1175 [DOI] [PubMed] [Google Scholar]
- 64. Potrykus K., Cashel M. (2008) (p)ppGpp: still magical? Annu. Rev. Microbiol. 62, 35–51 [DOI] [PubMed] [Google Scholar]
- 65. Comas I., Borrell S., Roetzer A., Rose G., Malla B., Kato-Maeda M., Galagan J., Niemann S., Gagneux S. (2012) Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 44, 106–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nudler E., (2009) RNA polymerase active center: the molecular engine of transcription. Annu. Rev. Biochem. 78, 335–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang D., Bushnell D. A., Westover K. D., Kaplan C. D., Kornberg R. D. (2006) Structural basis of transcription: role of the trigger loop in substrate specificity and catalysis. Cell 127, 941–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vassylyev D. G., Vassylyeva M. N., Zhang J., Palangat M., Artsimovitch I., Landick R. (2007) Structural basis for substrate loading in bacterial RNA polymerase. Nature 448, 163–168 [DOI] [PubMed] [Google Scholar]