

Background: SOD1-enriched protein inclusions and Ca2+ overload are hallmarks in ALS-affected motor neurons. Ca2+ burden correlates with SOD1 aggregation in cellular models.
Results: Ca2+ induces conformational changes that enhance and shift SOD1 aggregation from fibrils toward amorphous aggregates.
Conclusion: SOD1 aggregation is enhanced and modulated by Ca2+.
Significance: Ca2+ can behave as a pathogenic effector in the formation of ALS proteinaceous inclusions.
Keywords: Amyloid, Amyotrophic Lateral Sclerosis (Lou Gehrig's Disease), Biophysics, Calcium, Circular Dichroism (CD), Electron Microscopy (EM), Infrared Spectroscopy, Neurodegenerative Diseases, Protein Aggregation, Superoxide Dismutase (SOD)
Abstract
Imbalance in metal ion homeostasis is a hallmark in neurodegenerative conditions involving protein deposition, and amyotrophic lateral sclerosis (ALS) is no exception. In particular, Ca2+ dysregulation has been shown to correlate with superoxide dismutase-1 (SOD1) aggregation in a cellular model of ALS. Here we present evidence that SOD1 aggregation is enhanced and modulated by Ca2+. We show that at physiological pH, Ca2+ induces conformational changes that increase SOD1 β-sheet content, as probed by far UV CD and attenuated total reflectance-FTIR, and enhances SOD1 hydrophobicity, as probed by ANS fluorescence emission. Moreover, dynamic light scattering analysis showed that Ca2+ boosts the onset of SOD1 aggregation. In agreement, Ca2+ decreases SOD1 critical concentration and nucleation time during aggregation kinetics, as evidenced by thioflavin T fluorescence emission. Attenuated total reflectance FTIR analysis showed that Ca2+ induced aggregates consisting preferentially of antiparallel β-sheets, thus suggesting a modulation effect on the aggregation pathway. Transmission electron microscopy and analysis with conformational anti-fibril and anti-oligomer antibodies showed that oligomers and amyloidogenic aggregates constitute the prevalent morphology of Ca2+-induced aggregates, thus indicating that Ca2+ diverts SOD1 aggregation from fibrils toward amorphous aggregates. Interestingly, the same heterogeneity of conformations is found in ALS-derived protein inclusions. We thus hypothesize that transient variations and dysregulation of cellular Ca2+ levels contribute to the formation of SOD1 aggregates in ALS patients. In this scenario, Ca2+ may be considered as a pathogenic effector in the formation of ALS proteinaceous inclusions.
Introduction
Amyotrophic lateral sclerosis (ALS)2 is a fatal neurodegenerative disease characterized by the selective degeneration of motor neurons in the spinal cord, brainstem, and cerebral cortex (1). Most cases of ALS are sporadic with no known genetic linkage, whereas ∼10% are associated with familial causes (fALS). Both forms share a similar neurodegeneration pattern and are clinically and pathologically indistinguishable (2), thus suggesting a common molecular mechanism. One of the common features in all ALS patients is the presence of cytoplasmic proteinaceous aggregates in motor neurons, suggested to play a determinant role in neuron toxicity, degeneration, and cell death (3–7). In this respect, inclusions enriched in mutant variants of superoxide dismutase-1 (SOD1) are a well established hallmark of fALS-SOD1-associated forms of the disease (7–11). However, sporadic ALS patients also contain wild type SOD1 in proteinaceous inclusions (12–15), which share aberrant conformations of mutant SOD1 clinical variants that are known to cause aggregation (16–18). In fact, expression of recombinant peptide fragments of wild type SOD1 in cultured cells results in insoluble fALS-SOD1-like conformers, suggesting that SOD1 is inherently prone to aggregate (19). Therefore, fALS mutations and environmental adverse conditions are probably triggers of SOD1 toxic aggregation, which seems to be an intrinsic property of this protein. Thus, wild type SOD1 is suggested to be involved in ALS pathology (20, 21), inducing toxicity that degenerates motor neurons (22).
SOD1 is a highly stable homodimeric protein that holds in each monomer a disulfide bridge as well as a copper/zinc binuclear site. Interestingly, SOD1 aggregates from a transgenic fALS mouse model tend to be metal-deficient and/or lack the disulfide bond (23, 24), raising the possibility that disease-causing mutations may deregulate and increase levels of SOD1 immature conformers. In agreement, the folding pathway of SOD1 clinical mutants has been suggested to favor the accumulation of metal-depleted (apo) monomeric intermediates (25), which are prone to aggregate (26–28). In fact, post-translational modifications are major structural determinants for SOD1 stability (29, 30). Each SOD1 chain folds into a β-barrel that is flanked by two major loops, the zinc and the electrostatic loops, which together shape the active site. In the absence of metal coordination, the β-barrel and dimer interface remain intact, but the associated loops become highly disordered (31–33). Further, reduction of the native disulfide bridge promotes dimer dissociation, yielding monomers with a high level of conformational flexibility (34, 35).
Immature SOD1 states alone are, however, insufficient to explain SOD1-linked ALS pathogenesis; not all ALS-related SOD1 variants evidence increased destabilization of the apo state (36) and reduced apo state (37) with respect to the wild type form. Thus, additional chemical and biological factors must account for the toxicity of SOD1 in ALS. One lead arises from the fact that, although ubiquitously expressed in all tissues (38, 39), SOD1 only aggregates within specific motor neurons in ALS. This preferential vulnerability suggests that unique properties within the affected neurons, besides SOD1 mutations, are likely to be mandatory for the onset of SOD1 aggregation. One such characteristic within the affected motor neurons is a vulnerability to calcium overload; indeed, these cells express highly calcium-permeable AMPA receptors (40–44) with concurrent low calcium buffering capacity due to a lack of the Ca2+-buffering proteins parvalbumin and calbindin (45, 46). In agreement, calcium ions accumulate in the spinal and brain stem motor neurons of ALS patients as well as in animal and cell models of ALS-SOD1 (47–53). Moreover, studies on these models have shown that Ca2+ overload promotes and correlates with SOD1 aggregation (54, 55). Interestingly, oculo-motor neurons, which are not affected in ALS, have a 5–6-fold higher Ca2+ buffering capacity than the spinal and brain stem ALS-vulnerable motor neurons (56). This particular observation suggests that Ca2+ may be a key factor in modulating SOD1 toxicity in ALS; indeed, endogenous Ca2+ levels are systematically elevated in the specific motor neurons in which SOD1 proteinaceous aggregates are found. In this study, we have thus addressed in vitro the effect of this metal ion on the aggregation mechanism of SOD1, and the results obtained suggest a link between elevated Ca2+ levels and SOD1 aggregation in ALS.
MATERIALS AND METHODS
Chemicals and Sample Preparation
All reagents were of the highest grade commercially available. SOD1 was expressed in E. coli BL21(DE3) strain and grown and purified as described (57). All SOD1 experiments were performed using the demetallated form (apo-SOD1). The preparation of apo-SOD1 was obtained following published procedures (58). Metal content of apo-SOD1 was confirmed using the colorimetric reagent Zincon (59). A Chelex resin (Bio-Rad) was used to remove contaminant trace metals from all buffer solutions and to maintain apo-SOD1 in the demetallated form. Concentration of SOD1 was determined using the extinction coefficient 10,800 cm−1 m−1 at 280 nm. The broad range of concentrations used throughout biophysical experiments relates to the specific requirements and limitations of each of the different techniques used.
Circular Dichroism (CD)
Far UV CD analyses were performed using a Jasco J-815 spectropolarimeter equipped with a Peltier-controlled thermostated cell support. CD spectra were the average of eight scans obtained by collecting data at 0.1 nm intervals from 260 to 190 nm. The results were expressed as mean residue molar ellipticity [θ] with units of degrees cm2/dmol, as calculated from the equation,
where [θ]obs is the ellipticity measured in millidegrees, MRW is the mean residue molecular weight, c is the protein concentration in mg/ml, and l is the optical path length of the cell in cm. Spectra were recorded with 30 μm apo-SOD1 samples in 50 mm Tris, pH 7.5, that were previously incubated overnight with increasing concentrations of CaCl2 at 37 °C and 600 rpm.
ATR-FTIR
Infrared spectra were performed on a Bruker IFS 66/S spectrometer equipped with a mercury/cadmium/telluride (MCT) infrared detector and a thermostatized Harrick BioATR II cell. All measurements were obtained in an ATR cell with 150 μm apo-SOD1 and 300 μm SOD1 aggregates at pH 7.5 formed in the absence and presence of 300 and 600 μm CaCl2, respectively. Each spectrum comprises the mean of 150 scans taken at a resolution of 2 cm−1. Spectra were corrected for the buffer and water vapor. Difference absorption spectra are the average of independent subtractions between three data sets of Ca2+-incubated and control samples. The 1750–1700 cm−1 region is shown to demonstrate the absence of major contributions from noise or water vapor artifacts, thus validating the data. Region assignments were based on typical absorption regions for specific secondary structure elements (60).
ANS Binding Assay
ANS fluorescence emission enhancement was evaluated in a BMG Fluostar Optima fluorescence plate reader using a 370-nm excitation filter and a 480-nm emission filter. Samples of 15 μm apo-SOD1 were prepared as triplicates in 50 mm Tris, pH 7.5, that were previously incubated overnight with increasing concentrations of CaCl2 at 37 °C and 600rpm in black 96-well plates (Nunc, catalog no. 732-2701). The ANS fluorescence emission spectrum was recorded in a Cary Varian Eclipse instrument possessing a Peltier-thermostated cell support.
Dynamic Light Scattering (DLS)
DLS measurements were carried out in a Malvern Zetasizer Nano ZS instrument equipped with a 4-megawatt helium-neon laser (632 nm). 60 μm apo-SOD1 samples in 50 mm Tris, pH 7.5, were incubated with different concentration ratios of CaCl2/SOD1, filtered through a 0.45-μm filter, and incubated overnight at 37 °C and 600 rpm before DLS analyses. A CaCl2/SOD1 molar ratio of 2 was used to compare the mean light scattering intensity variation over time with control. After 24, 48, 68, and 90 h of incubation, 50-μl aliquots were collected and measured in a 45-μl quartz cuvette (Hellma) at 37 °C. The operating procedure was set to 17 runs, each being averaged for eight measurements. The resulting data were analyzed using DTS (version 6.32) software (Malvern).
Thioflavin T Fluorescence Binding
Real-time ThT fluorescence (480 nm) was recorded using a BMG Fluostar Optima fluorescence plate reader upon excitation at 440 nm. Readings were taken every 7 min, and the well plates were subjected to 5 min of agitation at 600 rpm prior to each fluorescence measurement. Assays were performed in black 96-well plates (Nunc, catalog no. 732-2701). Samples were prepared as triplicates at a wide range of concentrations of apo-SOD1 (20–120 μm) with and without adding CaCl2 up to a molar ratio of Ca2+/SOD1 = 2 and mixed with a 2× molar ratio of ThT in 50 mm Tris, pH 7.5, at 37 °C. Final sample volume in each lane was 200 μl, and all wells contained one Teflon bead (⅛-inch diameter). Aggregation curve analyses were performed by fitting the data to the equation,
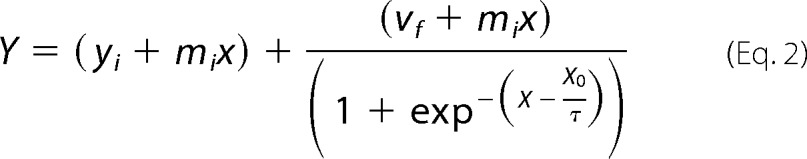 |
where Y corresponds to fluorescence intensity, x is the time, and X0 corresponds to the time of half-height of fluorescence intensity (t50). The lag phase is calculated by tlag = X0 − 2τ, and the apparent rate velocity (kapp) = 1/τ. Kinetic parameters have been determined from six independent determinations.
Dot Blotting Using Amyloid Conformational Antibodies
10 μl of SOD1 aggregates obtained at the plateau phase of each aggregation kinetic curve were dotted in triplicates onto PVDF membranes and probed with a 1:500 dilution of the anti-amyloid oligomer A11 antibody (AB9234, Merck Millipore) and 1:1000 for the anti-amyloid fibril OC antibody (AB2286, Merck Millipore) according to the manufacturer's instructions. Dots were visualized using a horseradish peroxidase-conjugated IgG secondary antibody with a chemiluminescence detection system (GE Healthcare). Images were recorded and analyzed using Quantity One analysis software from Bio-Rad.
Transmission Electron Microscopy
For visualization by TEM, 5 μl of 300 μm apo-SOD1 aggregates formed with and without adding CaCl2 up to a molar ratio of Ca2+/SOD1 = 2 were absorbed to carbon-coated collodion film supported on 400-mesh copper grids and negatively stained with 1% uranyl acetate. The grids were exhaustively visualized with a Jeol microscope (JEM-1400), operated at 80 kV.
RESULTS
Calcium Increases Apo-SOD1 β-Sheet Content and Hydrophobicity
We have started by investigating if Ca2+ has an effect on SOD1 native structure. This follows the evidence that protein aggregation does not necessarily require the formation of an unfolded conformer but may rather result from dynamic fluctuations around the native state, due to environmental variations, disruption of post-translation interactions, or mutations (61). In particular, we focused on effects on β-sheet structures and hydrophobicity changes because these properties are often altered in aggregation-prone conformers (62). The effects of increasing Ca2+ concentration on SOD1 secondary structure were investigated using far UV circular dichroism (Fig. 1A). We observed that incubation of the protein with CaCl2 at up to a molar ratio of Ca2+/SOD1 of 3 results in a roughly linear change of the signal of the negative band centered at 218 nm (Fig. 1A, inset), thus suggesting a direct correlation between the presence of Ca2+ and an increased content of SOD1 β-sheets. In a control experiment using NaCl, no change was observed. The impact of Ca2+ on SOD1 secondary structure was further analyzed by ATR-FTIR analysis. Indeed, the difference FTIR absorption spectrum in the amide I region, obtained from subtracting the Ca2+ incubated from the control SOD1 sample, also reveals secondary structure changes. An increase in β-sheet content (positive amplitude bands in the 1620–1630 cm−1 region) is observed alongside a decrease in randomness (negative amplitude bands in the 1642–1652 cm−1 region) (Fig. 1B). The observed variations are thus suggestive of conformational rearrangements taking place in the presence of Ca2+.
FIGURE 1.

Effect of Ca2+ on the secondary structure and hydrophobicity of apo-SOD1. A, far UV CD spectra of apo-SOD1 (−Ca2+) and upon overnight incubation with Ca2+ (+Ca2+) Inset, plot of the intensity of the SOD1-negative band centered at 218 nm, at increasing Ca2+/SOD1 ratios. B, ATR-FTIR difference spectrum in the amide I region of Ca2+-incubated SOD1 minus the control; positive peaks denote features increased in the presence of Ca2+. C, ANS fluorescence emission spectra of apo-SOD1 (−Ca2+) and upon incubation with Ca2+ (+Ca2+), overlaid with the emission spectrum of unbound ANS. See “Materials and Methods” for further details.
To verify if Ca2+ also promotes alterations on SOD1 hydrophobicity, we have used ANS, a fluorophore that interacts with exposed hydrophobic patches within β-sheeted structures (63). ANS is scarcely fluorescent when free in an aqueous neutral solution, emitting at 520 nm (Fig. 1C, trace a). However, a blue shift to around 480 nm as well as an increase in intensity of fluorescence emission are observed upon ANS binding to hydrophobic surfaces in proteins (64). In agreement with previous reports, apo-SOD1 binds ANS (Fig. 1C, trace b) (65); however, the presence of Ca2+ results in a ∼15% increase in ANS fluorescence emission (Fig. 1C, trace c). The intensity of ANS fluorescence emission was found to increase up to an excess molar ratio of Ca2+/SOD1 of 2. For higher molar ratios of Ca2+, the ANS emission tends to decrease, suggesting that fewer hydrophobic moieties are accessible (not shown). This could be an indication that intermolecular hydrophobic interactions are being formed at higher Ca2+ concentrations, thus decreasing the exposed hydrophobic regions available for ANS binding. This would be compatible with an oligomerization process through structural reorganization involving hydrophobic burial (66). Overall, the results indicate that in the presence of Ca2+, SOD1 adopts distinct conformational states that are β-sheet-enriched.
Calcium Promotes Oligomerization and Conformational Heterogeneity of Apo-SOD1
In order to investigate a possible link between the ANS results and the formation of intermolecular interactions at increasing Ca2+ concentrations, we have used DLS, a very sensitive technique for assessing changes in the oligomerization state of protein solutions (67). After 24 h of incubation at 37 °C and constant agitation, a monomodal size distribution profile by volume with a mean peak averaging at a hydrodynamic diameter of ∼5.4 ± 0.4 nm was observed for all samples, irrespective of the presence of Ca2+ up to a molar ratio of 4. However, the absence of a multimodal distribution by volume in DLS does not necessarily imply the absence of aggregates if the population of formed oligomeric species is very low relative to that of dimeric SOD1 (68, 69). We have thus probed for oligomeric species by comparing the mean hydrodynamic diameter (Z-average size), a parameter extremely sensitive to the presence of minor populations of larger oligomers, as well as the mean count rate parameter for evaluation of the scattering intensity. From this combined analysis, we determined an increase in the mean hydrodynamic diameter for the Ca2+-incubated SOD1 samples, from 6.5 ± 0.3 nm for the control to 7.9 ± 0.9, 10.1 ± 1.5, and 10.1 ± 2.5 nm for samples at Ca2+/SOD1 molar ratios of 1, 2, and 4, respectively. An increase in mean light scattering intensity was also observed upon increasing concentrations of added Ca2+, from 356 ± 18 nm for the control, to 377 ± 10, 439 ± 26, and 543 ± 31 nm for samples at Ca2+/SOD1 ratios of 1, 2, and 4, respectively. The results suggest a correlation between increasing Ca2+/SOD1 ratios and enhanced oligomerization (Fig. 2), in agreement with Ca2+ inducing aggregation and conformational heterogeneity on SOD1. The used Ca2+ concentrations (60–240 μm) bear some resemblance to physiological ones because intracellular micromolar transients of free Ca2+ are known to occur in vivo upon neuron stimulation (70–72).
FIGURE 2.

Impact of increasing Ca2+ concentrations on apo-SOD1 oligomerization. DLS analysis (mean size and count rate) of apo-SOD1 upon overnight incubation at 37 °C and stirring, at increasing Ca2+/SOD1 ratios. See “Materials and Methods” for further details. Error bars, S.D.
Calcium Enhances Apo-SOD1 Aggregation
In order to further characterize the effect of Ca2+ on the aggregation propensity of SOD1, we have analyzed the variation of the mean light scattering intensity over time (Fig. 3A). We determined that the presence of Ca2+ promotes an earlier formation of an aggregation nucleus (t < 48 h), whereas the onset of aggregation in the control (without Ca2+) occurs substantially later (t > 70 h). This suggests that the conformational changes induced by the presence of Ca2+ give rise to self-association-prone conformers that promote SOD1 oligomerization under physiological pH. The effect of Ca2+ on SOD1 aggregation kinetics was further investigated monitoring binding of thioflavin T, a fluorescent probe that recognizes diverse amyloid-like fibrils and amorphous aggregates (73–76) as well as oligomers and protofibers (77–79). In both cases a sigmoidal-type transition was observed, suggesting a nucleation-dependent aggregation mechanism (Fig. 3B). However, Ca2+ was shown to impact the aggregation profile of SOD1, namely the lag time and fibrillation rate (Table 1). This was evidenced by a decrease in the time required for the formation of the aggregation nucleus (which was decreased by ∼26 h in the presence of Ca2+) and by a decrease in the apparent elongation rate constants.
FIGURE 3.

Ca2+ enhances apo-SOD1 aggregation. A, aggregation profile of SOD1 monitored by mean light scattering intensity analysis over time, in the absence and in the presence of Ca2+. B, aggregation kinetics of SOD1 monitored by ThT fluorescence emission, in the absence and in the presence of Ca2+. −Ca2+, no CaCl2; +Ca2+, CaCl2/SOD1 = 2. See Table 1 for kinetic parameters and “Materials and Methods” for further details. Error bars, S.D.
TABLE 1.
Effect of Ca2+ on SOD1 aggregation kinetics under physiological pH
Apo-SOD1 aggregation (80 μm) was monitored by ThT fluorescence emission in 50 mm Tris, pH 7.5, with or without Ca2+ under 600 rpm agitation with a Teflon bead at 37 °C. (n = 6).
| Kinetic parameters | −Ca2+ | +Ca2+ |
|---|---|---|
| tlag (h) | 78 ± 12 | 52 ± 5 |
| t50 (h) | 98 ± 8 | 85 ± 8 |
| kapp (h−1) | 0.10 ± 0.03 | 0.058 ± 0.012 |
In order to obtain insight into SOD1 oligomers formed at the onset of the exponential phase (t = 48 h), we have compared the particle size distributions (Fig. 4A). In the control without calcium, although poorly populated, it is already possible to observe a broad distribution of oligomers (Fig. 4A, bottom). However, Ca2+ populates a pool of larger oligomers (Fig. 4A, top). Indeed, Ca2+ promotes the formation of an increased fraction of larger aggregates (>500 nm), whereas in the control, smaller aggregates (<500 nm) predominate (Fig. 4B).
FIGURE 4.

Ca2+ favors the formation of larger SOD1 aggregates. A, comparison of the particle size distributions of apo-SOD1 aggregates in the presence (top) and in the absence (bottom) of Ca2+ upon 48-h incubation at 37 °C and stirring. Note the broader distributions at lower particle diameters in the absence of Ca2+. B, relative distribution of total light scattering intensities arising from aggregated SOD1 species with a hydrodynamic diameter under and above 500 nm, for the Ca2+-incubated SOD1 and control. +Ca2+, CaCl2/SOD1 = 2. See “Materials and Methods” for further details. Error bars, S.D.
The effect of Ca2+ on SOD1 aggregation was also investigated under reducing conditions, a well established factor known to hasten SOD1 aggregation (28, 80) (Fig. 5). A similar effect is observed with respect to non-reducing conditions; further, with a reductant, Ca2+ also lowers SOD1 critical concentration for aggregation (Table 2). In summary, the presence of Ca2+ potentiates SOD1 aggregation, and this effect is observed irrespective of the redox status of the intramolecular disulfide bond (Cys57–Cys146).
FIGURE 5.

Ca2+ enhances apo-SOD1 aggregation also under reducing conditions. Aggregation kinetics of 80 μm apo-SOD1 with 10 mm tris(2-carboxyethyl)phosphine was monitored by ThT fluorescence emission, in the absence and in the presence of Ca2+. −Ca2+, no CaCl2; +Ca2+, CaCl2/SOD1 = 2. See Table 2 for kinetic parameters and “Materials and Methods” for further details.
TABLE 2.
Effect of Ca2+ on apo-SOD aggregation kinetics under reducing conditions
Apo-SOD1 aggregation was monitored by ThT fluorescence emission in 50 mm Tris, pH 7.5, plus 10 mm tris(2-carboxyethyl)phosphine with or without a 2 molar ratio concentration of Ca2Cl under agitation (600 rpm) with a Teflon bead at 37 °C. NF, no fibrillation observed. See “Materials and Methods” for further details.
| [SOD1] | −Ca2 |
+Ca2 |
||||
|---|---|---|---|---|---|---|
| tlag | t50 | kapp | tlag | t50 | kapp | |
| μm | h | h | h−1 | h | h | h−1 |
| 20 | NF | NF | NF | 16 ± 3 | 23 ± 5 | 0.29 ± 0.12 |
| 40 | 12 ± 4 | 23 ± 5 | 0.16 ± 0.08 | 7 ± 2 | 12 ± 3 | 0.4 ± 0.01 |
| 80 | 13 ± 4 | 17 ± 4 | 0.41 ± 0.11 | 6 ± 2 | 8 ± 4 | 0.73 ± 0.14 |
| 120 | 8 ± 2 | 12 ± 3 | 0.42 ± 0.02 | 11 ± 4 | 17 ± 6 | 0.33 ± 0.22 |
Calcium Influences the Secondary Structure of SOD1 Aggregates
We have also examined the difference ATR-FTIR spectra to probe for conformational changes on SOD1 aggregates formed in the presence of Ca2+ (Fig. 6). Ca2+ aggregates display prominent β-sheet components at 1630 and 1695 cm−1, likely to be associated with intermolecular antiparallel arrangements of the β-strands, because antiparallel β-sheets exhibit a strong band near ∼1630 cm−1 and a weaker band near ∼1990 cm−1 (81–83). On the other hand, in the control, a main β-sheet contribution at 1640 cm−1 is observed that can be attributed either to parallel β-sheets or to twisted antiparallel β-sheet structures, because both types of structure have absorption frequencies shifted to higher wave numbers than the corresponding antiparallel β-sheets and often exhibit a reduced/negligible high wave number side band (84). Moreover, Ca2+ aggregates have a high content of intermolecular β-sheets (band at 1612 cm−1) (85) as well as β-turns (bands at 1678 and 1664 cm−1); on the other hand, the control denotes a higher content of random structures, as evidenced by the 1655 cm−1 negative band.
FIGURE 6.

Ca2+ induces secondary structure changes on SOD1 aggregates. ATR-FTIR difference spectrum in the amide I region of SOD1 aggregates formed in the presence of Ca2+ minus in its absence. Positive peaks denote features that are enriched by the presence of Ca2+, whereas negative peaks denote structures preferentially formed in the absence of Ca2+. The analyzed aggregates were obtained at pH 7.5 after incubation under constant agitation with a Teflon bead at 37 °C for 150 h. −Ca2+, no CaCl2; +Ca2+, CaCl2/SOD1 = 2. See “Materials and Methods” for further details.
Calcium Influences the Morphology of SOD1 Aggregate
Having established that Ca2+ modulates the secondary structure of SOD1 aggregates, we moved to investigate their morphological differences. For this purpose, we have used conformational antibodies in combination with TEM. The conformational antibodies A11 and OC recognize generic epitopes of soluble oligomers and fibrils, within amyloid proteins, respectively (86, 87). Dot blot analysis of SOD1 aggregates obtained after the plateau phase of the aggregation kinetics was reached (t ∼200 h) showed that these are immunoreactive toward both antibodies, indicating that SOD1 aggregates comprise amyloid epitopes (Fig. 7). Interestingly, incubation with Ca2+ results in different binding patterns; the presence of Ca2+ increases the reactivity toward oligomers (A11) while simultaneously decreasing proportionally the immune response toward fibrils (OC). This result implies that Ca2+ decreases the extent of fibril formation, suggesting that the aggregation pathway is diverted toward amorphous aggregates.
FIGURE 7.
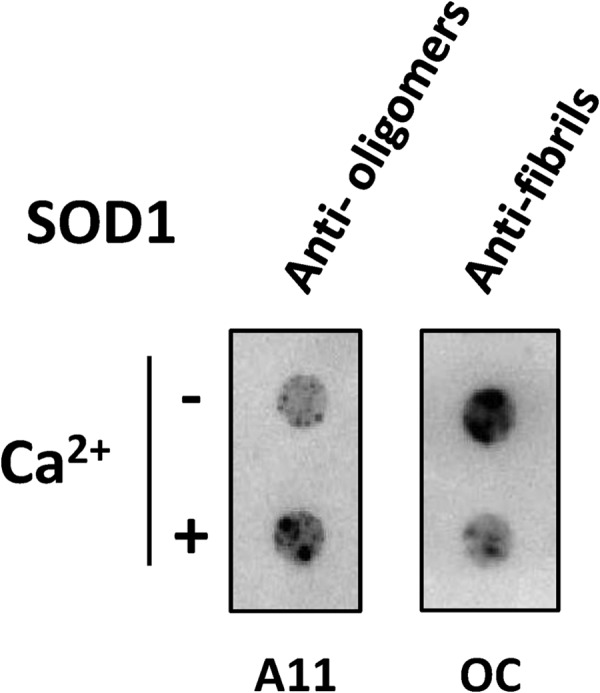
Ca2+ influences SOD1 aggregation pathway toward non-fibrillar aggregates. Shown is dot blot analysis using conformational antibodies. Anti-amyloid oligomer (A11) and anti-amyloid fibril (OC) antibodies were used to test conformers prevalent in SOD1 aggregates formed in the presence and absence of Ca2+ (n = 3). −Ca2+, no CaCl2; +Ca2+, CaCl2/SOD1 = 2. See “Materials and Methods” for further details.
The morphology of the aggregates was further investigated using TEM (Fig. 8). In the absence of Ca2+, SOD1 forms amyloid-like fibrils with characteristic ultrastructural properties with an approximate diameter of 10 nm and several μm in length. In the presence of Ca2+, fibrils still formed, but they were shorter and less abundant (Fig. 8, bottom panels). Furthermore, other species were present, namely amorphous aggregates (Fig. 8, bottom right panel, arrow) and oligomers of different sizes (Fig. 8, bottom panels, arrowheads), thus forming a more heterogeneous sample. These results fully agree with those obtained using conformational antibodies. TEM observations suggest that Ca2+ interfered with SOD1 fibrillogenesis but did not avoid the generation of intermediate amyloidogenic species, such as oligomers and fibrillar aggregates. Thus, the preferential non-fibrillar aggregates and oligomers that are formed by the presence of Ca2+ retain the ability to bind ThT and accounted for most of the intensity observed in this assay (Fig. 1C). Interestingly, electron microscopy of Lewy body-like SOD1-enriched inclusions from the spinal cord of an ALS mouse model revealed that its core consisted of a heterogeneous mass of tangled filamentous material covered with small granules (14, 88, 89). This suggests that SOD1 inclusions in vivo are composed of protein aggregates with heterogeneous morphologies, like the ones here found to be formed in vitro in the presence of Ca2+.
FIGURE 8.

Ca2+ modulates the morphology of SOD1 aggregates, as analyzed by TEM. In the absence of Ca2+, apo-SOD1 formed typical amyloid fibrils with diameters of ∼10 nm (top). In the presence of Ca2+, fibrils are less abundant; in particular, amorphous aggregates (bottom right panel, arrow) and oligomers of variable sizes (bottom panels, arrowheads) were often visualized. Analyzed aggregates derived from apo-SOD1 incubated at pH 7.5 with or without Ca2+ under constant agitation with a Teflon bead at 37 °C for 150 h. −Ca2+, no CaCl2; +Ca2+, CaCl2/SOD1 = 2. See “Materials and Methods” for further details.
DISCUSSION
Ca2+ is increased in ALS and is particularly abundant in the specific motor neurons affected in this disease, where SOD1-enriched proteinaceous inclusions are found. As a ligand, Ca2+ is particularly versatile because it binds sites with irregular geometry, thus facilitating its nonspecific association to proteins (90, 91). In agreement, a bound Ca2+ was described in a crystal structure of an SOD1 clinical mutant (92) although sequence analysis and bioinformatics failed to identify any canonical Ca2+ binding motif within SOD1. Here we have assayed the effect of Ca2+ on the SOD1 aggregation mechanism. Overall, the results obtained show that under physiological pH, Ca2+ induces conformational changes on the SOD1 fold that increase its propensity to aggregate. Specifically, Ca2+ increases SOD1 β-sheet content and hydrophobicity, which are features likely to result in favorable intramolecular interactions leading to the formation of aggregates (93, 94). Indeed, the presence of Ca2+ was shown to promote SOD1 aggregation, both under oxidizing and reducing conditions, diverting the aggregation pathway toward non-fibrillar aggregates and oligomers instead of fibrils. Recent literature suggests that amorphous rather than amyloid-type aggregates are found in SOD1-ALS patient tissue (95), and the reported effects of Ca2+ would go along these lines, but this is still a matter of debate in the field (96, 97). This modulation effect by a metal ion has also been demonstrated for other proteins involved in neurodegenerative disorders, highlighting the influence of the chemical neuronal environment with respect to metal ion homeostasis in pathologic aggregation processes (98). Interestingly, Ca2+ can also induce α-synuclein aggregation, as shown by cell culture and in vitro studies (99–101). This evidence is particularly relevant because pathologic α-synuclein accumulation in Parkinson disease selectively affects dopaminergic neurons that are particularly vulnerable to Ca2+ dyshomeostasis, correlating calcium overload with the onset of the disease (102–105). Also, in the case of SOD1, the observed effect of Ca2+ can have a particularly meaningful physiological relevance because SOD1 only undergoes aggregation within specific neuron cells of ALS patients (38, 39) that simultaneously show Ca2+ overload (47–53). Moreover, it was recently reported that calcium overload in the cytoplasm of cultured motor neurons expressing an ALS-associated mutant promotes SOD1 aggregation into inclusions. Importantly, Ca2+ increase is suggested not to be a consequence of SOD1 aggregation because the rise in Ca2+ levels in these cells occurs prior to the formation of SOD1 inclusions (54, 55). In any case, SOD1 aggregates promote a further increase in the levels of cytosolic Ca2+ (55). In agreement, increasing the Ca2+ load exacerbates the insolubility and toxicity of a SOD1 clinical variant (106–109). On the other hand, an opposite effect is observed when Ca2+ buffering in cellular and animal models of fALS is increased, which resulted in a protective effect toward aggregation (110–112). Our study provides in vitro evidence supporting the hypothesis that Ca2+ overload in ALS can actually play a role in SOD1 deposition. Nevertheless, there must be other factors involved in SOD1 aggregation because otherwise SOD1 would probably also aggregate on dopaminergic neurons, which are neurons that feature high levels of labile cytosolic calcium.
Acknowledgments
We thank M. J. Saraiva (IBMC, Portugal) and R. Fernandes (Histology and Electron Microscopy Service, Instituto Biologia Molecular e Celular, Portugal) for assistance with TEM analysis. We gratefully acknowledge M. Oliveberg (Stockholm University) for the SOD1 plasmid.
This work was supported by the Fundação para a Ciência e Tecnologia (FCT/MCTES, Portugal) through Research Grants PTDC/QUI-BIQ/117789/2010 and PTDC/EBB-BIO/117793/2010 (to C. M. G.), Postdoctoral Fellowship SFRH/BPD/47477/2008 (to S. S. L.), and Strategic Grant PEst-OE/EQB/LA0004/2011 (to the Instituto Tecnologia Química e Biológica Laboratório Associado).
- ALS
- amyotrophic lateral sclerosis
- fALS
- familial ALS
- ThT
- thioflavin T
- SOD
- superoxide dismutase
- ATR
- attenuated total reflectance
- ANS
- 1-anilino-8-naphthalenesulfonate
- DLS
- dynamic light scattering
- TEM
- transmission electron microscopy.
REFERENCES
- 1. Rowland L. P., Shneider N. A. (2001) Amyotrophic lateral sclerosis. N. Engl. J. Med. 344, 1688–1700 [DOI] [PubMed] [Google Scholar]
- 2. Talbot K., Ansorge O. (2006) Recent advances in the genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Common pathways in neurodegenerative disease. Hum. Mol. Genet. 15, R182–R187 [DOI] [PubMed] [Google Scholar]
- 3. Watanabe M., Dykes-Hoberg M., Culotta V. C., Price D. L., Wong P. C., Rothstein J. D. (2001) Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol. Dis. 8, 933–941 [DOI] [PubMed] [Google Scholar]
- 4. Bruijn L. I., Houseweart M. K., Kato S., Anderson K. L., Anderson S. D., Ohama E., Reaume A. G., Scott R. W., Cleveland D. W. (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281, 1851–1854 [DOI] [PubMed] [Google Scholar]
- 5. Johnston J. A., Dalton M. J., Gurney M. E., Kopito R. R. (2000) Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 97, 12571–12576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Prudencio M., Hart P. J., Borchelt D. R., Andersen P. M. (2009) Variation in aggregation propensities among ALS-associated variants of SOD1. Correlation to human disease. Hum. Mol. Genet. 18, 3217–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kato S., Takikawa M., Nakashima K., Hirano A., Cleveland D. W., Kusaka H., Shibata N., Kato M., Nakano I., Ohama E. (2000) New consensus research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 (SOD1) gene mutations. Inclusions containing SOD1 in neurons and astrocytes. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1, 163–184 [DOI] [PubMed] [Google Scholar]
- 8. Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J. P., Deng H. X. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 [DOI] [PubMed] [Google Scholar]
- 9. Valentine J. S., Doucette P. A., Zittin Potter S. (2005) Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 74, 563–593 [DOI] [PubMed] [Google Scholar]
- 10. Stieber A., Gonatas J. O., Gonatas N. K. (2000) Aggregates of mutant protein appear progressively in dendrites, in periaxonal processes of oligodendrocytes, and in neuronal and astrocytic perikarya of mice expressing the SOD1(G93A) mutation of familial amyotrophic lateral sclerosis. J. Neurol. Sci. 177, 114–123 [DOI] [PubMed] [Google Scholar]
- 11. Sasaki S., Warita H., Murakami T., Shibata N., Komori T., Abe K., Kobayashi M., Iwata M. (2005) Ultrastructural study of aggregates in the spinal cord of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 109, 247–255 [DOI] [PubMed] [Google Scholar]
- 12. Forsberg K., Jonsson P. A., Andersen P. M., Bergemalm D., Graffmo K. S., Hultdin M., Jacobsson J., Rosquist R., Marklund S. L., Brännström T. (2010) Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One 5, e11552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Forsberg K., Andersen P. M., Marklund S. L., Brännstrom T. (2011) Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 121, 623–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shibata N., Hirano A., Kobayashi M., Sasaki S., Kato T., Matsumoto S., Shiozawa Z., Komori T., Ikemoto A., Umahara T. (1994) Cu/Zn superoxide dismutase-like immunoreactivity in Lewy body-like inclusions of sporadic amyotrophic lateral sclerosis. Neurosci. Lett. 179, 149–152 [DOI] [PubMed] [Google Scholar]
- 15. Shibata N., Asayama K., Hirano A., Kobayashi M. (1996) Immunohistochemical study on superoxide dismutases in spinal cords from autopsied patients with amyotrophic lateral sclerosis. Dev. Neurosci. 18, 492–498 [DOI] [PubMed] [Google Scholar]
- 16. Guareschi S., Cova E., Cereda C., Ceroni M., Donetti E., Bosco D. A., Trotti D., Pasinelli P. (2012) An overoxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. U.S.A. 109, 5074–5079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bosco D. A., Morfini G., Karabacak N. M., Song Y., Gros-Louis F., Pasinelli P., Goolsby H., Fontaine B. A., Lemay N., McKenna-Yasek D., Frosch M. P., Agar J. N., Julien J. P., Brady S. T., Brown R. H., Jr. (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yates D. (2010) Motor neuron disease. Misfolded wild-type SOD1 may link sporadic and familial ALS. Nat. Rev. Neurol. 6, 645. [DOI] [PubMed] [Google Scholar]
- 19. Wang J., Slunt H., Gonzales V., Fromholt D., Coonfield M., Copeland N. G., Jenkins N. A., Borchelt D. R. (2003) Copper-binding-site-null SOD1 causes ALS in transgenic mice. Aggregates of non-native SOD1 delineate a common feature. Hum. Mol. Genet. 12, 2753–2764 [DOI] [PubMed] [Google Scholar]
- 20. Kabashi E., Valdmanis P. N., Dion P., Rouleau G. A. (2007) Oxidized/misfolded superoxide dismutase-1. The cause of all amyotrophic lateral sclerosis? Ann. Neurol. 62, 553–559 [DOI] [PubMed] [Google Scholar]
- 21. Gruzman A., Wood W. L., Alpert E., Prasad M. D., Miller R. G., Rothstein J. D., Bowser R., Hamilton R., Wood T. D., Cleveland D. W., Lingappa V. R., Liu J. (2007) Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 104, 12524–12529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haidet-Phillips A. M., Hester M. E., Miranda C. J., Meyer K., Braun L., Frakes A., Song S., Likhite S., Murtha M. J., Foust K. D., Rao M., Eagle A., Kammesheidt A., Christensen A., Mendell J. R., Burghes A. H., Kaspar B. K. (2011) Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jonsson P. A., Graffmo K. S., Andersen P. M., Brännström T., Lindberg M., Oliveberg M., Marklund S. L. (2006) Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain 129, 451–464 [DOI] [PubMed] [Google Scholar]
- 24. Lelie H. L., Liba A., Bourassa M. W., Chattopadhyay M., Chan P. K., Gralla E. B., Miller L. M., Borchelt D. R., Valentine J. S., Whitelegge J. P. (2011) Copper and zinc metallation status of copper-zinc superoxide dismutase from amyotrophic lateral sclerosis transgenic mice. J. Biol. Chem. 286, 2795–2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rumfeldt J. A., Lepock J. R., Meiering E. M. (2009) Unfolding and folding kinetics of amyotrophic lateral sclerosis-associated mutant Cu,Zn superoxide dismutases. J. Mol. Biol. 385, 278–298 [DOI] [PubMed] [Google Scholar]
- 26. Banci L., Bertini I., Durazo A., Girotto S., Gralla E. B., Martinelli M., Valentine J. S., Vieru M., Whitelegge J. P. (2007) Metal-free superoxide dismutase forms soluble oligomers under physiological conditions. A possible general mechanism for familial ALS. Proc. Natl. Acad. Sci. U.S.A. 104, 11263–11267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Teilum K., Smith M. H., Schulz E., Christensen L. C., Solomentsev G., Oliveberg M., Akke M. (2009) Transient structural distortion of metal-free Cu/Zn superoxide dismutase triggers aberrant oligomerization. Proc. Natl. Acad. Sci. U.S.A. 106, 18273–18278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chattopadhyay M., Durazo A., Sohn S. H., Strong C. D., Gralla E. B., Whitelegge J. P., Valentine J. S. (2008) Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc. Natl. Acad. Sci. U.S.A. 105, 18663–18668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Furukawa Y., O'Halloran T. V. (2006) Posttranslational modifications in Cu,Zn-superoxide dismutase and mutations associated with amyotrophic lateral sclerosis. Antioxid. Redox. Signal. 8, 847–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hörnberg A., Logan D. T., Marklund S. L., Oliveberg M. (2007) The coupling between disulphide status, metallation and dimer interface strength in Cu/Zn superoxide dismutase. J. Mol. Biol. 365, 333–342 [DOI] [PubMed] [Google Scholar]
- 31. Strange R. W., Antonyuk S., Hough M. A., Doucette P. A., Rodriguez J. A., Hart P. J., Hayward L. J., Valentine J. S., Hasnain S. S. (2003) The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J. Mol. Biol. 328, 877–891 [DOI] [PubMed] [Google Scholar]
- 32. Banci L., Bertini I., Cramaro F., Del Conte R., Viezzoli M. S. (2003) Solution structure of Apo Cu,Zn superoxide dismutase. Role of metal ions in protein folding. Biochemistry 42, 9543–9553 [DOI] [PubMed] [Google Scholar]
- 33. Assfalg M., Banci L., Bertini I., Turano P., Vasos P. R. (2003) Superoxide dismutase folding/unfolding pathway. Role of the metal ions in modulating structural and dynamical features. J. Mol. Biol. 330, 145–158 [DOI] [PubMed] [Google Scholar]
- 34. Lindberg M. J., Normark J., Holmgren A., Oliveberg M. (2004) Folding of human superoxide dismutase. Disulfide reduction prevents dimerization and produces marginally stable monomers. Proc. Natl. Acad. Sci. U.S.A. 101, 15893–15898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arnesano F., Banci L., Bertini I., Martinelli M., Furukawa Y., O'Halloran T. V. (2004) The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J. Biol. Chem. 279, 47998–48003 [DOI] [PubMed] [Google Scholar]
- 36. Rodriguez J. A., Shaw B. F., Durazo A., Sohn S. H., Doucette P. A., Nersissian A. M., Faull K. F., Eggers D. K., Tiwari A., Hayward L. J., Valentine J. S. (2005) Destabilization of apoprotein is insufficient to explain Cu,Zn-superoxide dismutase-linked ALS pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 102, 10516–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vassall K. A., Stubbs H. R., Primmer H. A., Tong M. S., Sullivan S. M., Sobering R., Srinivasan S., Briere L. A., Dunn S. D., Colón W., Meiering E. M. (2011) Decreased stability and increased formation of soluble aggregates by immature superoxide dismutase do not account for disease severity in ALS. Proc. Natl. Acad. Sci. U.S.A. 108, 2210–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cleveland D. W., Rothstein J. D. (2001) From Charcot to Lou Gehrig. Deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2, 806–819 [DOI] [PubMed] [Google Scholar]
- 39. Wang J., Xu G., Borchelt D. R. (2002) High molecular weight complexes of mutant superoxide dismutase 1. Age-dependent and tissue-specific accumulation. Neurobiol. Dis. 9, 139–148 [DOI] [PubMed] [Google Scholar]
- 40. Vandenberghe W., Robberecht W., Brorson J. R. (2000) AMPA receptor calcium permeability, GluR2 expression, and selective motoneuron vulnerability. J. Neurosci. 20, 123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shaw P. J., Eggett C. J. (2000) Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J. Neurol. 247, I17–I27 [DOI] [PubMed] [Google Scholar]
- 42. Williams T. L., Day N. C., Ince P. G., Kamboj R. K., Shaw P. J. (1997) Calcium-permeable α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors. A molecular determinant of selective vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 42, 200–207 [DOI] [PubMed] [Google Scholar]
- 43. Van Den Bosch L., Vandenberghe W., Klaassen H., Van Houtte E., Robberecht W. (2000) Ca2+-permeable AMPA receptors and selective vulnerability of motor neurons. J. Neurol. Sci. 180, 29–34 [DOI] [PubMed] [Google Scholar]
- 44. Guatteo E., Carunchio I., Pieri M., Albo F., Canu N., Mercuri N. B., Zona C. (2007) Altered calcium homeostasis in motor neurons following AMPA receptor but not voltage-dependent calcium channels' activation in a genetic model of amyotrophic lateral sclerosis. Neurobiol. Dis. 28, 90–100 [DOI] [PubMed] [Google Scholar]
- 45. Alexianu M. E., Ho B. K., Mohamed A. H., La Bella V., Smith R. G., Appel S. H. (1994) The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 36, 846–858 [DOI] [PubMed] [Google Scholar]
- 46. Palecek J., Lips M. B., Keller B. U. (1999) Calcium dynamics and buffering in motoneurones of the mouse spinal cord. J. Physiol. 520, 485–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Siklós L., Engelhardt J. I., Alexianu M. E., Gurney M. E., Siddique T., Appel S. H. (1998) Intracellular calcium parallels motoneuron degeneration in SOD-1 mutant mice. J. Neuropathol. Exp. Neurol. 57, 571–587 [DOI] [PubMed] [Google Scholar]
- 48. von Lewinski F., Fuchs J., Vanselow B. K., Keller B. U. (2008) Low Ca2+ buffering in hypoglossal motoneurons of mutant SOD1 (G93A) mice. Neurosci. Lett. 445, 224–228 [DOI] [PubMed] [Google Scholar]
- 49. Jaiswal M. K., Keller B. U. (2009) Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol. Pharmacol. 75, 478–489 [DOI] [PubMed] [Google Scholar]
- 50. Bilsland L. G., Nirmalananthan N., Yip J., Greensmith L., Duchen M. R. (2008) Expression of mutant SOD1 in astrocytes induces functional deficits in motoneuron mitochondria. J. Neurochem. 107, 1271–1283 [DOI] [PubMed] [Google Scholar]
- 51. Kawamata H., Manfredi G. (2010) Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech. Ageing Dev. 131, 517–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Siklós L., Engelhardt J., Harati Y., Smith R. G., Joó F., Appel S. H. (1996) Ultrastructural evidence for altered calcium in motor nerve terminals in amyotropic lateral sclerosis. Ann. Neurol. 39, 203–216 [DOI] [PubMed] [Google Scholar]
- 53. Grosskreutz J., Van Den Bosch L., Keller B. U. (2010) Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 47, 165–174 [DOI] [PubMed] [Google Scholar]
- 54. Tateno M., Sadakata H., Tanaka M., Itohara S., Shin R. M., Miura M., Masuda M., Aosaki T., Urushitani M., Misawa H., Takahashi R. (2004) Calcium-permeable AMPA receptors promote misfolding of mutant SOD1 protein and development of amyotrophic lateral sclerosis in a transgenic mouse model. Hum. Mol. Genet. 13, 2183–2196 [DOI] [PubMed] [Google Scholar]
- 55. Tradewell M. L., Cooper L. A., Minotti S., Durham H. D. (2011) Calcium dysregulation, mitochondrial pathology and protein aggregation in a culture model of amyotrophic lateral sclerosis. Mechanistic relationship and differential sensitivity to intervention. Neurobiol. Dis. 42, 265–275 [DOI] [PubMed] [Google Scholar]
- 56. Vanselow B. K., Keller B. U. (2000) Calcium dynamics and buffering in oculomotor neurones from mouse that are particularly resistant during amyotrophic lateral sclerosis (ALS)-related motoneurone disease. J. Physiol. 525, 433–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ahl I. M., Lindberg M. J., Tibell L. A. (2004) Coexpression of yeast copper chaperone (yCCS) and CuZn-superoxide dismutases in Escherichia coli yields protein with high copper contents. Protein Expr. Purif. 37, 311–319 [DOI] [PubMed] [Google Scholar]
- 58. Potter S. Z., Zhu H., Shaw B. F., Rodriguez J. A., Doucette P. A., Sohn S. H., Durazo A., Faull K. F., Gralla E. B., Nersissian A. M., Valentine J. S. (2007) Binding of a single zinc ion to one subunit of copper-zinc superoxide dismutase apoprotein substantially influences the structure and stability of the entire homodimeric protein. J. Am. Chem. Soc. 129, 4575–4583 [DOI] [PubMed] [Google Scholar]
- 59. Säbel C. E., Neureuther J. M., Siemann S. (2010) A spectrophotometric method for the determination of zinc, copper, and cobalt ions in metalloproteins using Zincon. Anal. Biochem. 397, 218–226 [DOI] [PubMed] [Google Scholar]
- 60. Barth A., Zscherp C. (2002) What vibrations tell us about proteins. Q. Rev. Biophys. 35, 369–430 [DOI] [PubMed] [Google Scholar]
- 61. Banci L., Bertini I., D'Amelio N., Gaggelli E., Libralesso E., Matecko I., Turano P., Valentine J. S. (2005) Fully metallated S134N Cu,Zn-superoxide dismutase displays abnormal mobility and intermolecular contacts in solution. J. Biol. Chem. 280, 35815–35821 [DOI] [PubMed] [Google Scholar]
- 62. Chiti F., Taddei N., Baroni F., Capanni C., Stefani M., Ramponi G., Dobson C. M. (2002) Kinetic partitioning of protein folding and aggregation. Nat. Struct. Biol. 9, 137–143 [DOI] [PubMed] [Google Scholar]
- 63. Semisotnov G. V., Rodionova N. A., Razgulyaev O. I., Uversky V. N., Gripas' A. F., Gilmanshin R. I. (1991) Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 31, 119–128 [DOI] [PubMed] [Google Scholar]
- 64. Cardamone M., Puri N. K. (1992) Spectrofluorimetric assessment of the surface hydrophobicity of proteins. Biochem. J. 282, 589–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tiwari A., Liba A., Sohn S. H., Seetharaman S. V., Bilsel O., Matthews C. R., Hart P. J., Valentine J. S., Hayward L. J. (2009) Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. J. Biol. Chem. 284, 27746–27758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bemporad F., Chiti F. (2012) Protein misfolded oligomers. Experimental approaches, mechanism of formation, and structure-toxicity relationships. Chem. Biol. 19, 315–327 [DOI] [PubMed] [Google Scholar]
- 67. Li Y., Lubchenko V., Vekilov P. G. (2011) The use of dynamic light scattering and brownian microscopy to characterize protein aggregation. Rev. Sci. Instrum. 82, 053106 [DOI] [PubMed] [Google Scholar]
- 68. Ferré-D'Amaré A. R., Burley S. K. (1994) Use of dynamic light scattering to assess crystallizability of macromolecules and macromolecular assemblies. Structure 2, 357–359 [DOI] [PubMed] [Google Scholar]
- 69. Nobbmann U., Connah M., Fish B., Varley P., Gee C., Mulot S., Chen J., Zhou L., Lu Y., Shen F., Yi J., Harding S. E. (2007) Dynamic light scattering as a relative tool for assessing the molecular integrity and stability of monoclonal antibodies. Biotechnol. Genet. Eng. Rev. 24, 117–128 [DOI] [PubMed] [Google Scholar]
- 70. Montero M., Alonso M. T., Carnicero E., Cuchillo-Ibáñez I., Albillos A., García A. G., García-Sancho J., Alvarez J. (2000) Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat. Cell Biol. 2, 57–61 [DOI] [PubMed] [Google Scholar]
- 71. Tamano H., Takeda A. (2011) Dynamic action of neurometals at the synapse. Metallomics 3, 656–661 [DOI] [PubMed] [Google Scholar]
- 72. Franks K. M., Sejnowski T. J. (2002) Complexity of calcium signaling in synaptic spines. BioEssays 24, 1130–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hawe A., Sutter M., Jiskoot W. (2008) Extrinsic fluorescent dyes as tools for protein characterization. Pharm. Res. 25, 1487–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. LeVine H., 3rd (1999) Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 309, 274–284 [DOI] [PubMed] [Google Scholar]
- 75. LeVine H., 3rd (1993) Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides. Detection of amyloid aggregation in solution. Protein Sci. 2, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Biancalana M., Koide S. (2010) Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 1804, 1405–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jan A., Gokce O., Luthi-Carter R., Lashuel H. A. (2008) The ratio of monomeric to aggregated forms of Aβ40 and Aβ42 is an important determinant of amyloid-β aggregation, fibrillogenesis, and toxicity. J. Biol. Chem. 283, 28176–28189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Maezawa I., Hong H. S., Liu R., Wu C. Y., Cheng R. H., Kung M. P., Kung H. F., Lam K. S., Oddo S., Laferla F. M., Jin L. W. (2008) Congo red and thioflavin-T analogs detect Aβ oligomers. J. Neurochem. 104, 457–468 [DOI] [PubMed] [Google Scholar]
- 79. Necula M., Kayed R., Milton S., Glabe C. G. (2007) Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 282, 10311–10324 [DOI] [PubMed] [Google Scholar]
- 80. Oztug Durer Z. A., Cohlberg J. A., Dinh P., Padua S., Ehrenclou K., Downes S., Tan J. K., Nakano Y., Bowman C. J., Hoskins J. L., Kwon C., Mason A. Z., Rodriguez J. A., Doucette P. A., Shaw B. F., Valentine J. S. (2009) Loss of metal ions, disulfide reduction and mutations related to familial ALS promote formation of amyloid-like aggregates from superoxide dismutase. PLoS One 4, e5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chirgadze Y. N., Nevskaya N. A. (1976) Infrared spectra and resonance interaction of amide-I vibration of the paraellel-chain pleated sheets. Biopolymers 15, 627–636 [DOI] [PubMed] [Google Scholar]
- 82. Krimm S., Bandekar J. (1986) Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 38, 181–364 [DOI] [PubMed] [Google Scholar]
- 83. van de Weert M., Haris P. I., Hennink W. E., Crommelin D. J. (2001) Fourier transform infrared spectrometric analysis of protein conformation. Effect of sampling method and stress factors. Anal. Biochem. 297, 160–169 [DOI] [PubMed] [Google Scholar]
- 84. Barth A. (2007) Infrared spectroscopy of proteins. Biochim. Biophys. Acta 1767, 1073–1101 [DOI] [PubMed] [Google Scholar]
- 85. Okuno A., Kato M., Taniguchi Y. (2006) The secondary structure of pressure- and temperature-induced aggregates of equine serum albumin studied by FT-IR spectroscopy. Biochim. Biophys. Acta 1764, 1407–1412 [DOI] [PubMed] [Google Scholar]
- 86. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 87. Kayed R., Head E., Sarsoza F., Saing T., Cotman C. W., Necula M., Margol L., Wu J., Breydo L., Thompson J. L., Rasool S., Gurlo T., Butler P., Glabe C. G. (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bruijn L. I., Becher M. W., Lee M. K., Anderson K. L., Jenkins N. A., Copeland N. G., Sisodia S. S., Rothstein J. D., Borchelt D. R., Price D. L., Cleveland D. W. (1997) ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338 [DOI] [PubMed] [Google Scholar]
- 89. Kato S., Nakashima K., Horiuchi S., Nagai R., Cleveland D. W., Liu J., Hirano A., Takikawa M., Kato M., Nakano I., Sakoda S., Asayama K., Ohama E. (2001) Formation of advanced glycation end-product-modified superoxide dismutase-1 (SOD1) is one of the mechanisms responsible for inclusions common to familial amyotrophic lateral sclerosis patients with SOD1 gene mutation, and transgenic mice expressing human SOD1 gene mutation. Neuropathology 21, 67–81 [DOI] [PubMed] [Google Scholar]
- 90. Berridge M. J., Bootman M. D., Roderick H. L. (2003) Calcium signalling. Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529 [DOI] [PubMed] [Google Scholar]
- 91. Carafoli E. (2005) Calcium. A universal carrier of biological signals. Delivered on 3 July 2003 at the Special FEBS Meeting in Brussels. FEBS J. 272, 1073–1089 [DOI] [PubMed] [Google Scholar]
- 92. Winkler D. D., Schuermann J. P., Cao X., Holloway S. P., Borchelt D. R., Carroll M. C., Proescher J. B., Culotta V. C., Hart P. J. (2009) Structural and biophysical properties of the pathogenic SOD1 variant H46R/H48Q. Biochemistry 48, 3436–3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kundu B., Guptasarma P. (2002) Use of a hydrophobic dye to indirectly probe the structural organization and conformational plasticity of molecules in amorphous aggregates of carbonic anhydrase. Biochem. Biophys. Res. Commun. 293, 572–577 [DOI] [PubMed] [Google Scholar]
- 94. Vetri V., Militello V. (2005) Thermal induced conformational changes involved in the aggregation pathways of β-lactoglobulin. Biophys. Chem. 113, 83–91 [DOI] [PubMed] [Google Scholar]
- 95. Kerman A., Liu H.-N., Croul S., Bilbao J., Rogaeva E., Zinman L., Robertson J., Chakrabartty A. (2010) Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol. 119, 335–344 [DOI] [PubMed] [Google Scholar]
- 96. Nordlund A., Oliveberg M. (2006) Folding of Cu/Zn superoxide dismutase suggests structural hotspots for gain of neurotoxic function in ALS. Parallels to precursors in amyloid disease. Proc. Natl. Acad. Sci. U.S.A. 103, 10218–10223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sábado J., Casanovas A., Hernandez S., Piedrafita L., Hereu M., Esquerda J. E. (2013) Immunodetection of disease-associated conformers of mutant Cu/Zn superoxide dismutase 1 selectively expressed in degenerating neurons in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 72, 646–661 [DOI] [PubMed] [Google Scholar]
- 98. Leal S. S., Botelho H. M., Gomes C. M. (2012) Metal ions as modulators of protein conformation and misfolding in neurodegeneration. Coordination Chem. Rev. 256, 2253–2270 [Google Scholar]
- 99. Nath S., Goodwin J., Engelborghs Y., Pountney D. L. (2011) Raised calcium promotes α-synuclein aggregate formation. Mol. Cell Neurosci. 46, 516–526 [DOI] [PubMed] [Google Scholar]
- 100. Nielsen M. S., Vorum H., Lindersson E., Jensen P. H. (2001) Ca2+ binding to α-synuclein regulates ligand binding and oligomerization. J. Biol. Chem. 276, 22680–22684 [DOI] [PubMed] [Google Scholar]
- 101. Lowe R., Pountney D. L., Jensen P. H., Gai W. P., Voelcker N. H. (2004) Calcium(II) selectively induces α-synuclein annular oligomers via interaction with the C-terminal domain. Protein Sci. 13, 3245–3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mosharov E. V., Larsen K. E., Kanter E., Phillips K. A., Wilson K., Schmitz Y., Krantz D. E., Kobayashi K., Edwards R. H., Sulzer D. (2009) Interplay between cytosolic dopamine, calcium, and α-synuclein causes selective death of substantia nigra neurons. Neuron 62, 218–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chan C. S., Guzman J. N., Ilijic E., Mercer J. N., Rick C., Tkatch T., Meredith G. E., Surmeier D. J. (2007) “Rejuvenation” protects neurons in mouse models of Parkinson's disease. Nature 447, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 104. Surmeier D. J., Guzman J. N., Sanchez-Padilla J. (2010) Calcium, cellular aging, and selective neuronal vulnerability in Parkinson's disease. Cell Calcium 47, 175–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Surmeier D. J., Guzman J. N., Sanchez-Padilla J., Schumacker P. T. (2011) The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson's disease. Neuroscience 198, 221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kim H. J., Im W., Kim S., Kim S. H., Sung J. J., Kim M., Lee K. W. (2007) Calcium-influx increases SOD1 aggregates via nitric oxide in cultured motor neurons. Exp. Mol. Med. 39, 574–582 [DOI] [PubMed] [Google Scholar]
- 107. Van Damme P., Braeken D., Callewaert G., Robberecht W., Van Den Bosch L. (2005) GluR2 deficiency accelerates motor neuron degeneration in a mouse model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 64, 605–612 [DOI] [PubMed] [Google Scholar]
- 108. Kuner R., Groom A. J., Müller G., Kornau H. C., Stefovska V., Bresink I., Hartmann B., Tschauner K., Waibel S., Ludolph A. C., Ikonomidou C., Seeburg P. H., Turski L. (2005) Mechanisms of disease. Motoneuron disease aggravated by transgenic expression of a functionally modified AMPA receptor subunit. Ann. N.Y. Acad. Sci. 1053, 269–286 [DOI] [PubMed] [Google Scholar]
- 109. Parone P. A., Da Cruz S., Han J. S., McAlonis-Downes M., Vetto A. P., Lee S. K., Tseng E., Cleveland D. W. (2013) Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J. Neurosci. 33, 4657–4671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Van Den Bosch L., Van Damme P., Bogaert E., Robberecht W. (2006) The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 1068–1082 [DOI] [PubMed] [Google Scholar]
- 111. Roy J., Minotti S., Dong L., Figlewicz D. A., Durham H. D. (1998) Glutamate potentiates the toxicity of mutant Cu/Zn-superoxide dismutase in motor neurons by postsynaptic calcium-dependent mechanisms. J. Neurosci. 18, 9673–9684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Beers D. R., Ho B. K., Siklós L., Alexianu M. E., Mosier D. R., Mohamed A. H., Otsuka Y., Kozovska M. E., McAlhany R. E., Smith R. G., Appel S. H. (2001) Parvalbumin overexpression alters immune-mediated increases in intracellular calcium, and delays disease onset in a transgenic model of familial amyotrophic lateral sclerosis. J. Neurochem. 79, 499–509 [DOI] [PubMed] [Google Scholar]