

Background: Human aminopeptidase A (APA) regulates central hypertension through its calcium-modulated preference to cleave N-terminal acidic residues from peptides.
Results: We determined the crystal structures of APA complexed with different ligands and calcium.
Conclusion: The S1 pocket of APA contains a calcium-binding site and accommodates only acidic residues.
Significance: This study elucidates the calcium-modulated substrate specificity of APA and will aid in the development of antihypertensive APA inhibitors.
Keywords: Aminopeptidase, Calcium-binding Proteins, Hypertension, Protein Structure, X-ray Crystallography
Abstract
Hypertension is regulated through both the central and systemic renin-angiotensin systems. In the central renin-angiotensin system, zinc-dependent aminopeptidase A (APA) up-regulates blood pressure by specifically cleaving the N-terminal aspartate, but not the adjacent arginine, from angiotensin II, a process facilitated by calcium. Here, we determined the crystal structures of human APA and its complexes with different ligands and identified a calcium-binding site in the S1 pocket of APA. Without calcium, the S1 pocket can bind both acidic and basic residues through formation of salt bridges with the charged side chains. In the presence of calcium, the binding of acidic residues is enhanced as they ligate the cation, whereas the binding of basic residues is no longer favorable due to charge repulsion. Of the peptidomimetic inhibitors of APA, amastatin has higher potency than bestatin by fitting better in the S1 pocket and interacting additionally with the S3′ subsite. These results explain the calcium-modulated substrate specificity of APA in central hypertension regulation and can guide the design and development of brain-targeting antihypertensive APA inhibitors.
Introduction
Approximately 1 billion people or ∼26% of the adult population of the world are affected by hypertension (1). Hypertension cannot be cured, but antihypertensive treatment can reduce cardiovascular morbidity and mortality. Despite the availability of many antihypertensive drugs, hypertension remains difficult to control. ∼20% of the hypertensive population are “resistant hypertension” patients, who do not respond well to three or more different antihypertensive drugs used at the same time (2). This proportion is even greater when patients who do not respond well to two antihypertensive drugs used concurrently are included. Additionally, current antihypertensive drugs have lower efficacy in patients of African ancestry (3, 4) or patients with renal impairment or diabetes (5, 6). Consequently, the demand to develop new antihypertensive drugs with different mechanisms of action remains high.
Blood pressure is regulated through both the central and systemic renin-angiotensin systems (RASs)2 (7–9). Some hypertensive patients show hyperactivity in the central RAS but low activity in the systemic RAS. Hence, these patients are resistant to treatments targeting the systemic RAS (10). The same set of peptidases play essential roles in both the central and systemic RASs (Fig. 1) (11, 12). Renin (also known as angiotensinogenase) cleaves the N-terminal 12 residues from the protein angiotensinogen to produce the peptide angiotensin I. Angiotensin-converting enzyme (ACE) then cleaves the C-terminal two residues from angiotensin I to produce angiotensin II. Aminopeptidase A (APA) cleaves the N-terminal aspartate from angiotensin II to produce angiotensin III. Finally, aminopeptidase N (APN) cleaves the N-terminal arginine from angiotensin III to produce angiotensin IV. Interestingly, angiotensin peptides play different roles in the central and systemic RASs: in the systemic RAS, angiotensin II is the main effector peptide that increases blood pressure, whereas in the central RAS, angiotensin III plays this role (13–15). Renin and ACE in the systemic RAS are among the main targets for antihypertensive drugs (11). Recently, APA in the central RAS has been suggested as a new target for antihypertensive drugs (16–21). In several types of hypertension animal models, brain-targeting APA inhibitors, either directly injected into the brain or administered orally followed by brain distribution, have been shown to be effective in lowering blood pressure (16–19). However, the design and development of this new class of antihypertensive drugs have been hindered by the lack of an atomic structure of APA. Like the crystal structures of renin and ACE (22, 23), the crystal structure of APA can inform therapeutic discoveries and aid in the control and treatment of hypertension.
FIGURE 1.
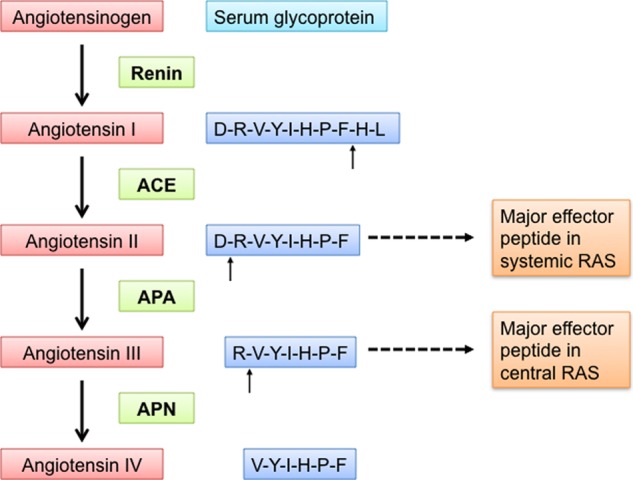
Central and systemic RASs that together regulate blood pressure. In both the central and systemic RASs, renin cleaves angiotensinogen to produce angiotensin I, ACE converts angiotensin I to angiotensin II, APA converts angiotensin II to angiotensin III, and APN converts angiotensin III to angiotensin IV. Angiotensin II is the major effector peptide in the systemic RAS, whereas angiotensin III is the major effector peptide in the central RAS.
Mammalian APA is a member of the M1 family of zinc metalloenzymes and is widely expressed in many tissues, including the brain (20, 24). As a cell surface-anchored ectoenzyme, it preferentially cleaves acidic residues, either glutamate or aspartate, from the N terminus of peptide substrates (25). The catalytic mechanism of APA is believed to be similar to that of another zinc-dependent aminopeptidase in the same family, APN, whose structure we reported recently (26). However, APA is unique among M1 family zinc metalloenzymes for favoring cleavage of acidic residues. Intriguingly, the substrate specificity of APA is modulated by calcium, a unique feature among all families of zinc metalloenzymes. More specifically, calcium enhances the enzymatic activity of APA for cleavage of acidic residues while reducing its activity with basic residues (27–31). The calcium-modulated substrate specificity of APA is deemed physiologically relevant because the concentrations of calcium that modulate APA activity are in the same range as those found in brain fluid (i.e. 1–2 mm) (32). Mutagenesis studies have identified several residues in APA that may be involved in calcium modulation (27–31). However, the structural mechanism for calcium-modulated substrate specificity of APA has remained a puzzle due to the lack of an atomic structure of APA. Understanding this mechanism will not only enrich our knowledge about the interesting APA enzymology but will also provide insights into central hypertension regulation by APA.
Here, we have determined the crystal structures of the human APA ectodomain by itself and in complex with amino acids or peptidomimetic inhibitors. These structures illustrate detailed interactions between APA and its ligands. We have also identified a calcium-binding site in APA and elucidated the structural basis for calcium-modulated APA activity. Additionally, this study reveals the structural basis for the different APA-inhibiting potencies of peptidomimetic inhibitors. Taken together, these results provide an understanding of the substrate specificity and calcium modulation of APA in central hypertension regulation and will guide the development of a new class of brain-targeting APA inhibitors to treat hypertension.
EXPERIMENTAL PROCEDURES
Reagents and Constructs
The synthetic substrates glutamyl-p-nitroanilide (pNA), aspartyl-pNA, arginine-pNA, phenylalanine-pNA, and leucine-pNA and the peptidomimetic inhibitors bestatin and amastatin were from Sigma-Aldrich. pFastBac (Invitrogen) was described previously (26).
Protein Preparation and Crystallization
The human APA ectodomain (residues 76–957) was expressed and purified as described previously for porcine APN (26). Briefly, the human APA ectodomain, containing an N-terminal honeybee melittin signal peptide and a C-terminal His6 tag, was expressed in insect cells using the Bac-to-Bac expression system (Invitrogen), secreted into cell culture medium, and subsequently purified on a nickel-nitrilotriacetic acid affinity column and a Superdex 200 gel filtration column (GE Healthcare). APA was concentrated to 10 mg/ml and stored in buffer containing 20 mm Tris (pH 7.4) and 200 mm NaCl. Crystallization of APA was set up using the sitting drop vapor diffusion method at 4 °C, with 2 μl of protein solution added to 2 μl of well buffer containing 1.6 m sodium citrate (pH 7.0). After 4 weeks, APA crystals were harvested in buffer containing 10% (v/v) glycerol and 1.6 m sodium citrate (pH 7.0) and flash-frozen in liquid nitrogen.
Heavy atom derivatives of APA crystals were prepared by soaking crystals overnight in buffer containing 1 mm heavy atom (HgCl2 or K2PtBr6), 10% (v/v) glycerol, and 1.6 m sodium citrate (pH 7.0). Crystals of APA-ligand complexes were prepared by soaking crystals overnight in buffer containing 8 mm ligand (amino acids or peptidomimetic inhibitors), 10% (v/v) glycerol, and 1.6 m sodium citrate (pH 7.0). Crystals of APA-ligand complexes in the presence of calcium were prepared by soaking crystals overnight in buffer containing 8 mm ligand, 20 mm CaCl2, 10% (v/v) glycerol, and 1.6 m sodium citrate (pH 7.0).
Data Collection and Structure Determination
Data were collected at Advanced Light Source beamline 4.2.2 at a wavelength of 1.0003 Å and at 100 K for native crystals. The crystal structure was determined with a mercury derivative and a platinum derivative by the multiple isomorphous replacement and anomalous scattering (MIRAS) method, with data collection conducted at wavelengths of 1.0003 and 1.06883 Å. X-ray diffraction data were processed using HKL2000 (33). The program SOLVE (34) was used to find heavy atom sites and for MIRAS phasing. From a mercury-derivatized crystal, 14 mercury sites were identified. From a platinum-derivatized crystal, 12 platinum sites were identified. The program RESOLVE was used for solvent flattening and automated model building (35). The model was improved by iterative cycles of manual building in Coot (36) and refinement with CNS (37) and Refmac (38). The similes files of ligands were loaded onto CCP4 Module Sketcher to generate CIF (crystallographic information file) files. The generated CIF files were used in CCP4 refinement of APA-ligand complex structures. The CIF files were also converted to topology and parameter files in CNS using module cns_import_cif. The generated topology and parameter files were used in CNS refinement of APA-ligand complex structures. Models of ligands were built based on Fo − Fc omit maps calculated in the absence of the ligands. For the APA native model, 97% of residues are in the favored regions of the Ramachandran plot, and 0.23% of residues are in the disallowed regions.
Catalysis and Inhibition Assays
APA catalytic activities were determined with 10 nm APA and 1 mm aminoacyl-pNA (amino acid-pNA) in 100 μl of 25 mm Tris-HCl buffer at pH 7.0 in the presence or absence of 2 mm CaCl2. The reactions were incubated at 37 °C for 30 min. Formation of product pNA was measured using a spectrophotometer and monitored at 405 nm.
APA inhibition assays were carried out using a concentration gradient of the peptidomimetic inhibitors. Formation of product pNA was measured every 10 min. The IC50 is defined as the concentration of each inhibitor that led to 50% of maximal APA turnover of Glu-pNA. The Ki values for the inhibition assay were determined from the IC50 using the Cheng-Prusoff equation: Ki = IC50/(1 + [S]/Km).
RESULTS
Structural Determinations of APA and APA-Ligand Complexes
Mammalian APA is a class II membrane protein with a short intracellular N-terminal tail, a single-pass transmembrane anchor, a small stalk, and a large C-terminal ectodomain (24). The human APA ectodomain (residues 76–957) was expressed, purified, and crystallized in space group P6422 (a = 142.3, b = 142.3, and c = 237.3 Å), with one molecule/asymmetric unit. The structure was determined by MIRAS using one mercury derivative and one platinum derivative. The final structural model was refined at 2.15 Å resolution (Fig. 2, A and B). It contains residues 76–954 of the APA ectodomain (except for disordered loop 608–611) and 11 N-linked glycans. APA-ligand complexes were prepared by soaking each of the ligands (amino acids or peptidomimetic inhibitors) into APA crystals in the presence or absence of calcium. Their structures were determined by molecular replacement using the unliganded APA structure as the search template and were subsequently refined. Data and model statistics are listed in Table 1.
FIGURE 2.
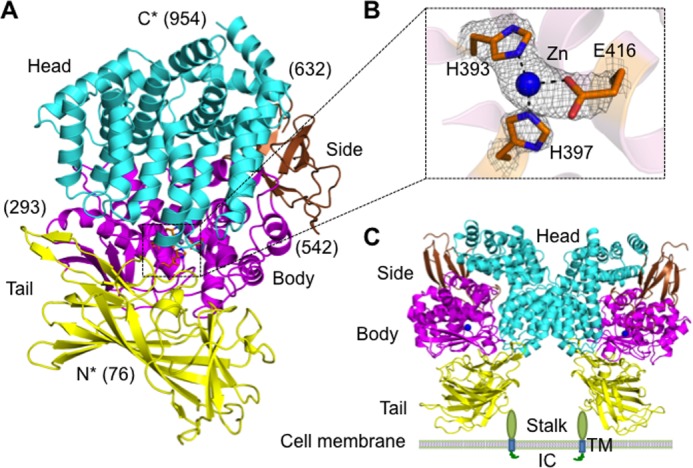
Overall structure of the human APA ectodomain. A, the human APA ectodomain contains four domains: head (cyan), side (brown), body (magenta), and tail (yellow). Residue numbers indicating domain boundaries are in parentheses. B, 2Fo − Fc electron density map of the zinc-binding site (contoured at 1.5σ). C, structure of the dimeric APA ectodomain and model of dimeric full-length APA on the cell surface. Zinc is shown as a blue sphere. IC, intracellular tail; TM, transmembrane anchor.
TABLE 1.
Data collection and refinement statistics
Values in parentheses are for the highest resolution shell. r.m.s.d., root mean square deviation.
| Native APA | APA-glutamate complex | APA-Ca-glutamate complex | APA-Ca-aspartate complex | APA-arginine complex | APA-amastatin complex | APA-bestatin complex | |
|---|---|---|---|---|---|---|---|
| Data collection | |||||||
| Space group | P6422 | P6422 | P6422 | P6422 | P6422 | P6422 | P6422 |
| Cell dimensions | |||||||
| a, c (Å) | 142.3, 237.3 | 142.2, 237.1 | 142.2, 237.1 | 142.1, 237.4 | 142.3, 237.2 | 142.7, 237.8 | 141.9, 237.1 |
| γ | 120° | 120° | 120° | 120° | 120° | 120° | 120° |
| Resolution (Å) | 50–2.05 | 50–2.4 | 50–2.15 | 50–2.4 | 50–2.25 | 50–2.4 | 50–2.4 |
| Total reflections | 534,840 | 336,976 | 525,986 | 403,261 | 481,998 | 609,019 | 334,475 |
| Unique reflections | 76,159 | 49,269 | 74,275 | 54,380 | 67,214 | 53,672 | 55,096 |
| Wilson B-factor | 35.90 | 42.53 | 31.44 | 45.48 | 32.96 | 34.62 | 43.00 |
| Rsym | 0.080 (0.573) | 0.085 (0.537) | 0.083 (0.454) | 0.077 (0.613) | 0.095 (0.439) | 0.126 (0.425) | 0.086 (0.639) |
| Rp.i.m. (41, 42) | 0.035 (0.274) | 0.033 (0.264) | 0.031 (0.196) | 0.032 (0.271) | 0.038 (0.200) | 0.039 (0.131) | 0.038 (0.278) |
| I/σ | 22.0 (2.9) | 17.5 (2.6) | 19.16 (3.67) | 25.7 (2.3) | 21.8 (3.9) | 14.9 (7.18) | 21.4 (2.4) |
| Completeness (%) | 99.4 (99.6) | 88.7 (90.8) | 97.3 (93.9) | 97.9 (98.9) | 99.8 (98.3) | 98.2 (100.0) | 99.7 (99.7) |
| Redundancy | 6.0 (5.2) | 6.8 (5.1) | 7.0 (5.6) | 7.3 (7.1) | 7.1 (5.5) | 11.0 (10.5) | 6.0 (5.7) |
| Refinement | |||||||
| Resolution (Å) | 38.9–2.15 | 38.8–2.4 | 45.6–2.15 | 48.6–2.4 | 45.6–2.25 | 41.2–2.4 | 33.8–2.4 |
| Test set reflections | 3832 | 2518 | 3742 | 2750 | 3403 | 2713 | 2793 |
| Rwork/Rfree | 0.173/0.218 | 0.167/0.226 | 0.183/0.248 | 0.152/0.224 | 0.167/0.236 | 0.210/0.276 | 0.170/0.252 |
| No. of atoms | 7993 | 7854 | 7945 | 7901 | 8011 | 7956 | 7853 |
| Protein | 7186 | 7153 | 7159 | 7159 | 7160 | 7151 | 7159 |
| Carbohydrate | 210 | 238 | 224 | 224 | 224 | 238 | 224 |
| Ligand | 0 | 10 | 10 | 9 | 12 | 33 | 22 |
| Ion | 1 | 1 | 2 | 2 | 1 | 1 | 1 |
| Water | 595 | 452 | 550 | 507 | 614 | 533 | 445 |
| B-factors (Å2) | 58.7 | 61.5 | 56.9 | 64.6 | 59.7 | 56.1 | 65.4 |
| Protein | 56.9 | 59.6 | 55.2 | 62.8 | 57.5 | 54.2 | 63.6 |
| Ligand | 79.7 | 67.1 | 96.3 | 94.9 | 53.4 | 74.4 | |
| Ion | 80.8 | 74.1 | 59.2 | 77.0 | 85.0 | 76.5 | 74.2 |
| Water | 58.8 | 60.6 | 58.6 | 62.8 | 61.9 | 55.2 | 64.4 |
| r.m.s.d. | |||||||
| Bond lengths (Å) | 0.007 | 0.008 | 0.007 | 0.007 | 0.008 | 0.009 | 0.009 |
| Bond angles | 1.239° | 1.297° | 1.265° | 1.201° | 1.245° | 1.313° | 1.282° |
| Ramachandran plot | |||||||
| Favored (%) | 97 | 96 | 96 | 96 | 96 | 97 | 95 |
| Outliers (%) | 0.23 | 0.57 | 0.69 | 0.57 | 0.57 | 0.69 | 0.34 |
Overall Architecture
The human APA ectodomain has the same domain structures and arrangement as other M1 family zinc metalloenzymes (Fig. 2, A and C, and Fig. 3). It consists of four distinct domains: head (residues 633–954), side (residues 543–632), body (residues 294–542), and tail (residues 76–293) (Fig. 2A). The zinc-binding active site is located in a cavity created by the head, side, and body domains (Fig. 2B). The active site cavity of M1 family zinc metalloenzymes can alternate between an open conformation for substrate loading or product unloading and a closed conformation for catalysis. These conformational changes are through a hinge-opening movement between the head domain and the other domains, accompanied by positional changes of catalytic residues (26). All of the catalytic residues in the current APA structure are in their catalytically active positions (Fig. 4), suggesting that the APA structure captured in this study is in a closed conformation. However, the active site cavity of APA in the closed conformation is significantly narrower than that of mammalian APN in the closed conformation (Fig. 3A), suggesting that APA excludes large peptide substrates from accessing its active site and hence has a more restricted range of substrates compared with APN.
FIGURE 3.
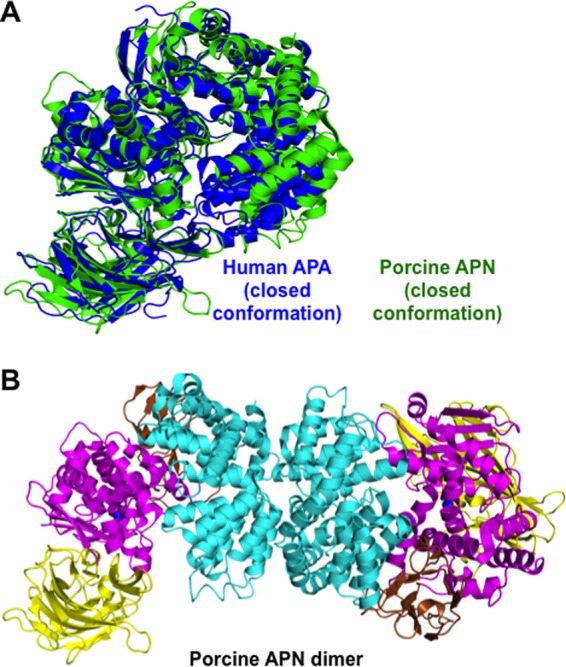
Structural comparisons of the conformation and dimer formation of porcine APN and human APA. A, structural alignment of APA (blue) and APN (green; Protein Data Bank code 4FKE), both of which are in the catalytically active and closed conformation. B, structure of the dimeric porcine APN ectodomain.
FIGURE 4.
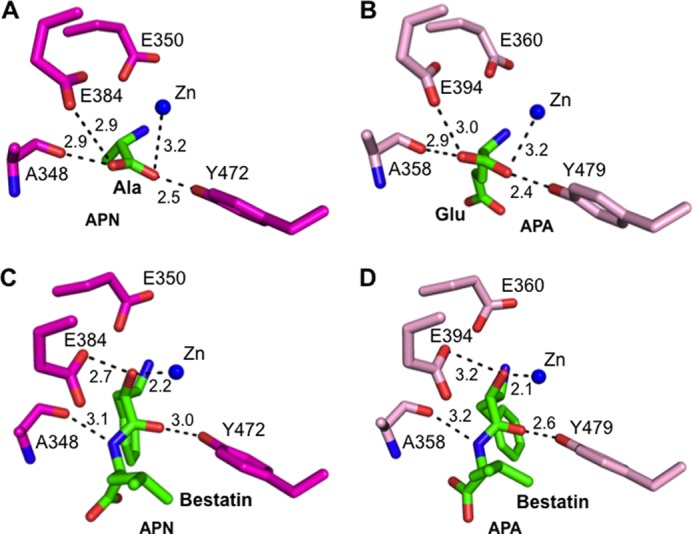
Structural comparisons of the active sites of porcine APN and human APA. A, interaction between the catalytic residues of APN (magenta) and the main chain of bound alanine (green; Protein Data Bank code 4FKH). B, interaction between the catalytic residues of APA (pink) and the main chain of bound glutamate (green). C, interaction between the catalytic residues of APN and the main chain of bound bestatin (green; Protein Data Bank code 4FKK). D, interaction between the catalytic residues of APA and the main chain of bound bestatin (green).
The APA ectodomain forms a novel homodimer. In the crystal, there is only one molecule of the APA ectodomain in each asymmetric unit. However, two molecules from adjacent asymmetric units form the homodimer through their head domains (Fig. 2C), burying ∼1600 Å2 of accessible surface between them. These structural data are consistent with the previous observation that full-length mammalian APA forms a homodimer on cell surfaces (25). Interestingly, although APA and APN both form a homodimer through their head domains, different regions in their head domains are involved in dimer formation, leading to completely different dimer interfaces (Figs. 2C and 3B). Dimer formation can increase the stability of APA and APN, both of which are ectoenzymes and face harsher environments than monomeric intracellular zinc metalloenzymes in the same family. Furthermore, the head-to-head dimer arrangements in both APA and APN can facilitate the closed-to-open conformational changes, during which the dimer interfaces are maintained and the membrane anchors move along the cell membrane. The different dimer arrangements of APA and APN suggest that a conserved dimer interface is not a prerequisite for the above functional benefits associated with dimer formation.
Substrate Specificity
To investigate the substrate specificity of APA, we determined the crystal structures of APA complexed with amino acids or peptidomimetic inhibitors (Fig. 5, A and B, and Fig. 6, A and B). APA and APN form the same interactions with the main chain of the N-terminal residue (P1 residue) of their ligands (Fig. 4), suggesting that the two enzymes share a common catalytic mechanism despite their modest sequence similarity. However, the S1 pocket that accommodates the P1 side chain of ligands differs markedly between APA and APN, consistent with the different substrate specificities of the two enzymes. In general, the S1 pocket of APN is largely hydrophobic and prefers to accommodate hydrophobic amino acid side chains. In contrast, the S1 pocket of APA is polar and prefers to accommodate polar P1 side chains. In this study, we focused on the interactions between the S1 pocket of APA and the P1 side chains of ligands that are major determinants of the substrate specificity of APA.
FIGURE 5.
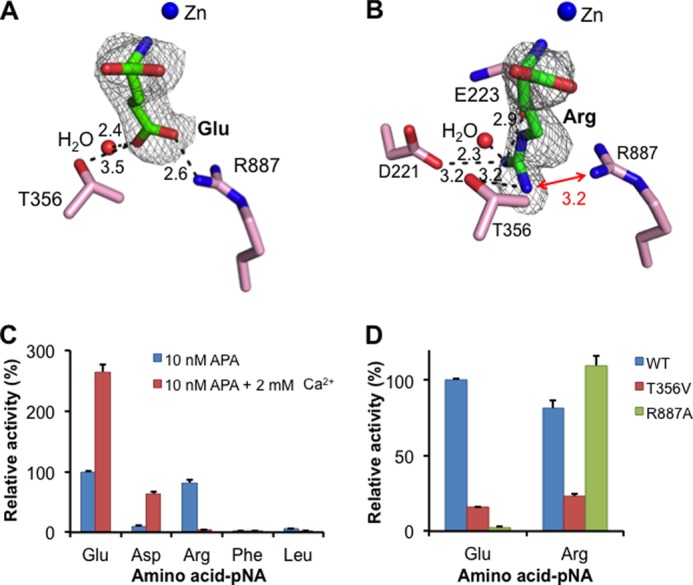
Substrate specificity of APA in the absence of calcium. A, detailed interactions between a glutamate side chain and the S1 pocket of APA. B, detailed interactions between an arginine side chain and the S1 pocket of APA. APA residues are in pink, and ligands are in green. Electron density maps of ligands correspond to Fo − Fc omit maps (contoured at 2.5σ) that were calculated in the absence of ligands. Models of APA-bound ligands were built based on these maps. Unit of distances is angstrom. C, relative catalytic activities of APA on different substrates (amino acid-pNA) in the absence or presence of calcium. The catalytic activity of APA on Glu-pNA in the absence of calcium was taken as 100%. Each bar shows the mean ± S.E. (n = 3). D, relative catalytic activities of APA mutants on substrates. The catalytic activity of wild-type APA on Glu-pNA was taken as 100%.
FIGURE 6.
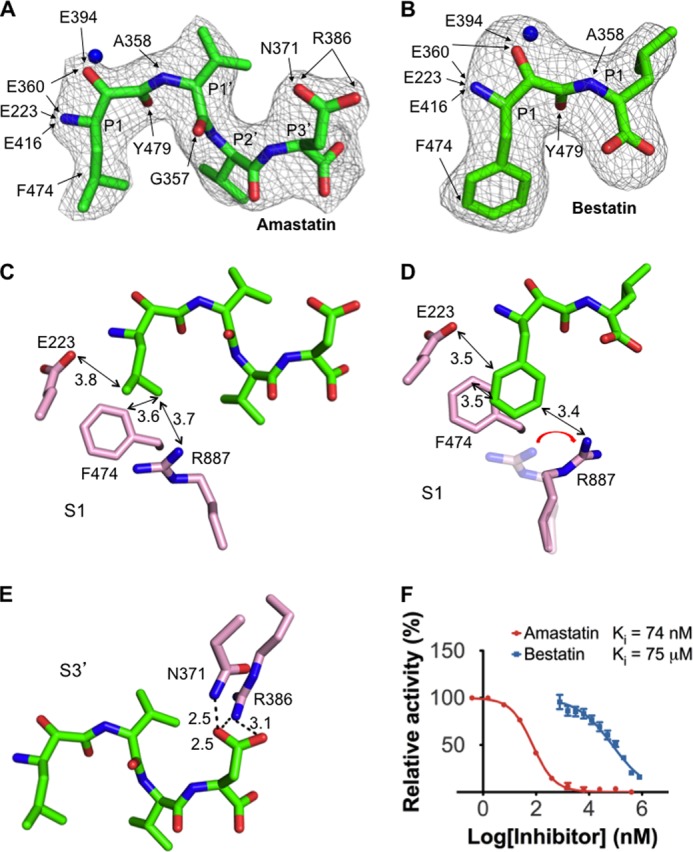
APA-inhibiting mechanisms of peptidomimetic inhibitors. A, interactions between the inhibitor amastatin and APA. B, interactions between the inhibitor bestatin and APA. Detailed interactions between the inhibitors and APA residues are shown by arrows. Electron density maps of inhibitors correspond to Fo − Fc omit maps (contoured at 2.5σ) that were calculated in the absence of inhibitors. Models of APA-bound inhibitors were built based on these maps. C, detailed interactions between the P1 side chain of amastatin and the S1 pocket of APA. D, detailed interactions between the P1 side chain of bestatin and the S1 pocket of APA. E, detailed interactions between the P3′ side chain of amastatin and the S3′ subsite of APA. F, inhibition of APA catalytic activity by amastatin and bestatin. The catalytic activity of APA on Glu-pNA in the absence of inhibitor was taken as 100%. All values are mean ± S.E. (n = 3).
The S1 pocket of APA is well suited to accommodate the side chains of acidic residues. The carboxylate side chain of bound glutamate forms a strong salt bridge with Arg-887 and a hydrogen bond with Thr-356 in the S1 pocket (Fig. 5A). These energetically favorable interactions are consistent with high APA activity on substrates with a P1 glutamate (Fig. 5C). Mutation R887A causes a dramatic decrease in APA activity on substrates with a P1 glutamate through removal of the salt bridge (Fig. 5D). Mutation T356V also decreases APA activity on substrates with a P1 glutamate through removal of the hydrogen bond. In the absence of calcium, aspartate could not be soaked into the APA crystals, consistent with the relatively low APA activity on substrates with a P1 aspartate in the absence of calcium. Presumably, the relatively short side chain of aspartate would form a weak salt bridge with Arg-887, and thus, aspartate is less favored than glutamate by APA at the P1 substrate site.
The S1 pocket also accommodates the side chains of basic residues (Fig. 5C). The guanidinium group of bound arginine forms a salt bridge with the side chain of Asp-221, a hydrogen bond with the main chain carbonyl of Glu-223, and another hydrogen bond with the side chain of Thr-356 (Fig. 5B). Mutation T356V decreases APA activity on substrates with a P1 arginine through removal of the hydrogen bond with the Thr-356 side chain. Interestingly, compared with bound glutamate, the side chain of bound arginine points in a different direction, which reduces, but does not eliminate, charge repulsion with Arg-887 in the S1 pocket. Mutation R887A increases APA activity on substrates with a P1 arginine through elimination of the charge repulsion (Fig. 5D). These results are consistent with the observation that in the absence of calcium, arginine is less favored than glutamate, but more favored than aspartate, by APA at the P1 substrate site (Fig. 5C).
The S1 pocket discriminates against the side chains of neutral residues, as evidenced by low APA activity on substrates with a P1 leucine or phenylalanine (Fig. 5C). Accordingly, the amino acids leucine and phenylalanine were not observed bound to APA when soaked into the APA crystals. Instead, two peptidomimetic inhibitors, amastatin and bestatin, which contain a P1 leucine and phenylalanine, respectively, were successfully soaked into the APA crystals (Fig. 6, A and B). The side chains of leucine and phenylalanine both form hydrophobic interactions with the side chains of Phe-474 in the S1 pocket but also have unfavorable steric interactions with the polar residues in the S1 pocket (Fig. 6, C and D). Steric hindrance from the P1 phenylalanine of bestatin even forces Arg-887 to adopt a different conformation (Fig. 6D). Therefore, residues with large neutral side chains are disfavored by APA at the P1 substrate site.
Calcium Modulation
To investigate the calcium-modulated substrate specificity of APA, we soaked calcium into the crystals of APA complexed with glutamate. The Fo − Fc map showed clear additional electron density in the S1 pocket of APA, which we interpreted to be a calcium ion and two calcium-coordinating water molecules (Fig. 7A). Our interpretation was based on the following observations. First, in the absence of calcium, the proposed calcium-binding site is occupied by a water molecule (Fig. 7B). In the presence of calcium, the structural model was first refined with the water molecule occupying the proposed calcium-binding site, and the Fo − Fc map was then calculated. The Fo − Fc map contained significant positive electron density at this site, indicating a species more electron-rich than water. Second, in the absence of calcium, the water molecule occupying the proposed calcium-binding site is four-coordinate, forming hydrogen bonds with another water molecule, the Asp-221 side chain, the Glu-223 main chain carbonyl, and the bound glutamate (which is likely protonated due to the strong bifurcated salt bridge with Arg-887) (Fig. 7B). In the presence of calcium, two additional water molecules are observed, increasing the coordination number to six, consistent with calcium replacing the water (Fig. 7A). Third, the geometry of the coordinated calcium is octahedral, consistent with the geometry for calcium coordination in other calcium-binding proteins (39, 40). Finally, previous biochemical data show that mutation of Asp-221 to other residues (e.g. alanine or asparagine) abolishes calcium modulation of APA activity (30), which is consistent with our structural data. Therefore, we conclude that the calcium-binding site is located in the S1 pocket of APA adjacent to the P1 side chain of its ligands.
FIGURE 7.
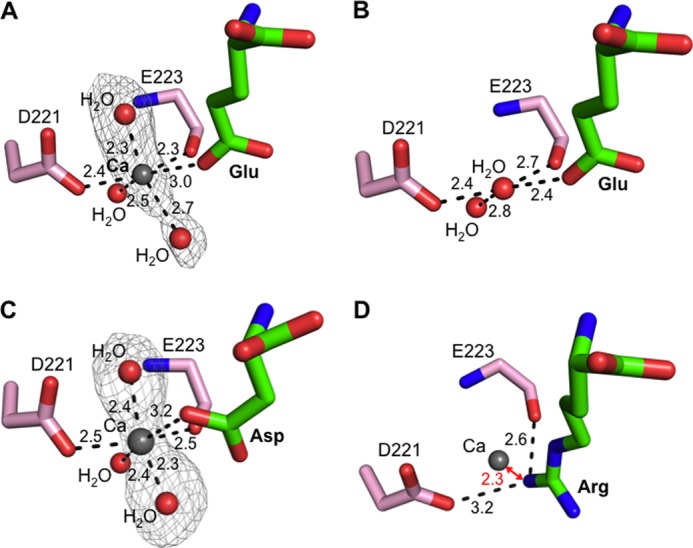
Calcium-modulated substrate specificity of APA. A, observed calcium-binding site in the S1 pocket of glutamate-bound APA. The electron density map of calcium and two additional calcium-coordinating water molecules corresponds to the Fo − Fc omit map (contoured at 3.5σ) that was calculated with a water molecule occupying the calcium-binding site and in the absence of the two additional water molecules. B, in the absence of calcium, a water molecule occupies the calcium-binding site in the S1 pocket of glutamate-bound APA. C, observed calcium-binding site in the S1 pocket of aspartate-bound APA. The electron density map was calculated in the same way as described for A, except that it was contoured at 3.0σ. D, modeled calcium-binding site in the S1 pocket of arginine-bound APA. In the presence of calcium, arginine was not observed upon soaking into APA crystals. Instead, a structural model was constructed in which calcium replaced the water molecule occupying the calcium-binding site as in Fig. 5B.
As discussed above, in the absence of calcium, aspartate could not be observed in soaked APA crystals due to the relatively low binding affinity of aspartate. In contrast, in the presence of calcium, aspartate was observed following soaking into the APA crystals, consistent with the observation that calcium enhances the APA activity on substrates with a P1 aspartate (Fig. 5C). Using the same strategies that we used to identify the calcium-binding site in the glutamate-bound APA structure, we again identified the same calcium-binding site in the aspartate-bound APA structure (Fig. 7C).
The identified calcium-binding site provides a structural basis for the calcium-modulated APA activity. Calcium enhances the APA-binding affinities of glutamate and aspartate by ligating their carboxylate side chains (Fig. 7, A and C). In contrast, arginine could not be observed following soaking into calcium-bound APA crystals. The positively charged side chain of arginine cannot bind effectively due to charge repulsion with calcium, although it could be observed in soaked APA crystals in the absence of calcium (Figs. 5B and 7D). Therefore, the calcium ion that binds to the S1 pocket of APA enhances the binding of acidic residues at the P1 substrate site through coordination of their side chains but reduces the binding of basic residues at the P1 substrate site due to charge repulsion with their positively charged side chains.
APA Inhibitors
Two peptidomimetic inhibitors, amastatin and bestatin, are universal inhibitors of M1 family zinc metalloenzymes. Here, we show that amastatin is a much more potent APA inhibitor than bestatin (Fig. 6F). By determining the crystal structures of APA complexed with each inhibitor, we elucidated the structural details of APA inhibition by these two inhibitors. First, as discussed above, the hydrophobic P1 side chains of both of the inhibitors have unfavorable steric interactions with the polar S1 pocket of APA. Due to its bulkier side chain, the P1 phenylalanine of bestatin has more severe steric clashes with the polar S1 pocket, forcing Arg-887 in the S1 pocket to adopt a different conformation (Fig. 6D). Second, amastatin contains four residues, whereas bestatin contains only two. The side chain of the C-terminal residue of amastatin (i.e. P3′ position) fits nicely into the S3′ subsite of APA, forming a strong bifurcated salt bridge with Arg-386 and a hydrogen bond with Asn-371 (Fig. 6E). In contrast, the shorter bestatin does not interact with the S3′ subsite of APA. These structural differences between the two inhibitors account for their different APA-inhibiting potencies.
Our study also has implications for the mechanisms of action of APA inhibitors that were developed prior to the structure determination of APA. For example, RB150 is a prodrug for the potent APA inhibitor EC33 (18). RB150 is orally available, can pass the blood-brain barrier, and is subsequently converted to EC33 in the brain (Fig. 8). EC33 has been shown to be an effective antihypertensive compound in animal models (18). Interestingly, EC33 and glutamate share common structural features by containing a free N-terminal amine group and a negatively charged side chain of the same length. The free N-terminal amine group of EC33 likely binds to the APA active site in the same way as those of peptide substrates and analogs (Fig. 8). Although the side chains of EC33 and glutamate contain a sulfonic acid group and a carboxylate group, respectively, the former likely binds APA in the same way as the latter by forming salt bridges with Arg-887 and calcium in the S1 pocket. In addition, EC33 also contains a thiol group on the opposite end of the molecule to the amine group, in contrast to the carboxylate group of the C terminus of glutamate. It was hypothesized that the thiol group of EC33 directly interacts with the active site zinc (18), but this hypothesis will need to be tested by crystallographic studies of APA complexed with EC33. Nevertheless, EC33 likely functions as a substrate analog and competitively binds to the APA active site.
FIGURE 8.
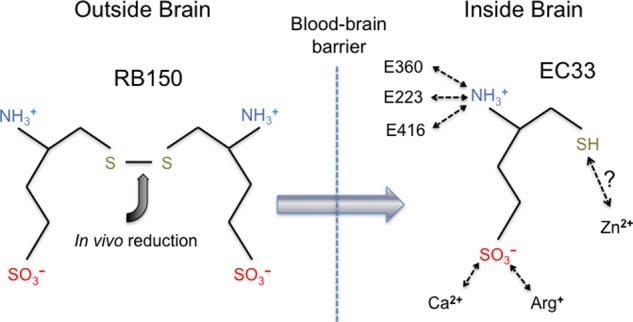
Possible mechanism of action of RB150, a brain-targeting APA inhibitor. RB150 is an orally available prodrug, can pass the blood-brain barrier, and is converted to its active form (EC33) inside the brain (18). Potential interactions between EC33 and human APA are shown schematically.
DISCUSSION
Hyperactivity in the central RAS has been implicated in the development and maintenance of hypertension (7–9). In particular, a high-level central RAS, accompanied by a low-level systemic RAS, has been described in some hypertensive patients who are resistant to treatments targeting the systemic RAS (10). The major effector peptide for blood pressure regulation in the brain is angiotensin III (13–15). Angiotensin III is produced by the action of APA on angiotensin II and is converted to angiotensin IV by APN (Fig. 1) (11, 12). To achieve central hypertension regulation, it is critical that the two different enzymes control these two angiotensin-converting steps. Thus, the substrate specificity of APA is essential for central hypertension regulation. How APA specifically cleaves only the N-terminal aspartate from angiotensin II but does not additionally cleave the subsequent arginine following the formation of angiotensin III has been a mystery. More intriguingly, the substrate specificity of APA is modulated by calcium at physiologically relevant concentrations. This study has elucidated the structural mechanisms for calcium-modulated high APA activity on angiotensin II and low APA activity on angiotensin III, providing structural insights into central hypertension regulation by human APA.
In this study, we determined the crystal structure of human APA by itself and in complex with a variety of ligands in the absence and presence of calcium. In the absence of calcium, the polar S1 pocket of APA forms salt bridges and hydrogen bonds with the charged side chains of both acidic and basic residues at the N terminus of ligands, but it has unfavorable steric interactions with the hydrophobic side chains of neutral residues. Thus, in the absence of calcium, the S1 pocket of APA has evolved to accommodate both acidic and basic residues, but not neutral residues, at the N terminus of ligands. In fact, in the absence of calcium, APA has higher activity on substrates with an N-terminal arginine than on those with an N-terminal aspartate. We further identified a calcium-binding site in the S1 pocket. When bound to APA, calcium is coordinated by the carboxylate side chain of glutamate or aspartate but leads to charge repulsion with the guanidinium group of arginine. Thus, APA-bound calcium enhances the binding of glutamate and aspartate to APA and reduces the binding of arginine to APA. Consequently, in the presence of calcium, APA has much higher activity on substrates with an N-terminal aspartate (e.g. angiotensin II) than on those with an N-terminal arginine (e.g. angiotensin III). These unique and elegant structural mechanisms ensure that under physiological conditions, APA cleaves only angiotensin II, but not angiotensin III, providing regulation to central hypertension.
This study also provides a structural platform for the design and development of novel APA inhibitors to treat hypertension. In several types of experimental and genetic hypertension animal models, orally active and brain-targeting APA inhibitors can modulate central RAS activity and reduce blood pressure without changing systemic RAS activity (16–19). Furthermore, these APA inhibitors do not alter blood pressure in normotensive animals. The crystal structures of renin and ACE, two other peptidases in the RAS, have been successfully used in guiding rational drug design. The crystal structures of APA and its complexes with ligands can also be used in the rational design and development of novel antihypertensive drugs. More specifically, on the basis of the structural information of APA, we can design novel APA inhibitors with functional groups that explore and fit into different subsites in APA. In addition, using the structural framework of APA, we can also optimize and further develop existing APA inhibitors to enhance their potencies. To conclude, the structural information contained in this study regarding APA holds promise for the design and development of a new class of antihypertensive drugs.
Acknowledgments
We thank Carrie Wilmot, Erik Yukl, Hiroshi Hiasa, and Robert Geraghty for discussion and comments and the staff at Advance Light Source beamline 4.2.2 for assistance in x-ray data collection. Computer resources were provided by the Basic Sciences Computing Laboratory of the University of Minnesota Supercomputing Institute.
- RAS
- renin-angiotensin system
- ACE
- angiotensin-converting enzyme
- APA
- aminopeptidase A
- APN
- aminopeptidase N
- pNA
- p-nitroanilide
- MIRAS
- multiple isomorphous replacement and anomalous scattering.
REFERENCES
- 1. Kearney P. M., Whelton M., Reynolds K., Muntner P., Whelton P. K., He J. (2005) Global burden of hypertension: analysis of worldwide data. Lancet 365, 217–223 [DOI] [PubMed] [Google Scholar]
- 2. Calhoun D. A., Jones D., Textor S., Goff D. C., Murphy T. P., Toto R. D., White A., Cushman W. C., White W., Sica D., Ferdinand K., Giles T. D., Falkner B., Carey R. M. (2008) Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 51, 1403–1419 [DOI] [PubMed] [Google Scholar]
- 3. Radevski I., Skudicky D., Candy G., Sathekge S., Strugo V., Sareli P. (1999) Antihypertensive monotherapy with nisoldipine CC is superior to enalapril in black patients with severe hypertension. Am. J. Hypertens. 12, 194–203 [DOI] [PubMed] [Google Scholar]
- 4. Saunders E., Weir M. R., Kong B. W., Hollifield J., Gray J., Vertes V., Sowers J. R., Zemel M. B., Curry C., Schoenberger J., Wright J. T., Kirkendall W., Conradi E. C., Jenkins P., McLean B., Massie B., Berenson G., Flamenbaum W. (1990) A comparison of the efficacy and safety of a β-blocker, a calcium channel blocker, and a converting enzyme inhibitor in hypertensive blacks. Arch. Intern. Med. 150, 1707–1713 [PubMed] [Google Scholar]
- 5. Matchar D. B., McCrory D. C., Orlando L. A., Patel M. R., Patel U. D., Patwardhan M. B., Powers B., Samsa G. P., Gray R. N. (2008) Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers for treating essential hypertension. Ann. Intern. Med. 148, 16–29 [DOI] [PubMed] [Google Scholar]
- 6. Saine D. R., Ahrens E. R. (1996) Renal impairment associated with losartan. Ann. Intern. Med. 124, 775. [DOI] [PubMed] [Google Scholar]
- 7. Basso N., Ruiz P., Mangiarua E., Taquini A. C. (1981) Renin-like activity in the rat brain during the development of DOC-salt hypertension. Hypertension 3, 11–14 [DOI] [PubMed] [Google Scholar]
- 8. Ganten D., Hermann K., Bayer C., Unger T., Lang R. E. (1983) Angiotensin synthesis in the brain and increased turnover in hypertensive rats. Science 221, 869–871 [DOI] [PubMed] [Google Scholar]
- 9. Guyenet P. G. (2006) The sympathetic control of blood pressure. Nat. Rev. Neurosci. 7, 335–346 [DOI] [PubMed] [Google Scholar]
- 10. Bakris G., Bursztyn M., Gavras I., Bresnahan M., Gavras H. (1997) Role of vasopressin in essential hypertension: racial differences. J. Hypertens. 15, 545–550 [DOI] [PubMed] [Google Scholar]
- 11. Zaman M. A., Oparil S., Calhoun D. A. (2002) Drugs targeting the renin-angiotensin-aldosterone system. Nat. Rev. Drug Discov. 1, 621–636 [DOI] [PubMed] [Google Scholar]
- 12. Oparil S., Haber E. (1974) The renin-angiotensin system (first of two parts). N. Engl. J. Med. 291, 389–401 [DOI] [PubMed] [Google Scholar]
- 13. Zini S., Fournié-Zaluski M. C., Chauvel E., Roques B. P., Corvol P., Llorens-Cortès C. (1996) Identification of metabolic pathways of brain angiotensin II and III using specific aminopeptidase inhibitors: predominant role of angiotensin III in the control of vasopressin release. Proc. Natl. Acad. Sci. U.S.A. 93, 11968–11973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Réaux A., de Mota N., Zini S., Cadel S., Fournié-Zaluski M. C., Roques B. P., Corvol P., Llorens-Cortès C. (1999) PC18, a specific aminopeptidase N inhibitor, induces vasopressin release by increasing the half-life of brain angiotensin III. Neuroendocrinology 69, 370–376 [DOI] [PubMed] [Google Scholar]
- 15. Zini S., Demassey Y., Fournié-Zaluski M. C., Bischoff L., Corvol P., Llorens-Cortès C., Sanderson P. (1998) Inhibition of vasopressinergic neurons by central injection of a specific aminopeptidase A inhibitor. Neuroreport 9, 825–828 [DOI] [PubMed] [Google Scholar]
- 16. Bodineau L., Frugière A., Marc Y., Inguimbert N., Fassot C., Balavoine F., Roques B., Llorens-Cortès C. (2008) Orally active aminopeptidase A inhibitors reduce blood pressure: a new strategy for treating hypertension. Hypertension 51, 1318–1325 [DOI] [PubMed] [Google Scholar]
- 17. Reaux A., Fournié-Zaluski M. C., David C., Zini S., Roques B. P., Corvol P., Llorens-Cortès C. (1999) Aminopeptidase A inhibitors as potential central antihypertensive agents. Proc. Natl. Acad. Sci. U.S.A. 96, 13415–13420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fournié-Zaluski M. C., Fassot C., Valentin B., Djordjijevic D., Reaux-Le Goazigo A., Corvol P., Roques B. P., Llorens-Cortès C. (2004) Brain renin-angiotensin system blockade by systemically active aminopeptidase A inhibitors: a potential treatment of salt-dependent hypertension. Proc. Natl. Acad. Sci. U.S.A. 101, 7775–7780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marc Y., Gao J., Balavoine F., Michaud A., Roques B. P., Llorens-Cortès C. (2012) Central antihypertensive effects of orally active aminopeptidase A inhibitors in spontaneously hypertensive rats. Hypertension 60, 411–418 [DOI] [PubMed] [Google Scholar]
- 20. Zini S., Masdehors P., Lenkei Z., Fournié-Zaluski M. C., Roques B. P., Corvol P., Llorens-Cortès C. (1997) Aminopeptidase A: distribution in rat brain nuclei and increased activity in spontaneously hypertensive rats. Neuroscience 78, 1187–1193 [DOI] [PubMed] [Google Scholar]
- 21. Mizutani S., Ishii M., Hattori A., Nomura S., Numaguchi Y., Tsujimoto M., Kobayshi H., Murohara T., Wright J. W. (2008) New insights into the importance of aminopeptidase A in hypertension. Heart Fail. Rev. 13, 273–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Natesh R., Schwager S. L. U., Sturrock E. D., Acharya K. R. (2003) Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature 421, 551–554 [DOI] [PubMed] [Google Scholar]
- 23. Sielecki A. R., Hayakawa K., Fujinaga M., Murphy M. E. P., Fraser M., Muir A. K., Carilli C. T., Lewicki J. A., Baxter J. D., James M. N. G. (1989) Structure of recombinant human renin, a target for cardiovascular-active drugs, at 2.5 Å resolution. Science 243, 1346–1351 [DOI] [PubMed] [Google Scholar]
- 24. Li L., Wu Q., Wang J., Bucy R. P., Cooper M. D. (1993) Widespread tissue distribution of aminopeptidase A, an evolutionarily conserved ectoenzyme recognized by the BP-1 antibody. Tissue Antigens 42, 488–496 [DOI] [PubMed] [Google Scholar]
- 25. Benajiba A., Maroux S. (1980) Purification and characterization of an aminopeptidase A from hog intestinal brush-border membrane. Eur. J. Biochem. 107, 381–388 [DOI] [PubMed] [Google Scholar]
- 26. Chen L., Lin Y.-L., Peng G., Li F. (2012) Structural basis for multifunctional roles of mammalian aminopeptidase N. Proc. Natl. Acad. Sci. U.S.A. 109, 17966–17971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rozenfeld R., Iturrioz X., Maigret B., Llorens-Cortès C. (2002) Contribution of molecular modeling and site-directed mutagenesis to the identification of two structural residues, Arg-220 and Asp-227, in aminopeptidase A. J. Biol. Chem. 277, 29242–29252 [DOI] [PubMed] [Google Scholar]
- 28. Rozenfeld R., Iturrioz X., Okada M., Maigret B., Llorens-Cortès C. (2003) Contribution of molecular modeling and site-directed mutagenesis to the identification of a new residue, glutamate 215, involved in the exopeptidase specificity of aminopeptidase A. Biochemistry 42, 14785–14793 [DOI] [PubMed] [Google Scholar]
- 29. Claperon C., Rozenfeld R., Iturrioz X., Inguimbert N., Okada M., Roques B., Maigret B., Llorens-Cortès C. (2008) Asp218 participates with Asp213 to bind a Ca2+ atom into the S1 subsite of aminopeptidase A: a key element for substrate specificity. Biochem. J. 416, 37–46 [DOI] [PubMed] [Google Scholar]
- 30. Goto Y., Hattori A., Mizutani S., Tsujimoto M. (2007) Aspartic acid 221 is critical in the calcium-induced modulation of the enzymatic activity of human aminopeptidase A. J. Biol. Chem. 282, 37074–37081 [DOI] [PubMed] [Google Scholar]
- 31. Goto Y., Hattori A., Ishii Y., Mizutani S., Tsujimoto M. (2006) Enzymatic properties of human aminopeptidase A. Regulation of its enzymatic activity by calcium and angiotensin IV. J. Biol. Chem. 281, 23503–23513 [DOI] [PubMed] [Google Scholar]
- 32. Jones H. C., Keep R. F. (1988) Brain fluid calcium concentration and response to acute hypercalcaemia during development in the rat. J. Physiol. 402, 579–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 34. Terwilliger T. C., Berendzen J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Terwilliger T. C. (2000) Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 37. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 38. Murshudov G. N., Vagin A. A., Lebedev A., Wilson K. S., Dodson E. J. (1999) Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. D Biol. Crystallogr. 55, 247–255 [DOI] [PubMed] [Google Scholar]
- 39. Toyoshima C., Nakasako M., Nomura H., Ogawa H. (2000) Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 405, 647–655 [DOI] [PubMed] [Google Scholar]
- 40. Findeisen F., Minor D. L., Jr. (2010) Structural basis for the differential effects of CaBP1 and calmodulin on CaV1.2 calcium-dependent inactivation. Structure 18, 1617–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 42. Weiss M. S. (2001) Global indicators of x-ray data quality. J. Appl. Crystallogr. 34, 130–135 [Google Scholar]