

Abstract
Decreased β-cell mass reflects a shift from quiescence/proliferation into apoptosis, it plays a crucial role in the pathophysiology of diabetes. A major attempt to restore β-cell mass and normoglycemia is to improve β-cell survival. Here we show that switching off the Fas pathway using Fas apoptotic inhibitory protein (Faim/TOSO), which regulates apoptosis upstream of caspase 8, blocked β-cell apoptosis and increased proliferation in human islets. TOSO was clearly expressed in pancreatic β-cells and down-regulated in T2DM. TOSO expression correlated with β-cell turnover; at conditions of improved survival, TOSO was induced. In contrast, TOSO downregulation induced β-cell apoptosis. Although TOSO overexpression resulted in a 3-fold induction of proliferation, proliferating β-cells showed a very limited capacity to undergo multiple rounds of replication. Our data suggest that TOSO is an important regulator of β-cell turnover and switches β-cell apoptosis into proliferation.
Keywords: TOSO, Fas, Islets, β-cell proliferation, Diabetes
Introduction
In both T1DM and T2DM, the major mechanism leading to decreased β-cell mass is increased β-cell apoptosis [1–3]. In T1DM, β-cell destruction occurs through immune mediated processes; mononuclear cell infiltration in the pancreatic islets and interaction between antigen presenting cells and T-cells lead to high local concentrations of inflammatory cytokines, chemokines, reactive oxygen species (ROS) and other inflammatory products [4,5]. Cytokines and chemokines produced and secreted by activated macrophages, adipocytes and also by pancreatic β-cells have been suggested to initiate β-cell apoptosis [6]. Two major pathways trigger the onset of T1DM; the perforin/granzyme and the Fas/FasL system [7]. The Fas receptor (CD95), a 45-kDa type I transmembrane protein, is activated through interactions between antigen presenting β-cell and T-cells as well as through local expression of inflammatory mediators, i.e. cytokines, chemokines and other inflammatory compounds [5,7]. Fas binds to its ligand (FasL) and thus, initiates β-cell apoptosis [5].
FasL, a type II transmembrane protein of 40 kDa that can be secreted in a soluble form (26 kDa), is expressed primarily on activated T-lymphocytes [8] and also on β-cells [9,10]. Fas and Fas ligand are expressed in inflamed islets in pancreas sections of patients with recent-onset T1DM [11] as well as in patients with poorly controlled T2DM [9]. Triggering of Fas leads to the recruitment of Fas-associated death domain protein (FADD) and caspase-8 to the receptor (see Fig. 4). Binding of caspase-8 results in its activation by autoproteolytic cleavage and the release of the active subunits. The remaining caspase-8 prodomain is replaced by uncleaved procaspase-8, which then starts a new activation cycle.
Fig. 4.
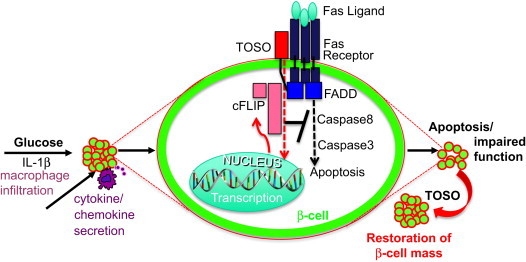
Model of the role of TOSO in protecting the β-cell from Fas/FasL induced apoptosis. Elevated glucose levels and IL-1β, which is consequently secreted by β-cells or by infiltrating macrophages in diabetes, induce apoptosis and impair function through activation of the Fas receptor. Binding of the receptor to FasL, which is constitutively expressed by β-cells, leads to recruitment of the Fas associated death domain (FADD) to the receptor complex followed by cleavage of caspases and apoptosis. Direct binding of TOSO to FADD via its C-terminal tail at the cell membrane leads to increased levels of cFLIP which binds to FADD and inhibits further cleavage of Caspases. FLIP is also able to induce β-cell proliferation and thus may lead to the restoration of β-cell mass and function.
A NOD mouse strain with a mutation in the Fas receptor, NOD lpr/lpr, did not develop diabetes [12]. β-cells from NOD mice that express a dominant-negative form of the Fas-associated death domain protein (FADD) were resistant to Fas induced cell death in vitro and in vivo. Furthermore, deletion of Fas protects islets from the toxic effects of cytokines [13] and of islet amyloid polypeptide (hIAPP) [14], which is suggested to be a major contributor to β-cell failure in T2DM [15].
In line with this observation, there are numerous studies on isolated rodent and human islets showing cytokine-induced Fas upregulation in the β-cell [10,16–18]. On the other hand, islets from NOD lpr/lpr were not protected against the autoimmune attack when transplanted into diabetic wild type recipients [19]; and only very few Fas-expressing β-cells were detected in islets of NOD mice at the onset of hyperglycemia [20]. Also, Fas signaling is needed for insulin secretion as shown in mice, pointing to a physiological role of the Fas receptor in β-cells. In human islets, an inhibitor of Fas-induced apoptosis, termed cellular FLICE (caspase-8)-inhibitory protein (FLIP) [21], was able to protect β-cells from cell death and restored β-cell function even under hyperglycemic conditions and in the presence of Fas. FLIP structurally resembles caspase-8 and thus interferes with its recruitment to the death-inducing signaling complex (DISC) and hence plays a critical role as an endogenous modulator of apoptosis [22]. Moreover, Fas signals do not always result in apoptosis but can also trigger a pathway that leads to proliferation [23]. Thereby, FLIP is pivotal in turning signals from cell death into those for cell survival/proliferation [24]. In β-cells, FLIP switched Fas activation from a death signal into a proliferation signal, and this may potentially expand β-cell mass [25]. The antagonistic anti-Fas antibody ZB4 inhibited the beneficial effect of FLIP at elevated glucose, demonstrating that Fas receptor activation is required for FLIP mediated proliferation. FLIP is also protective against cytokine-induced activation of caspase-8-dependent apoptosis [26].
A further upstream regulator of Fas is the cell surface protein TOSO, also named Fas apoptotic inhibitory protein (Faim3). It is expressed in activated T-cells [27,28]. TOSO negatively regulates FasL- and TNFα-induced apoptosis in lymphoma cell lines [29]. Also, a TOSO antibody potentiates TNFα induced apoptosis [29]. TOSO overexpressing Jurkat cells are resistant to Fas induced apoptosis through expression of FLIP [27]. FLIP expression levels are down-regulated in TOSO-deficient mice, causing these mice to be highly sensitive to Fas triggered apoptosis [30].
Thus, TOSO would provide a promising tool to block Fas induced apoptosis in β-cells, and its presence and function in human islets was investigated in the present study. The advantage of TOSO would be to regulate endogenous FLIP levels. These physiological FLIP levels are often not achieved by FLIP overexpression, and higher FLIP levels could even reverse its effect by induction of cell death. In the present study we provide evidence for constitutive expression of TOSO in the human β-cell and suggest a novel approach to prevent and treat diabetes by switching Fas signaling from apoptosis to proliferation. However, multiple rounds of self-duplication could not be achieved in human β-cells, confirming previous observations, which show that human β-cells have only a very limited capacity to self-duplicate [31].
Material and methods
Islet culture
Human islets were isolated from pancreata of 8 healthy organ donors at the University of Lille or University of Chicago and cultured in CMRL-1066 medium as described previously [32]. Islets were cultured on extracellular matrix coated dishes derived from bovine corneal endothelial cells (Novamed Ltd., Jerusalem, Israel) for 4 days, allowing the cells to attach to the dishes and spread [33] and exposed to 5.5, 11.1, or 33.3 mM glucose or 5.5 mM plus recombinant human IL-1β (0.02–2 ng/ml, R&D Systems, Minneapolis, MN) or IFNγ (1000 U/ml, PeproTec, Rocky Hill, NJ, USA).
Transfection
At 2 days post-isolation and culture on extracellular matrix coated dishes, isolated islets were exposed to transfection using Ca2+–KRH medium (KCl 4.74 mM, KH2PO4 1.19 mM, MgCl26H2O 1.19 mM, NaCl 119 mM, CaCl2 2.54 mM, NaHCO3 25 mM, HEPES 10 mM). After 1 h incubation lipoplexes (Lipofectamine2000, Invitrogen, Carlsbad, CA, USA)/DNA ratio 2.5:1, 3 μg CMV-TOSO, RIP-TOSO, or CMV-GFP control plasmid DNA/100 islets or 50 nM siRNA to TOSO (RNAs of 21 nucleotides, designed to target human TOSO; ON-TARGETplus SMARTpool human FAIM3), (Dharmacon, Lafayette CO, USA) and scramble siRNA (Dharmacon) were added to transfect the cells as described previously [34,35]. After additional 6 h incubation, CMRL 1066 medium containing 20% FCS and l-Glutamine were added to the transfected islets. Transfection efficiency was determined using RT PCR, immunocytochemistry and western blotting. TOSO plasmid was obtained from the Full-Length Mammalian Gene Collection (invitrogen) and cloned into the pIRES2-AcGFP1 vector (invitrogen) named CMV-TOSO or into the pIT-HindIII-A-myc vector (kindly provided by T. Trüb, University Hospital Zurich), named RIP-TOSO.
Cell culture and thymidine analog administration
Post-transfection, the culture medium was supplemented with 10 μmol/L of CldU (Sigma, St. Louis, MO, USA, cat#C6891) for 1 or 2 days followed by 10 μmol/L of IdU (MP Biomedicals, Illkirch, France, cat#100357) for same time duration.
Glucose stimulated insulin secretion
Islets used to perform glucose-stimulated insulin secretion experiments were kept in culture medium on matrix-coated plates. For acute insulin release in response to glucose, islets were washed and pre-incubated (30 min) in Krebs-Ringer bicarbonate buffer (KRB) containing 2.8 mM glucose and 0.5% BSA. KRB was then replaced by KRB 2.8 mM glucose for 1 h (basal), followed by an additional 1 h in KRB 16.7 mM glucose (stimulated). Islets were extracted with 0.18 N HCl in 70% ethanol for determination of insulin content. Islet insulin was determined using mouse insulin ELISA (ALPCO, Salem, NH, USA).
RNA extraction and RT-PCR analysis
Total RNA was isolated from cultured human islets as described previously [32]. For quantitative analysis, we used the Applied Biosystems StepOne Real-Time PCR system (Applied Biosystems, Carlsbad, CA, USA) with a commercial kit (Power SYBR Green PCR Master Mix; Applied Biosystems). Primers used: 5′CATGAACACAGACCGGG3′/5′GAACTGGAGGGACCTTG-3′ (human TOSO), 5′GTTGGCCAGGCTGGTGTCCAG3′/5′CTGTGATGAGCTGCTCAGGGTGG3′ (human tubulin), and 5′TCACCCACACTGTGCCCATCTACGA3′/5′CAGCGGAACCGCTCATTGCCAATGG3′ (β-actin).
Western blot analysis
At the end of the incubation periods, islets were washed in ice-cold PBS and lysed for 40 min on ice in 40 μl lysis buffer containing 20 mM Tris acetate, 0.27 M sucrose, 1 mM EDTA, 1 mM EGTA, 50 mM NaF, 1% Triton X-100, 5 mM sodium pyrophosphate and 10 mM β-glycerophosphate. Prior to use, the lysis buffer was supplemented with Protease- and Phosphatase-inhibitors (Pierce, Rockford, IL, USA). Equivalent amounts of protein from each treatment group were run on a NuPAGE 4–12% Bis-Tris gel (Invitrogen) and electrically transferred onto PVDF membranes. Membranes were incubated with rabbit anti-TOSO and rabbit anti-FLIPL (C-19), (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), and rabbit anti-β-actin (Cell Signaling Technology, Danvers, MA, USA) antibodies, followed by horseradish-peroxidase-linked anti-rabbit IgG.
Immunocytochemistry
Pancreatic sections from 8 healthy controls (4 lean and 4 obese) and from 7 patients (3 lean and 4 obese) with T2DM were obtained from the National Disease Research Interchange (NDRI), approval for the studies were granted by the Ethical Commission of Bremen University. For detection of β-cell TOSO expression insulin and TOSO staining were performed, 3 sections/pancreas were incubated in blocking buffer containing 0.2% Tween 20, 3% IgG-free Bovine serum albumin (BSA), 0.5% Triton X-100 for 1 h RT and overnight at 4 °C with rabbit anti-TOSO (Santa Cruz). Subsequently, all sections were double-stained for insulin and detected by donkey anti-guinea pig FITC-conjugated antibody (Dako, Hamburg, Germany). To evaluate TOSO antibody specificity, the antibody was pre-incubated for 1 h at 37 °C with TOSO blocking peptide (Santa Cruz). Intensity and saturation of the staining was measured using Adobe Photoshop© Extended analysis software after an adapted model used by Pham et al. [36], expressed per islet area and normalized to the background signal.
For lineage tracing experiments, cultured islets were fixed in 4% paraformaldehyde for 30 min RT; permeabilized by 0.5% Triton-X-100 for 4 min RT and incubated in freshly diluted 1.5 N HCl for 40 min RT. After blocking for 1 h RT, staining for IdU and insulin was carried with by incubating with mouse anti-BrdU antisera (1:100) (Dako) and guinea pig anti-insulin (1:100) (Cat#18-0067; Zymed Laboratories Inc., San Francisco, CA, USA) overnight at 4 °C. The dishes were washed in a low salt TBST Buffer (36 mM Tris, 50 mM NaCl, 0.5% Tween-20; pH 8.0) and then in PBS. For CldU staining, dishes were incubated with Rat anti BrdU (Dako) (1:100; diluted in antibody dilution buffer) overnight at 4 °C. Incubation with secondary antibodies was carried out using AMCA donkey anti-Guinea Pig (1:100), Cy3 donkey anti-rat (1:100 and FITC donkey anti-mouse (1:100) at RT for 1 h).
For detection of β-cell apoptosis and proliferation, insulin and TUNEL or Ki67 staining (In Situ Cell Death Detection Kit, TMR red; Roche Diagnostics, Mannheim, Germany) were performed as described previously [37]. Fluorescent dishes were analyzed using Nikon MEA53200 (Nikon GmbH, Dusseldorf, Germany) microscope and images were acquired using NIS-Elements software (Nikon).
Statistical analysis
Samples were evaluated in a randomized manner by 2 investigators (G.D, S.L.) who were blinded to the treatment conditions. Data are presented as means±SE and were analyzed by Student's t-tests. To account for multiplicity in the treated cells in vitro, a Bonferroni correction was used.
Results
TOSO is down-regulated in diabetes and correlates positively with β-cell proliferation and negatively with β-cell apoptosis
To identify cell type specific TOSO localization in the human pancreas, we assessed TOSO expression in pancreatic sections from autopsy from lean and obese non-diabetic patients and from patients with T2DM. The specificity of the antibody was confirmed by pre-incubation with TOSO blocking peptide as a negative control (Fig. 1A, right panel). Constitutive expression of TOSO was detected in insulin producing β-cells in human pancreatic sections. TOSO was expressed in both lean and obese non-diabetic patients but depleted in lean and obese patients with T2DM (Fig. 1A). Quantification of the staining showed a 2.1-fold and 8.6-fold decrease in intensity and saturation in lean patients with T2DM vs. lean controls and a 3.5- and 9.9-fold decrease in intensity and saturation in obese patients with T2DM vs. obese controls, respectively (Fig. 1B).
Fig. 1.
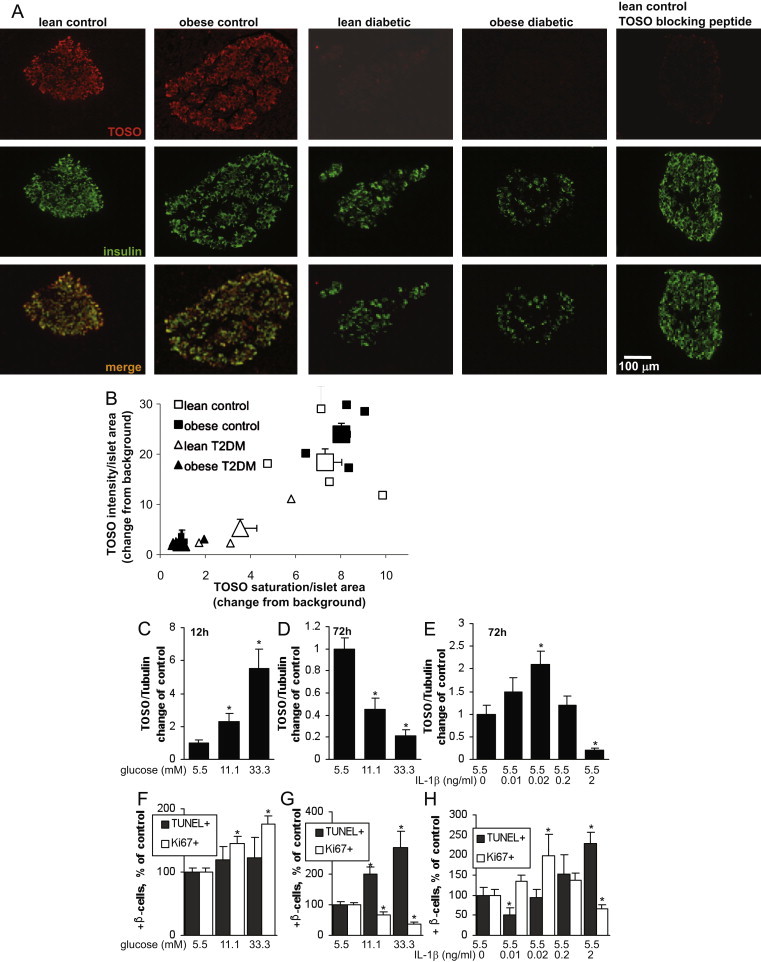
TOSO is downregulated in T2DM and correlates positively with β-cell proliferation and negatively with β-cell apoptosis. (A,B) Representative co-staining for TOSO in red and insulin in green in human pancreatic sections from poorly controlled lean (n=4) and obese (n=3) patients with T2DM and lean (n=4) and obese (n=4) non-diabetic controls. From each pancreas, 3 sections were stained and analyzed and measurements for intensity and saturation are given as signal divided by islet area and normalized to background. Small symbols show means of all analyzed islets/pancreas, large symbols show means±SE of all pancreases/group. (C–H) Human pancreatic islets were cultured on extracellular matrix-coated dishes and exposed to increasing concentrations of glucose (5.5–33.3 mM) for 12 (C,F) and 72 (D,G) h or to IL-1β (0.01–2 ng/ml) for 72 h (E,H). TOSO mRNA expression was analyzed by RT-PCR and expressed as relative changes of control and compared to Tubulin levels (C–E). In parallel, proliferation was measured by the Ki67 antibody stained in red and apoptosis analyzed by the TUNEL assay (F–H). Islets were triple-stained for insulin and counterstained for DAPI (not shown). Results are means±SE of the percentage of Ki67- and TUNEL-positive β-cells. The average number of β-cells counted was 8150 for each treatment group in 3 separate experiments from 3 different organ donors.
To investigate whether such changes in TOSO expression occur during the process of the switch from proliferation into apoptosis, we analyzed TOSO expression in isolated human islets in response to short-term (12 h, Fig. 1C) and long-term (72 h, Fig. 1D) incubation with elevated glucose concentrations, 2 distinct conditions when glucose induced proliferation (Fig. 1F) or apoptosis (Fig. 1G).
TOSO was up-regulated dependent on glucose concentrations during 12 h of exposure; in contrast, TOSO was down regulated to almost undetectable levels after long-term incubation of the islets for 72 h (Fig. 1D), a condition where glucose induced β-cell apoptosis (Fig. 1G). Previously we have found that the cytokine IL-1β has also a dual physiological role on the regulation of β-cell function and survival [38,39], it induces proliferation at low and apoptosis at high concentrations (Fig. 1H). In line with these findings, low-dose IL-1β induced TOSO expression while high-dose almost depleted TOSO mRNA expression (Fig. 1E).
TOSO improves β-cell survival and function under diabetogenic conditions
To understand the physiological role of TOSO depletion in diabetes on the β-cell, the expression of TOSO was silenced in isolated human islets by using specific siRNAs. TOSO levels resulting from its overexpression and depletion were analyzed by immunocytochemistry (Fig. 2A), RT-PCR (Fig. 2D) and Western blotting (Fig. 2E). Protein was almost undetectable in islets upon siRNA treatment and RNA was 4.3-fold down-regulated (Fig. 2D), while overexpression was achieved in almost all islet cells even under elevated glucose or IL-1β/IFNγ exposure (Fig. 2A).
Fig. 2.
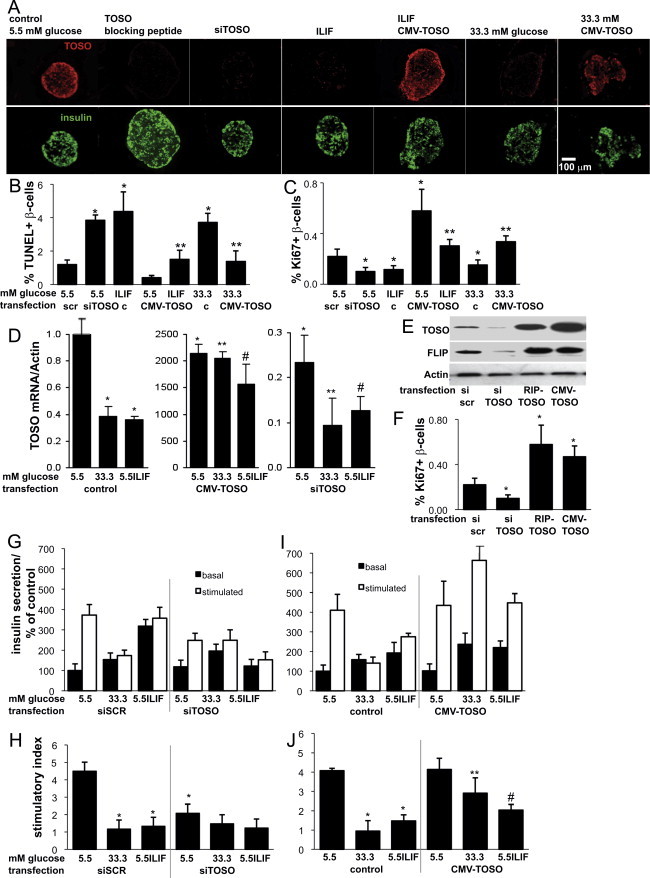
TOSO improves β-cell survival and function under diabetogenic conditions. Human pancreatic islets were cultured on extracellular matrix-coated dishes and exposed to increasing concentrations of glucose (5.5–33.3 mM) or the cytokine mixture IL-1β (2 ng/ml) and IFNγ (1000 U/ml) for 72 h with or without depletion of TOSO by siTOSO or overexpression by CMV-TOSO transfection. TOSO was analyzed in each condition by immunostaining of Bouin-fixed paraffin-embedded islet pellets. As control, TOSO peptide was incubated with TOSO antibody before staining. Representative co-staining for TOSO in red and insulin in green are shown (A). Apoptosis was analyzed by the TUNEL assay in islet sections (B) and proliferation by the Ki67 antibody (C). Islets were triple-stained for insulin and counterstained for DAPI (not shown). Results are means±SE of the percentage of Ki67- and TUNEL-positive β-cells. (D) TOSO mRNA expression was analyzed by RT-PCR and expressed as relative changes of control and compared to Tubulin levels. (E) Western blot analysis was performed 3 days after transfection with siRNA specific for TOSO, RIP-TOSO or CMV-TOSO plasmids. The same blot was analyzed for TOSO, c-FLIP (full length) and actin after stripping. (F) In parallel experiments, β-cell proliferation was assessed in islet pellets by double-staining for Ki67 and insulin. (B,C,F). The average number of β-cells counted was 7450 for each treatment group in 3 separate experiments from 3 different organ donors. (G–J) Glucose stimulated insulin secretion assays were performed after the 72 h culture period. (G,I) Basal (2.8 mM) and glucose stimulated (16.7 mM) insulin secretion was normalized to whole islet insulin content, respectively and expressed as percent change of basal conditions at 5.5 mM glucose. (H,J) Stimulatory index denotes the amount of glucose stimulated (16.7 mM glucose) divided by the amount of basal insulin secretion. Data are shown as mean±SE from 3 islet isolations from 3 different donors. p<0.05 to 5.5 mM glucose control,⁎⁎p<0.05 to 33.3 mM glucose control, #p<0.05 to IL/IF control.
TOSO depletion resulted in a 3.3-fold induction of β-cell apoptosis, similar to cytokine exposure for 4 days (3.5-fold induction, Fig. 2A and B).
Also, β-cell apoptosis was 4-fold increased by the cytokine mix IL-1β/IFNγ and 3.5-fold by 33.3 mM glucose, which was prevented by CMV-TOSO overexpression.
On the other hand, TOSO overexpression induced a 2.6-fold increase in β-cell proliferation, as assessed by the mitotic marker Ki67 (Fig. 2C). β-cell proliferation decreased upon cytokine and elevated glucose treatment, which was back to basal levels upon TOSO overexpression. The pro-survival effect of TOSO expression was hypothesized to be due to the regulation of cFLIP, which leads to inhibition of apoptotic Fas signaling and acts as a switch to proliferation. Hence, we analyzed the levels of FLIP upon depletion and overexpression of TOSO. To address the effect of paracrine signaling, overexpression was carried out either specifically in the β-cells by using a rat insulin promoter driven expression plasmid (RIP-TOSO), or in all islet cell types using a cytomegalovirus driven plasmid (CMV-TOSO). Western blot analysis confirmed the efficient silencing and overexpression of TOSO at the protein level (Fig. 2E). Depletion of TOSO caused reduced FLIP expression; and, similarly, the increased amount of TOSO induced FLIP expression (Fig. 2E). β-cell proliferation was analyzed using the Ki67 antibody as shown in Fig. 2C, and was confirmed to be increased upon both β-cell specific and universal induction of TOSO expression, thus emphasizing its signaling in β-cells themselves (Fig. 2F).
The effect of the varying levels of TOSO expression on β-cell function was analyzed by carrying out glucose stimulated insulin secretion (GSIS) after depletion or overexpression of TOSO in presence and absence of diabetic stimuli. Elevated glucose and cytokines completely abolished GSIS, and also TOSO depletion resulted in a 2.2-fold (p<0.05) reduction in the stimulatory index at basal conditions, but had no additive effect in islets exposed to diabetic milieu (Fig. 2G and H) at elevated glucose (33.3 mM) or the cytokine mixture. In contrast, TOSO overexpression improved GSIS 3.1-fold at elevated glucose levels and 1.4-fold (p<0.05) at cytokine treatment (Fig. 2I and J), while at basal condition at 5.5 mM glucose, it had no effect.
TOSO overexpression induces early proliferation in β-cells
Sequential thymidine analog labeling is a recent and effective method for lineage tracing of proliferating cells, previously used in vivo to show the lack of any specialized β-cell progenitors in mice [40]. We optimized the protocol to be used to detect multiple rounds of proliferation in vitro using immunocytochemistry.
To assess the nature of increased proliferation of β-cells upon TOSO overexpression, sequential thymidine analog labeling was carried out after overexpression of TOSO driven by the rat insulin promoter (RIP-TOSO) or cytomegalovirus promoter (CMV-TOSO). On day 1 and 2 after the RIP/CMV-TOSO transfection, we incorporated 10 μmol/L of the thymidine analogs 5-chloro-2-deoxyuridine (CldU) and 5-iodo-2-deoxyuridine (IdU), respectively for 1 day (Fig. 3A) into the islet culture medium. To exclude an effect of the expression changes of TOSO after overexpression and to take into account the number of days after transfection, analogs were also incorporated on day 2 and day 4 after transfection in a parallel experiment (Fig. 3B). The incorporation of the analogs, and thus the proliferation, was visualized using immunocytochemistry.
Fig. 3.
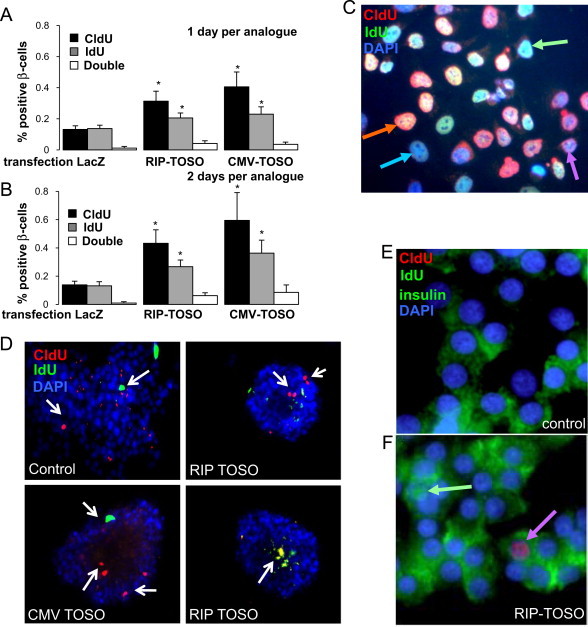
TOSO over expression induces early proliferation in β-cells. Human pancreatic islets were cultured on extracellular matrix-coated dishes and TOSO overexpression was achieved by transfection with RIP-TOSO or CMV-TOSO. LacZ transfection was used as positive control. On day 1 (A) or day 2 (B) after the RIP/CMV-TOSO transfection, 10 μmol/L of the thymidine analog 5-chloro-2-deoxyuridine (CldU) and on day 2 (A) or day 4 (B) 5-iodo-2-deoxyuridine (IdU) were added into the islet culture medium. Thereafter, islets were fixed and labeled for thymidine incorporation by using specific antibodies against CldU and IdU (see material and methods). Results are means±SE of the percentage of CldU/IdU-positive β-cells. The average number of β-cells counted was 7156 for each treatment group in 3 separate experiments from 3 different organ donors. (C,D) CldU/IdU and DAPI staining is shown in HeLa cells and human islets. CldU-positive cells are shown in red, IdU-positive cells are shown in green, and double positive cells are shown in orange (merged). Arrows show positive cells. In islets, only few double-positive cells were detected (D, panel d). (E,F) Triple staining for CldU in red, IdU in green (nucleus) and insulin in green (cytosol). Single-positive cells are shown (arrows). ⁎p<0.05 to 5.5 mM glucose lacZ transfected control.
After quantification, it was observed that under control conditions, the extent of proliferation during administration of both the analogs was similar (Fig. 3A and B). In the TOSO transfected islets, there was an about 2-fold induction in β-cell proliferation (2.4-fold and 3.0-fold by RIP- and CMV-TOSO, respectively when CldU was administered over 1 day, Fig. 3A, and 3.1-fold and 4.3-fold by RIP- and CMV-TOSO, respectively when CldU was administered over 2 days, Fig. 3B). The percentage of β-cells undergoing proliferation during the administration of the second analog IdU was significantly less than that during the first CldU administration (1.5-fold and 1.7-fold decrease during IdU compared to CldU by RIP- and CMV-TOSO, respectively when analogs were administered over 1 day, Fig. 3A, and 2.0-fold and 2.8-fold increase during IdU administration by RIP- and CMV-TOSO, respectively when analogs were administered over 2 days, Fig. 3B). A very small percentage (0.04–0.08%) of co-labeled cells were observed in the TOSO transfected samples as compared to 0.01% in the control (Fig. 3A and B), suggesting a very limited capacity of β-cells to undergo multiple rounds of proliferation. To eliminate the possibility of preferential analog uptake, the sequence of analogs was reversed and the data obtained was found to be consistent (data not shown). Also, for taking into account the number of days after transfection, 2 different administration time durations were implemented, 2 days or 1 day per analog, which consistently showed that TOSO overexpression led to an induction of an early round of proliferation in the β-cells (Fig. 3A and B).
As a proof of principle, lineage tracing was carried out in HeLa cells and proliferating differentially labeled cells could be observed (Fig. 3C).
Fig. 3D–F shows fluorescent microscopic images of a very limited number of cells inside the islets carrying the thymidine analog labels. Co-staining for insulin confirms the analysis in β-cells (Fig. 3E and F).
Discussion
More than a decade ago, Hitoshi et al. cloned and characterized a novel immunoglobulin domain-containing type I transmembrane protein, TOSO, exhibiting potent pathway-specific anti-apoptotic effects in hematopoietic cells [27]. TOSO inhibited apoptosis induced by Fas-, TNFα-, FADD-, and PMA/ionomycin; but not staurosporine- or ceramide-induced apoptosis. The mechanism of inhibition and its specificity were hypothesized to be due to inhibition of caspase-8 processing through induction of cFLIP expression. The name TOSO was given after a Japanese liquor that is drunk on New Year's Day to celebrate long life and eternal youth [27], reflecting its pro-survival effect.
The role of TOSO in maintaining cFLIP expression during Fas mediated apoptosis of lymphocytes and hepatocytes has been elucidated using TOSO−/− mice [30]. B-cells and thymocytes from these mice show increased sensitivity to Fas-triggered apoptosis, and these mice suffer greater mortality and exhibit exacerbated liver damage in response to Fas engagement in vivo. TOSO modulates Fas-mediated apoptosis by influencing the expression of c-FLIP and regulating the physical binding of caspase-8 to Fas receptor.
The anti-apoptotic function of TOSO depends on ubiquitination of an adapter kinase, RIP1, and involves the recruitment of the death adapter FADD to a TOSO/RIP1 protein complex. Upon activation by FasL and TNFα, TOSO promotes the activation of pro-survival signaling pathways and protects from liver damage [29]. TLR activation leads to decreased TOSO expression shown in leukemic B-cells [41].
The activation of inflammatory pathways has been discovered as a causal event for β-cell destruction in diabetes [42,43], which is also mediated by TLR activation [32,44].
Thereby, not only pro-inflammatory compounds are activated (e.g. IL-1β, Fas), but also the anti-inflammatory cytokine interleukin-1 receptor antagonist and the anti-apoptotic cFLIP are down-regulated in β-cells in diabetes [9,42,45], and also TOSO downregulation was reported in response to TLR activation [41].
Thus, attempts to block Fas-induced apoptosis and the activation of pro-inflammatory cytokines could be a strategy to prevent diabetes. Here we show down-regulation of another protective factor in T2DM, and its loss promotes β-cell destruction.
TOSO expression correlated positively with β-cell proliferation; both the proliferation-inducing treatments i.e. acute high glucose and low dose IL-1β positively regulated TOSO expression. On the other hand, pro-apoptotic stimuli i.e. chronic elevated glucose and high dose IL-1β almost depleted TOSO mRNA expression. Thus, depletion of TOSO coincides with activation of β-cell apoptosis under conditions of stress. TOSO, when overexpressed, not only rescued β-cells from apoptosis but also triggered proliferation. This pro-survival signaling has also been observed in TOSO overexpressing Jurkat cells in which CD95L- and TNFα stimulation readily induced the activation of Erk1/2 [29]. While TNFα potentiates cell death, it is insufficient to cause apoptosis in β-cells [46]. Since TOSO promotes the TNFα induced signaling cascade, it is possible, that TNFα would induce β-cell apoptosis in the absence of TOSO, but there is no experimental proof for this hypothesis so far.
Intact Fas receptor signaling is also necessary for β-cell secretory function [47]. Hence, we investigated whether TOSO expression can also contribute to glucose stimulated insulin secretion. While TOSO overexpression was not effective at basal glucose levels, our results show that TOSO protected the inhibition of insulin secretion by elevated glucose and cytokines and restored β-cell function.
Mature human β-cells have only a very limited capacity to undergo proliferation [31]. To control and to foster β-cell proliferation has become a long-term goal in β-cell research. With the goal to find a subpopulation of β-cells with a higher proliferative capacity, we overexpressed TOSO and investigated the nature of the induced proliferation by using the technique of sequential thymidine analog labeling, previously developed in mice by Teta et al. [40].
We optimized the method so that it could be used in a similar fashion in vitro in human islets, to detect multiple rounds of proliferation. TOSO overexpression induced an early round of proliferation in the isolated human islets, irrespective of any effect of sequence of analog administration or time after transfection, however, only in a very limited number of islets (maximum 0.08%), more than one round of proliferation was observed during the 6-d culture period. Such limited proliferation capacity is in confirmation with the results from mouse β-cells in vivo [40,48].
Conclusions
In summary, TOSO is a novel anti-apoptotic protein, which interferes with the Fas triggered apoptosis by regulating FLIP and thereby initiating a pro-survival signaling cascade (see Fig. 4 for our model). Thus, TOSO hints to be a promising therapeutic target to rescue β-cells from apoptosis induced by elevated glucose and cytokines and, hence, intervening in the progression of diabetes. However, with the means of TOSO, we were unable to develop a β-cell, which would undergo multiple rounds of replication during culture.
Conflict of interest
None declared.
Acknowledgments
This work was supported by the Competence network diabetes mellitus funded by the Federal Ministry of Education and Research (BMBF), the German Research Foundation (DFG, Emmy Noether Program MA4172/1-1), the European Research Council (ERC 260336) and the Stein-Oppenheimer Foundation. We thank the National Disease Research Interchange (NDRI) for providing human pancreatic sections. Human islets were provided through the Islet Cell Resource Consortium, administered by the Administrative and Bioinformatics Coordinating Center (ABCC) and supported by the National Center for Research Resources (NCRR), The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), the Juvenile Diabetes Research Foundation (JDRF), the Chicago Diabetes Project and through the European Consortium for Islet Transplantation (ECIT), Islets for Research Distribution Program.
References
- 1.Kurrer M.O., Pakala S.V., Hanson H.L., Katz J.D. Beta cell apoptosis in T cell-mediated autoimmune diabetes. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:213–218. doi: 10.1073/pnas.94.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butler A.E., Janson J., Bonner-Weir S., Ritzel R., Rizza R.A. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 3.Meier J.J., Bhushan A., Butler A.E., Rizza R.A., Butler P.C. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia. 2005;48:2221–2228. doi: 10.1007/s00125-005-1949-2. [DOI] [PubMed] [Google Scholar]
- 4.Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14:619–633. doi: 10.2337/diab.14.10.619. [DOI] [PubMed] [Google Scholar]
- 5.Eizirik D.L., Mandrup-Poulsen T. A choice of death—the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia. 2001;44:2115–2133. doi: 10.1007/s001250100021. [DOI] [PubMed] [Google Scholar]
- 6.Donath M.Y., Storling J., Maedler K., Mandrup-Poulsen T. Inflammatory mediators and islet beta-cell failure: a link between type 1 and type 2 diabetes. Journal of Molecular Medicine. 2003;81:455–470. doi: 10.1007/s00109-003-0450-y. [DOI] [PubMed] [Google Scholar]
- 7.Mathis D., Vence L., Benoist C. beta-Cell death during progression to diabetes. Nature. 2001;414:792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- 8.Krammer P.H. CD95's deadly mission in the immune system. Nature. 2000;407:789–795. doi: 10.1038/35037728. [DOI] [PubMed] [Google Scholar]
- 9.Maedler K., Spinas G.A., Lehmann R., Sergeev P., Weber M. Glucose induces beta-cell apoptosis via upregulation of the Fas-receptor in human islets. Diabetes. 2001;50:1683–1690. doi: 10.2337/diabetes.50.8.1683. [DOI] [PubMed] [Google Scholar]
- 10.Loweth A.C., Williams G.T., James R.F., Scarpello J.H., Morgan N.G. Human islets of Langerhans express Fas ligand and undergo apoptosis in response to interleukin-1beta and Fas ligation. Diabetes. 1998;47:727–732. doi: 10.2337/diabetes.47.5.727. [DOI] [PubMed] [Google Scholar]
- 11.Moriwaki M., Itoh N., Miyagawa J., Yamamoto K., Imagawa A. Fas and Fas ligand expression in inflamed islets in pancreas sections of patients with recent-onset Type I diabetes mellitus. Diabetologia. 1999;42(11):1332–1340. doi: 10.1007/s001250051446. [40] [DOI] [PubMed] [Google Scholar]
- 12.Chervonsky A.V., Wang Y., Wong F.S., Visintin I., Flavell R.A. The role of Fas in autoimmune diabetes. Cell. 1997;89:17–24. doi: 10.1016/s0092-8674(00)80178-6. [DOI] [PubMed] [Google Scholar]
- 13.Allison J., Thomas H.E., Catterall T., Kay T.W., Strasser A. Transgenic expression of dominant-negative Fas-associated death domain protein in beta cells protects against Fas ligand-induced apoptosis and reduces spontaneous diabetes in nonobese diabetic mice. Journal of Immunology. 2005;175:293–301. doi: 10.4049/jimmunol.175.1.293. [DOI] [PubMed] [Google Scholar]
- 14.Park Y.J., Lee S., Kieffer T.J., Warnock G.L., Safikhan N. Deletion of Fas protects islet beta cells from cytotoxic effects of human islet amyloid polypeptide. Diabetologia. 2012 doi: 10.1007/s00125-012-2451-2. [DOI] [PubMed] [Google Scholar]
- 15.Clark A., Wells C.A., Buley I.D., Cruickshank J.K., Vanhegan R.I. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Research. 1988;9:151–159. [PubMed] [Google Scholar]
- 16.Liu D., Pavlovic D., Chen M.C., Flodstrom M., Sandler S. Cytokines induce apoptosis in beta-cells isolated from mice lacking the inducible isoform of nitric oxide synthase (iNOS−/−) Diabetes. 2000;49:1116–1122. doi: 10.2337/diabetes.49.7.1116. [DOI] [PubMed] [Google Scholar]
- 17.Yamada K., Takane-Gyotoku N., Yuan X., Ichikawa F., Inada C. Mouse islet cell lysis mediated by interleukin-1-induced Fas. Diabetologia. 1996;39:1306–1312. doi: 10.1007/s001250050574. [DOI] [PubMed] [Google Scholar]
- 18.Riachy R., Vandewalle B., Moerman E., Belaich S., Lukowiak B. 1,25-Dihydroxyvitamin D3 protects human pancreatic islets against cytokine-induced apoptosis via down-regulation of the Fas receptor. Apoptosis. 2006;11:151–159. doi: 10.1007/s10495-006-3558-z. [DOI] [PubMed] [Google Scholar]
- 19.Allison J., Strasser A. Mechanisms of beta cell death in diabetes: a minor role for CD95. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:13818–13822. doi: 10.1073/pnas.95.23.13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas H.E., Darwiche R., Corbett J.A., Kay T.W. Evidence that beta cell death in the nonobese diabetic mouse is Fas independent. Journal of Immunology. 1999;163:1562–1569. [PubMed] [Google Scholar]
- 21.Irmler M., Thome M., Hahne M., Schneider P., Hofmann K. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388(6638):190–195. doi: 10.1038/40657. [Jul 10: 5] [DOI] [PubMed] [Google Scholar]
- 22.Tschopp J., Irmler M., Thome M. Inhibition of fas death signals by FLIPs. Current Opinion in Immunology. 1998;5:552–558. doi: 10.1016/s0952-7915(98)80223-9. [8] [DOI] [PubMed] [Google Scholar]
- 23.Aggarwal B.B., Singh S., LaPushin R., Totpal K. Fas antigen signals proliferation of normal human diploid fibroblast and its mechanism is different from tumor necrosis factor receptor. FEBS Letters. 1995;364:5–8. doi: 10.1016/0014-5793(95)00339-b. [DOI] [PubMed] [Google Scholar]
- 24.Kataoka T., Budd R.C., Holler N., Thome M., Martinon F. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Current Biology. 2000;10:640–648. doi: 10.1016/s0960-9822(00)00512-1. [DOI] [PubMed] [Google Scholar]
- 25.Maedler K., Fontana A., Ris F., Sergeev P., Toso C. FLIP switches Fas-mediated glucose signaling in human pancreatic β cells from apoptosis to cell replication. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8236–8241. doi: 10.1073/pnas.122686299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cottet S., Dupraz P., Hamburger F., Dolci W., Jaquet M. cFLIP Protein prevents tumor necrosis factor-alpha-mediated induction of caspase-8-dependent apoptosis in insulin-secreting betaTc-Tet cells. Diabetes. 2002;51:1805–1814. doi: 10.2337/diabetes.51.6.1805. [DOI] [PubMed] [Google Scholar]
- 27.Hitoshi Y., Lorens J., Kitada S.I., Fisher J., LaBarge M. Toso, a cell surface, specific regulator of Fas-induced apoptosis in T cells. Immunity. 1998;8:461–471. doi: 10.1016/s1074-7613(00)80551-8. [DOI] [PubMed] [Google Scholar]
- 28.Song Y., Jacob C.O. The mouse cell surface protein TOSO regulates Fas/Fas ligand-induced apoptosis through its binding to Fas-associated death domain. The Journal of Biological Chemistry. 2005;280:9618–9626. doi: 10.1074/jbc.M413609200. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen X.H., Lang P.A., Lang K.S., Adam D., Fattakhova G. Toso regulates the balance between apoptotic and nonapoptotic death receptor signaling by facilitating RIP1 ubiquitination. Blood. 2011;118:598–608. doi: 10.1182/blood-2010-10-313643. [DOI] [PubMed] [Google Scholar]
- 30.Huo J., Xu S., Guo K., Zeng Q., Lam K.P. Genetic deletion of faim reveals its role in modulating c-FLIP expression during CD95-mediated apoptosis of lymphocytes and hepatocytes. Cell Death and Differentiation. 2009;16:1062–1070. doi: 10.1038/cdd.2009.26. [DOI] [PubMed] [Google Scholar]
- 31.Rutti S., Sauter N.S., Bouzakri K., Prazak R., Halban P.A. In vitro proliferation of adult human Beta-cells. PLoS One. 2012;7:e35801. doi: 10.1371/journal.pone.0035801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schulthess F.T., Paroni F., Sauter N.S., Shu L., Ribaux P. CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell Metabolism. 2009;9:125–139. doi: 10.1016/j.cmet.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Kaiser N., Corcos A.P., Sarel I., Cerasi E. Monolayer culture of adult rat pancreatic islets on extracellular matrix: modulation of B-cell function by chronic exposure to high glucose. Endocrinology. 1991;129:2067–2076. doi: 10.1210/endo-129-4-2067. [DOI] [PubMed] [Google Scholar]
- 34.Ardestani A., Sauter N.S., Paroni F., Dharmadhikari G., Cho J.H. Neutralizing IL-1{beta} induces {beta}-cell survival by maintaining PDX1 nuclear localization. Journal of Biological Chemistry. 2011 doi: 10.1074/jbc.M110.210526. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Le Bacquer O., Shu L., Marchand M., Neve B., Paroni F. TCF7L2 splice variants have distinct effects on {beta}-cell turnover and function. Human Molecular Genetics. 2011;20:1906–1915. doi: 10.1093/hmg/ddr072. [DOI] [PubMed] [Google Scholar]
- 36.Pham N.A., Morrison A., Schwock J., Aviel-Ronen S., Iakovlev V. Quantitative image analysis of immunohistochemical stains using a CMYK color model. Diagnostic Pathology. 2007;2:8. doi: 10.1186/1746-1596-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shu L., Sauter N.S., Schulthess F.T., Matveyenko A.V., Oberholzer J. Transcription factor 7-like 2 regulates beta-cell survival and function in human pancreatic islets. Diabetes. 2008;57:645–653. doi: 10.2337/db07-0847. [DOI] [PubMed] [Google Scholar]
- 38.Maedler K., Schumann D.M., Sauter N., Ellingsgaard H., Bosco D. Low concentration of interleukin-1{beta} induces FLICE-inhibitory protein-mediated {beta}-cell proliferation in human pancreatic islets. Diabetes. 2006;55:2713–2722. doi: 10.2337/db05-1430. [DOI] [PubMed] [Google Scholar]
- 39.Meier J.J., Ritzel R.A., Maedler K., Gurlo T., Butler P.C. Increased vulnerability of newly forming beta cells to cytokine-induced cell death. Diabetologia. 2006;49:83–89. doi: 10.1007/s00125-005-0069-3. [DOI] [PubMed] [Google Scholar]
- 40.Teta M., Rankin M.M., Long S.Y., Stein G.M., Kushner J.A. Growth and regeneration of adult beta cells does not involve specialized progenitors. Developmental Cell. 2007;12:817–826. doi: 10.1016/j.devcel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 41.Vire B., David A., Wiestner A. TOSO, the Fcmicro receptor, is highly expressed on chronic lymphocytic leukemia B cells, internalizes upon IgM binding, shuttles to the lysosome, and is downregulated in response to TLR activation. Journal of Immunology. 2011;187:4040–4050. doi: 10.4049/jimmunol.1100532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maedler K., Sergeev P., Ris F., Oberholzer J., Joller-Jemelka H.I. Glucose-induced beta-cell production of interleukin-1beta contributes to glucotoxicity in human pancreatic islets. The Journal of Clinical Investigation. 2002;110:851–860. doi: 10.1172/JCI15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donath M.Y., Boni-Schnetzler M., Ellingsgaard H., Halban P.A., Ehses J.A. Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends in Endocrinology and Metabolism. 2010;21:261–267. doi: 10.1016/j.tem.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 44.Ehses J.A., Meier D.T., Wueest S., Rytka J., Boller S. Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high-fat diet. Diabetologia. 2010;53:1795–1806. doi: 10.1007/s00125-010-1747-3. [DOI] [PubMed] [Google Scholar]
- 45.Maedler K., Sergeev P., Ehses J.A., Mathe Z., Bosco D. Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8138–8143. doi: 10.1073/pnas.0305683101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Omori K., Mitsuhashi M., Ishiyama K., Nair I., Rawson J. mRNA of the pro-apoptotic gene BBC3 serves as a molecular marker for TNF-alpha-induced islet damage in humans. Diabetologia. 2011;54:2056–2066. doi: 10.1007/s00125-011-2183-8. [DOI] [PubMed] [Google Scholar]
- 47.Schumann D.M., Maedler K., Franklin I., Konrad D., Storling J. The Fas pathway is involved in pancreatic {beta} cell secretory function. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:2861–2866. doi: 10.1073/pnas.0611487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teta M., Long S.Y., Wartschow L.M., Rankin M.M., Kushner J.A. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]