

Abstract
Electron transfer (ET) reactions in biomolecular systems represent an important class of processes at the interface of physics, chemistry and biology. The theoretical description of these reactions constitutes a huge challenge because extensive systems require a quantum-mechanical treatment and a broad range of time scales are involved. Thus, only small model systems may be investigated with the modern density functional theory techniques combined with non-adiabatic dynamics algorithms. On the other hand, model calculations based on Marcus's seminal theory describe the ET involving several assumptions that may not always be met. We review a multi-scale method that combines a non-adiabatic propagation scheme and a linear scaling quantum-chemical method with a molecular mechanics force field in such a way that an unbiased description of the dynamics of excess electron is achieved and the number of degrees of freedom is reduced effectively at the same time. ET reactions taking nanoseconds in systems with hundreds of quantum atoms can be simulated, bridging the gap between non-adiabatic ab initio simulations and model approaches such as the Marcus theory. A major recent application is hole transfer in DNA, which represents an archetypal ET reaction in a polarizable medium. Ongoing work focuses on hole transfer in proteins, peptides and organic semi-conductors.
Keywords: electron transfer, density functional theory, multi-scale method
1. Introduction
Electron transfer (ET) constitutes an important class of chemical reactions in biomolecular complexes and has been an object of intensive research [1]. These reactions are found at the heart of fundamental biological processes, such as respiration [2], photosynthesis [3], and radiative damage and repair of DNA [4]. The biochemical ET occurs over large distances, often in a heterogeneous medium composed of different kinds of molecules and on a long time scale. All of these features make both the experimental and the computational studies of ET particularly demanding.
The classical tool to describe the ET in complex molecular and biomolecular systems is the Marcus theory [5,6] and its extensions [7–11]. To evaluate the rate of transfer between an electron donor and an acceptor, three parameters are needed: the reaction free energy ΔG, the electronic coupling V and the reorganization energy (RE) λ. V can be calculated with quantum chemistry (QC) [12], and, nowadays, also with highly accurate methods [13]. Particularly cost-efficient methods are based on the fragment molecular orbital (FMO) approaches [14–18] and on constrained density functional theory (DFT) [19,20]. These allow V to be calculated along nanosecond molecular dynamics (MD) trajectories [21,22]. In addition, effective sampling of the configuration space is needed to evaluate ΔG and λ, which are thermodynamic quantities. A rather accurate estimation of these parameters is required because of their appearance in the argument of an exponential, and the RE is particularly challenging to determine. It can be decomposed into two parts: the inner sphere RE λi, which is also sensitive to quantum effects [23], and the solvent contribution to the outer sphere RE λs, which can be calculated with classical MD simulations [24–27]; the effects of several structural and dynamic factors on λs have been discussed previously [28,29].
The application of the standard Marcus theory is based on several assumptions, as follows. (i) The electronic structures of the electron donor and of the electron acceptor are known, including the information on the de/localization of the charge. (ii) ET occurs significantly more slowly than the structural dynamics of the molecular system.1 (iii) The mechanism of transfer is known (adiabatic or non-adiabatic, solvent-controlled, etc.). A good indicator may be the relation of V and λ [35], although the distinction may not always be easy to make use of [23].
An alternative solution is to perform an unbiased simulation of ET using a non-adiabatic MD scheme and high-level electronic structure calculations [36], yet such methods are limited to several tens of atoms and time intervals of up to a picosecond. Stepping down the ladder of accuracy of QC methods, the DFT may be able to describe slightly larger systems, still being restricted to a picosecond time scale [37–39]. Even worse, semi-empirical QC methods cannot treat systems with hundreds of atoms on nanosecond time scales either, and this has been possible only with more approximate methods [40] so far.
In order to make a rigorous description of such processes practicable, we have developed a novel multi-scale computational scheme, where the number of degrees of freedom is reduced carefully yet an unbiased description of the ET reaction in the spirit of non-adiabatic QC-MD schemes is still provided. The computational scheme is based on a twofold combination of quantum-mechanical (QM) methods with empirical force fields and linear scaling approaches: (i) A QM region is defined, and the remainder of the system will be described with molecular mechanics (MM), with interaction between the regions to be treated with a standard QM/MM coupling. (ii) An FMO approach is applied in the QM region, which leads to a linear scaling of the computational time with the system size. (iii) Only the frontier orbitals (highest-occupied molecular orbitals (HOMOs) or lowest-unoccupied molecular orbitals (LUMOs)) are considered in the QM treatment, while the remainder of the electronic structure is covered by MM; this is similar to the simple Hamiltonians in the Pariser–Parr–Pople (PPP) or Hückel molecular orbital (HMO) methods. Since an approximate total energy expression for the combined system is derived, the atomic forces can be computed and the standard semi-classical techniques can be applied to propagate the nuclear and electronic degrees of freedom simultaneously, such as the mean-field (MF) Ehrenfest or surface-hopping (SH) methods.
This resulting computational scheme is cost-efficient and allows the electronic and nuclear degrees of freedom to be propagated for several hundreds of QM atoms simultaneously. We have applied this method to the problems of hole/ET in DNA, proteins, peptides and organic materials, which will be presented briefly after reviewing the methodology.
2. Methodology
The computational scheme for the simulation of ET is derived from an expression for total energy within DFT. A favourable computational efficiency is achieved by the introduction of several approximations, among which the following ones are prominent:
— use of a linear scaling ansatz for the QC problem, by means of an FMO approach;
— efficient calculation of the FMOs with a semi-empirical DFT scheme, self-consistent charge/density functional tight binding (SCC-DFTB) [41];
— application of a hybrid QM/MM scheme to treat the influence of the environment; and
— a combination of QC and MM calculations on yet another level, treating only selected (frontier) orbitals quantum mechanically while the remainder of the system is covered by a force field.
These approximations are discussed in the first section below. The following sections concern the total energy expression, the calculation of energy derivatives, the effects that are described effectively or neglected in the approach, and the QM propagation schemes applied.
2.1. Linear combination of fragment orbitals
The QC problem can be solved very efficiently, provided it is possible to decompose the molecular system naturally into a set of M spatially non-overlapping fragments, for which separate QC calculations can be performed. This idea is close to that of the FMO approach of Kitaura et al. [42]. An example is the purine nucleobases for the hole transfer in DNA (figure 1). Then, the computational problem is reduced to the determination of the molecular orbitals on each of the M fragments (indexed as m)
| 2.1 |
where the ith molecular orbital (FMO) of fragment m is expressed in an atomic basis set χμ with a set of FMO coefficients 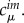 In the case of DNA, these fragments are the nucleobases over which the excess charge is considered to travel. In the case of proteins, these may be important amino acid side chains and parts of the protein backbone. The methodology may also be applied to organic materials as these consist of individual molecules, which can be treated as fragments.
In the case of DNA, these fragments are the nucleobases over which the excess charge is considered to travel. In the case of proteins, these may be important amino acid side chains and parts of the protein backbone. The methodology may also be applied to organic materials as these consist of individual molecules, which can be treated as fragments.
Figure 1.
Application of the fragment orbital approach for hole transfer in DNA. Purine nucleobases are considered as the charge-carrying fragments (coloured by atom), while the remaining components of the system constitute the MM environment (DNA backbones in black, water molecules in red/dark grey, counter-ions in yellow/light grey). (Online version in colour.)
To solve for the FMOs in equation (2.1), Hartree–Fock (HF) or DFT methods can be used. However, the application of semi-empirical QC methods can speed up these calculations by as much as three orders of magnitude. This is an essential step as it allows the FMOs to be calculated along the MD trajectories in the nanosecond regime, whereas the HF and DFT approaches would be limited to a picosecond range. In our work, the QC calculations are based on the SCC-DFTB method [41]. Evidently, the use of such approximate methods requires a thorough benchmarking, which has been done for hole transfer in DNA in our initial work [16]. It was shown that SCC-DFTB provides orbitals in close agreement with full DFT calculations. Also, the charge-transfer (CT) parameters—the on-site energies ɛm and electronic couplings Hmn (see below)—agree with those provided by the higher level DFT and wave function methods.
To save computer time, only the region where the charge can localize is treated with QC, while the entire remainder of the system is described with MM. In the case of hole transfer in DNA, the QC part contains the purine nucleobases (guanines and adenines), while the MM part contains the other nucleobases, the sugar–phosphate backbones as well as the solvent (water molecules and counter-ions). The effect of the MM environment is taken into account using a QM/MM approach. To this end, the calculation of FMOs 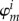 in equation (2.1) includes the electric field induced by the MM environment, represented by point charges, as an external potential. Also, the electrostatic interactions between the individual fragments (i.e. between the purine nucleobases) is only included via point-charge electrostatics. Thus, the MOs are constrained to be localized in the pre-determined region, and their spatial support cannot change in the course of the simulation, regardless of any geometry changes.
in equation (2.1) includes the electric field induced by the MM environment, represented by point charges, as an external potential. Also, the electrostatic interactions between the individual fragments (i.e. between the purine nucleobases) is only included via point-charge electrostatics. Thus, the MOs are constrained to be localized in the pre-determined region, and their spatial support cannot change in the course of the simulation, regardless of any geometry changes.
Actually, only the excess electron or hole is treated quantum mechanically, while the entire remainder of the system (i.e. the charge-neutral variant of the molecular system) is described with an MM force field. This is comparable to the HMO [43,44] and PPP [45,46] models, in which a π – σ separation is assumed: only the π-electrons are treated with QC while the σ-electrons are considered to be condensed to the nuclei with classical force-field terms. The approximation introduced here proceeds even further, by considering the quantum system as consisting of a single electron or hole. The basis set for the expansion of the wave function of excess electron is constituted by the FMOs. In the simplest form, the wave function of the excess electron consists of a superposition of the LUMOs of the fragments (i = LUMO in 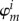 ), and the wave function Ψ of the hole is built from the HOMOs
), and the wave function Ψ of the hole is built from the HOMOs 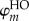
| 2.2 |
where am are the expansion coefficients in the basis of the HOMOs. Thus, it is assumed that the lowest unoccupied orbitals of the complex are linear combinations of the LUMOs of the individual fragments, or that the highest occupied orbitals are linear combinations of the HOMOs of fragments (for hole transfer). This assumption is confirmed for hole transfer in DNA in figure 2: the energies of the highest occupied orbitals in a stack of adenine molecules obtained with a diagonalization of the ‘coarse-grained’ Hamiltonian in the FMO approach (introduced below in equation (2.9)) are compared with those yielded by full atomistic SCC-DFTB calculations. These sets of orbital energies match convincingly, and the deviations do not exceed 0.01 eV. Furthermore, a visual inspection of the orbitals reveals that the five highest occupied orbitals of the stacked complex of six adenines represent superpositions of the weakly coupled HOMOs of the individual adenines.
Figure 2.
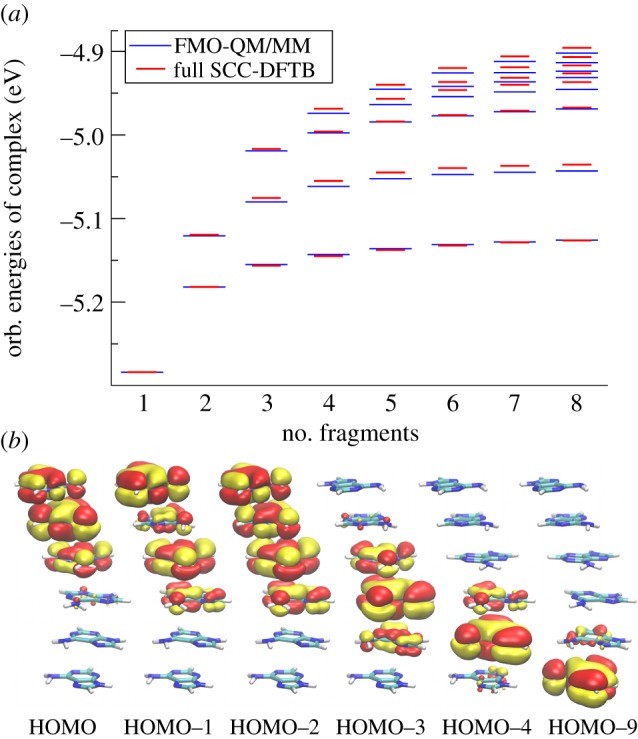
(a) Energies of n highest occupied orbitals in complexes of n adenine molecules (n = 1 … 8) in the idealized B-DNA configuration, yielded by full SCC-DFTB (red/dark) and FMO calculations (blue/light). (b) Highest occupied orbitals of the stack of six adenine molecules in the idealized B-DNA conformation obtained with SCC-DFTB. Shown are orbitals that correspond to linear combinations of HOMO of the individual molecules; five of them represent the five highest occupied orbitals, HOMO through HOMO–4. (Online version in colour.)
Note that also further orbitals of the fragments (HOMO–1, HOMO–2, etc. or LUMO+1, LUMO+2, etc.) can be included if required for a more accurate description.
2.2. Approximate total energy expression for the hybrid FMO/MM approach
The starting point is the expression for the DFT total energy of the system with N – 1 electrons, i.e. a system which contains a hole (radical cation),2 with electron density ρ. The first step towards an efficient method is to expand this energy around the density ρ0 of the neutral system with N electrons up to the second order, using the differential density δρ = ρ − ρ0
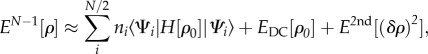 |
2.3 |
where ni are the occupation numbers of the Kohn–Sham orbitals Ψi (with nHOMO = 1 and ni = 2 otherwise), EDC[ρ0] is the DFT double-counting contribution [47], and E2nd is a contribution that is of second order in the differential density and will be analysed in detail later. Here, the aim is to express the energy explicitly as a sum of the form
| 2.4 |
which will require further approximations.
2.2.1. The Koopmans theorem and the zeroth-order term
If we assume the orbitals of the charged system Ψi to be identical to those of the neutral system 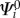
| 2.5 |
then we may rearrange the energy expression to obtain
| 2.6 |
where 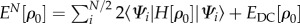 is the energy of the N-electron, charge-neutral system. This expression has the form of equation (2.4).
is the energy of the N-electron, charge-neutral system. This expression has the form of equation (2.4).
Equation (2.5) looks like a drastic approximation at first sight. For discussion, let us assume the wave function of hole to be localized on the fragment m completely so that ΨHO = φm. We may expect three kinds of changes upon ionization: (a) the shape of the orbital φm will change, (b) the orbitals on the fragment m (indexed as k) 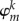 will mix (rotate), and (c) the orbital energies
will mix (rotate), and (c) the orbital energies 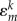 will change.
will change.
Regarding (a), it has to be kept in mind that relatively large molecular fragments are usually considered. Then, the excess charge is distributed over many atoms as the HOMO/LUMO is quite delocalized most likely. In such a case, the frontier orbitals are unlikely to change much upon ionization. Whenever the excess charge is localized on a few atoms in a fragment, the approximation of preserved frontier orbitals may become problematic; then, a third-order expansion may account, for example, for the spatial expansion of the electron density in small anions with respect to the neutral molecules, as discussed for SCC-DFTB recently [48]. Also, it was shown that, after removing an electron from the HOMO, this orbital remains static and the charge migration is driven by lower lying orbitals [49].
We have tested this approximation for the nucleobase guanine, which is the dominant hole-carrying fragment in DNA. As a measure of similarity of orbitals in the radical cation molecule and those in the neutral one, the overlap of these orbitals obtained on the DFT-level B3LYP/def2-TZVP was evaluated (see figure 3 for the results). The HOMO is the same in the neutral and ionized molecule (the overlap is 0.99 and 0.999 for the α- and β-orbitals of the radical cation, respectively). On a few occasions, there is some rotation of lower lying orbitals and changes of their energy order. Therefore, the response to taking an electron out of the HOMO occurs in lower lying orbitals as expected (b). The effect of charging on the orbital energy will be covered by the second-order term, as discussed later, while this term will include also any effects of the kinds (b) and (c) in an effective way. Also, note that the electronic structure obtained with SCC-DFTB agrees well with higher level results [16].
Figure 3.
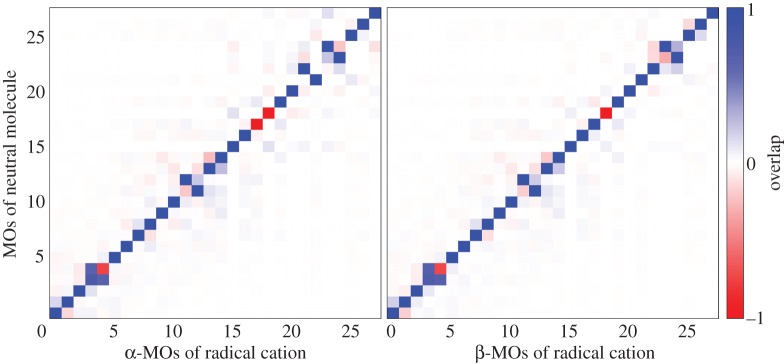
Analysis of the electronic structure of the radical cation guanine on the DFT-level B3LYP/def2-TZVP. The overlap of occupied valence orbitals of the radical cation with those of the neutral molecule. (The HOMO of the neutral molecule is no. 27. The β-MO no. 27 of the radical cation is unoccupied.) (Online version in colour.)
Equation (2.6) can be interpreted as follows. The (N – 1)-electron energy is approximated to the zeroth order by the N-electron energy EN[ρ0], corrected to the first order by subtracting the energy of one electron occupying the HOMO. This is in the spirit of the Koopmans theorem completely. The second-order term represents the change of electron–electron interaction upon the removal of the electron, discussed further below, and it will take the form of a Hubbard-type contribution to the on-site energy owing to the change of charge on the fragment. In this way, it will cover the electronic relaxation effects, i.e. points (a), (b) and (c), effectively.
Since EN[ρ0] denotes the energy of the charge-neutral, N-electron system, it can be computed using HF or DFT. But, it can be represented by any other total energy expression, and, aiming at a computationally efficient scheme, this energy will be obtained with an empirical force field
| 2.7 |
Here, we see the importance of passing from equation (2.3) to the form of equation (2.4): we can represent a large part of the energy by the energy of the empirical force field.3
2.2.2. First-order term
The first-order term in equation (2.6) is expanded in the FMO functions according to equation (2.2)
| 2.8 |
leading to the FMO Hamiltonian matrix 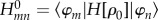 . From now on, we consider one orbital φm (HOMO or LUMO) per fragment, instead of
. From now on, we consider one orbital φm (HOMO or LUMO) per fragment, instead of 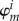 . It is possible (and required for certain applications) to include multiple orbitals per fragment, and this will be discussed later.
. It is possible (and required for certain applications) to include multiple orbitals per fragment, and this will be discussed later.
The Hamiltonian matrix is evaluated easily using the FMO expansion (equation (2.1)) as
| 2.9 |
where the double sum runs over the relevant atomic-orbital-like basis functions, and 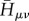 is the Hamiltonian in this atomic orbital (AO) basis, which is built for the current geometry of the molecular system. This matrix
is the Hamiltonian in this atomic orbital (AO) basis, which is built for the current geometry of the molecular system. This matrix 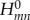 is easy to set up, as it requires only SCC-DFTB calculations to be performed on the fragments m and n to determine
is easy to set up, as it requires only SCC-DFTB calculations to be performed on the fragments m and n to determine 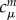 and
and 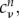 respectively, which then enter the sum in equation (2.9) simply. As described earlier, the AO Hamiltonian
respectively, which then enter the sum in equation (2.9) simply. As described earlier, the AO Hamiltonian 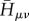 is set up with SCC-DFTB for the current geometry of the molecular system. These integrals are interpolated from values that have been pre-calculated and tabulated, therefore the set-up of
is set up with SCC-DFTB for the current geometry of the molecular system. These integrals are interpolated from values that have been pre-calculated and tabulated, therefore the set-up of 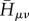 is not time-consuming at all. Furthermore, the AO Hamiltonian contains the interaction with the environment via a QM/MM coupling; the whole approach is described in [16].
is not time-consuming at all. Furthermore, the AO Hamiltonian contains the interaction with the environment via a QM/MM coupling; the whole approach is described in [16].
Then, the set of FMOs is orthogonalized with the scheme proposed by Löwdin [50], simplifying the following propagation of the wave function, as described later. The Hamiltonian matrix corresponding to these orthogonalized orbitals is considered in all what follows.
In the study by Kubař & Elstner [21], the time series of the on-site energies 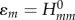 and of the couplings
and of the couplings 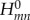 (with m≠n) was computed along classical MD trajectories for DNA. Here, the dynamics of solvated DNA was simulated using a classical force field, and the CT parameters ɛm and
(with m≠n) was computed along classical MD trajectories for DNA. Here, the dynamics of solvated DNA was simulated using a classical force field, and the CT parameters ɛm and 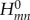 were recorded as described earlier. Every DNA base was treated with SCC-DFTB, and the electrostatic interaction with the point charges on the sugars, phosphate groups, counter-ions and water, as well as the charges on all of the other DNA bases, were included using the QM/MM scheme.
were recorded as described earlier. Every DNA base was treated with SCC-DFTB, and the electrostatic interaction with the point charges on the sugars, phosphate groups, counter-ions and water, as well as the charges on all of the other DNA bases, were included using the QM/MM scheme.
The off-diagonal elements 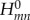 (m≠n) are smaller than 0.1 eV on average and fluctuate in time owing to the structural dynamics of DNA. It is only the relative orientation of the fragments (DNA bases) that induces this variation, while the solvent has only a minor impact. The opposite is true for the diagonal matrix elements
(m≠n) are smaller than 0.1 eV on average and fluctuate in time owing to the structural dynamics of DNA. It is only the relative orientation of the fragments (DNA bases) that induces this variation, while the solvent has only a minor impact. The opposite is true for the diagonal matrix elements 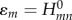 . The fluctuations driven by the molecular vibrations of the fragments lead to oscillations with a period of ca 20 fs and an amplitude of ca 0.1 eV. The solvent, however, introduces much larger fluctuations of 0.3–0.4 eV, and these fluctuations are the major driving force for the CT dynamics, because these fluctuations are of the same magnitude as the energy differences between different fragment energies; note that the difference in ionization potentials (IPs) of guanine and adenine is ca 0.4 eV [21]. The magnitude of the fluctuations due to the internal dynamics and due to the environment can be compared visually in figure 4 for the nucleobases in the double-stranded DNA.
. The fluctuations driven by the molecular vibrations of the fragments lead to oscillations with a period of ca 20 fs and an amplitude of ca 0.1 eV. The solvent, however, introduces much larger fluctuations of 0.3–0.4 eV, and these fluctuations are the major driving force for the CT dynamics, because these fluctuations are of the same magnitude as the energy differences between different fragment energies; note that the difference in ionization potentials (IPs) of guanine and adenine is ca 0.4 eV [21]. The magnitude of the fluctuations due to the internal dynamics and due to the environment can be compared visually in figure 4 for the nucleobases in the double-stranded DNA.
Figure 4.
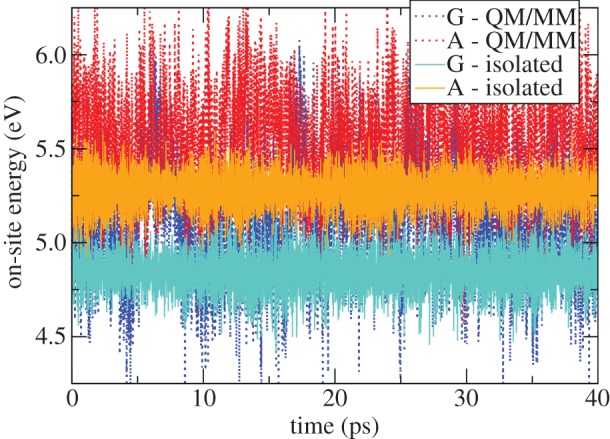
Time course of instantaneous IPs for a guanine and an adenine in a solvated DNA oligonucleotide, calculated with SCC-DFTB twice: considering isolated molecules (light lines) and taking into account the environment by means of a QM/MM implementation (dark lines). (Online version in colour.)
There is an issue related to the application of classical MD simulation, namely whether the amplitudes of molecular vibrations of the fragments, which may be of a quantum character, are in agreement with reality. (Importantly, these amplitudes drive the magnitude of oscillations of CT parameters, above all the instantaneous IPs or electron affinities.) This problem was analysed in the context of excitation energies of nucleobases recently [51], and it was concluded that the intra-molecular motion due to the zero-point vibrations—an effect of a purely QM character—determined the shape of electronic spectra. The point is actually that most of the internal modes of motion of a molecule of the size of the nucleobases are in the vibrational ground state, so that the amplitude of motion is given by the zero-point vibrations.4 The crucial questions are then (i) how this ‘zero-point dynamics’ compares with the classical dynamics and (ii) how the instantaneous IP depends on the internal structure of the fragments. Notably, the IP of the nucleobases in the solvated double-stranded DNA is driven mainly by the electric field induced by the environment—the solvent; the related modes of motion possess lower frequencies, thus the quantum effects may be of lesser importance in these systems. However, this need not be the case in all of the CT systems. In organic semi-conductors, for instance, the environment is non-polar and does not induce any strong electric field, so that the fluctuations in the internal structure of charge-carrying fragments are the decisive factor. Then, the (possibly quantum) character of the internal modes of motion and the applicability of classical mechanics require close investigation.
2.2.3. Second-order terms
The second-order term in equation (2.6) contains the second derivatives of Hartree and exchange–correlation (XC) energy
| 2.10 |
The differential density δρ = ρ − ρ0 is decomposed into contributions located on the individual molecular fragments m,
| 2.11 |
Inserting equation (2.11) into equation (2.10), an expression is obtained which describes the electron–electron interaction between the differential densities δρm(r) and δρn(r), and the second-order term is obtained as a sum of pairwise contributions
| 2.12 |
Two cases may be distinguished here: for large distances (greater than 3 Å) between the fragments m and n, the XC term can be neglected and only the Hartree interaction prevails; this is shown for the atomistic SCC-DFTB method, e.g. fig. 1 in [48]. Note that the actual inter-fragment distances in our applications are always in this range, being ca 3.4 Å for stacked organic molecules (DNA bases, molecules of organic semi-conductors) and larger in the other cases. Furthermore, the Hartree interaction may be approximated by the Coulomb interaction of fragment charges ΔQm
| 2.13 |
Here, the differential charge on the fragment m due to the presence of the excess charge is obtained from the wave function expansion coefficient am introduced in equation (2.2) as 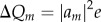 (e is the elementary charge), and Rmn is the centre-of-mass distance of the fragments. In the other limit m = n,
(e is the elementary charge), and Rmn is the centre-of-mass distance of the fragments. In the other limit m = n, 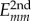 describes the on-site charge self-interaction. This can be obtained with the effective Hubbard model, and the diagonal term becomes
describes the on-site charge self-interaction. This can be obtained with the effective Hubbard model, and the diagonal term becomes
| 2.14 |
where Um is the Hubbard parameter of the molecular fragment m, which is a constant that can be calculated as the second derivative of the total energy of the molecule with respect to its charge. Note that the applied approximations are basically the same as those involved in the derivation of the second-order term in SCC-DFTB [41]. The second-order term can be written in a condensed form as
| 2.15 |
with Γmm = e2·Um and, for m ≠ n, Γmn = e2/Rmn.
DFT calculations of radical systems are prone to the self-interaction error (SIE), which would lead to a drastic over-delocalization of the excess charge in the system. In SCC-DFTB, which is formally identical to the formalism derived here, it can be shown that the SIE occurs as a result of the second-order terms E2nd [52]. Since it would be too involved to subtract the electron self-interaction orbital by orbital as proposed earlier [53], a better (but, nevertheless, approximate) option is to consider only the spin density in the correction scheme [37,54]; the spin density corresponds to the density of the excess charge in the current computational method. Following this approach, the second-order term is reduced by a scaling with a constant empirical factor C = 0.2 [47].
With these approximations, the total energy (equation (2.6)) reads
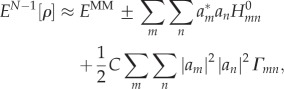 |
2.16 |
where the plus sign applies for the transfer of excess electron, and the minus sign shall be used for the hole transfer.
2.3. Total energy and its derivatives
To allow for MD simulation, it is crucial to have a total energy expression as derived in equation (2.16). This involves a few approximations, but it still enables the same equations as for full DFT to be derived formally. The total energy is a function of the expansion coefficients of the wave function of excess charge am (equation (2.2)) and of the spatial coordinates Rα of atoms α,
| 2.17 |
To perform a simulation within the Born–Oppenheimer (BO) approximation, the derivatives of EN−1 with respect to am can be considered, to arrive at a coarse-grained version of the Kohn–Sham equations in a form equivalent to those obtained in full DFT, with the matrix elements of the Hamiltonian taking the form
| 2.18 |
(again, with plus and minus signs for electron and hole transfer, respectively). These coarse-grained Kohn–Sham equations have to be solved self-consistently.
At this point, it is apparent how the second-order energy term corrects for the approximations due to the Koopmans theorem (issues (a), (b) and (c) in §2.2.1) effectively. The matrix elements 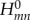 are evaluated according to equation (2.9) for the electro-neutral fragments. For instance, the diagonal terms
are evaluated according to equation (2.9) for the electro-neutral fragments. For instance, the diagonal terms 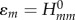 are the Kohn–Sham energies of the HOMOs of neutral fragments. Then, the on-site energy
are the Kohn–Sham energies of the HOMOs of neutral fragments. Then, the on-site energy 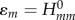 is corrected by an additional term (containing
is corrected by an additional term (containing 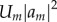 as the major contribution), which is the change of on-site energy due to the removal or addition of one electron from/to the system. Here, the deformation and rotation of the fragment orbitals as well as the change of orbital energy due to the different electron–electron interactions are included in an effective way. The shift of energy is described by the fragment-specific Hubbard parameter Um, which is determined for each individual fragment as the second derivative of total energy with respect to the charge.
as the major contribution), which is the change of on-site energy due to the removal or addition of one electron from/to the system. Here, the deformation and rotation of the fragment orbitals as well as the change of orbital energy due to the different electron–electron interactions are included in an effective way. The shift of energy is described by the fragment-specific Hubbard parameter Um, which is determined for each individual fragment as the second derivative of total energy with respect to the charge.
Kohn–Sham equations with the Hamiltonian in equation (2.18) determine the coefficients am that minimize the total energy for a given geometry Rα. Once the minimum of electronic energy is determined, the forces on atoms can be computed by taking the derivatives
| 2.19 |
Here, EN−1 contains the three parts discussed earlier. The derivatives of E0 are simply the forces obtained from the MM force field, and they are implemented in the applied MM package already. The first-order term E1 consists of the kinetic energy (T), electron–nucleus (vel−nuc), Hartree (vH) and XC (vXC) contributions
| 2.20 |
The derivatives of kinetic energy and XC originating from E1 cannot be obtained in a practicable way and are neglected; the consequences will be discussed in §2.4. The derivatives due to the Hartree contribution to E1 together with the Coulomb interaction in E2 are approximated by the interaction of atom-centred point charges according to the Coulomb law.
This ansatz, which connects the quantum system (the excess electron or hole) with the classical (atomic) system, is implemented in the following way: the charge on fragment m due to the excess electron hole, ΔQm, is distributed among the atoms of the fragment, providing additional contributions to atomic charges 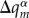 . Then, the Coulomb interaction energy between all of the point charges
. Then, the Coulomb interaction energy between all of the point charges 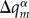 and
and 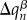 (the sum running over all fragments m and atoms α) corresponds to the Hartree term in E1 plus the contributions from E2. To compute the derivatives of this energy term with respect to Rα, it suffices to update the point charges of the MM atoms
(the sum running over all fragments m and atoms α) corresponds to the Hartree term in E1 plus the contributions from E2. To compute the derivatives of this energy term with respect to Rα, it suffices to update the point charges of the MM atoms 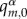 with the additional atom-centred contributions due to the excess charge,
with the additional atom-centred contributions due to the excess charge,
| 2.21 |
The contributions 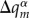 are obtained by a linear interpolation
are obtained by a linear interpolation
| 2.22 |
between the atomic charges of the charged molecule (a cation for hole transfer, an anion for excess ET) obtained prior to the simulation for the neutral (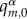 ) and for the charged
) and for the charged 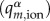 molecular fragments. These sets of atomic charges are obtained with restrained electrostatic potential (RESP) fitting [55] prior to the simulation. The use of RESP charges for MD simulation is an established standard, and it was reported that the choice of atomic charge sets may affect the features of excess charge in simulations [56].
molecular fragments. These sets of atomic charges are obtained with restrained electrostatic potential (RESP) fitting [55] prior to the simulation. The use of RESP charges for MD simulation is an established standard, and it was reported that the choice of atomic charge sets may affect the features of excess charge in simulations [56].
The resulting simulation scheme is conceptually simple: the solution of equation (2.18) gives coefficients am, which are projected to the differential atomic charges 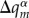 . These are used to update the atomic charges in the MM part of the calculation. Then, cost-efficient MM forces are computed, considering these updated atomic charges in the QM region. The influence of the excess charge on the MD is covered fully in this way. The presence of the excess charge induces a polarization of the environment, i.e. a ‘polaron’ accompanies the transferring charge, and this is the determining factor for ET in molecular systems immersed in an aqueous environment. A remaining problem is the application of a non-polarizable force field, and this will be discussed later in detail.
. These are used to update the atomic charges in the MM part of the calculation. Then, cost-efficient MM forces are computed, considering these updated atomic charges in the QM region. The influence of the excess charge on the MD is covered fully in this way. The presence of the excess charge induces a polarization of the environment, i.e. a ‘polaron’ accompanies the transferring charge, and this is the determining factor for ET in molecular systems immersed in an aqueous environment. A remaining problem is the application of a non-polarizable force field, and this will be discussed later in detail.
Note that the only part of the system treated actually with quantum mechanics is the excess charge—a single electron or hole. This is done with a QM/MM approach so that the interaction with the molecular system is accounted for. Still, the entire remainder of the system is described classically, with MM. Thus, there is an implicit underlying assumption that the studied system is described adequately with an available MM force field; this assumption is typically accepted for solvated DNA, peptides and proteins.
Based on the described approximations, a very efficient MD scheme is implemented, making use of BO dynamics and updating the MM atomic charges in every time step as directed by the coarse-grained quantum mechanics. For instance, eight QC calculations have to be performed in every time step of the simulation of hole transfer in a DNA octanucleotide, one for each CT-active nucleobase. Each fragment contains about 15 atoms, and a calculation of such a system using the semi-empirical SCC-DFTB method takes less than 100 ms on one CPU core of a main-stream desktop computer. As these calculations on fragments, which may be performed in parallel, are the most time-consuming part of the calculation, an efficiency of as much as 1 ns per day is obtained.
2.4. Effects not taken into account
The computational scheme involves several notable approximations: classical non-polarizable MM calculation is applied to obtain the energy of the charged system. Also, the kinetic energy and XC contributions to the first-order energy term (T and vXC in equation (2.20)) are neglected in the MM calculation of forces on atoms due to the excess electron. Consequently, several kinds of interactions are not described with the total energy in equation (2.16), and they are discussed in this section in detail.
2.4.1. Intra-fragment interactions
Although the change of Coulomb interactions upon the charging of the system is covered by the scheme formally, its portion corresponding to the interactions within a fragment is missing. The reason for this is the way in which the commonly used empirical force fields are constructed—the non-bonded interaction between a pair of atoms within the distance of fewer than three covalent bonds is not computed, as they are accounted for by the bonded terms. In addition, the non-bonded interaction between atoms connected by three bonds (1–4 interaction) is scaled down by a certain factor. As a matter of fact, most of the atom pairs within a fragment of typical size (a nucleobase or an amino acid side chain) are within this distance. Therefore, the MM energy of the fragment does not actually change upon the arrival of excess charges, thus no change of forces occurs, and the energy cost of the structural deformation of the fragment due to the varying charge cannot be described.
In the Marcus theory, this effect is covered by the inner sphere RE λi. Following an earlier study [57], we introduce λi by means of an additional empirical contribution to the total energy
| 2.23 |
There have been several estimates of the parameter 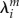 , which reflects the response of the molecular geometry of the fragment m to the varying charge. Applied here are values obtained from QC calculations with an implicit solvent; see the discussion in [47];
, which reflects the response of the molecular geometry of the fragment m to the varying charge. Applied here are values obtained from QC calculations with an implicit solvent; see the discussion in [47]; 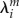 amounts to 0.23 eV for both adenine and guanine, which are considered for hole transfer in DNA. Supplying this additional energy term to the total energy, the intra-fragment contributions are corrected for effectively.
amounts to 0.23 eV for both adenine and guanine, which are considered for hole transfer in DNA. Supplying this additional energy term to the total energy, the intra-fragment contributions are corrected for effectively.
Such an effective, empirical description of the energy cost of the molecular geometry change shows an additional advantage over the application of classical MM. As found recently [23], the rearrangement of molecular geometry caused by the change of the charge state may occur via tunnelling—an exclusively QM mechanism. If that is the case then the usual classical way of calculating λi as mentioned earlier would yield values overestimated by a factor of as much as 2. For this reason, it is appropriate to describe the inner sphere reorganization by means of an additional, empirical term in the expression for the total energy, instead of aiming at an explicit account for the changes in molecular geometry.
2.4.2. Inter-fragment interactions
Since the charges in the QM region are updated according to the location of the excess charge, the additional Coulomb interaction between the fragments relative to the neutral system is covered by the present formalism. This is the main contribution from the off-diagonal terms in equation (2.16). However, the van der Waals interaction between the fragments in the charged state may also differ from that in the neutral state. Now, this effect is missing in the description because the XC contribution to the forces on atoms was neglected in the derivative of first-order energy in equation (2.20). The possible consequences are of two kinds: (i) Owing to the change of charge density, the exchange repulsion may differ between the charged and the neutral species. This may result in a slightly different van der Waals distance of the molecular fragments. In principle, an additional charge-dependent term could be introduced into the classical force field to take this interaction into account (as suggested in [47], supplementary information). A possible effect would be a modified distortion of the structure of DNA, i.e. a small compression or expansion of the region where the excess charge is localized. This effect is expected to be small, as discussed for DNA in [58] and in general in [59]. Such a minor change of structure will be overwritten completely by the dynamics of the system, which introduces much larger fluctuations than the geometry change induced by the charge-dependent van der Waals interactions. (ii) Another effect may be the change of dispersive interactions. To inspect this for hole transfer in DNA, we computed the isotropic polarizability of the neutral and the cationic forms of both purine bases on the DFT/PBE/TZVP level. Very similar values were obtained—for A, 13.62 and 13.61 Å, respectively, and for G, 14.58 and 14.67 Å, respectively. Therefore, the effect seems negligible for the hole transfer in DNA, and no correction has to be added in the current scheme.
2.4.3. Electronic polarization of the environment
The electric-charge density of the molecular environment is represented by fixed point charges in the standard force fields, as applied here as well. This approximation inherent to MM-based methods is a source of errors whenever the electronic polarization is changing during the process being simulated. Obviously, the transfer of electric charge is such a process, and it has been established that the solvent RE is overestimated severely if evaluated with a non-polarized force field [60–63].5 While this is the most prominent effect, other relevant quantities may be affected as well, such as the magnitude of fluctuation of on-site energies ɛm; as an example, note again the large fluctuations in IP of nucleobases in DNA (figure 4).
The factor by which the energy-related quantities are overestimated is related to the optical dielectric constant of the medium [62,64]. Then, if it is not practicable to pass to a polarizable MM force field, the most straightforward way to correct for the overestimated electrostatic interaction is to scale the relevant interactions down by a suitable factor [64–66]. The effect on the energy barrier can be illustrated by considering the relation between the RE λs (representing the barrier) and the magnitude of fluctuation of one of the energy quantities describing the system; available choices are the energy gap expressed as the difference in IP (ΔIP) [26],
| 2.24 |
the IP itself [24] and the total electronic energy [30]. The relation of the (possibly scaled) electric potential induced by the MM environment to λs becomes clear by noting that the IP is nearly directly proportional to the electric potential [21]. While no such quantity as RE is defined in the developed simulation scheme, the reasoning presented here applies to the effect of scaling of the MM electric field on the energy barrier to ET. Simply speaking, the reduced energy barrier leads to an increased rate of transfer.
Blumberger & Lamoureux [67] described the more complex character of the problem, finding the RE in an aqueous medium to be overestimated by 26–34% when the TIP3P water model was used. Based on this observation is the suggested choice of the value of the scaling factor of 1/1.4–1/1.5. Within the current framework, this approach is used in the QM/MM calculations of the CT-active molecular fragments, where the MM charges of the molecular environment are divided by a factor of 1.5 in the application to hole transfer in DNA. In other recent work [68,69], it was proposed to scale the charge of charged residues and ions in classical MD simulations with the common biomolecular force fields by a factor of ca 0.7, to correct for the energetics of ion solvation. While this procedure is clearly debatable, we wish to point out that the introduction of the scaling factor of 1/1.4–1/1.5 in our method as well as our motivation by the overestimated solvent RE is consistent with this reasoning completely. Effectively, the excess charge interacting with the MM environment is reduced by a factor of 0.67–0.71 as a result of the scaling.
The scaling of MM electric field also influences the energy landscape of the simulated ET reaction, which is determined by the differences in IP of the charge-carrying fragments in the case of hole transfer. Whenever this difference is induced by the different character of the molecular environments of the individual fragments, it decreases as a consequence of the scaling of the MM electric field. Based on the analysis in [68,69], where correct solvation energetics was obtained with the scaling of atomic charges, it can be expected that the MM scaling also improves the energetics of ET in our simulations. Note that this applies in the case of hole transfer in Escherichia coli DNA photolyase, where the energy landscape will be due to the different MM environment of the charge-carrying fragments, and thus it will be affected by the MM scaling. Contrary to that, this point will not occur in applications where all of the fragments experience the same or a similar environment because of symmetry, as in the case of hole transfer in DNA where the energy differences are due to the different chemical characters of adenine and guanine.
2.5. Non-adiabatic propagation schemes
As indicated in §2.3, the computational scheme described up to this point can be used readily to perform a multi-scale simulation of ET within the BO approximation. Note that QM/MM-BO simulations of ET were also performed by other researchers previously [37–40]. Since the validity of BO for the systems of interest is ambiguous or even unlikely, it is preferable to resort to a non-adiabatic representation. Our work makes use of the extensive development in this field, and both main approaches, the MF (Ehrenfest) and the SH method, have been implemented. For a discussion of the general features and applicability of MF and SH, we refer to previous publications [70,71].
2.5.1. Mean-field (Ehrenfest) method
The energy within the MF method [72,73] is obtained by averaging over all (adiabatic) states weighted by their populations. Then, all of the states interact with a common potential as a result of the classical environment, which in turn interacts with the combination of states in the quantum system. The quantum system is propagated with a straightforward numerical solution of the time-dependent Schrödinger equation (TDSE). Figure 5a shows schematically how the wave function, which corresponds to one of the adiabatic states at the start of the simulation, is driven into a combination of adiabatic states as soon as these become coupled via the atomic dynamics.
Figure 5.
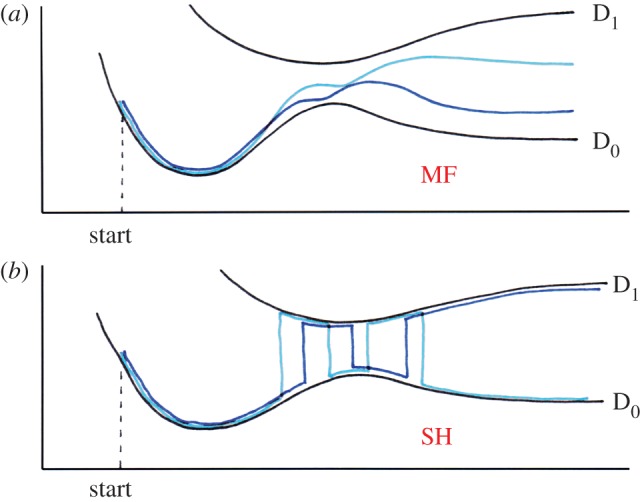
Schematic of the behaviour of the wave function in the MF ((a) Ehrenfest) and the surface hopping ((b) SH) simulation. In each case, the course of two simulations is drawn. In SH, note that the system may leave the high-coupling region (in the centre) in the D0 state (light) or in the D1 state (dark). (Online version in colour.)
The current MF implementation [47] is in the framework of time-dependent DFT, representing a coarse-grained version of the previous work by Niehaus et al. [74]. The Lagrangian formalism is used to derive the equations of motion from the total energy expression presented in the previous sections. The potential energy is taken from the above derivation, and there are kinetic energy contributions related both to the coordinates of atoms and to the expansion coefficients of the wave function for excess charge. Two sets of equations of motion are obtained: the electronic system evolves according to the TDSE with the Hamiltonian Hmn containing the second-order contribution (equation (2.18))
| 2.25 |
For the classical (MM) system, Newton's laws of motion are obtained
| 2.26 |
Note that these equations of motion contain atomic charges on the CT-active fragments augmented by the differential charges corresponding to the distribution of excess electron, 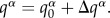
These two sets of equations are coupled to each other, so that they have to be propagated simultaneously. The MM equations involve the (time-dependent) charges of the (N – 1)-electron system qα, which are obtained by a mapping (projection) of the excess charge onto the classical MM system. On the other hand, the time-dependent electronic Hamiltonian depends on the coordinates of all atoms parametrically. Practically, the coarse-grained electronic Hamiltonian is calculated first, and a step of the quantum propagation of the excess charge is performed, followed by a projection of the excess charge to the atomic charges and then a step of classical dynamics of the atoms is done. The following iteration (time step) continues with another calculation of the Hamiltonian and a quantum propagation step, and so on. By performing the projection of the excess charge onto the MM system, it is ensured that the environment (e.g. solvent) is being polarized according to the position and de/localization of the excess charge, and it turns out that this is an important factor for the understanding of the CT process.
2.5.2. Surface hopping
Within an SH scheme [75], the quantum system is only allowed to reside in one of a certain set of states, either adiabatic or diabatic. The principle of SH is to propagate the populations of states in time, while the system occupies one of the states at a time, and a transition to another state (surface hop) is possible in every step of the simulation. This is depicted in figure 5b. The key component of the algorithm is the calculation of the probability of such a surface hop. Accurate SH algorithms are known, such as the fewest switches by Tully [76]. Following a recent study [36], the SH algorithm based on a local diabatization of the adiabatic states [77] is implemented in this work. We present a brief outline of the method here, and refer to the original work for details [78].
In every step of the simulation, the eigenproblem for the quantum system—the excess charge—is solved, providing the adiabatic states, and the current wave function of the excess charge is expressed as their combination. Then, the set of diabatic states is redefined to be identical to the adiabatic ones at the beginning of the time step, and a step of propagation of the wave function (population of the states) expressed in this locally diabatic basis is performed according to the TDSE. The newly obtained populations are used to calculate the probabilities of surface hops from the current state to each of the other states, and a random number is drawn to determine whether a transition to another state (surface hop) occurs in this step.
This procedure is an alternative to the direct numerical integration of the TDSE, which is performed in the MF approach; see above. All of the remaining parts of the simulation protocol are the same. The charge distribution of the excess electron or hole, determined by the currently occupied adiabatic state, is projected onto the MM atomic charges of the participating molecular fragments, and the charges of the corresponding atoms are updated in the MM engine. Then, a step of the classical dynamics of the atoms is performed.
It should be noted that the current SH scheme is a particularly simple one. For instance, explicit calculation of non-adiabatic couplings 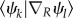 is not involved, and the overlap of the adiabatic state vectors
is not involved, and the overlap of the adiabatic state vectors 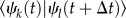 is considered instead. Furthermore, the velocities of atoms are not rescaled after a completed surface hop, which is common with other SH implementations. This way, sudden changes in the dynamics of the system are avoided, but also the total energy of the system is not conserved; this is probably a minor issue because of the application of a thermostat to conserve the temperature rather than the energy. Also, the so-called classically forbidden transitions [79] are not treated in any particular way, so that the classical system is assumed always to contain enough energy to support the surface hop, effectively. These two topics in the coarse-grained QC context are the object of current work in our laboratory. The same is true about the implementation of a possibly necessary decoherence correction [80–82].
is considered instead. Furthermore, the velocities of atoms are not rescaled after a completed surface hop, which is common with other SH implementations. This way, sudden changes in the dynamics of the system are avoided, but also the total energy of the system is not conserved; this is probably a minor issue because of the application of a thermostat to conserve the temperature rather than the energy. Also, the so-called classically forbidden transitions [79] are not treated in any particular way, so that the classical system is assumed always to contain enough energy to support the surface hop, effectively. These two topics in the coarse-grained QC context are the object of current work in our laboratory. The same is true about the implementation of a possibly necessary decoherence correction [80–82].
2.5.3. Which quantum-mechanical propagator?
Now, it is necessary to choose the propagation scheme to be used. It shall be noted here that while every propagation scheme may have its own distinct issues, as will be discussed later in this section, there are also problems that stem from the introduction of the semi-classical representation. One of them is the too slow quantum decoherence, which is caused by the division of the system into a quantum part and a classical part, and thus this problem is shared by both MF and SH schemes [83]. (Still, it is possible that the problem will manifest itself to a different extent in MF and SH.)
The simplest propagation scheme is the MF (Ehrenfest) method, which does not require the coarse-grained Hamiltonian to be diagonalized. The TDSE is integrated explicitly with a numerical method, including all of the adiabatic states in the calculation implicitly. The current wave function of the hole is transferred directly into the propagation of atomic coordinates (i.e. MD) by means of the mapping onto the atomic point charges. The conceptual simplicity comes at the cost of a potentially severe MF error, increasing the delocalization of the wave function largely. This is evident above all in applications where ET proceeds along a flat energy landscape, as is the case in hole transfer in homogeneous DNA sequences.
The BO approximation is involved in most of the main-stream QM/MM simulations and may be plugged into the current hybrid methodology as well. The coarse-grained electronic eigenproblem has to be solved by a diagonalization of the coarse-grained Hamiltonian, and already this step has turned out to be critical on rare occasions, in the simulations of hole transfer along flat energy landscapes. Still, the most important limitation is that only the lowest adiabatic state—the ground state—is considered. As shown recently [36], a proper description of elementary ET events may require higher adiabatic states to be considered, and so the application of BO is restricted to systems where this is not the case. Another possible problem concerns the fact that artificial transfer over long distances may be obtained with BO; this would happen when the instantaneous IP of a distant fragment is decreased by an accidental environmental fluctuation, even though there is no electronic coupling between the involved molecular fragments whatsoever.
The SH approach seems to be the only one free of such principal limitations. In spite of its complex character and the fact that it is still an approximation, it is possible to construct an implementation of SH that is suitable for the application to ET processes. SH involves the higher adiabatic states naturally, and it suffers neither from the delocalization issue (as a single adiabatic state is mapped to the MD engine at any given instant) nor from the possible artificial transfer when there is no coupling of the involved fragments. This makes it perhaps the best choice of the presented approaches. We note that it is not possible to rule out that further issues will emerge in future applications of the current simulation method to new molecular systems. A conceivable example may be a too large number of fragment orbitals that would need to be considered in the application to organic semiconductor systems (see below), which would imply a too large number of adiabatic states to be considered for SH.
2.6. Implementation details and simulation protocols
The described methodology has been implemented in a local version of the Gromacs simulation package [84,85], v. 4.0.3 and (newly) v. 4.6. The most time-consuming procedure in the simulation is the SCC-DFTB calculations of the molecular fragments. The SCC-DFTB method has been included as an integral part of the Gromacs MD engine mdrun. Since the calculations of the different fragments are independent of each other, they may be performed at the same time on different processing units, and this simple parallelization has been implemented. Favourable computational efficiency is achieved as a result of full integration of the QC calculations in the programme and of the described parallelization. A multi-scale simulation of ET in a system such as the DNA sequences described below takes typically 1 day per nanosecond, provided the number of charge-carrying molecular fragments in the system does not exceed the number of available CPU cores.
Production simulations have been performed for DNA and protein systems so far, and the simulation protocols follow the standards established for classical MD simulation closely. The Amber parm99 force field is used [86,87], including the usual corrections specific for DNA [88] and proteins [89]. The biomolecule is placed in a rectangular box filled with TIP3P water molecules, and Na+ or Cl– counter-ions are added to neutralize the system. The smooth particle–mesh Ewald method is applied to evaluate the Coulomb interactions while the Lennard–Jones interactions are cut off at 1 nm. The simulations are performed at a constant temperature of 300 K and a pressure of 1 bar, maintained by the Nosé–Hoover thermostat and the Parrinello–Rahman barostat, respectively, with a characteristic time of 0.5 ps. The lengths of bonds involving a hydrogen atom are constrained to the equilibrium lengths with LINCS or SHAKE. The time step is usually 1 fs; for the initial DNA simulations presented below, the time step was 0.2 fs. The number of steps made, and thus the total time simulated differs by application.
Prior to each production simulation, an equilibration is performed with a stationary excess charge placed on a selected molecular fragment. To this end, modified MM charges of the atoms in this fragment are used; the magnitude of these charges is estimated beforehand according to equation (2.22) with ΔQm = 1. The equilibration procedure starts with a steepest-descents minimization of 100 steps, and it continues with a short dynamics (of 20 ps typically) where the solvent is heated up to 300 K by means of a Berendsen thermostat while the solute is kept at 10 K. The next step is a simulation (of 20 ps again) where the entire system is brought to a temperature of 300 K with the Berendsen thermostat. Finally, a simulation (of 100 ps to 1 ns, differing by application) is performed with the same set-up as described for the production phase.
3. Applications
3.1. Hole transfer in DNA
The original motivation to develop the presented methodology was to describe the transfer of a radical cation in double-stranded DNA. There has been a long debate about the mechanism, and all of the issues of localization of charge, adiabaticity of transfer and time-scale separation have been raised. While it is established that the mechanism is not band-like because the disorder due to dynamics and solvent prevents the formation of extended orbitals [90], initial studies concentrated on the distinction [91] between one-step superexchange tunnelling and what was interpreted as multi-step hopping [92–94] mechanisms. Static DNA molecules were considered here, and models involving the dynamics of the system—except model-based work, e.g. by Jortner et al. [95]—appeared later. A prominent role is played by the idea of the polaron, the deformation or polarization of the molecular environment in the vicinity of the transferring hole [96]. It constitutes the basis of, for example, the phonon-assisted polaron-gated [97] and polaron drift [98] models, and its existence was supported by the studies of sequence-dependent transfer [99].
There is a question that is both crucial and interesting but that is by no means answered unambiguously yet: is the hole in DNA delocalized over several nucleobases or rather confined to a single one? The delocalization of the hole charge would be a prerequisite of band transport or, for example, a conformational gating [100] mechanism, while other experimental work was interpreted in terms of the hopping mechanism assuming a localized hole [101–103]. Generally, the quantum delocalizing effect competes with the localizing forces of the solvent [56]. A certain consensus of a spatially localized hole has been reached recently in the theoretical and computational community [39,40,104,105]. It is possible to understand how peculiar the question of delocalization is by noticing the qualitative change over time of the results reported by several researchers. For instance, while Conwell [98] previously considered delocalized holes, she concluded more recently that the hole is quite localized [39]. Our own previous work [106] involved a numerical integration of TDSE for the hole, without taking into account the polarization of the environment due to the excess charge. Thus, the localizing forces of the environment were not included in the simulation, and a delocalized hole was observed. This has changed upon critical analysis and has passed to the method reviewed here, and a rather localized hole is observed now, as described later.
The hole transfer in DNA was studied by means of atomistic simulation in several laboratories over the last decade. Several studies relied on the post-processing of MD trajectories performed for a system that did not contain the excess charge. Here, the various approaches used a Monte Carlo formalism [107], calculation of the state of the hole for individual snapshots [108], or involved the integration of the TDSE in a simple way [106,109]. Principally, none of this work could describe the polarization response of the environment to the presence of the hole, and so the agreement of the obtained rates of transfer with the experimental results obtained in these studies may have been accidental to a considerable extent. The response of the environment was taken into account in the DFT-based work in Mantz et al. [37], but the observation of a localized hole was actually forced by the biased simulation protocol. So, the only work from that time which involved most of the interactions between the excess charge and the environment seems to be that in the study by Steinbrecher et al. [40], in which a parametrized model of the electronic structure was used, making nanosecond simulations possible. Still, the model was not quite correct, as it left out the description of inner sphere reorganization. The addition of this term would have made the model complete and self-contained; also, however, the rate of transfer would have decreased considerably. Thus, the good agreement with experimental reports seems to have resulted from a compensation of errors brought about by the approximations in the model. We note that there is also newer work that describes the discussed interactions [39], but the application of full DFT calculations brings an increased computational cost, restricting the available time scale to the picosecond range.
The parts of DNA that exhibit the lowest IP are the purine nucleobases, guanine (G) and adenine (A). Therefore, G and A are the molecular fragments that are considered in the FMO calculations. The pyrimidine nucleobases, the DNA backbones and the solvent compose the MM environment, being represented by point electric charges. In a simulation, the transferring hole (radical cation) evolves via the purines along the DNA double strand. We note that the efficiency of hole transfer is not limited by the possible distribution of purines between both DNA strands as the electronic coupling can be sufficient in such a case as well [21]. The first application deals with two archetypical DNA species [78], one with a homogeneous sequence AAAA in which the IP of all of the considered fragments are equal, and one with a sequence GAG in which the transfer from one G to the other has to overcome a higher energy region represented by the A.
3.1.1. Oligonucleotides with a homogeneous sequence
The different non-adiabatic propagation schemes, MF and SH, provide markedly different pictures of hole transfer in the DNA sequence AAAA; the amount of delocalization and a measure of the mobility of the hole are shown in table 1. The wave function of the hole delocalizes over multiple As within picoseconds after an MF simulation is started, even though the initial conditions are those of a perfectly localized hole. As soon as such delocalization has occurred, there is no way for the wave function to go back into a localized state. This is a consequence of the MF description, where the MM environment interacts with the excess charge assuming a state that corresponds to a combination of adiabatic states. The amount of delocalization is large and probably dependent on the size of the system, i.e. the number of purines considered.
Table 1.
The features of hole transfer in the DNA oligonucleotide AAAA (homogeneous sequence). The delocalization of the wave function (given as the number of fragments, parameter Srec), the mobility of the hole (parameter L)a and the number of elementary transfer events in a simulation of 1 ns. MF, mean-field; SH, surface-hopping; BO, Born–Oppenheimer simulations. Data taken from [78].
| MF | SH | BO | |
|---|---|---|---|
| delocalization | 2.42 ± 0.64 | 1.07 ± 0.17 | 1.17 ± 0.28 |
| mobility | 2149 | 696 | 326 |
| rate of hopping | — | 109 | 110 |
aDelocalization is given as 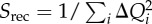 (
(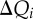 are the hole occupations of the individual fragments). Mobility L is the sum of elementary ‘distances’ travelled by the centre of charge of the wave function in each time step of the simulation; the base-pair step is a unit of distance here. See [78] for details.
are the hole occupations of the individual fragments). Mobility L is the sum of elementary ‘distances’ travelled by the centre of charge of the wave function in each time step of the simulation; the base-pair step is a unit of distance here. See [78] for details.
The large delocalization observed in MF simulations leads to very fast transfer. It was shown previously that a much lower RE applies in the transfer of a delocalized hole than in the transfer of a spatially confined hole [110]. Then, the increased transfer rate is a direct consequence. Actually, in the MF simulations of hole dynamics in AAAA, it is merely possible to speak about the mobility of the wave function rather than about the rate of transfer because the ‘position’ of the delocalized hole is difficult to determine.
For this reason, it seems to be inappropriate to use the (conceptually simple) MF scheme to simulate hole transfer in systems such as the homogeneous DNA sequences. Instead, an implementation of the SH method is a good alternative, in spite of the disadvantages of SH, similar to the hopping algorithm, not being unambiguous, etc. [83]. In the SH simulations of hole transfer in AAAA, the wave function of the hole remains localized to a single A for most of the time, which is the same appearance that is observed in BO simulations. The rate of next-neighbour hole transfer can be determined in such a case and amounts to ca 100 ns–1, which is roughly an order of magnitude larger than the experimental estimates [101,102]. This discrepancy may be caused by the preliminary character of the SH implementation, which seems to require additional attention regarding the calculation of hopping probabilities and decoherence behaviour. Development along these lines is underway in our laboratory.
3.1.2. Oligonucleotides with a heterogeneous sequence
The DNA species with the nucleobase sequence GAG represents a qualitatively different system with regard to hole transfer. The energy landscape is not flat anymore because of the IP of A being ca 0.4 eV larger than that of G. Consequently, a spatial extension of the hole spanning this high-energy region is unfavourable. The wave function of the hole is localized considerably even in MF simulations, and the MF error does not manifest itself notably. Although there are still isolated instants when the wave function is spread over two nucleobases, the wave function always returns to a rather spatially confined state, which is observed for most of the simulation time. The amount of delocalization and the rate of transfer of the hole are shown in table 2.
Table 2.
The features of hole transfer in the DNA oligonucleotide GAG (heterogeneous sequence). The delocalization of the wave function (Srec as defined in table 1) and the number of transfer events in a simulation of 1 ns, averaged over 20 simulations. Abbreviations are the same as given in the legend to table 1. Data taken from [78].
| MF | SH | BO | |
|---|---|---|---|
| delocalization | 1.36 ± 0.31 | 1.00 ± 0.02 | 1.00 ± 0.00 |
| number of transfers | 3.4 ± 2.3 | 3.3 ± 1.8 | 0 |
The appearance of the CT events is similar in MF and SH simulations, perhaps as a consequence of similar localization of the hole. Also, the rate of transfer does not differ between MF and SH simulations, and three or four transfer events are observed per nanosecond on average.6 We note that this nearly perfect agreement may be the result of a sort of cancellation of errors to a certain extent as the MF error cannot be excluded entirely and the SH scheme is not in a final form yet. These results will have to be checked as soon as the SH algorithm has been upgraded (see the section on the non-adiabatic dynamics above). It can be expected that slower rates will be obtained with the corrected methodology.
3.1.3. Conclusion on hole transfer in DNA
The hole transfer in homogeneous DNA sequences does not seem to be a great problem for the Marcus theory. The hole is likely to be localized well. On the basis of spectroscopic measurements, the relaxation of water was reported to occur with two significant time scales of 1.4 and 19 ps [111], and similar observations were made in MD simulation studies [112]. Thus, the relaxation occurs just faster than the hole transfer. Lastly, the calculation of the adiabaticity parameter [35], as performed in [78], predicts a solvent-controlled adiabatic mechanism of transfer.
The hole transfer in heterogeneous sequences represents a more complex problem. The simple model of thermally induced hopping [94] may not be sensitive enough to reproduce the difference between various DNA sequences. Notably, Schuster and co-workers [99,113] observed a more intricate dependence of hole transfer efficiency on the exact nucleobase sequence, which was attributed to the so-called phonon-assisted polaron transport involving a delocalized hole. The particular topics of distance dependence and sequence dependence of hole transfer are the prospective applications of the reviewed simulation scheme.
3.2. Note on charge transport in DNA
The presented simulation approach is aimed at the temporal evolution of an excess electron in a complex molecular system, following an injection of this electron or hole into the system.7 Apart from this ‘chemical’ process, it is possible to consider the charge transport through the molecular system, which is considered rather from a point of view of physics and can also be called conductivity. We would like to mention our recent work in this area briefly.
To this end, our FMO scheme for the calculation of the CT Hamiltonian is combined with the Landauer–Büttiker formalism [114] to compute the electrical conductivity. The structural dynamics of the molecular system is reflected by the time-dependent Hamiltonian, which enters the calculation of electric current under the approximation of instantaneous electron transport by means of tunnelling. The variation of the Hamiltonian in time has a huge impact on the electron transport, and the description of transport improves dramatically in comparison with static models, which were often considered previously.
This flavour of our simulation approach was introduced in the work on charge transport in double-stranded DNA [115], where the effect of structural fluctuations was analysed in detail. Subsequent applications dealt with another class of DNA molecules, G4-DNA [116], double-stranded DNA containing mismatches [117], under mechanical stress [118] and in a micro-hydrated environment [119]. This multi-scale scheme has been developed further, by taking into account the dissipative character of the environment [120,121], and by passing to the time-dependent Green's function approach [122].
3.3. Hole transfer in a protein
ET is the most frequently occurring type of enzyme-catalysed reactions in biochemistry. Although many of the ET reactions taking place in proteins, often requiring the presence of a catalytic metal centre, can be described with the Marcus theory with success, examples can be found where this is not so straightforward. This is the case with E. coli DNA photolyase [123], in which the photo-activation process proceeds via a sequential transfer of a hole from the flavin adenine dinucleotide (FAD) cofactor via two tryptophan side chains (Trp1 and Trp2) to a terminal tryptophan (Trp3) at the surface of the protein [124] (figure 6). The hole transfer in the subsystem composed of three tryptophan side chains was considered in our work [34], and the main findings will be presented in this review because another type of energy relations in a CT system is observed here.
Figure 6.
The photo-activation of E. coli DNA photolyase by means of hole transfer from the FAD cofactor to a tryptophan side chain on the surface of the protein via two other tryptophan residues. (The CT system is shown as thick tubes). (Online version in colour.)
The fundamental difference between the charge transfer in DNA and in this enzyme concerns the properties of the molecular environment rather than the charge-carrying molecular fragments themselves. Much like in the DNA species AAAA, all of the charge-carrying fragments are chemically identical (all of them are tryptophan residues), and so they possess equal inherent IP. But, the highly heterogeneous structure of the protein affects the energy landscape of the transfer: Trp1 is buried in the interior of the protein molecule whereas Trp3 is located at the surface, exposed to the solvent; Trp2 is moderately solvent-accessible. These structural features have consequences for hole transfer.
A first point is that the solvent is effectively more polarizable than the protein. Consequently, when the hole is located on Trp2, it is stabilized more strongly than on Trp1, and the stabilization is the strongest on Trp3; the energy difference amounts to ca 0.6 eV for each of the steps. This fact provides the hole transfer reaction with a driving force, so that the lowest energy state has the hole located on Trp3 and a re-transfer to Trp2 or Trp1 is unlikely. Another point concerns the dynamics of the system. Upon a completed hole transfer event, the environment adjusts—re-polarizes—according to the new location of the hole. The applicability of the classical theory of CT requires this re-polarization to occur substantially faster than the transfer itself. While the component of polarization that corresponds to the solvent meets this criterion, the relaxation of the (albeit smaller) component due to protein takes hundreds of picoseconds, interfering with the time scale of transfer.
The hybrid methodology for ET simulations is an ideal means to describe the transfer under such complex conditions as seen in this example. The rate of hole transfer obtained from simulations in [34] is in reasonable accordance with the experimental reports, deviating by less than an order of magnitude, while the calculations with the Marcus theory predict a rate for the second step Trp2 → Trp3 that is three orders of magnitude too slow (table 3). Interestingly, when a hole transfer simulation is started from the system equilibrated with the hole localized on Trp2—so that the environment is relaxed fully—the elementary transfer Trp2 → Trp3 is much slower than observed in the simulations performed without the artificial relaxation (see fig. 14 in [34]). The artificial relaxation of the intermediate state performed in this test simulation is equivalent to the assumptions of fully relaxed reactants in the Marcus theory. Therefore, the reason for the second elementary transfer occurring so fast is most probably that the energy barrier for this step increases slowly only after the first step Trp1 → Trp2, allowing for the second step to take place well before the environment has relaxed. Note that very slow rates of relaxation, on time scales longer than 100 ps, were indeed reported in protein systems [125]. Such a sequence of events is not in accordance with the assumptions of the Marcus theory, which considers an instantaneous relaxation of the environment. The example of hole transfer in E. coli DNA photolyase demonstrates the advantage of an explicit simulation of ET over the classical theory of transfer, in cases where certain assumptions of the classical theory are not fulfilled.
Table 3.
Rate (per nanosecond) of elementary transfer steps of hole transfer along three tryptophan side chains in E. coli DNA photolyase. Data obtained with MF multi-scale simulation and with the Marcus theory (using several sets of values for the Marcus parameters), as well as the experimental reference. All data taken from [34].
| transfer step | our work | Marcus theory | experiment |
|---|---|---|---|
| Trp1 → Trp2 | 12 | 5–290 | 110 |
| Trp2 → Trp3 | 15 | <0.0003 | 33 |
3.4. Hole transfer in peptides
Peptides have been a popular class of systems to study ET, and the studies by Giese and co-workers [126,127] concentrated on the hole transfer between two amino acid side chains via a relay amino acid (see figure 7 for an example of such a system). The characteristics of these ET systems differ from both DNA and the E. coli DNA photolyase in several respects.
Figure 7.
One of Giese's peptide systems to study hole transfer. Three optionally methoxy-functionalized tyrosine side chains (orange in online version) are the charge-carrying fragments. (Online version in colour.)
The conformational flexibility of small peptides in an aqueous solution is much larger, making the sampling of conformational space difficult. The consequences for ET become clear by noting the straightforward connection between the structure and the electronic coupling between the individual electron-carrying fragments. Concerning the fragments that are involved in the transfer, it may be necessary to consider not only the amino acid side chains but also the π-electron systems of the peptide bonds [128], and this represents another technical challenge because of the difficult definition of the QM/MM boundary.
Our preliminary study of hole transfer in these peptides focused on the basic characterization of the transfer and the calculation of parameters of the Marcus theory [129]. An interesting result is the outer sphere RE for the sequential transfer 1 → 2 → 3: for the entire transfer 1 → 3, it is very similar to that obtained for the elementary steps 1 → 2 and 2 → 3. This is different from the situation for the hole transfer in DNA, where strong distance dependence was observed [110]. The reason is perhaps the relatively large distance between the individual amino acid side chains involved in the transfer, which are not immediate neighbours on the peptide chain—rather, there are two or three other amino acids in between. Then, the RE is already saturated for the elementary transfer steps. In the current work on this topic in our laboratory, the QM region has involved the π-electron systems of the peptide backbone explicitly, and use has been made of a Green function approach to compute the electronic couplings [130] to account for the super-exchange mechanism on long time scales correctly. Also, we plan to perform direct simulations of hole transfer in these systems, in spite of the presumably extensive computational cost due to the long time scale of transfer exceeding nanoseconds.
3.5. Hole transfer in organic semi-conductors
Organic semi-conductors are a class of ET systems that are of great interest currently. At the same time, they possess ET characteristics that are markedly different from those observed in the previously mentioned applications [131]. For these reasons, we are working to extend the presented hybrid methodology, to describe the ET in systems such as hexabenzocoronene (HBC) derivatives. Owing to their self-organizing capabilities [132], these materials form phases such as liquid crystals and self-assembling monolayers on surfaces; an example of the latter structures is shown in figure 8.
Figure 8.
A monolayer of alkylated HBC deposited on a gold surface (10 identical molecules; surface on the bottom side, not shown). (Online version in colour.)
The difference between these systems and those presented above is clear—there is no aqueous or generally polar solvent around the electron-carrying molecules. Therefore, there is little orientational polarizability in the system that would oppose the transfer of electron, and merely electron polarizability is conceivable. Also, the inner sphere RE seems to be much smaller than in DNA bases or amino acids. On the other hand, the electronic couplings between the individual molecular fragments can be quite large. As pointed out recently [133], the reversed relation of RE and electron coupling probably leads to an entirely different mechanism of transfer—adiabatic in this case. An additional computational challenge is represented by the extended, highly symmetric aromatic character of the electronic systems of these molecules. Two HOMOs are degenerated actually, and another orbital has only a slightly lower energy, so that at least three orbitals per molecular fragment have to be considered for the hole transfer.
Since the charge will probably be rather delocalized over several molecular fragments, it may not be meaningful to describe the transfer as consisting of hopping events. On the other hand, the delocalization will be limited owing to the disorder caused by the structural fluctuations. This point can be inferred from the results of preliminary calculations performed on the complex of six HBC fragments. The electronic structure of the complex was computed along an MD trajectory, and the delocalization of the HOMO was obtained as  where ΔQi is the occupation of each fragment by the HOMO (figure 9). The delocalization amounts to 2.3 on average, which is distinctly smaller than the value of 3.3 obtained for the (static) ideally ordered structure, illustrating the disordering effect of the dynamics, which prevents the formation of extended electronic bands.
where ΔQi is the occupation of each fragment by the HOMO (figure 9). The delocalization amounts to 2.3 on average, which is distinctly smaller than the value of 3.3 obtained for the (static) ideally ordered structure, illustrating the disordering effect of the dynamics, which prevents the formation of extended electronic bands.
Figure 9.
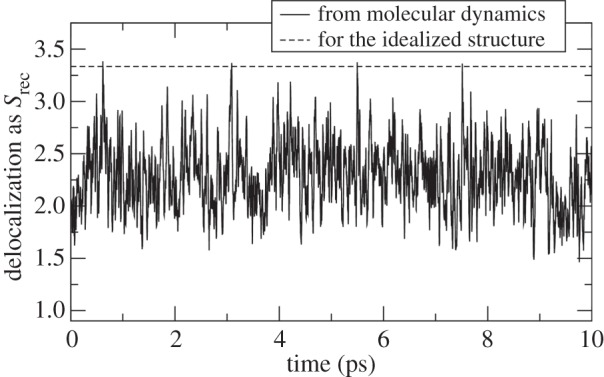
Delocalization of the HOMO in the complex of six HBC units. Observation in an MD simulation (running averages over 20 fs) compared with the value for the ideally ordered structure.
Apparently, the hole transfer in organic semi-conductors is an application with particularly high requirements for the flexibility of the theoretical or computational approach that would describe it, and it seems that, unlike the Marcus theory, our hybrid methodology may meet these requirements.
4. Discussion and conclusion
We have developed a multi-scale computational scheme that allows us to simulate the electron and hole transfer processes in QM regions containing several hundreds of atoms, embedded in extended non-reactive environments, on a time scale of up to tens or hundreds of nanoseconds. The scheme relies on an approximated expression for the total energy, which allows us to implement state-of-the-art non-adiabatic propagation methods. Therefore, the simulation does not rely on any assumptions about the localization of the electronic structure, the nuclear and electronic time scales and the mechanism of the transport process. The adiabatic as well as the non-adiabatic effects are covered naturally, and the hopping transfer and the band transport are described as well as the super-exchange tunnelling, although the accessible time scales may be exceeded for the latter. The quantum description is reduced to the frontier orbitals, and it is easy to draw parallels with the parameters used in the Marcus theory—in terms of the electronic coupling, the driving force and the RE.
The method makes use of a QM/MM coupling scheme. The reactive region in which the charge can be localized is treated with a QC method. It is split further into the frontier orbitals, which are treated with QC explicitly, while the remainder of this region is described with MM, much like in the HMO and PPP approaches. The main approximations are the following:
— The wave function of the excess electron or hole is considered as a superposition of orbitals of the molecular fragments. This makes the QC calculations, which are the most time-consuming component of the simulation scheme, scale linearly with the system size.
— The total energy to the first order is the energy of the neutral system E0 plus the energy of the excess electron E1 (or its negative in the case of hole transfer), according to the Koopmans approximation. We showed that the basic assumption, a similar shape of the frontier orbitals of the neutral and the ionized fragments, holds well.
— All of the energy change due to the different on-site electron–electron interactions as well as that due to the relaxation and rotation of orbitals is covered by the second-order term E2 by using a Hubbard model effectively. These effects are evaluated for isolated fragments and are assumed to hold in the weakly interacting complex system as well.
— In the calculation of forces on the atoms, the contributions of kinetic energy and XC to E1 have to be neglected further. Consequently, two kinds of processes taking place upon the charging of the system are not covered: the geometry relaxation of the fragments and the change of van der Waals interactions between the fragments. While the latter effect is small, the former is significant and is taken into account by an additional inner sphere reorganization contribution to the total energy. This may even be a superior approximation because quantum effects can be taken into consideration, which would be missing in the standard semi-classical dynamics schemes.
The derived approximate expression for the total energy allows us to compute the forces on the atoms, as in the standard QC methods. This is the basis for the application of a non-adiabatic semi-classical scheme, MF (Ehrenfest) or SH. The testing revealed that the application of the MF method is problematic because it leads to a pronounced, overestimated delocalization of the wave function of the excess charge. This resulted in the wrong description of the transfer because the outer sphere reorganization energies were underestimated. This issue does not apply in the SH scheme.
The framework has been applied to the hole transfer in DNA, in E. coli DNA photolyase, and in model peptides and organic materials. We summarize the main findings here.
While there has been no real consensus on the fundamental features of hole transfer in DNA, our results showed that this can be resolved on the basis of direct ET simulations. The hole is localized nearly completely as confirmed by non-adiabatic SH simulations, and reasonable hopping rates were obtained, albeit slightly faster than in the experiments. In DNA, the time scale of transfer not shorter than tens of picoseconds is perhaps long enough so that no problems related to the relaxation effects would occur, and it is possible to apply the Marcus theory. Also, RE is much larger than the electronic coupling, so that the hole is localized on a single nucleobase predominantly, and the transfer proceeds with the solvent-controlled adiabatic mechanism in homogeneous sequences. The detailed sequence dependence of transfer can be readily studied with direct simulations.
For the consecutive hole transfer reaction in the protein E. coli DNA photolyase, we find the second elementary step to be fast, in accordance with the experiments. This fast transfer cannot be predicted on the basis of the standard Marcus theory because of the time-scale separation problem: the time scale of transfer interferes with that of the structural relaxation. Generally, long time-scale relaxation effects ranging from pico- to nanoseconds [112] can be observed in biomolecular complexes whenever a sudden change of the electrostatics in the system has taken place. It seems to be common that there are slow relaxation modes in proteins, on a time scale exceeding 100 ps [125], which are not present in DNA for instance.8 In this case it does not seem possible to construct a simple model since the dynamics of the system seems to depend on structural details of the molecule. Rather, an atomistic approach is needed, particularly regarding the environment, which may be pronouncedly heterogeneous.
The study of hole transfer in model peptides relies on the Marcus theory, which is justified as the transfer is slow. The focus is the calculation of electronic couplings for the transfer between the amino acid side chains, involving Green's function methods and taking into account the π-electron systems in the backbone.
An application of the multi-scale simulation scheme for organic semi-conductors faces an additional challenge as it is necessary to include multiple molecular orbitals per fragment. The preliminary results show that the ET in these materials is anything but similar to that in the aqueous biomolecular complexes. RE may be much smaller owing to the non-polar character of the system, being dominated by the inner sphere component λi. The relation of RE to the electronic coupling may not be as clear as in DNA or in the protein, or may even be reversed. Consequently, considerable delocalization of charge is observed, making it possible for the transfer to proceed with an entirely different mechanism from that in the previous cases, similar to band transport. Then, direct dynamics simulations would be a method with clear advantage over the Marcus theory. This particular case demonstrates that our methodology seems to be well suited for the description of fast CT processes for which the detailed mechanism is still in question, in particular when the application of a hopping scheme may not be possible. The organic electronics is perhaps a typical example [133–135].
These entirely different characters of ET in the various molecular systems make it difficult to develop a unified phenomenological model of transfer. Then, a multi-scale simulation scheme as reviewed here is a viable alternative to characterize the features and predict the rate of the ET reaction.
Acknowledgements
We thank Alexander Heck for valuable discussions and Florian Weigend for help with QC calculations. The reviewed work was supported by the Deutsche Forschungsgemeinschaft under contracts EL 206/5-1, EL 206/5-2 and EL 206/5-3 within the priority programme Quantum Transport at the Molecular Scale (SPP 1243).
Endnotes
While this separation of time scales [30,31] may not be valid in some fast processes [32–34], an extension of the Marcus model to account for this effect was proposed [32].
The derivation for the system with an excess electron (radical anion) is analogous.
Also, it may be noted that only the relative energetics of the molecular conformations is described by the zeroth-order term.
The typical frequency of skeletal C–C vibrations is 1600 cm–1, and 1 kT = 209 cm–1 at 300 K.
The atomic charges are overestimated in the commonly used biomolecular force fields to compensate for the missing electronic polarization in polar environments such as aqueous solutions.
Unfortunately, no experimental work is known to the authors that could be interpreted in terms of the absolute rate of hole transfer in this or similarly simple heterogeneous DNA sequences.
The injection of charge itself cannot be simulated with the current methodology.
Whereas the change in electrostatics concerns the dipole moment of the chromophore in spectroscopic studies, the same is expected when an electron is transferring in the molecular system.
References
- 1.Marcus RA. 1999. Electron transfer—past and future. Adv. Chem. Phys. 106, 1–6. ( 10.1002/9780470141656.ch1) [DOI] [Google Scholar]
- 2.Gray HB, Winkler JR. 1996. Electron transfer in proteins. Annu. Rev. Biochem. 65, 537–561. ( 10.1146/annurev.bi.65.070196.002541) [DOI] [PubMed] [Google Scholar]
- 3.Hervas M, Navarro JA, De La Rosa MA. 2003. Electron transfer between membrane complexes and soluble proteins in photosynthesis. Acc. Chem. Res. 36, 798–805. ( 10.1021/ar020084b) [DOI] [PubMed] [Google Scholar]
- 4.Bixon M, Jortner J. 1999. Electron transfer—from isolated molecules to biomolecules. Adv. Chem. Phys. 106, 35–202. ( 10.1002/9780470141656.ch3) [DOI] [Google Scholar]
- 5.Marcus RA. 1956. On the theory of oxidation-reduction reactions involving electron transfer. I. J. Chem. Phys. 24, 966–978. ( 10.1063/1.1742723) [DOI] [Google Scholar]
- 6.Marcus RA, Sutin N. 1985. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 811, 265–322. ( 10.1016/0304-4173(85)90014-X) [DOI] [Google Scholar]
- 7.Levich VG, Dogonadze RR. 1959. Theory of non-radiation electron transitions from ion to ion in solutions. Dokl. Akad. Nauk SSSR 124, 123–126. [Google Scholar]
- 8.Hush NS. 1961. Adiabatic theory of outer sphere electron-transfer reactions in solution. Trans. Faraday Soc. 57, 557–580. ( 10.1039/tf9615700557) [DOI] [Google Scholar]
- 9.Hopfield JJ. 1974. Electron-transfer between biological molecules by thermally activated tunneling. Proc. Natl Acad. Sci. USA 71, 3640–3644. ( 10.1073/pnas.71.9.3640) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jortner J. 1976. Temperature-dependent activation-energy for electron-transfer between biological molecules. J. Chem. Phys. 64, 4860–4867. ( 10.1063/1.432142) [DOI] [Google Scholar]
- 11.Small DW, Matyushov DV, Voth GA. 2003. The theory of electron transfer reactions: what may be missing? J. Am. Chem. Soc. 125, 7470–7478. ( 10.1021/ja029595j) [DOI] [PubMed] [Google Scholar]
- 12.Newton MD. 1991. Quantum chemical probes of electron-transfer kinetics: the nature of donor-acceptor interactions. Chem. Rev. 91, 767–792. ( 10.1021/cr00005a007) [DOI] [Google Scholar]
- 13.Blancafort L, Voityuk AA. 2005. CASSCF/CAS-PT2 study of hole transfer in stacked DNA nucleobases. J. Phys. Chem. A 110, 6426–6432. ( 10.1021/jp061184s) [DOI] [PubMed] [Google Scholar]
- 14.Troisi A, Orlandi G. 2001. The hole transfer in DNA: calculation of electron coupling between close bases. Chem. Phys. Lett. 344, 509–518. ( 10.1016/S0009-2614(01)00792-8) [DOI] [Google Scholar]
- 15.Senthilkumar K, Grozema FC, Bickelhaupt FM, Siebbeles LDA. 2003. Charge transport in columnar stacked triphenylenes: effects of conformational fluctuations on charge transfer integrals and site energies. J. Chem. Phys. 119, 9809–9817. ( 10.1063/1.1615476) [DOI] [Google Scholar]
- 16.Kubař T, Woiczikowski PB, Cuniberti G, Elstner M. 2008. Efficient calculation of charge-transfer matrix elements for hole transfer in DNA. J. Phys. Chem. B 112, 7937–7947. ( 10.1021/jp801486d) [DOI] [PubMed] [Google Scholar]
- 17.Oberhofer H, Blumberger J. 2010. Insight into the mechanism of the Ru2+–Ru3+ electron self-exchange reaction from quantitative rate calculations. Angew. Chem. Int. Ed. 49, 3631–3634. ( 10.1002/anie.200906455) [DOI] [PubMed] [Google Scholar]
- 18.Oberhofer H, Blumberger J. 2012. Revisiting electronic couplings and incoherent hopping models for electron transport in crystalline C60 at ambient temperatures. Phys. Chem. Chem. Phys. 14, 13 846–13 852. ( 10.1039/c2cp41348e) [DOI] [PubMed] [Google Scholar]
- 19.Oberhofer H, Blumberger J. 2010. Electronic coupling matrix elements from charge constrained density functional theory calculations using a plane wave basis set. J. Chem. Phys. 133, 244105 ( 10.1063/1.3507878) [DOI] [PubMed] [Google Scholar]
- 20.Van Voorhis T, Kowalczyk T, Kaduk B, Wang L-P, Cheng C-L, Wu Q. 2010. The diabatic picture of electron transfer, reaction barriers, and molecular dynamics. Annu. Rev. Phys. Chem. 61, 149–170. ( 10.1146/annurev.physchem.012809.103324) [DOI] [PubMed] [Google Scholar]
- 21.Kubař T, Elstner M. 2008. What governs the charge transfer in DNA? The role of DNA conformation and environment. J. Phys. Chem. B 112, 8788–8798. ( 10.1021/jp803661f) [DOI] [PubMed] [Google Scholar]
- 22.Oberhofer H, Blumberger J. 2009. Charge constrained density functional molecular dynamics for simulation of condensed phase electron transfer reactions. J. Chem. Phys. 131, 064101 ( 10.1063/1.3190169) [DOI] [PubMed] [Google Scholar]
- 23.Tesar SL, Leveritt JM, III, Kurnosov AA, Burin AL. 2012. Temperature dependence for the rate of hole transfer in DNA: nonadiabatic regime. Chem. Phys. 393, 13–18. ( 10.1016/j.chemphys.2011.11.017) [DOI] [Google Scholar]
- 24.VandeVondele J, Sulpizi M, Sprik M. 2006. From solvent fluctuations to quantitative redox properties of quinones in methanol and acetonitrile. Angew. Chem. Int. Ed. 45, 1936–1938. ( 10.1002/anie.200503581) [DOI] [PubMed] [Google Scholar]
- 25.VandeVondele J, Sulpizi M, Sprik M. 2007. Electron transfer properties from atomistic simulations and density functional theory. CHIMIA 61, 155–158. ( 10.2533/chimia.2007.155) [DOI] [Google Scholar]
- 26.Blumberger J. 2008. Free energies for biological electron transfer from QM/MM calculation: method, application and critical assessment. Phys. Chem. Chem. Phys. 10, 5651–5667. ( 10.1039/b807444e) [DOI] [PubMed] [Google Scholar]
- 27.Kowalczyk T, Wang L-P, Van Voorhis T. 2011. Simulation of solution phase electron transfer in a compact donor–acceptor dyad. J. Phys. Chem. B 115, 12 135–12 144. ( 10.1021/jp204962k) [DOI] [PubMed] [Google Scholar]
- 28.Muegge I, Qi PX, Wand AJ, Chu ZT, Warshel A. 1997. The reorganization energy of cytochrome c revisited. J. Phys. Chem. B 101, 825–836. ( 10.1021/jp962478o) [DOI] [Google Scholar]
- 29.Parson WW, Chu ZT, Warshel A. 1998. Reorganization energy of the initial electron-transfer step in photosynthetic bacterial reaction centers. Biophys. J. 74, 182–191. ( 10.1016/S0006-3495(98)77778-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beratan DN, Skourtis SS, Balabin IA, Balaeff A, Keinan S, Venkatramani R, Xiao D. 2009. Steering electrons on moving pathways. Acc. Chem. Res. 42, 1669–1678. ( 10.1021/ar900123t) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skourtis SS, Waldeck DH, Beratan DN. 2010. Fluctuations in biological and bioinspired electron-transfer reactions. Annu. Rev. Phys. Chem. 61, 461–485. ( 10.1146/annurev.physchem.012809.103436) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, Lin S, Allen JP, Williams JC, Blankert S, Laser C, Woodbury NW. 2007. Protein dynamics control the kinetics of initial electron transfer in photosynthesis. Science 316, 747–750. ( 10.1126/science.1140030) [DOI] [PubMed] [Google Scholar]
- 33.Skourtis SS, Beratan DN. 2007. Photosynthesis from the protein's perspective. Science 316, 703–704. ( 10.1126/science.1142330) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woiczikowski PB, Steinbrecher T, Kubař T, Elstner M. 2011. Nonadiabatic QM/MM simulations of fast charge transfer in Escherichia coli DNA photolyase. J. Phys. Chem. B 115, 9846–9863. ( 10.1021/jp204696t) [DOI] [PubMed] [Google Scholar]
- 35.Rips I, Jortner J. 1987. Dynamic solvent effects on outer-sphere electron transfer. J. Chem. Phys. 87, 2090–2104. ( 10.1063/1.453184) [DOI] [Google Scholar]
- 36.Plasser F, Lischka H. 2011. Semiclassical dynamics simulations of charge transport in stacked π-systems. J. Chem. Phys. 134, 034309 ( 10.1063/1.3526697) [DOI] [PubMed] [Google Scholar]
- 37.Mantz YA, Gervasio FL, Laino T, Parrinello M. 2007. Solvent effects on charge spatial extent in DNA and implications for transfer. Phys. Rev. Lett. 99, 058104 ( 10.1103/PhysRevLett.99.058104) [DOI] [PubMed] [Google Scholar]
- 38.Kinz-Thompson CD, Conwell EM. 2010. Proton transfer in adenine-thymine radical cation embedded in B-form DNA. J. Phys. Chem. Lett. 1, 1403–1407. ( 10.1021/jz100214h) [DOI] [PubMed] [Google Scholar]
- 39.Kravec SM, Kinz-Thompson CD, Conwell EM. 2011. Localization of a hole on an adenine-thymine radical cation in B-form DNA in water. J. Phys. Chem. B 115, 6166–6171. ( 10.1021/jp110062y) [DOI] [PubMed] [Google Scholar]
- 40.Steinbrecher T, Koslowski T, Case DA. 2008. Direct simulation of electron transfer reactions in DNA radical cations. J. Phys. Chem. B 112, 16 935–16 944. ( 10.1021/jp8076134) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elstner M, Porezag D, Jungnickel G, Elsner J, Haugk M, Frauenheim T, Suhai S, Seifert G. 1998. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 58, 7260–7268. ( 10.1103/PhysRevB.58.7260) [DOI] [Google Scholar]
- 42.Kitaura K, Ikeo E, Asada T, Nakano T, Uebayasi M. 1999. Fragment molecular orbital method: an approximate computational method for large molecules. Chem. Phys. Lett. 313, 701–706. ( 10.1016/S0009-2614(99)00874-X) [DOI] [Google Scholar]
- 43.Hückel E. 1931. Quantentheoretische Beiträge zum Benzolproblem. Z. Phys. 70, 204–286. ( 10.1007/BF01339530) [DOI] [Google Scholar]
- 44.Hückel E. 1932. Quantentheoretische Beiträge zum Problem der aromatischen und ungesättigten Verbindungen. III. Z. Phys. 76, 628–648. ( 10.1007/BF01341936) [DOI] [Google Scholar]
- 45.Pariser R, Parr RG. 1953. A semi-empirical theory of the electronic spectra and electronic structure of complex unsaturated molecules. 1. and 2. J. Chem. Phys. 21, 466–471, 767–776 ( 10.1063/1.1698929) [DOI] [Google Scholar]
- 46.Pople JA. 1953. Electron interaction in unsaturated hydrocarbons. Trans. Faraday Soc. 49, 1375–1385. ( 10.1039/tf9534901375) [DOI] [Google Scholar]
- 47.Kubař T, Elstner M. 2010. Coarse-grained time-dependent density functional simulation of charge transfer in complex systems: application to hole transfer in DNA. J. Phys. Chem. B 114, 11 221–11 240. ( 10.1021/jp102814p) [DOI] [PubMed] [Google Scholar]
- 48.Gaus M, Cui Q, Elstner M. 2011. DFTB3: Extension of the self-consistent-charge density-functional tight-binding method (SCC-DFTB). J. Chem. Theory Comput. 7, 931–948. ( 10.1021/ct100684s) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuleff AI, Dreuw A. 2009. Theoretical description of charge migration with a single Slater-determinant and beyond. J. Chem. Phys. 130, 034102 ( 10.1063/1.3058899) [DOI] [PubMed] [Google Scholar]
- 50.Löwdin PO. 1950. On the non-orthogonality problem connected with the use of atomic wave functions in the theory of molecules and crystals. J. Chem. Phys. 18, 365–375. ( 10.1063/1.1747632) [DOI] [Google Scholar]
- 51.Plasser F, Aquino JAA, Hase WL, Lischka H. 2012. UV absorption spectrum of alternating DNA duplexes. Analysis of excitonic and charge transfer interactions. J. Phys. Chem. A 116, 11 151–11 160. ( 10.1021/jp304725r) [DOI] [PubMed] [Google Scholar]
- 52.Lundberg M, Nishimoto Y, Irle S. 2012. Delocalization errors in a Hubbard-like model: consequences for density-functional tight-binding calculations of molecular systems. Int. J. Quantum Chem. 112, 1701–1711. ( 10.1002/qua.23178) [DOI] [Google Scholar]
- 53.Perdew JP, Zunger A. 1981. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 23, 5048–5079. ( 10.1103/PhysRevB.23.5048) [DOI] [Google Scholar]
- 54.Mantz YA, Gervasio FL, Laino T, Parrinello M. 2007. Charge localization in stacked radical cation DNA base pairs and the benzene dimer studied by self-interaction corrected density-functional theory. J. Phys. Chem. A 111, 105–112. ( 10.1021/jp063080n) [DOI] [PubMed] [Google Scholar]
- 55.Bayly CI, Cieplak P, Cornell WD, Kollman PA. 1993. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97, 10 269–10 280. ( 10.1021/j100142a004) [DOI] [Google Scholar]
- 56.Kurnikov IV, Tong GSM, Madrid M, Beratan DN. 2002. Hole size and energetics in double helical DNA: competition between quantum delocalization and solvation localization. J. Phys. Chem. B 106, 7–10. ( 10.1021/jp0132329) [DOI] [Google Scholar]
- 57.Olofsson J, Larsson S. 2001. Electron hole transport in DNA. J. Phys. Chem. B 105, 10 398–10 406. ( 10.1021/jp011052t) [DOI] [Google Scholar]
- 58.Alexandre SS, Artacho E, Soler J, Chacham H. 2003. Small polarons in dry DNA. Phys. Rev. Lett. 91, 108105 ( 10.1103/PhysRevLett.91.108105) [DOI] [PubMed] [Google Scholar]
- 59.Hou G, Zhu X, Elstner M, Cui Q. 2012. A modified QM/MM Hamiltonian with the self-consistent-charge density-functional-tight-binding theory for highly charged QM regions. J. Chem. Theory Comput. 8, 4293–4304. ( 10.1021/ct300649f) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuharski RA, Bader JS, Chandler D, Sprik M, Klein ML, Impey RW. 1998. Molecular model for aqueous ferrous-ferric electron transfer. J. Chem. Phys. 89, 3248–3257. ( 10.1063/1.454929) [DOI] [Google Scholar]
- 61.King G, Warshel A. 1990. Investigation of the free energy functions for electron transfer reactions. J. Chem. Phys. 93, 8682–8692. ( 10.1063/1.459255) [DOI] [Google Scholar]
- 62.Schulten K, Tesch M. 1991. Coupling of protein motion to electron transfer: molecular dynamics and stochastic quantum mechanics study of photosynthetic reaction centers. Chem. Phys. 158, 421–446. ( 10.1016/0301-0104(91)87081-6) [DOI] [Google Scholar]
- 63.Alden RG, Parson WW, Chu ZT, Warshel A. 1995. Calculations of electrostatic energies in photosynthetic reaction centers. J. Am. Chem. Soc. 117, 12 284–12 298. ( 10.1021/ja00154a031) [DOI] [Google Scholar]
- 64.Marchi M, Gehlen JN, Chandler D, Newton M. 1993. Diabatic surfaces and the pathway for primary electron transfer in a photosynthetic reaction center. J. Am. Chem. Soc. 115, 4178–4190. ( 10.1021/ja00063a041) [DOI] [Google Scholar]
- 65.Warshel A, Hwang J-K. 1986. Simulation of the dynamics of electron transfer reactions in polar solvents: semiclassical trajectories and dispersed polaron approaches. J. Chem. Phys. 84, 4938–4957. ( 10.1063/1.449981) [DOI] [Google Scholar]
- 66.Sulpizi M, Raugei S, VandeVondele J, Carloni P, Sprik M. 2007. Calculation of redox properties: understanding short- and long-range effects in rubredoxin. J. Phys. Chem. B 111, 3969–3976. ( 10.1021/jp067387y) [DOI] [PubMed] [Google Scholar]
- 67.Blumberger J, Lamoureux G. 2008. Reorganization free energies and quantum corrections for a model electron self-exchange reaction: comparison of polarizable and non-polarizable solvent models. Mol. Phys. 106, 1597–1611. ( 10.1080/00268970802220112) [DOI] [Google Scholar]
- 68.Leontyev IV, Stuchebrukhov AA. 2010. Electronic continuum model for molecular dynamics simulations of biological molecules. J. Chem. Theory Comput. 6, 1498–1508. ( 10.1021/ct9005807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Igor Leontyev V, Stuchebrukhov AA. 2011. Accounting for electronic polarization in non-polarizable force fields. Phys. Chem. Chem. Phys. 13, 2613–2626. ( 10.1039/c0cp01971b) [DOI] [PubMed] [Google Scholar]
- 70.Prezhdo OV, Rossky PJ. 1997. Mean-field molecular dynamics with surface hopping. J. Chem. Phys. 107, 825–834. ( 10.1063/1.474382) [DOI] [Google Scholar]
- 71.Tully JC. 1998. Mixed quantum-classical dynamics. Faraday Discus. 110, 407–419. ( 10.1039/a801824c) [DOI] [Google Scholar]
- 72.Ehrenfest P. 1927. Bemerkung über die angenäherte Gültigkeit der klassischen Mechanik innerhalb der Quantenmechanik. Z. Phys. A 45, 455–457. [Google Scholar]
- 73.McLachlan AD. 1964. A variational solution of the time-dependent Schrodinger equation. Mol. Phys. 8, 39–44. ( 10.1080/00268976400100041) [DOI] [Google Scholar]
- 74.Niehaus TA, Heringer D, Torralva B, Frauenheim T. 2005. Importance of electronic self-consistency in the TDDFT based treatment of nonadiabatic molecular dynamics. Eur. Phys. J. D 35, 467–477. ( 10.1140/epjd/e2005-00079-7) [DOI] [Google Scholar]
- 75.Tully JC, Preston RK. 1971. Trajectory surface hopping approach to nonadiabatic molecular collisions: the reaction of H+ with D2. J. Chem. Phys. 55, 562–572. ( 10.1063/1.1675788) [DOI] [Google Scholar]
- 76.Tully JC. 1990. Molecular dynamics with electronic transitions. J. Chem. Phys. 93, 1061–1071. ( 10.1063/1.459170) [DOI] [Google Scholar]
- 77.Granucci G, Persico M, Toniolo A. 2001. Direct semiclassical simulation of photochemical processes with semiempirical wave functions. J. Chem. Phys. 114, 10 608–10 615. ( 10.1063/1.1376633) [DOI] [Google Scholar]
- 78.Kubař T, Elstner M. 2013. Efficient algorithms for the simulation of non-adiabatic electron transfer in complex molecular systems: application to DNA. Phys. Chem. Chem. Phys. 15, 5794–5813. ( 10.1039/c3cp44619k) [DOI] [PubMed] [Google Scholar]
- 79.Fang JY, Hammes-Schiffer S. 1999. Comparison of surface hopping and mean field approaches for model proton transfer reactions. J. Chem. Phys. 110, 11 166–11 175. ( 10.1063/1.479058) [DOI] [Google Scholar]
- 80.Zhu C, Nangia S, Jasper AW, Truhlar DG. 2004. Coherent switching with decay of mixing: an improved treatment of electronic coherence for non-Born–Oppenheimer trajectories. J. Chem. Phys. 121, 7658–7670. ( 10.1063/1.1793991) [DOI] [PubMed] [Google Scholar]
- 81.Granucci G, Persico M, Zoccante A. 2010. Including quantum decoherence in surface hopping. J. Chem. Phys. 133, 134111 ( 10.1063/1.3489004) [DOI] [PubMed] [Google Scholar]
- 82.Subotnik JE, Shenvi N. 2011. A new approach to decoherence and momentum rescaling in the surface hopping algorithm. J. Chem. Phys. 134, 024105 ( 10.1063/1.3506779) [DOI] [PubMed] [Google Scholar]
- 83.Doltsinis NL. 2002. Nonadiabatic dynamics: mean-field and surface hopping. In Quantum simulations of complex many-body systems: from theory to algorithms (eds Grotendorst J, Marx D, Muramats A.), pp. 377–397. Jülich, Germany: John von Neumann Institute for Computing. [Google Scholar]
- 84.van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. 2005. GROMACS: fast, exible, free. J. Comput. Chem. 26, 1701–1718. ( 10.1002/jcc.20291) [DOI] [PubMed] [Google Scholar]
- 85.Hess B, Kutzner C, van der Spoel D, Lindahl E. 2008. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447. ( 10.1021/ct700301q) [DOI] [PubMed] [Google Scholar]
- 86.Cornell WD, et al. 1995. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J. Am. Chem. Soc. 117, 5179–5197. ( 10.1021/ja00124a002) [DOI] [Google Scholar]
- 87.Wang J, Cieplak P, Kollman PA. 2000. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 21, 1049–1074. () [DOI] [Google Scholar]
- 88.Pérez A, Marchán I, Svozil D, Šponer J, Cheatham TE, III, Laughton CA, Orozco M. 2007. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys. J. 92, 3817–3829. ( 10.1529/biophysj.106.097782) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. 2006. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725. ( 10.1002/prot.21123) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lewis J, Cheatham T, Starikov EB, Wang H, Sankey OF. 2003. Dynamically amorphous character of electronic states in poly(dA)-poly(dT) DNA. J. Phys. Chem. B 107, 2581–2587. ( 10.1021/jp026772u) [DOI] [Google Scholar]
- 91.Meggers E, Michel-Beyerle ME, Giese B. 1998. Sequence dependent long range hole transport in DNA. J. Am. Chem. Soc. 120, 12 950–12 955. ( 10.1021/ja983092p) [DOI] [Google Scholar]
- 92.Bixon M, Giese B, Wessely S, Langenbacher T, Michel-Beyerle ME, Jortner J. 1999. Long-range charge hopping in DNA. Proc. Natl Acad. Sci. USA 96, 11 713–11 716. ( 10.1073/pnas.96.21.11713) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berlin YA, Burin AL, Ratner MA. 2001. Charge hopping in DNA. J. Am. Chem. Soc. 123, 260–268. ( 10.1021/ja001496n) [DOI] [PubMed] [Google Scholar]
- 94.Bixon M, Jortner J. 2001. Charge transport in DNA via thermally induced hopping. J. Am. Chem. Soc. 123, 12 556–12 567. ( 10.1021/ja010018p) [DOI] [PubMed] [Google Scholar]
- 95.Jortner J, Bixon M, Langenbacher T, Michel-Beyerle ME. 1998. Charge transfer and transport in DNA. Proc. Natl Acad. Sci. USA 95, 12 759–12 765. ( 10.1073/pnas.95.22.12759) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Conwell EM, Rakhmanova SV. 2000. Polarons in DNA. Proc. Natl Acad. Sci. USA 97, 4556–4560. ( 10.1073/pnas.050074497) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Henderson PT, Jones D, Hampikian G, Kan Y, Schuster GB. 1999. Long-distance charge transport in duplex DNA: the phonon-assisted polaron-like hopping mechanism. Proc. Natl Acad. Sci. USA 96, 8353–8358. ( 10.1073/pnas.96.15.8353) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Conwell EM. 2005. Charge transport in DNA in solution: the role of polarons. Proc. Natl Acad. Sci. USA 102, 8795–8799. ( 10.1073/pnas.0501406102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu C-S, Schuster GB. 2003. Base sequence effects in radical cation migration in duplex DNA: support for the polaron-like hopping model. J. Am. Chem. Soc. 125, 6098–6102. ( 10.1021/ja029333h) [DOI] [PubMed] [Google Scholar]
- 100.O'Neill MA, Barton JK. 2004. DNA charge transport: conformationally gated hopping through stacked domains. J. Am. Chem. Soc. 126, 11 471–11 483. ( 10.1021/ja048956n) [DOI] [PubMed] [Google Scholar]
- 101.Takada T, Kawai K, Cai X, Sugimoto A, Fujitsuka M, Majima T. 2004. Charge separation in DNA via consecutive adenine hopping. J. Am. Chem. Soc. 126, 1125–1129. ( 10.1021/ja035730w) [DOI] [PubMed] [Google Scholar]
- 102.Conron SMM, Thazhathveetil AK, Wasielewski MR, Burin AL, Lewis FD. 2010. Direct measurement of the dynamics of hole hopping in extended DNA G-tracts. An unbiased random walk. J. Am. Chem. Soc. 132, 14 388–14 390. ( 10.1021/ja106991f) [DOI] [PubMed] [Google Scholar]
- 103.Thazhathveetil AK, Trifonov A, Wasielewski MR, Lewis FD. 2011. Increasing the speed limit for hole transport in DNA. J. Am. Chem. Soc. 133, 11 485–11 487. ( 10.1021/ja204815d) [DOI] [PubMed] [Google Scholar]
- 104.Burin AL, Uskov DB. 2008. Strong localization of positive charge in DNA induced by its interaction with environment. J. Chem. Phys. 129, 025101 ( 10.1063/1.2953693) [DOI] [PubMed] [Google Scholar]
- 105.Blancafort L, Duran M, Poater J, Salvador P, Simon S, Solà M, Voityuk AA. 2009. Excess charge delocalization in organic and biological molecules: some theoretical notions. Theor. Chem. Acc. 123, 29–40. ( 10.1007/s00214-009-0538-8) [DOI] [Google Scholar]
- 106.Kubař T, Kleinekathöfer U, Elstner M. 2009. Solvent fluctuations drive the hole transfer in DNA: a mixed quantum–classical study. J. Phys. Chem. B 113, 13 107–13 117. ( 10.1021/jp9073587) [DOI] [PubMed] [Google Scholar]
- 107.Řeha D, Barford W, Harris S. 2008. A multi-scale method for the calculation of charge transfer rates through the ∏-stack of DNA: application to DNA dynamics. Phys. Chem. Chem. Phys. 10, 5436–5444. ( 10.1039/b719619a) [DOI] [PubMed] [Google Scholar]
- 108.Hatcher E, Balaeff A, Keinan S, Venkatramani R, Beratan DN. 2008. PNA versus DNA: effects of structural fluctuations on electronic structure and hole-transport mechanisms. J. Am. Chem. Soc. 130, 11 752–11 761. ( 10.1021/ja802541e) [DOI] [PubMed] [Google Scholar]
- 109.Grozema FC, Tonzani S, Berlin YA, Schatz GC, Siebbeles LDA, Ratner MA. 2008. Effect of structural dynamics on charge transfer in DNA hairpins. J. Am. Chem. Soc. 130, 5157–5166. ( 10.1021/ja078162j) [DOI] [PubMed] [Google Scholar]
- 110.Kubař T, Elstner M. 2009. Solvent reorganization energy of hole transfer in DNA. J. Phys. Chem. B 113, 5653–5656. ( 10.1021/jp901888r) [DOI] [PubMed] [Google Scholar]
- 111.Pal SK, Zhao L, Zewail AH. 2003. Water at DNA surfaces: ultrafast dynamics in minor groove recognition. Proc. Natl Acad. Sci. USA 100, 8113–8118. ( 10.1073/pnas.1433066100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Furse KE, Corcelli SA. 2010. Molecular dynamics simulations of DNA solvation dynamics. J. Phys. Chem. Lett. 1, 1813–1820. ( 10.1021/jz100485e) [DOI] [Google Scholar]
- 113.Liu C-S, Hernandez R, Schuster GB. 2004. Mechanism for radical cation transport in duplex DNA oligonucleotides. J. Am. Chem. Soc. 126, 2877–2884. ( 10.1021/ja0378254) [DOI] [PubMed] [Google Scholar]
- 114.Büttiker M, Landauer R. 1982. Traversal time for tunneling. Phys. Rev. Lett. 49, 1739–1742. ( 10.1103/PhysRevLett.49.1739) [DOI] [Google Scholar]
- 115.Woiczikowski PB, Kubař T, Gutiérrez R, Caetano RA, Cuniberti G, Elstner M. 2009. Combined density functional theory and Landauer approach for hole transfer in DNA along classical molecular dynamics trajectories. J. Chem. Phys. 130, 215104 ( 10.1063/1.3146905) [DOI] [PubMed] [Google Scholar]
- 116.Woiczikowski PB, Kubař T, Gutiérrez R, Cuniberti G, Elstner M. 2010. Structural stability versus conformational sampling in biomolecular systems: why is the charge transfer efficiency in G4-DNA better than in double-stranded DNA? J. Chem. Phys. 133, 035103 ( 10.1063/1.3460132) [DOI] [PubMed] [Google Scholar]
- 117.Lee MH, Avdoshenko S, Gutiérrez R, Cuniberti G. 2010. Charge migration through DNA molecules in the presence of mismatches. Phys. Rev. B 82, 155455 ( 10.1103/PhysRevB.82.155455) [DOI] [Google Scholar]
- 118.Wolter M, Woiczikowski PB, Elstner M, Kubař T. 2012. Response of the electric conductivity of double-stranded DNA on moderate mechanical stretching stresses. Phys. Rev. B 85, 075101 ( 10.1103/PhysRevB.85.075101) [DOI] [Google Scholar]
- 119.Wolter M, Elstner M, Kubař T. Submitted. Charge transport in desolvated DNA. [DOI] [PubMed]
- 120.Gutiérrez R, Caetano RA, Woiczikowski PB, Kubař T, Elstner M, Cuniberti G. 2009. Charge transport through biomolecular wires in a solvent: bridging molecular dynamics and model Hamiltonian approaches. Phys. Rev. Lett. 102, 208102 ( 10.1103/PhysRevLett.102.208102) [DOI] [PubMed] [Google Scholar]
- 121.Gutiérrez R, Caetano R, Woiczikowski PB, Kubař T, Elstner M, Cuniberti G. 2010. Structural fluctuations and quantum transport through DNA molecular wires: a combined molecular dynamics and model Hamiltonian approach. New J. Phys. 12, 023022 ( 10.1088/1367-2630/12/2/023022) [DOI] [Google Scholar]
- 122.Popescu B, Woiczikowski PB, Elstner M, Kleinekathöfer U. 2012. Time-dependent view of sequential transport through molecules with rapidly fluctuating bridges. Phys. Rev. Lett. 109, 176802 ( 10.1103/PhysRevLett.109.176802) [DOI] [PubMed] [Google Scholar]
- 123.Park HW, Kim ST, Sancar A, Deisenhofer J. 1995. Crystal structure of DNA photolyase from Escherichia coli. Science 268, 1866–1872. ( 10.1126/science.7604260) [DOI] [PubMed] [Google Scholar]
- 124.Brettel K, Byrdin M. 2010. Reaction mechanisms of DNA photolyase. Curr. Opin. Struct. Biol. 20, 693–701. ( 10.1016/j.sbi.2010.07.003) [DOI] [PubMed] [Google Scholar]
- 125.Golosov AA, Karplus M. 2007. Probing polar solvation dynamics in proteins: a molecular dynamics simulation analysis. J. Phys. Chem. B 111, 1482–1490. ( 10.1021/jp065493u) [DOI] [PubMed] [Google Scholar]
- 126.Wang M, Gao J, Müller P, Giese B. 2009. Electron transfer in peptides with cysteine and methionine as relay amino acids. Angew. Chem. Int. Ed. 48, 4232–4234. ( 10.1002/anie.200900827) [DOI] [PubMed] [Google Scholar]
- 127.Gao J, Müller P, Wang M, Eckhardt S, Lauz M, Fromm KM, Giese B. 2011. Electron transfer in peptides: the influence of charged amino acids. Angew. Chem. Int. Ed. 50, 1926–1930. ( 10.1002/anie.201003389) [DOI] [PubMed] [Google Scholar]
- 128.Lauz M, Eckhardt S, Fromm KM, Giese B. 2012. The influence of dipole moments on the mechanism of electron transfer through helical peptides. Phys. Chem. Chem. Phys. 14, 13 785–13 788. ( 10.1039/c2cp41159h) [DOI] [PubMed] [Google Scholar]
- 129.Heck A, Woiczikowski PB, Kubař T, Giese B, Elstner M, Steinbrecher TB. 2012. Charge transfer in model peptides: obtaining Marcus’ parameters from molecular simulation. J. Phys. Chem. B 116, 2284–2293. ( 10.1021/jp2086297) [DOI] [PubMed] [Google Scholar]
- 130.Priyadarshy S, Skourtis SS, Risser SM, Beratan DN. 1996. Bridge-mediated electronic interactions: differences between Hamiltonian and Green function partitioning in a nonorthogonal basis. J. Chem. Phys. 104, 9473–9481. ( 10.1063/1.471690) [DOI] [Google Scholar]
- 131.Bredas JL, Calbert JP, da Silva DA, Cornil J. 2002. Organic semiconductors: a theoretical characterization of the basic parameters governing charge transport. Proc. Natl Acad. Sci. USA 99, 5804–5809. ( 10.1073/pnas.092143399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hoeben FJM, Jonkheijm P, Meijer EW, Schenning APHJ. 2005. About supramolecular assemblies of π-conjugated systems. Chem. Rev. 105, 1491–1546. ( 10.1021/cr030070z) [DOI] [PubMed] [Google Scholar]
- 133.Gajdos F, Oberhofer H, Dupuis M, Blumberger J. 2013. On the inapplicability of electron-hopping models for the organic semiconductor phenyl-C61-butyric acid methyl ester (PCBM). J. Phys. Chem. Lett. 4, 1012–1017. ( 10.1021/jz400227c) [DOI] [PubMed] [Google Scholar]
- 134.McMahon DP, Troisi A. 2010. Organic semiconductors: impact of disorder at different timescales. Chem. Phys. Chem 11, 2067–2074. ( 10.1002/cphc.201000182) [DOI] [PubMed] [Google Scholar]
- 135.Troisi A. 2011. The speed limit for sequential charge hopping in molecular materials. Org. Electron. 12, 1988–1991. ( 10.1016/j.orgel.2011.08.020) [DOI] [Google Scholar]