

Crystals of the V. cholerae HigBA2 complex and of the C-terminal domain of HigA2 diffracted to 2.2 and 1.8 Å resolution, respectively.
Keywords: toxin–antitoxin modules, ribonucleases, intrinsic disorder, macromolecular complexes, persistence
Abstract
The genome of Vibrio cholerae encodes two higBA toxin–antitoxin (TA) modules that are activated by amino-acid starvation. Here, the TA complex of the second module, higBA2, as well as the C-terminal domain of the corresponding HigA2 antitoxin, have been purified and crystallized. The HigBA2 complex crystallized in two crystal forms. Crystals of form I belonged to space group P21212, with unit-cell parameters a = 129.0, b = 119.8, c = 33.4 Å, and diffracted to 3.0 Å resolution. The asymmetric unit is likely to contain a single complex consisting of two toxin monomers and one antitoxin dimer. The second crystal form crystallized in space group P3221, with unit-cell parameters a = 134.5, c = 55.4 Å. These crystals diffracted to 2.2 Å resolution and probably contain a complex with a different stoichiometry. Crystals of the C-terminal domain of HigA2 belonged to space group C2, with unit-cell parameters a = 115.4, b = 61.2, c = 73.8 Å, β = 106.7°, and diffracted to 1.8 Å resolution.
1. Introduction
Toxin–antitoxin (TA) modules are stress-response units that are found in almost all free-living bacteria and in some archaea (for reviews, see Gerdes et al., 2005 ▶; Buts et al., 2005 ▶; Yamaguchi et al., 2011 ▶). They were originally discovered on plasmids, where they are associated with plasmid maintenance through post-segregational killing (Gerdes et al., 1986 ▶). TA modules are widespread in bacterial and archaeal chromosomes, where a variety of TA families can be present in multiple members per chromosome (Makarova et al., 2009 ▶; Leplae et al., 2011 ▶; Sevin & Barloy-Hubler, 2007 ▶; Pandey & Gerdes, 2005 ▶). The physiological role of chromosomal TA modules is still a matter of extensive research. However, it has now been recognized that TA modules are strongly involved in stress response and probably promote cell survival in unfavourable growth conditions such as nutrient depletion or antibiotic and hypoxic stress (Gerdes et al., 2005 ▶; Yamaguchi et al., 2011 ▶). For several TA modules a direct link to cell dormancy, persistence and biofilm formation has been shown (Tripathi et al., 2012 ▶; Maisonneuve et al., 2011 ▶; González-Barrios et al., 2006 ▶; Keren et al., 2004 ▶).
A typical TA module consists of an operon where a gene encoding a ‘toxin’ is preceded by a gene encoding a neutralizing ‘antitoxin’. Antitoxins usually have a modular structure consisting of an intrinsically disordered toxin-neutralizing domain and a globular DNA-binding and dimerization domain (Li et al., 2008 ▶; Oberer et al., 2007 ▶; Madl et al., 2006 ▶; Loris et al., 2003 ▶). The intrinsically disordered domain is observed to fold upon binding its cognate toxin (Kamada et al., 2003 ▶; Kamada & Hanaoka, 2005 ▶; Garcia-Pino et al., 2008 ▶, 2010 ▶; Li et al., 2009 ▶; De Jonge et al., 2009 ▶; Brown et al., 2009 ▶; Drobnak et al., 2013 ▶), resulting in a nontoxic complex. Another important functional feature of this intrinsically disordered domain is its high susceptibility to proteolytic degradation, which results in shorter half-lives of the antitoxins compared with their toxin counterparts (Lehnherr & Yarmolinsky, 1995 ▶; Van Melderen et al., 1996 ▶; Camacho et al., 2002 ▶).
The higBA (host inhibition of growth) module was first discovered on plasmid Rts1 and has a gene organization that is inverted compared with most other TA operons (Tian et al., 1996 ▶) but that is shared with mqsRA (Yamaguchi et al., 2009 ▶; Kasari et al., 2010 ▶) and hicAB modules (Jørgensen et al., 2009 ▶). Two higBA modules (termed higBA1 and higBA2 hereafter; see §2 for exact definitions) that are activated by amino-acid starvation are found in a large integron on the Vibrio cholerae chromosome II (Christensen-Dalsgaard & Gerdes, 2006 ▶; Christensen-Dalsgaard et al., 2010 ▶). Both the higBA1 and the higBA2 modules from V. cholerae efficiently stabilize test plasmids in Escherichia coli (Christensen-Dalsgaard et al., 2010 ▶), giving support to a hypothesis in which TA modules contribute to the genetic stability of the V. cholerae integron.
Based on sequence similarities, HigB toxins belong to the RelE superfamily, which further includes the YoeB, YafQ and YhaV toxins (Anantharaman & Aravind, 2003 ▶). This group of toxins form part of a larger group of microbial endoribonucleases that also includes barnase (Mauguen et al., 1982 ▶), RNase T1 (Heinemann & Saenger, 1982 ▶) and restrictocin (Yang & Moffat, 1996 ▶). Chromosomal and plasmid-borne HigB toxins have been shown to stall protein translation in V. cholerae and E. coli (Budde et al., 2007 ▶; Christensen-Dalsgaard & Gerdes, 2006 ▶; Christensen-Dalsgaard et al., 2010 ▶). HigB associates with ribosomes and is a ribosome-dependent endoribonuclease with a cleavage pattern similar to that of RelE (Hurley & Woychik, 2009 ▶; Christensen-Dalsgaard et al., 2010 ▶).
The C-terminal domain of HigA antitoxins is predicted to belong to the cro-type helix–turn–helix (HTH) family of transcription factors. Interestingly, in most other cases DNA-binding domains are found at the N-termini of antitoxins. HigA1 from V. cholerae acts as an autorepressor in vivo (Budde et al., 2007 ▶). Whether the HigB toxin is a co-repressor or not is currently unclear.
In this paper, we report the purification, crystallization and preliminary characterization by small-angle X-ray scattering and X-ray crystallography of HigBA2, the complex formed by the toxin and antitoxin genes (gene names VCA0468 and VCA0469) encoded by the second higBA operon from V. cholerae, as well as of the C-terminal domain of the HigA2 antitoxin. This makes V. cholerae higBA2 the first member of the higBA family for which structural information will become available for the full TA complex.
2. Materials and methods
2.1. Nomenclature
In this paper, the term higBA corresponds to any operon encoding members of the HigA and HigB protein family. When referring to the two V. cholerae higBA modules, they are always termed higBA1 and higBA2, respectively, as defined by Christensen-Dalsgaard & Gerdes (2006 ▶). Their corresponding toxins and antitoxins are termed HigB1 and HigA1 and HigB2 and HigA2, respectively. The corresponding complexes are referred to as HigBA1 and HigBA2, respectively.
2.2. Expression and purification of the HigBA2 complex
The higBA2 locus (gene names VCA0468 and VCA0469) of V. cholerae strain N16961 (Heidelberg et al., 2000 ▶) was amplified from chromosomal DNA with the primers H6_MBP_NdeI_higBA2-f (CCCCCATATGAAAAGTGTATTTGTCGAATCAAC) and higA2-down3 (CCCCCGGATCCGTTATAGCTCGGCTATGTGTG). The PCR product was digested with NdeI and BamHI and inserted into pOPTHM, resulting in plasmid pMCD103. Once the fragment had been cloned, its sequence was confirmed by dye dideoxynucleotide sequencing. The fragment containing higBA2 was then isolated from pMCD103 by digestion with NdeI and BamHI and ligated into plasmid pET15b+ (Novagen) also digested with NdeI and BamHI. In this construct, a His tag followed by a thrombin cleavage site (with sequence MGSSHHHHHHSSGLVPRGSH) is placed at the N-terminus of HigB2, which otherwise consists of its full-length wild-type sequence including the N-terminal methionine. HigA2 corresponds to the full-length wild-type protein without any modifications or tags (Supplementary Fig. S11).
The construct was transformed into E. coli BL21 (DE3) cells using the calcium chloride method. Cell cultures were grown in LB medium supplemented with ampicillin (100 mg l−1) at 310 K with aeration. Expression of the complex was induced by adding 1 mM IPTG when the OD600 nm reached 0.6. 4 h post-induction, the cells were harvested by centrifugation and resuspended in lysis buffer containing protease inhibitors (200 mM NaCl, 50 mM Tris–HCl pH 8.0, 0.1 g l−1 AEBSF, 1 mg l−1 leupeptin, 1 mM EDTA). The cells were lysed using a cell cracker and the lysate was centrifuged (40 min at 25 000g) to remove cell debris.
The supernatant was loaded onto a 5 ml Ni–NTA column (Qiagen) equilibrated with 500 mM NaCl, 50 mM Tris–HCl pH 8.0. The column was washed with five column volumes of 1 M NaCl, 10% ethylene glycol, 50 mM Tris–HCl pH 8.0 to elute nonspecifically bound proteins. The HigBA2 complex was eluted with a linear gradient of imidazole (0.0–1.0 M in ten column volumes; 50 ml) in 200 mM NaCl, 50 mM Tris–HCl pH 8.0. Elution of the complex was observed at a concentration of 250 mM imidazole. The fractions containing the HigBA2 complex were pooled, concentrated and loaded onto a Superdex 75 HR gel-filtration column equilibrated with 200 mM NaCl, 20 mM Tris–HCl pH 8.0. The purity of the complex was analyzed by SDS–PAGE.
2.3. Purification of the antitoxin HigA2 from the HigBA2 complex
The HigBA2 complex from the cell lysate was bound to an Ni–NTA column and the column was subsequently washed to elute nonspecifically bound impurities as described above. The column was then washed with 5 M guanidine–HCl, 0.5 M NaCl, 50 mM Tris–HCl pH 8.0 to disrupt the HigBA2 complex. Surprisingly, little if any protein was eluted at this point. Column-bound proteins were refolded by washing the column with 5% glycerol, 25 mM NaCl, 25 mM Tris–HCl pH 8.0 followed by the same buffer with a lower (1%) glycerol concentration. Finally, proteins were eluted using a linear gradient of imidazole (0.0–1.0 M in ten column volumes; 50 ml) in 200 mM NaCl, 50 mM Tris–HCl pH 8.0. This resulted in three separate peaks corresponding to antitoxin, toxin and some non-separated complex. Each of these peaks was further purified on a Superdex 75 HR gel-filtration column in 200 mM NaCl, 20 mM Tris–HCl pH 8.0 buffer. The purity of the proteins was analyzed by SDS–PAGE. The identities of the toxin and antitoxin proteins in the bands that migrated with the expected molecular weights were further confirmed by N-terminal sequencing of the first five residues (performed by AltaBioscience, Birmingham, England). Toxin (HigB2) and antitoxin (HigA2) samples were both concentrated to 5 mg ml−1 in 200 mM NaCl, 20 mM Tris–HCl pH 8.0.
2.4. Small-angle X-ray scattering (SAXS)
SAXS experiments were performed during two sessions on the SWING beamline at the SOLEIL synchrotron, Gif-sur-Yvette, France in the HPLC mode (David & Pérez, 2009 ▶). The HigBA2 complex was concentrated to 10 mg ml−1 in 50 mM HEPES pH 7.5, 100 mM NaCl, while the HigB2 toxin and the HigA2 antitoxin were concentrated to 5 mg ml−1 in 20 mM Tris pH 8, 200 mM NaCl. In each case, 80 µl protein sample was injected into a Shodex KW404-4F column which had been pre-equilibrated with the same buffer as used for the protein samples. Data were measured for 500 ms at 1 s intervals, with buffer data collected at the beginning of the chromatogram and data for the sample collected during peak elution, which enables the acquisition of data at different protein concentrations. The data were processed and analysed using the ATSAS package (Konarev et al., 2006 ▶). The SAXS MoW application was used for estimation of the molecular weight of the proteins and protein complexes (Fischer et al., 2010 ▶).
2.5. Crystallization
Crystallization conditions were screened by hanging-drop vapour diffusion at 293 K for all protein samples (toxin, antitoxin and complex). Hanging drops consisting of 1 µl protein solution and 1 µl precipitant solution were equilibrated against 110 µl precipitant solution in the reservoir. Crystallization conditions were tested using various commercially available screens: Crystal Screen and Crystal Screen 2 (Hampton Research), Morpheus, PACT premier and ProPlex (Molecular Dimensions) and Jena Classic (Jena Bioscience). The HigBA2 complex was used at 10 or 15 mg ml−1 in 200 mM NaCl, 20 mM Tris–HCl pH 8.0, while both the antitoxin HigA2 and the toxin HigB were used at 5 mg ml−1 in 200 mM NaCl, 20 mM Tris–HCl pH 8.0. The concentrations of the protein solutions were determined spectrophotometrically from the absorbance at 280 nm using extinction coefficients obtained from the method introduced by Gill & von Hippel (1989 ▶). For the HigBA2 complex an extinction coefficient of 42 860 M −1 cm−1 was used, thereby assuming a 2:2 stoichiometry. In the case of HigA2, the extinction coefficient used of 11 000 M −1 cm−1 corresponds to an antitoxin dimer. These assumptions are based on molecular-weight estimates derived from analytical gel-filtration (data not shown) and SAXS experiments (see below).
Crystals of the HigBA2 complex were optimized using the microseeding method. 1 µl of the hanging-drop solution containing small crystals was diluted in 50 µl precipitant solution, vortexed and serially diluted (tenfold steps) to a maximum dilution of 10−7. Next, 1 µl of these seed dilutions was mixed with 1 µl protein solution and equilibrated against mother liquor.
2.6. Data collection and analysis
All data were measured on the PROXIMA1 beamline at the SOLEIL synchrotron (Gif-sur-Yvette, France) using a PILATUS 6M detector. Crystals were vitrified in liquid nitrogen by a brief soak in the cryosolutions listed in Table 1 ▶. For HigBA2 crystal form I, X-ray diffraction data were measured using a wavelength of 0.9790 Å with a crystal-to-detector distance of 679.6 mm. By rotating the crystal through 180° in 0.2° increments, a total of 900 images were collected. Data for the second crystal form of the HigBA2 complex and for the HigA2 antitoxin crystals were collected in the same session using a wavelength of 0.9801 Å and a crystal-to-detector distance of 368.8 mm. These crystals were rotated through 120° in 0.2° increments and 600 diffraction images were collected in both cases. All data were indexed, integrated and scaled with XDS (Kabsch, 2010 ▶). Analysis of the unit-cell contents and calculation of Matthews coefficients (Matthews, 1968 ▶; Kantardjieff & Rupp, 2003 ▶) was performed with the program MATTHEWS_COEF, which is part of the CCP4 package (Winn et al., 2011 ▶).
Table 1. Crystallization conditions.
Crystals of the same crystal form but obtained from different conditions were generally of similar quality. The ‘best’ data are represented in Table 2 ▶.
| Protein solution | Reservoir solution | Temperature (K) | Cryoprotection | |
|---|---|---|---|---|
| HigBA2 form I | 10 mg ml−1 in 20 mM Tris pH 8, 200 mM NaCl | 100 mM bicine/Tris pH 8.5, 12.5%(w/v) PEG 1000, 12.5%(w/v) PEG 3350, 12.5%(w/w) MPD, 30 mM NaF, 30 mM NaBr, 30 mM NaI | 293 | None |
| 10 mg ml−1 in 20 mM Tris pH 8, 200 mM NaCl | 20%(v/v) ethanol, 100 mM Tris–HCl pH 8.5 | 293 | 35% glycerol | |
| 10 mg ml−1 in 20 mM Tris pH 8, 200 mM NaCl | 2.0 M ammonium phosphate monobasic, 100 mM Tris–HCl pH 8.5 | 293 | 25% glycerol | |
| 15 mg ml−1 in 50 mM Tris pH 7.5, 150 mM NaCl | 50 mM Tris–HCl pH 8.5, 200 mM magnesium acetate, 22–28%(w/v) PEG 8000 | 293 | Increase PEG to 35% | |
| HigBA2 form II | 10 mg ml−1 in 20 mM Tris pH 8, 200 mM NaCl | 200 mM ammonium sulfate, 100 mM sodium cacodylate trihydrate pH 6.5, 30%(w/v) PEG 8000 | 293 | Increase PEG to 40% |
| 10 mg ml−1 in 20 mM Tris pH 8, 200 mM NaCl | 2.0 M ammonium sulfate, 2%(w/v) PEG 400, 100 mM HEPES pH 7.5 | 293 | 20% glycerol | |
| HigA2 C-terminal domain† | 5 mg ml−1 in 20 mM Tris pH 8, 200 mM NaCl | 100 mM Tris–HCl pH 8.5, 200 mM calcium chloride, 20%(w/v) PEG 4000 | 293 | Increase PEG to 36.6% |
| 5 mg ml−1 in 20 mM Tris pH 8, 200 mM NaCl | 20%(w/v) PEG 3350, 200 mM potassium thiocyanate, 100 mM bis-tris propane | 277 | 10% glycerol | |
| 5 mg ml−1 in 20 mM Tris pH 8, 200 mM NaCl | 30%(w/v) PEG 4000, 100 mM Tris pH 8.5, 200 mM sodium acetate trihydrate | 293 | Increase PEG to 40% |
The protein concentration corresponds to the full-length protein.
2.7. Mass spectrometry
The protein contents of the different crystals were analyzed by mass spectrometry. Crystals were first washed three times with reservoir solution and once with distilled water. The washed crystals were then dissolved in distilled water (approximately 300 pmol protein per 100 µl) and these samples were further desalted on a C18 spin column (Perbio Science). Desalted samples were analyzed by ESI ion trap mass spectrometry (LTQ XL, Thermo Scientific) using direct infusion and the minimal ion source energy (ion surface-induced dissociation) to promote the efficient declustering of water molecules and salt adducts. Raw data were processed using the ProMass software (Thermo Scientific) to obtain deconvoluted spectra.
3. Results and discussion
The HigBA2 complex was purified to homogeneity from the cell lysate as shown in Fig. 1 ▶. After disruption of the complex with high concentrations of denaturants, the toxin HigB2 and the antitoxin HigA2 were separated and successfully refolded on the column based on SDS–PAGE analysis and circular-dichroism (CD) spectroscopy (Fig. 1 ▶; CD spectra for the HigB2 toxin and the HigA2 antitoxin are shown in Supplementary Fig. S2). Secondary-structure prediction based on the CD spectra using the K2D2 server (Perez-Iratxeta & Andrade-Navarro, 2008 ▶) suggested the presence of 12% α-helix and 28% β-strand for HigB2, while HigA2 was predicted to contain 50% α-helix and 7% β-strand. The hydrodynamic properties of the purified proteins and their complex were characterized with SAXS (Fig. 2 ▶). The radius of gyration was estimated by Guinier analysis of the scattering curve at low angles (Guinier, 1939 ▶). For all three proteins the Guinier plots were linear and the calculated radii of gyration were concentration-independent, indicating the absence of aggregation in the samples. The antitoxin HigA2 has a radius of gyration of 23.9 Å, and its estimated molecular weight of 27 kDa suggests that it is a dimer in solution (the expected molecular weight for a dimer is 23.4 kDa). This was expected since the HigA2 antitoxin has an HTH DNA-binding domain which is known to bind operator DNA as a dimer. The toxin HigB2 has a smaller radius of gyration of 19 Å and the molecular-weight estimate of 15.2 kDa corresponds almost exactly to the theoretical value for a monomer. The Kratky plots for both the antitoxin and the toxin show a prominent parabola followed by a linear increase at higher angles (Figs. 2 ▶ a and 2 ▶ b). This suggests that both proteins are globular with some flexibility present. In the case of HigB2 this flexibility is likely to reflect the presence of an N-terminal His tag (20 residues in total). The flexibility in the antitoxin HigA2, on the other hand, is expected to result from the intrinsically disordered region of its toxin-neutralizing N-terminal domain.
Figure 1.

Expression and purification of proteins encoded by the higBA2 module. Lane 1, molecular-weight standards (labelled in kDa). Lane 2, soluble fraction before induction. Lane 3, soluble fraction after 4 h of induction with IPTG. Lane 4, purified HigBA2 complex. Lane 5, purified HigB2 toxin. Lane 6, purified HigA2 antitoxin.
Figure 2.

SAXS curves for the HigA2 antitoxin (a), the HigB2 toxin (b) and the HigBA2 complex (c). The experimental data are drawn in black and the error margins in grey. The insets show the corresponding Kratky plots.
The HigBA2 complex has a radius of gyration of 31.2 Å with a molecular-weight estimate of 59 kDa. It is most likely that this complex is a heterotetramer formed by the association of one antitoxin dimer with two toxin monomers, which gives a theoretical molecular weight of 53 kDa. Kratky analysis of the complex was not performed owing to the low signal-to-noise ratio at higher scattering angles in this data set.
For the three samples screened, crystals could be obtained for the HigBA2 complex and for the HigA2 antitoxin. No crystals have been obtained to date for the HigB2 toxin. The first crystal form of the HigBA2 complex appeared after one week in several conditions, as listed in Table 1 ▶. The crystals initially appeared as short needles and could be further optimized by microseeding to obtain large needle-shaped crystals (Fig. 3 ▶ a). These crystals consistently diffracted to between 3.1 and 3.0 Å resolution (Fig. 4 ▶ a) and belonged to space group P21212, with unit-cell parameters a = 129.0, b = 119.8, c = 33.4 Å. Data-collection statistics are given in Table 2 ▶. The calculation of Matthews coefficients further supported the presence of one complex with stoichiometry HigA22:HigB22 in the asymmetric unit (V M = 2.43 Å3 Da−1, corresponding to 49% solvent content). SDS–PAGE analysis of these crystals revealed that they contained both toxin and antitoxin (Fig. 3 ▶ b, lane 1). This was further confirmed by mass-spectrometric analysis, where the determined experimental masses corresponded to the full-length HigA2 and HigB2 proteins (Fig. 5 ▶ a).
Figure 3.

Crystals of HigBA2 and HigA2. (a) Crystals of form I of the HigBA2 complex obtained after microseeding. (b) Zinc-stained SDS–PAGE of redissolved crystals (lane 1, form I crystals of HigBA2; lane 2, form II crystals of HigBA2; lane 3, crystals of HigA2; leftmost lane, molecular-mass markers labelled in kDa). The green arrows indicate bands corresponding to HigB2 (upper) and HigA2 (lower). The red arrows correspond to likely degradation products. The putative HigB2 degradation product is also visible in the mass spectrum shown in Fig. 5 ▶(b) as a peak corresponding to a mass of 13 897 Da. (c) Crystals of form II of the HigBA2 complex. (d) Crystals of the C-terminal domain of the antitoxin. The black bar corresponds to 0.1 mm and the scale is identical in (a), (c) and (d).
Figure 4.

Typical diffraction patterns of HigBA2 crystal form I (a), HigBA2 crystal form II (b) and crystals of the C-terminal domain of HigA2 (c). The insets show a magnification of the boxed parts of the diffraction images with rings that indicate the resolution limit for each crystal form.
Table 2. Data-collection statistics.
Values in parentheses are for the outermost resolution shell.
| HigBA2 form I | HigBA2 form II | HigA2 C-terminal domain | |
|---|---|---|---|
| Crystallization condition | 100 mM bicine/Tris pH 8.5, 12.5%(w/v) PEG 1000, 12.5%(w/v) PEG 3350, 12.5%(w/w) MPD, 30 mM NaF, 30 mM NaBr, 30 mM NaI | 200 mM ammonium sulfate, 100 mM sodium cacodylate trihydrate pH 6.5, 30%(w/v) PEG 8000 | 100 mM Tris–HCl pH 8.5, 200 mM calcium chloride, 20%(w/v) PEG 4000 |
| Resolution range (Å) | 40.5–3.0 (3.10–3.00) | 40.1–2.2 (2.48–2.19) | 38.5–1.8 (1.91–1.80) |
| Unit-cell parameters | a = 129.0, b = 119.8, c = 33.4 | a = 134.5, c = 55.4 | a = 115.4, b = 61.2, c = 73.9, β = 106.7 |
| Space group | P21212 | P3221 | C2 |
| Mosaicity (°) | 0.075 | 0.092 | 0.560 |
| R merge † | 0.078 (0.691) | 0.114 (1.63) | 0.053 (0.589) |
| No. of measured reflections | 41100 (6495) | 206547 (36513) | 179186 (21013) |
| No. of unique reflections | 10916 (1010) | 25120 (4612) | 44925 (6741) |
| Multiplicity | 3.8 (6.4) | 8.2 (7.9) | 4.0 (3.1) |
| 〈I/σ(I)〉 | 10.88 (1.64) | 12.51 (1.25) | 12.90 (2.30) |
| Completeness (%) | 98.5 (94.9) | 99.3 (95.8) | 97.5 (91.4) |
R
merge = 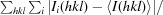
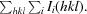
Figure 5.

Mass-spectrometric analysis of crystal contents. (a) ESI ion trap mass spectrum of crystal form I of the HigBA2 complex. A first major peak at 11 566 Da corresponds to the mass of the HigA2 antitoxin within 1 Da experimental error. The second major peak corresponds to a mass of 15 039 Da, again within 1 Da experimental error of the theoretical mass of the HigB2 toxin. (b) Identical ESI ion trap mass spectrum for crystal form II. Note the different ratio of peak intensities suggesting a different stoichiometry of the complex and the presence of a peak at 13 897 Da corresponding to a putative HigB2 degradation product.
A second crystal form of HigBA2 was observed after approximately eight months in a condition consisting of 200 mM ammonium sulfate, 100 mM sodium cacodylate trihydrate pH 6.5, 30%(w/v) PEG 8000 (Fig. 3 ▶ c). These crystals belonged to space group P3221, with unit-cell parameters a = 134.5, c = 55.4 Å, and diffracted to 2.2 Å resolution (Fig. 4 ▶ b). Data-collection statistics are given in Table 2 ▶. Analysis of the crystal content by SDS–PAGE shows multiple bands corresponding to the toxin, the antitoxin, a possible toxin-degradation product and an unknown contaminant with higher molecular weight (Fig. 3 ▶ b, lane 2). Mass-spectrometric analysis of these crystals confirmed these results and showed the presence of mainly full-length toxin and antitoxin (Fig. 5 ▶ b). A smaller peak at approximately 13.9 kDa is likely to correspond to the toxin-degradation product, which lacks eight amino-acid residues at the C-terminus. In contrast to the mass spectrum of crystal form I, the intensity of the antitoxin peak in crystal form II is much higher than the corresponding intensity of the toxin peak. This suggests that this crystal form might contain complexes with different stoichiometry, likely HigA22:HigB2. It is possible that the toxin is partially degraded during the long period of crystal growth (Figs. 3 ▶ b and 5 ▶ b), thereby enabling a complex with a higher antitoxin:toxin ratio to crystallize. The calculation of Matthews coefficients assuming a HigA22:HigB2 stoichiometry shows that either one (V M = 3.81 Å3 Da−1, corresponding to 67.7% solvent content) or two (V M = 1.90 Å3 Da−1, corresponding to 35.4% solvent content) such complexes can be accommodated in the asymmetric unit.
The crystallization of antitoxins is often nontrivial because of their intrinsically disordered region that is involved in toxin neutralization. Rhomboid-shaped crystals of HigA2 appeared in 100 mM Tris–HCl pH 8.5, 200 mM calcium chloride, 20%(w/v) PEG 4000 after a few months (Fig. 3 ▶ d). The crystals belonged to space group C2, with unit-cell parameters a = 115.4, b = 61.2, c = 73.8 Å, β = 106.7°, and diffracted to 1.8 Å resolution (Fig. 4 ▶ c). Data-collection statistics are given in Table 2 ▶. SDS–PAGE analysis of redissolved crystals showed them to contain a degradation product of around 10 kDa (Fig. 3 ▶ b), which is likely to correspond to the C-terminal HTH domain, as the N-terminal domain is predicted to be intrinsically disordered and therefore very unlikely to crystallize.
There has been continued interest in the structural biology of toxin–antitoxin modules in recent years, and structures of the complexes of most major type II TA modules have become available (Bøggild et al., 2012 ▶; Brown et al., 2009 ▶; Dalton & Crosson, 2010 ▶; De Jonge et al., 2009 ▶; Kamada et al., 2003 ▶; Kamada & Hanaoka, 2005 ▶; Khoo et al., 2007 ▶; Li et al., 2009 ▶; Maté et al., 2012 ▶; Mattison et al., 2006 ▶; Meinhart et al., 2003 ▶; Schumacher et al., 2009 ▶; Takagi et al., 2005 ▶). A notable exception here is the higBA family, for which only the structure of the HigA antitoxin from Coxiella burnetii CBU_1490 (PDB entry 3trb; 15% sequence identity to V. cholerae HigA2; J. Cheung, M. Franklin, M. Rudolph, M. Cassidy, E. Gary, F. Burshteyn & J. Love, unpublished work) is known. Interestingly, the HTH DNA-binding domain of HigA from C. burnetii is at the N-terminus of the protein and not at the C-terminus as in HigA2 from V. cholerae and other HigA members. This is also observed in the structure from E. coli CFT073 (PDB entry 2ict; Arbing et al., 2010 ▶) that was first identified as HigA and later reassigned as YddM from the VapA/VapI family. A search for similar structures in the Protein Data Bank also identifies the uncharacterized HTH-type transcriptional regulator YbaQ (PDB entry 2eby; RIKEN Structural Genomics/Proteomics Initiative, unpublished work) and a putative antidote protein of a plasmid-maintenance system (PDB entry 3cec; Joint Center for Structural Genomics, unpublished work). All of these structures are similar to each other but differ from HigA2 from V. cholerae in the location of the HTH DNA-binding domain at the N-terminus, which is followed by a long α-helix. It is very likely that these structures all belong to the VapA/VapI family of antitoxins. Thus, V. cholerae HigA2 remains the first true HigA family member for which structural information has become available.
The crystallization of the V. cholerae HigBA complex constitutes an important step forward towards the understanding of this important yet understudied TA family. As neither HigA2 nor HigB2 share sufficient sequence identity with other proteins present in the Protein Data Bank to attempt molecular replacement, the structures will be determined by SAD/MAD using crystals of selenomethionine-incorporated proteins.
Supplementary Material
Supplementary material file. DOI: 10.1107/S1744309113021490/pg5021sup1.pdf
Acknowledgments
This work was supported by grants from FWO-Vlaanderen, OZR-VUB, the Hercules Foundation, VIB and the Agency for Research of the Republic of Slovenia. SH acknowledges the receipt of a doctoral scholarship from OZR-VUB. AGP received a post-doctoral fellowship from FWO. The authors thank Andrew Thompson for beamline support. We are grateful to Kristina Dešman for assistance in manuscript preparation.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: PG5021).
References
- Anantharaman, V. & Aravind, L. (2003). Genome Biol. 4, R81. [DOI] [PMC free article] [PubMed]
- Arbing, M. A. et al. (2010). Structure, 18, 996–1010. [DOI] [PMC free article] [PubMed]
- Bøggild, A., Sofos, N., Andersen, K. R., Feddersen, A., Easter, A. D., Passmore, L. A. & Brodersen, D. E. (2012). Structure, 20, 1641–1648. [DOI] [PMC free article] [PubMed]
- Brown, B. L., Grigoriu, S., Kim, Y., Arruda, J. M., Davenport, A., Wood, T. K., Peti, W. & Page, R. (2009). PLoS Pathog. 5, e1000706. [DOI] [PMC free article] [PubMed]
- Budde, P. P., Davis, B. M., Yuan, J. & Waldor, M. K. (2007). J. Bacteriol. 189, 491–500. [DOI] [PMC free article] [PubMed]
- Buts, L., Lah, J., Dao-Thi, M.-H., Wyns, L. & Loris, R. (2005). Trends Biochem. Sci. 30, 672–679. [DOI] [PubMed]
- Camacho, A. G., Misselwitz, R., Behlke, J., Ayora, S., Welfle, K., Meinhart, A., Lara, B., Saenger, W., Welfle, H. & Alonso, J. C. (2002). Biol. Chem. 383, 1701–1713. [DOI] [PubMed]
- Christensen-Dalsgaard, M. & Gerdes, K. (2006). Mol. Microbiol. 62, 397–411. [DOI] [PubMed]
- Christensen-Dalsgaard, M., Jørgensen, M. G. & Gerdes, K. (2010). Mol. Microbiol. 75, 333–348. [DOI] [PMC free article] [PubMed]
- Dalton, K. M. & Crosson, S. (2010). Biochemistry, 49, 2205–2215. [DOI] [PMC free article] [PubMed]
- David, G. & Pérez, J. (2009). J. Appl. Cryst. 42, 892–900.
- De Jonge, N., Garcia-Pino, A., Buts, L., Haesaerts, S., Charlier, D., Zangger, K., Wyns, L., De Greve, H. & Loris, R. (2009). Mol. Cell, 35, 154–163. [DOI] [PubMed]
- Drobnak, I., De Jonge, N., Haesaerts, S., Vesnaver, G., Loris, R. & Lah, J. (2013). J. Am. Chem. Soc. 135, 1288–1294. [DOI] [PubMed]
- Fischer, H., de Oliveira Neto, M., Napolitano, H. B., Polikarpov, I. & Craievich, A. F. (2010). J. Appl. Cryst. 43, 101–109.
- Garcia-Pino, A., Balasubramanian, S., Wyns, L., Gazit, E., De Greve, H., Magnuson, R. D., Charlier, D., van Nuland, N. A. & Loris, R. (2010). Cell, 142, 101–111. [DOI] [PubMed]
- Garcia-Pino, A., Christensen-Dalsgaard, M., Wyns, L., Yarmolinsky, M., Magnuson, R. D., Gerdes, K. & Loris, R. (2008). J. Biol. Chem. 283, 30821–30827. [DOI] [PMC free article] [PubMed]
- Gerdes, K., Christensen, S. K. & Løbner-Olesen, A. (2005). Nature Rev. Microbiol. 3, 371–382. [DOI] [PubMed]
- Gerdes, K., Rasmussen, P. B. & Molin, S. (1986). Proc. Natl Acad. Sci. USA, 83, 3116–3120. [DOI] [PMC free article] [PubMed]
- Gill, S. C. & von Hippel, P. H. (1989). Anal. Biochem. 182, 319–326. [DOI] [PubMed]
- González Barrios, A. F., Zuo, R., Hashimoto, Y., Yang, L., Bentley, W. E. & Wood, T. K. (2006). J. Bacteriol. 188, 305–316. [DOI] [PMC free article] [PubMed]
- Guinier, A. (1939). Ann. Phys. 12, 161–237.
- Heidelberg, J. F. et al. (2000). Nature (London), 406, 477–483. [DOI] [PMC free article] [PubMed]
- Heinemann, U. & Saenger, W. (1982). Nature (London), 299, 27–31. [DOI] [PubMed]
- Hurley, J. M. & Woychik, N. A. (2009). J. Biol. Chem. 284, 18605–18613. [DOI] [PMC free article] [PubMed]
- Jørgensen, M. G., Pandey, D. P., Jaskolska, M. & Gerdes, K. (2009). J. Bacteriol. 191, 1191–1199. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kamada, K. & Hanaoka, F. (2005). Mol. Cell, 19, 497–509. [DOI] [PubMed]
- Kamada, K., Hanaoka, F. & Burley, S. K. (2003). Mol. Cell, 11, 875–884. [DOI] [PubMed]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci. 12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Kasari, V., Kurg, K., Margus, T., Tenson, T. & Kaldalu, N. (2010). J. Bacteriol. 192, 2908–2919. [DOI] [PMC free article] [PubMed]
- Keren, I., Shah, D., Spoering, A., Kaldalu, N. & Lewis, K. (2004). J. Bacteriol. 186, 8172–8180. [DOI] [PMC free article] [PubMed]
- Khoo, S. K., Loll, B., Chan, W. T., Shoeman, R. L., Ngoo, L., Yeo, C. C. & Meinhart, A. (2007). J. Biol. Chem. 282, 19606–19618. [DOI] [PubMed]
- Konarev, P. V., Petoukhov, M. V., Volkov, V. V. & Svergun, D. I. (2006). J. Appl. Cryst. 39, 277–286.
- Lehnherr, H. & Yarmolinsky, M. B. (1995). Proc. Natl Acad. Sci. USA, 92, 3274–3277. [DOI] [PMC free article] [PubMed]
- Leplae, R., Geeraerts, D., Hallez, R., Guglielmini, J., Drèze, P. & Van Melderen, L. (2011). Nucleic Acids Res. 39, 5513–5525. [DOI] [PMC free article] [PubMed]
- Li, G.-Y., Zhang, Y., Inouye, M. & Ikura, M. (2008). J. Mol. Biol. 380, 107–119. [DOI] [PubMed]
- Li, G.-Y., Zhang, Y., Inouye, M. & Ikura, M. (2009). J. Biol. Chem. 284, 14628–14636. [DOI] [PMC free article] [PubMed]
- Loris, R., Marianovsky, I., Lah, J., Laeremans, T., Engelberg-Kulka, H., Glaser, G., Muyldermans, S. & Wyns, L. (2003). J. Biol. Chem. 278, 28252–28257. [DOI] [PubMed]
- Madl, T., Van Melderen, L., Mine, N., Respondek, M., Oberer, M., Keller, W., Khatai, L. & Zangger, K. (2006). J. Mol. Biol. 364, 170–185. [DOI] [PubMed]
- Maisonneuve, E., Shakespeare, L. J., Jørgensen, M. G. & Gerdes, K. (2011). Proc. Natl Acad. Sci. USA, 108, 13206–13211. [DOI] [PMC free article] [PubMed] [Retracted]
- Makarova, K. S., Wolf, Y. I. & Koonin, E. V. (2009). Biol. Direct, 4, 19. [DOI] [PMC free article] [PubMed]
- Maté, M. J., Vincentelli, R., Foos, N., Raoult, D., Cambillau, C. & Ortiz-Lombardía, M. (2012). Nucleic Acids Res. 40, 3245–3258. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Mattison, K., Wilbur, J. S., So, M. & Brennan, R. G. (2006). J. Biol. Chem. 281, 37942–37951. [DOI] [PubMed]
- Mauguen, Y., Hartley, R. W., Dodson, E. J., Dodson, G. G., Bricogne, G., Chothia, C. & Jack, A. (1982). Nature (London), 297, 162–164. [DOI] [PubMed]
- Meinhart, A., Alonso, J. C., Sträter, N. & Saenger, W. (2003). Proc. Natl Acad. Sci. USA, 100, 1661–1666. [DOI] [PMC free article] [PubMed]
- Oberer, M., Zangger, K., Gruber, K. & Keller, W. (2007). Protein Sci. 16, 1676–1688. [DOI] [PMC free article] [PubMed]
- Pandey, D. P. & Gerdes, K. (2005). Nucleic Acids Res. 33, 966–976. [DOI] [PMC free article] [PubMed]
- Perez-Iratxeta, C. & Andrade-Navarro, M. A. (2008). BMC Struct. Biol. 8, 25. [DOI] [PMC free article] [PubMed]
- Schumacher, M. A., Piro, K. M., Xu, W., Hansen, S., Lewis, K. & Brennan, R. G. (2009). Science, 323, 396–401. [DOI] [PMC free article] [PubMed]
- Sevin, E. W. & Barloy-Hubler, F. (2007). Genome Biol. 8, R155. [DOI] [PMC free article] [PubMed]
- Takagi, H., Kakuta, Y., Okada, T., Yao, M., Tanaka, I. & Kimura, M. (2005). Nature Struct. Mol. Biol. 12, 327–331. [DOI] [PubMed]
- Tian, Q. B., Ohnishi, M., Tabuchi, A. & Terawaki, Y. (1996). Biochem. Biophys. Res. Commun. 220, 280–284. [DOI] [PubMed]
- Tripathi, A., Dewan, P. C., Barua, B. & Varadarajan, R. (2012). Proc. Natl Acad. Sci. USA, 109, 12497–12502. [DOI] [PMC free article] [PubMed]
- Van Melderen, L., Dao Thi, M. H., Lecchi, P., Gottesman, S., Couturier, M. & Maurizi, M. R. (1996). J. Biol. Chem. 271, 27730–27738. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yamaguchi, Y., Park, J.-H. & Inouye, M. (2009). J. Biol. Chem. 284, 28746–28753. [DOI] [PMC free article] [PubMed]
- Yamaguchi, Y., Park, J.-H. & Inouye, M. (2011). Annu. Rev. Genet. 45, 61–79. [DOI] [PubMed]
- Yang, X. & Moffat, K. (1996). Structure, 4, 837–852. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material file. DOI: 10.1107/S1744309113021490/pg5021sup1.pdf