

Abstract
Although one dimensional (1D) Pt nanostructures with well-defined sizes and shapes have fascinating physiochemical properties, their preparation remains a great challenge. Here we report an easy and novel synthesis of 1D Pt nanostructures with controllable morphologies, through the combination of designer self-assembling I3K and phage-displayed P7A peptides. The nanofibrils formed via I3K self-assembly acted as template. Pt precursors ((PtCl4)2− and (PtCl6)2−) were immobilized by electrostatic interaction on the positively charged template surface and subsequent reduction led to the formation of 1D Pt nanostructures. P7A was applied to tune the continuity of the Pt nanostructures. Here, the electrostatic repulsion between the deprotonated C-terminal carboxyl groups of P7A molecules was demonstrated to play a key role. We finally showed that continuous and ordered 1D Pt morphology had a significantly improved electrochemical performance for the hydrogen and methanol electro-oxidation in comparison with either 1D discrete Pt nanoparticle assemblies or isolated Pt nanoparticles.
Platinum (Pt) nanoparticles (NPs) are widely employed as electrocatalysts in fuel cells1. However, there are several obstacles associated with isolated nanoparticles, such as a large number of lattice boundaries, surface defect sites, interfaces between NPs and uncertainties associated with migration, aggregation and Ostwald ripening, all of which are detrimental to their catalytic performance and utilization efficiency. To address these issues, one-dimensional (1D) Pt nanostructures have recently been explored, and they exhibited improved catalytic activity, durability, and CO tolerance2,3,4,5,6. In fact, as a connection between nanometer-scale objects and the macro-scale world, the 1D anisotropic nanostructures possess some peculiar structural features and fascinating physicochemical properties, which are usually inaccessible to NPs and bulk materials7,8,9,10. However, the difficulties associated with the preparation of 1D metal nanostructures with well-controlled sizes, shapes, and composition have slowed down their advancement as compared with isolated isotropic nanostructures7,8,9.
Among documented strategies for achieving 1D inorganic growth, template-directed synthesis provides a good control over the dimensions and uniformity8. Due to their structural diversity, specific binding to metal ions, and biodegradability, self-assembled peptide nanostructures have been explored in recent years as promising templates for the fabrication of metal nanostructures11,12,13,14,15,16. For practical applications, an ideal peptide template should (1) be self-assembled from short peptides that can be synthesized on a large scale and (2) be well dispersed in aqueous solution to enable efficient and uniform immobilization of metal NPs onto the template surface under mild conditions10. However, the peptides reported so far for templating 1D Pt nanostructures are rather limited4,6,17,18,19 and more importantly, they are either long peptides (e.g. insulin6) and even viral particles (e.g. tobacco mosaic virus4) or extremely hydrophobic ones that usually contain the core sequences of amyloid peptides (e.g. FF17,18). These peptides have shown poor morphological control over 1D Pt nanostructures. In contrast, de novo design of short peptides offers a range of advantages in the effective control of Pt nanostructure and function. To date, however, little progress has been made towards the development of easy, controllable and cost-effective preparation of 1D Pt nanostructures.
To exploit the main advantages of both easy synthesis and direct control of self-assembled nanostructures of short designed peptides, we recently synthesized an ultrashort surfactant-like peptide I3K (Ac-IIIK-CONH2) that readily self-assembles into long and uniform nanofibrils in aqueous solution, driven by molecular amphiphilicity and β-sheet hydrogen bonding20,21. The I3K nanofibrils show excellent aqueous dispersion at higher concentrations (more than 10 mM) without detectable precipitation after storage in water for several months under ambient conditions, whereas, for example, amyloid fibrils readily precipitate in aqueous solution. In addition we have demonstrated that the I3K assemblies template the formation of 1D silica nanostructures with controllable morphologies and dimensions21,22,23. Based on these I3K nanofibrils, we here report a simple template-directed method to prepare 1D Pt nanostructures, and by coupling another short peptide P7A (TLHVSSY) that serves as a capping agent of the Pt NPs, the morphologies of the 1D Pt nanostructures on the I3K template surface can be finely tuned, with the ability to be switched between ordered and discrete 1D Pt NP superstructures. Our 1D continuous Pt nanostructures exhibited significantly improved electrocatalytic performances over the existing NP based systems.
Results
Synthesis and characterization
As shown schematically in Fig. 1, the route consists of two consecutive steps, peptide self-assembly and the synthesis of the Pt nanostructures on the assembled template. After one-week incubation at 4 mM in water (far above the peptide's critical aggregation concentration), I3K self-assembles into stable fibrils with lengths of several μm and a width of 10.0 ± 1.3 nm (Fig. 2). P7A is also a short peptide but selected through phage display technology, which specifically binds to the Pt crystal surface, showing good stabilization for small Pt NPs under 5 nm (Supplementary Fig. S1a)24,25. To obtain 1D Pt nanostructures, the aged I3K stock solution was mixed firstly with a fresh aqueous solution containing P7A and K2PtCl4, followed by the addition of a fresh NaBH4 solution. The final concentrations of I3K, P7A, K2PtCl4 and NaBH4 in the mixture were 2, 1, 1 and 1 mM, respectively. After reaction for 24 hr at ambient temperature, the mixture became a black suspension. Transmission electron microscopy (TEM) imaging indicated the formation of 1D discrete Pt NP assemblies with a width of 9.4 ± 1.2 nm (Fig. 3a and Supplementary Fig. S1b). The sizes of the assembled Pt NPs within such a 1D superstructure are 1.8 ± 0.3 nm, comparable to those of their free counterparts obtained without the template (Supplementary Fig. S1a). The edge-to-edge distance between the assembled Pt NPs is 0.7 ± 0.2 nm. High-resolution TEM (HRTEM) revealed a lattice spacing of 0.223/0.224 nm, characteristic of the interplane spacing of Pt (111) planes (the bottom panel of Fig. 3a).
Figure 1. Schematic synthesis strategy of the 1D Pt nanostructures.
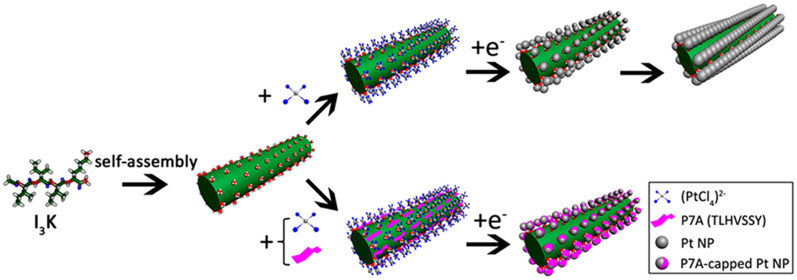
The first step is I3K self-assembly, leading to nanofibril formation (green column). The second step is anchoring of Pt NPs on the I3K nanofibril surface in the presence or absence of P7A, leading to the formation of a well ordered array or a discrete distribution of the Pt nanostructures. P7A, a peptide capping agent, clearly alters the morphological patterns of the Pt nanostructures on the template surface.
Figure 2. TEM micrograph of I3K nanofibrils.

The peptide solution (4 mM in water) was incubated for 1 week at pH of around 7 and the sample was negatively stained with 2% uranyl acetate before TEM imaging.
Figure 3. Structural characterization of 1D Pt nanostructures.

(a) TEM (top) and HRTEM (bottom) images of 1D assemblies of discrete Pt NPs produced by reducing K2PtCl4 in the presence of 2 mM I3K and 1 mM P7A; (b) TEM (top) and HRTEM (bottom) images of 1D assemblies of continuous Pt NPs produced by reducing K2PtCl4 in the presence of 2 mM I3K but without P7A; (c) Scanning electron microscopy (SEM) image of 1D ordered continuous Pt NP assemblies; (d) TEM images of Pt NP double and single helices produced by reducing K2PtCl4 in the presence of 2 mM I3K and 2 mM P7A.
Similar synthesis in the absence of P7A generated 1D ordered continuous Pt NP assemblies with a width of 10.2 ± 1.5 nm (Fig. 3b and 3c and Supplementary Fig. S1c). Although the lateral interparticle spacing is still discernible, the longitudal fusion of NPs along the peptide template is highly evident. In spite of such morphological changes, the lattice spacing of 0.223 nm still dominates for the 1D continuous Pt NP assemblies (the bottom panel of Fig. 3b).
When the amount of P7A was low (0.24 mM), NPs attached on the I3K template surface were close packed and some interparticle fusion was still evident along the longitudal direction (Supplementary Fig. S1d). When the amount of P7A increased to 2 mM, Pt NP double and single helices (Fig. 3d) were formed. For the two superstructures, the edge-to-edge distance between Pt NPs along the helix is 1.1 ± 0.2 nm and 1.6 ± 0.5 nm, respectively. Although the underlying mechanism associated with the formation of such helical superstructures remains elusive, it is likely to be associated with the twisting of β-sheets that comprise the peptide nanofibrils26. On the other hand, it is possible to make P7A stabilized Pt NPs separately from the template and then add them to the I3K solution. In this situation, Pt NPs adsorbed discretely onto the I3K nanofibrils, in a much looser arrangement than the 1D Pt nanostructures prepared as described above (Supplementary Fig. S2).
Mechanism
The I3K template surface is rich in hydrophilic lysine residues and, in the case of silica nanostructures, the interfacial electrostatic attraction between the positively charged lysine residues and negatively charged silica precursors is the driving force for the templated synthesis21,22. In the present case (the solution pH was in the range of 6.0–9.0, depending on reaction time), we hypothesize that the formation of the 1D Pt nanostructures is similarly driven by the electrostatic attraction, and proceeds with the binding of the negatively charged (PtCl4)2− ions to the positively charged surface of the template, followed by the reduction of Pt (II) to Pt (0) and the nanocrystal growth.
To confirm this hypothesis, two comparative experiments were carried out. In the first one, we used the nanofibrils (Fig. 4a) self-assembled from negatively charged I3D (Ac-IIID-CONH2) instead of the I3K nanofibrils as the template. Only random Pt NP aggregates were obtained, consistent with the result from the blank reaction without any additives (Fig. 4b). Subsequently, we substituted K2PtCl6 for K2PtCl4 as the precursor. 1D continuous Pt NP assemblies with a width of 13.6 ± 0.9 nm formed (Fig. 4c). However, the faster reduction rate associated with (PtCl6)2− ions made this type of 1D Pt nanostructure much rougher.
Figure 4. Micrographs of I3D nanofibrils and the Pt NP assemblies produced in comparative experiments.

(a) AFM image of I3D nanofibrils formed at 4 mM in water after one-week incubation at pH of 7. (b) TEM image of Pt NP aggregates synthesized by reducing K2PtCl4 in the presence of 2 mM I3D and 1 mM P7A after reaction for 24 hr. (c) TEM image of 1D continuous Pt NP assemblies produced by reducing K2PtCl6 in the presence of 2 mM I3K but without P7A after 24 hr reaction.
Through its specific and strong binding to the Pt crystal surface, P7A probably favors the formation of ultrasmall Pt NPs and helps to ensure the complete dispersion of the resulting Pt NPs in water for several months24. To examine the stabilization mechanism of this type of aqueous colloidal dispersion, we adjusted the pH from around 7.0 to 2.0 and precipitation immediately followed, together with clear NP aggregates observable in TEM (Supplementary Fig. S3a). When the solution pH was reversed to 7.0 or higher values, the black precipitates redispersed well in water and the solution became clear again (Supplementary Fig. S3b). As the pKa of the C-terminal carboxyl group of the peptide is around 2.2, this indicates that the electrostatic repulsion between the deprotonated C-terminal carboxyl groups of the attached P7A molecules is responsible for the stability of the Pt NP aqueous solution. Consistent with this result, when we used the C-terminal capping of P7A (TLHVSSY-CONH2) instead of the uncapped P7A, aggregation occurred during the very early stages of the reaction (Supplementary Fig. S3c). These results strongly suggest that the electrostatic repulsion between bound P7A is responsible for the formation of the 1D discrete Pt NP assemblies on the I3K template and for the rather uniform interparticle spacing. Without the peptide, Pt NPs tend to fuse along the I3K template, resulting in the formation of the well aligned and continuous 1D Pt NP assemblies schematically shown in Fig. 1.
Electrochemical performance
To probe the structure-activity relationships of the various Pt nanostructures, we conducted electrochemical measurements on a CHI 660D electrochemical workstation. To remove residual precursors, the as-synthesized Pt nanostructures were centrifuged and the collected precipitates were redispersed in water. After repeating the treatment five times, the concentration of Pt in solution was determined by atomic absorbance spectroscopy. The resulting Pt nanostructure solutions were mixed with VXC-72R carbon black in a 1:4 weight ratio of Pt to C and sonicated for a while to obtain homogenous suspensions. The Pt/C aqueous suspensions were dropped onto newly polished, cleaned glassy carbon electrodes (GCEs), dried and immobilized with Nafion solution. Cyclic voltammetry (CV) curves of hydrogen oxidation were recorded at ambient temperature in a N2-saturated 0.5 M H2SO4 solution at a scan rate of 50 mV s−1 for 1D Pt NP assemblies, isolated Pt NPs, and commercial Pt black. Note that the amounts of Pt loadings mounted on the working electrode (GCE) were 13.5 ± 0.2 μg cm−2 in all cases. All CV curves display hydrogen adsorption and desorption regions (−0.22 to 0.15 V versus SCE (saturated calomel reference electrode)) and Pt surface oxide formation and reduction regions (0.15 to 1.15 V), with representative ones given in Fig. 5a.
Figure 5. Comparison of electrocatalytic properties of different Pt nanostructures.
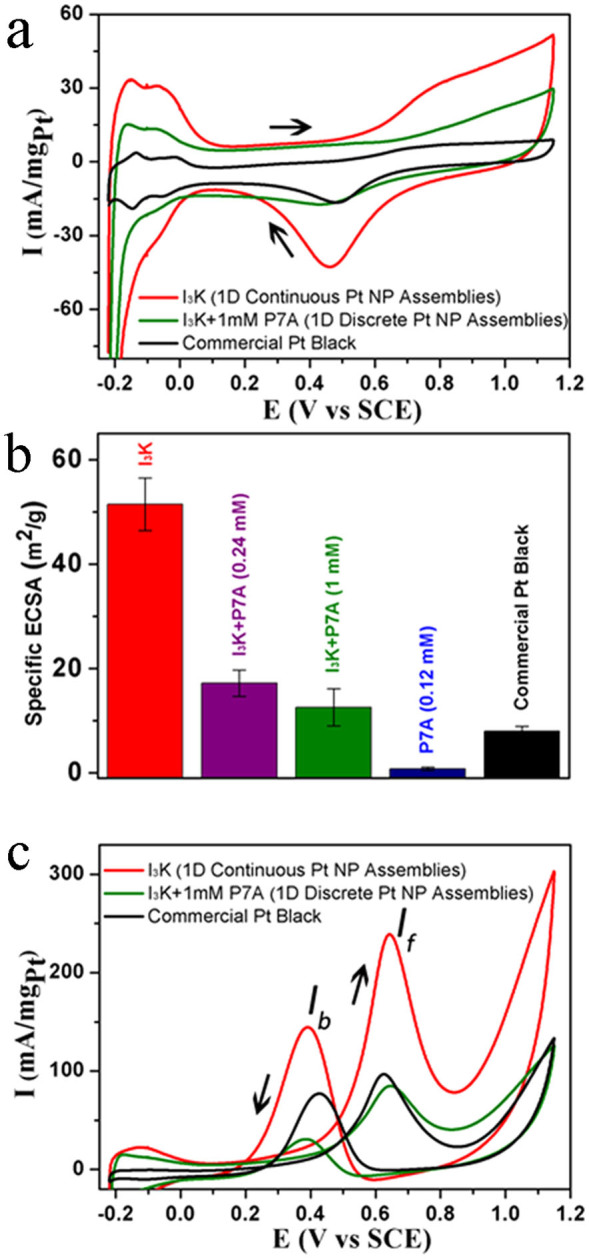
(a) CV curves in a N2-saturated 0.5 M H2SO4 solution with a sweep rate of 50 mV s−1; the indicated Pt nanostructures include 1D continuous Pt NP assemblies synthesized in the presence of only I3K (denoted as “I3K”), 1D discrete Pt NP assemblies synthesized in the presence of 2 mM I3K and 1 mM P7A (denoted as “I3K + 1mM P7A”) and commercial Pt black. (b) Specific ECSAs; note that in this case, specific ECSAs of the 1D Pt NP nanostructure produced in the presence of 2 mM I3K and 0.24 mM P7A and the isolated Pt NPs in the presence of 0.12 mM P7A are also given for comparison. (c) CV curves in a N2-saturated 0.5 M H2SO4 solution containing 1 M methanol.
The specific electrochemical active surface area (ECSA) was calculated by the integration of the hydrogen desorption region (Fig. 5b). It is evident that 1D Pt nanostructures have higher specific ECSA values than isolated NPs (commercial Pt black and Pt NPs produced in the presence of 0.12 mM P7A). For the various 1D Pt nanostructures, the electrochemical activity is proportional to their order and continuity along the axial direction. The 1D ordered and continuous Pt NP assemblies produced in the presence of only I3K showed an exceptionally high specific ESCA of 51.5 m2 g−1 and the 1D discrete Pt NP assemblies (produced in the presence of the I3K template and 1 mM P7A) had a specific ESCA of 12.6 m2 g−1, which is slightly greater than that of the commercial Pt black of 8.0 m2 g−1. In contrast, the 1D Pt NP assemblies with limited continuity, which were produced in the presence of the I3K template and 0.24 mM P7A, displayed a specific ESCA of 17.2 m2 g−1.
CV curves for the methanol oxidation of various Pt nanostructures were also recorded under the same conditions except the electrolyte solution contained 0.5 M H2SO4 and 1 M CH3OH. Note that the Pt loading was still at 13.5 ± 0.2 μg cm−2 in this case. As shown in Fig. 5c, the two anodic current peaks in the forward and backward sweeps (around 0.64 and 0.4 V) represent the methanol oxidation and the removal of the incompletely oxidized carbonaceous species from the Pt surface, respectively. Again, it is the 1D continuous Pt NP assemblies that display an exceptionally high activity for methanol oxidation, with a forward peak current (If) of 239 mA mg−1 at 0.64 V, which is significantly greater than those of the 1D discrete Pt NP assemblies (85 mA mg−1 at 0.65 V) and Pt black (97 mA mg−1 at 0.63 V). Furthermore, the ratios of If/Ib (Ib: the backward peak current) for the 1D continuous and discrete Pt NP assemblies and Pt black are about 1.6, 2.4, and 1.1, suggesting that the 1D Pt NP nanostructures have the better oxidation ability for the poisoning species (e.g. CO)3,4,27.
For the 1D continuous Pt nanostructures, the significant improvements in available surface area, electrocatalytic activity and CO tolerance can be explained by the peculiar structural properties associated with 1D continuous morphology. Specifically, the continuous feature of NP assemblies obviously favors charge transfer by reducing the number of interfaces between NPs3,4,5. The 1D structural anisotropy not only reduces surface defects but also allows for the preferential display of certain crystal facets such as (100) and (111), favoring high methanol oxidation activity and CO tolerance5,6. The higher electric conductivity of the 1D continuous Pt NP assemblies was verified by the SEM characterization in which high resolution images could be readily obtained without gold coating (Fig. 3C). In contrast, it is hard to obtain clear SEM images without gold coating for the discrete Pt nanostructures, because of their limited electric conductivity. While this is not a quantitative measurement of the electrical conductivity, it strongly indicates the isolation of the particles in discrete structures.
Discussion
We have demonstrated a new and easy route to produce 1D Pt nanostructures with controllable morphologies through the combination of designed I3K and phage-displayed P7A peptides. The I3K self-assembled nanofibrils function as a template and P7A serves as a capping agent to achieve switching between ordered and discrete 1D Pt NP superstructures. It was also demonstrated that double and single helical Pt NP assemblies can be fabricated at a higher P7A concentration of 2 mM. Benefitting from these special structural properties, the continuous and ordered 1D Pt morphology showed significantly improved electrochemical performance for the hydrogen and methanol electro-oxidation as compared to 1D discrete Pt NP assemblies and isolated Pt NPs. Its overall performance challenges that of the 1D continuous Pt nanostructures templated on insulin fibrils and TMV nanotubes4,6 in a number of respects including the low synthesis cost, ease of handling and clear structure-function relation of the short peptides. The present study provides a potential methodology for the large scale production of 1D Pt nanostructures with controllable morphologies and electrocatalytic properties. Detailed investigations on these 1D Pt nanostructures are underway for better control of morphology and systematic evaluation of their electrochemical performance as electrocatalysts for fuel cells.
Methods
Materials
Protected amino acids, coupling reagents, resins, cleavage reagents, and solvents for peptide synthesis were purchased from GL Biochem (Shanghai) Ltd. Potassium tetrachloroplatinate (K2PtCl4) and hexachloroplatinate (K2PtCl6) were obtained from Acros. All reagents were used as received except dichloromethane and N, N-dimethyl formamide that were redistilled prior to use. All water used was processed by a Millipore purification system with a minimum resistivity of 18.2 MΩ·cm.
Peptide synthesis and characterization
Ac-IIIK-CONH2 (I3K), TLHVSSY (P7A), Ac-IIID-CONH2 (I3D) and TLHVSSY-CONH2 were synthesized from the C terminus to the N terminus on a CEM Liberty microwave synthesizer, based on the standard Fmoc solid phase synthesis strategy. The Rink amide MBHA resin and Wang resin were used to allow the C-terminus capped (amidated) and uncapped, respectively. The N-terminus of I3K was capped with acetic anhydride prior to cleavage from resin. Detailed synthesis and purification procedures have been described in our previous work20,21. The final products were subjected to mass spectrometry (MALDI-ToF) and reversed-phase HPLC analyses (Supplementary Fig. S4 and S5), indicating high purities (95%).
Preparation of Pt nanostructures
0.4 mL of 5 mM P7A aqueous stock solution, 0.2 mL of freshly prepared 10 mM K2PtCl4 in water and 0.35 mL of water were added into 1 mL of 4 mM I3K well-aged aqueous stock solution (1 week, pH of ~7), followed by addition of 0.05 mL of freshly prepared 40 mM NaBH4 in water. The final concentrations of I3K, P7A, K2PtCl4 and NaBH4 were 2, 1, 1 and 1 mM, respectively. The mixture was sealed tightly in a plastic vial and stirred strongly for a while, then left to stand for 24 h at room temperature to insure complete reaction. To get different morphologies of 1D Pt nanostructures, the final concentration of P7A in the reaction mixture was adjusted to be 2, 0.24, and 0 mM, respectively, with other reaction conditions being the same. For comparative experiments, we used I3D instead of I3K or K2PtCl6 instead of K2PtCl4, also with other reaction conditions unchanged.
For isolated Pt NPs, the reaction was performed in the presence of 0.12 mM P7A or C-terminal capped P7A and in the absence of I3K, with other reaction condition being the same as above.
Instrumental characterization of Pt nanostructures
For TEM characterization, 30 μL of the above reaction solutions were placed on 300-mesh copper grids covered with carbon-stabilized Formvar film, followed by quick drying with filter paper. The grids were then observed with a JEOL JEM 2100UHR TEM operating at 200 kV. For SEM characterization, 20 μL of the reaction solutions were placed on a carefully cleaned SEM stub, vacuum dried at room temperature and then subjected to imaging with a Hitachi S-4800 field emission SEM at 10 kV.
Electrochemical experiments
CV measurements were carried out with a CHI 660D electrochemical workstation (ChenHua, Shanghai) in a standard three-electrode cell consisting of a glassy carbon electrode (GCE) with a diameter of 3 mm as the working electrode, a saturated calomel reference electrode (SCE) and a Pt wire counter electrode.
To remove residual precursors, the above reaction solutions were centrifuged at 10000 rpm for 6 min and the collected precipitates were redispersed in 1 mL water through sonication. After repeating this treatment five times, the concentration of Pt in solution was determined with an Analytic Jena ContrAA 700 high-resolution atomic absorbance spectrometer. The purified Pt solutions were mixed with VXC-72R carbon black in a 1:4 weight ratio of Pt to C and sonicated for a while to obtain homogenous Pt/C suspensions.
Before Pt loading, the GCE was polished successively with 1.0, 0.3, and 0.05 μm Al2O3 powders to a mirror finish, followed by successive sonication in ethanol, acetone, 50% (v/v) nitric acid solution, and water. After electrochemical cleaning in 0.5 M H2SO4 aqueous solution and copious rinsing with water, the electrode was dried with nitrogen purging.
4 μL of the Pt/C aqueous suspensions were dropped onto newly polished, cleaned glassy carbon electrodes (GCEs) and dried with an infrared lamp. To reach immobilization, 10 μL of Nafion ethanol solution (0.1 wt%, DuPont) was then dropped on these GCEs and air dried. The loading amounts of Pt were fixed at 13.5 ± 0.2 μg cm−2 for all Pt nanostructures.
The electrodes were immersed in 10 ml N2-saturated 0.5 M H2SO4 solution, and the potential was scanned from −0.22 to 1.15 V at a scan rate of 50 mV s−1 with a sample interval of 0.001 V to obtain the CV curves of hydrogen oxidation. The CV curves of methanol oxidation reaction (MOR) were recorded in 10 ml nitrogen-saturated solution containing 0.5 M H2SO4 and 1 M methanol. Note that 10 cycles were firstly performed and the 11th cycle was typically used for attaining reproducible signals in both cases.
After subscribing the double-layer current, the hydrogen desorption region (−0.2 ~ 0.15 V) of the CV curves of hydrogen oxidation was integrated to estimate the electrochemical active surface area (ECSA), according to the following formulas:
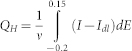 |
Where QH is the total charge; I and Idl are the current and double-layer current, respectively; E is the potential (versus SCE); and ν is the sweeping rate (50 mV s−1). The specific ECSA was calculated assuming:
where θm is the electric charge density for hydrogen adsorption and as a constant (210 μC cm−2), m is the loading amount of Pt.
Author Contributions
K.T. and J.W. contributed equally to this work. H.X., J.W. and J.L. conceived and designed the experiments; K.T., J.W., Y.L. and D.X. performed the experiments and analyzed the data. H.X., J.L. and H.S. wrote the manuscript. All authors discussed and commented on the manuscript.
Supplementary Material
Supplementary Information
Acknowledgments
The authors are grateful for the National Natural Science Foundation of China (21033005 and 21076231), the Natural Science Funds of Shandong Province of China (JQ201105 and ZR2010BQ028), and the UK Engineering and Physical Sciences Research Council (EPSRC). H.X. acknowledges the support by Program for New Century Excellent Talents in University (NCET-11-0735).
References
- Steel B. C. H. & Heinzel A. Materials for fuel-cell technologies. Nature 414, 345–352 (2001). [DOI] [PubMed] [Google Scholar]
- Chen Z., Waje M., Li W. & Yan Y. Supportless Pt and PtPd nanotubes as electrocatalysts for oxygen-reduction reactions. Angew. Chem. Int. Ed. 46, 4060–4063 (2007). [DOI] [PubMed] [Google Scholar]
- Choi S. M., Kim J. H., Jung J. Y., Yoon E. Y. & Kim W. B. Pt nanowires prepared via a polymer template method: its promise toward high Pt-loaded electrocatalysts for methanol oxidation. Electrochim. Acta 53, 5804–5811 (2008). [Google Scholar]
- Górzny M. Ł., Walton A. S. & Evans S. D. Synthesis of high-surface-area platinum nanotubes using a viral template. Adv. Funct. Mater. 20, 1295–1300 (2010). [Google Scholar]
- Koenigsmann C. & Wong S. S. One dimensional noble metal electrocatalysts: a promising structural paradigm for direct methanol fuel cells. Energy Environ. Sci. 4, 1161–1176 (2011). [Google Scholar]
- Zhang L., Li N., Gao F., Hou L. & Xu Z. Insulin amyloid fibrils: an excellent platform for controlled synthesis of ultrathin superlong platinum nanowires with high electrocatalytic activity. J. Am. Chem. Soc. 134, 11326–11329 (2012). [DOI] [PubMed] [Google Scholar]
- Xia Y. & Yang P. Chemistry and physics of nanowires. Adv. Mater. 13, 351–352 (2003). [Google Scholar]
- Xia Y. et al. One-dimensional nanostructures: synthesis, characterization, and applications. Adv. Mater. 15, 353–388 (2003). [Google Scholar]
- Tang Z. & Kotov N. A. One-dimensional assemblies of nanoparticles preparation, properties, and promise. Adv. Mater. 17, 951–962 (2005). [Google Scholar]
- Liang H. W., Liu J. W., Qian H. S. & Yu S. H. Multiplex templating process in one-dimensional nanoscale: controllable synthesis, macroscopic assemblies, and applications. Acc. Chem. Res. 46, 1450–1461 (2013). [DOI] [PubMed] [Google Scholar]
- Gao X. & Matsui H. Peptide-based nanotubes and their applications in bionanotechnology. Adv. Mater. 17, 2037–2050 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazit E. Use of biomolecular templates for the fabrication of metal nanowires. FEBS J. 274, 317–322 (2007). [DOI] [PubMed] [Google Scholar]
- Dickerson M. B., Sandhage K. H. & Naik R. R. Protein- and peptide-directed syntheses of inorganic materials. Chem. Rev. 108, 4935–4978 (2008). [DOI] [PubMed] [Google Scholar]
- Chen C. L. & Rosi N. L. Peptide-based method for the preparation of nanostructured inorganic materials. Angew. Chem. Int. Ed. 49, 1924–1942 (2010). [DOI] [PubMed] [Google Scholar]
- Yan X., Blacklock J., Li J. & Möhwald H. One-pot synthesis of polypeptide-gold nanoconjugates for in vitro gene transfection. ACS Nano 6, 111–117 (2012). [DOI] [PubMed] [Google Scholar]
- Yan X., Li J. & Möhwald H. Templating assembly of multifunctional hybrid colloidal spheres. Adv. Mater. 24, 2663–2667 (2012). [DOI] [PubMed] [Google Scholar]
- Song Y. et al. Synthesis of peptide-nanotube platinum-nanoparticle composites. Chem. Comm. 1044–1045 (2004). [DOI] [PubMed] [Google Scholar]
- Biswas K. & Rao C. N. R. Nanostructured peptide fibrils formed at the organic-aqueous interface and their use as templates to prepare inorganic nanostructures. ACS Appl. Mater. Interface 1, 811–815 (2009). [DOI] [PubMed] [Google Scholar]
- Kasotakis E. et al. Design of metal-binding sites onto self-assembled peptide fibrils. Biopolymer (Pept. Sci.) 92, 164–172 (2009). [DOI] [PubMed] [Google Scholar]
- Han S. et al. Self-assembly of short peptide amphiphiles: the cooperative effect of hydrophobic interaction and hydrogen bonding. Chem. Eur. J. 17, 13095–13102 (2011). [DOI] [PubMed] [Google Scholar]
- Xu H. et al. Twisted nanotubes formation from ultrashort amphiphilic peptide I3K and their templating for the fabrication of silica nanotubes. Chem. Mater. 22, 5165–5175 (2010). [Google Scholar]
- Wang S. et al. Mechanistic process underlying biomimetic synthesis of silica nanotubes from self-assembled ultrashort peptide templates. Chem. Mater. 23, 2466–2474 (2011). [Google Scholar]
- Wang S. et al. Biomimetic synthesis of silica nanostructures with controllable morphologies and sizes through tuning interfacial interactions. Chem. Comm. 48, 9415–9417 (2012). [DOI] [PubMed] [Google Scholar]
- Li Y., Whyburn G. P. & Huang Y. Specific peptide regulated synthesis of ultrasmall platinum nanocrystals. J. Am. Chem. Soc. 131, 15998–15999 (2009). [DOI] [PubMed] [Google Scholar]
- Li Y. & Huang Y. Morphology-controlled synthesis of platinum nanocrystals with specific peptides. Adv. Mater. 22, 1–5 (2010). [DOI] [PubMed] [Google Scholar]
- Aggeli A. et al. Hierarchical self-assembly of chiral rod-like molecules as a model for peptide β-sheet tapes, ribbons, fibrils, and fibers. Proc. Natl. Acad. Sci. USA 98, 11857–11862 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A. & Holt-Hindle P. Platinum-based nanostructured materials: synthesis, properties, and applications. Chem. Rev. 110, 3767–3804 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information