

Abstract
We took advantage of the ability of human telomeres to silence neighboring genes (telomere position effect or TPE) to design a high-throughput screening assay for drugs altering telomeres. We identified, for the first time, that two dietary flavones, acacetin and chrysin, are able to specifically alleviate TPE in human cells. We further investigated their influence on telomere integrity and showed that both drugs drastically deprotect telomeres against DNA damage response. However, telomere deprotection triggered by shelterin dysfunction does not affect TPE, indicating that acacetin and chrysin target several functions of telomeres. These results show that TPE-based screening assays represent valuable methods to discover new compounds targeting telomeres.
Keywords: DNA damage response, flavanoid, polyphenol, telomere, telomeric position effect, telomere-induced foci
Introduction
Telomeres are key features of linear chromosomes that preserve genome stability.1 They are composed of short tandemly repeated DNA sequences, and various telomere-associated proteins, which are essential to preserve chromosome stability by controlling telomere length, recombination, and DNA damage checkpoint.2,3 The maintenance of telomere length depends on telomerase, a specialized reverse transcriptase that uses its RNA template to add G-rich telomeric repeats to the terminal 3′ overhang.4
During aging, the somatic down-regulation of telomerase leads to progressive telomere shortening, which correlates with tissue dysfunction and loss of renewal capacity. In human cancer, telomerase overexpression is required to bypass senescence and for long-term cellular proliferation.5,6 Consequently, inhibition of telomerase was proposed as a powerful strategy for cancer therapy. Immunological and pharmacological inhibition of telomerase is currently evaluated in a series of clinical protocols. A possible limitation of this therapeutical approach is that telomere shortening can also drive some aspects of normal and pathological aging and can cooperate with p53 deficiency to favor carcinogenesis in aged mice.7 However, with many but not all telomerase therapeutic approaches, growth arrest has been observed only when telomeres reach a critically short length, which might also impair the function of neighboring normal cells. Conversely, components directly targeting the telomeric capping proteins or other molecular components of telomeres are expected to have more immediate and profound effects on tumorigenicity.8,9 As a proof of concept that a capping protein can be an attractive target, TRF2 inhibition markedly inhibits tumorigenicity in a xenografted mouse model while its overexpression reinforces tumor formation.10,11 Interestingly, telomeric proteins components also have extratelomeric roles,12,13 which opens the possibility to target specific pro-oncogenic properties of telosomes, independently of their ability to protect against genomic instability. There is also mounting evidence that telomeres are involved in several other diseases such as progeroid syndromes, idiopathic pulmonary fibrosis, and cardiovascular disorders.9 Overall, telomeres constitute an attractive target for the development of novel small-molecule therapeutic drugs against cancer and a variety of age-related pathologies. Therefore, there is an urgent demand for novel types of screening assays able to discriminate different features of telomere function.
In this context, the ability of heterochromatic telomeres to silence gene expression, a mechanism named telomeric position effect (TPE),14,15,16 could be an interesting readout to identify active drugs targeting telomeres with a radically different mechanism of action than the one targeted nowadays, which mainly inhibits telomerase activity. In particular, TPE may reflect important functions of telomeres that are not directly related to their role in chromosome stability. For instance, TPE may contribute to a fast adaptation to stress and environmental changes such as nonoptimal growth conditions in several yeast species and is often evoked as a mechanism leading to the changes in transcriptional program occurring in senescent and cancer cells.16
Compelling evidence for transcriptional silencing in the vicinity of human telomeres was provided experimentally by using transgenes inserted adjacent to telomeres, similar to the approach used in yeast after telomere fragmentation.14,15 By a telomere seeding procedure, natural telomeric regions have been replaced by artificial ones containing a reporter gene. Using this method, reporter genes in the vicinity of telomeric repeats were found expressed on average tenfold less than reporters at nontelomeric sites. Overexpression of the human telomerase reverse transcriptase (hTERT) in the telomeric clones resulted in telomere extension and decrease in transgene expression,14 while overexpression of the telosome component TRF1, involved in telomere length regulation, induces transgene re-expression,15 indicating the involvement of both telomere length and telosome in TPE as observed in yeast. In addition, treatment of cells with trichostatin A (TSA), an inhibitor of class I and II histone deacetylases antagonizes TPE. In human cells, TPE is not sensitive to DNA methylation15 while hypermethylation of telomeric transgenes is a secondary event in the telomeric-silencing process in mouse ES cells.17 Overall; these data suggest that both telomere-specific mechanisms and classical changes in the chromatin architecture are implicated in TPE.
Since TPE in human cells depends on telosome structure, we took advantage of the reporter system designed in our laboratory to uncover TPE in human cells15 to search for molecules able to specifically target TPE in an automated screening approach. Here we demonstrate the feasibility of this approach through the identification of two new antitelomere components leading to massive telomere deprotection and TPE alleviation. Thus, these compounds might represent hits for the subsequent development of new antitelomere molecules for cancer treatment.
Results
The two flavonoids acacetin and chrysin alleviate TPE
In order to identify molecules that are able to modulate TPE in human cells, we developed a cell-based assay suitable for automation with a C33A cancer cell line carrying a luciferase reporter gene in the vicinity of de novo formed telomeres (Figure 1a). TSA, which was previously described as an inhibitor of TPE15 was used as a positive control and DMSO as a bioinactive compound to estimate the Z′ (0.71) factor (Supplementary Figure 1a,b online) as defined by Zhang et al. 18 automated screen was performed in duplicate with the 1,120 molecules of the Prestwick library19 (Figure 1a). A total of 80 molecules were first identified as potential active compounds in duplicated primary screening experiments based on the increase in luminescence values and a Z′ factor > 0.5 (Figure 1a, Supplementary Figure 1c online). Data were then normalized using a trim polish procedure20 with polish set to 10%. Compounds were then considered hits if the mean trim polish of the duplicates was ≥2. Using this rule, 60 compounds were considered as hits. In a secondary screening on the automated workstation, these 60 potential active compounds, as well as 100 potential inactive compounds selected randomly from the 1,060 inactive molecules, were tested again in four independent wells. Data were normalized by the trim polish procedure with polish set to 50% (median polish) rather than the 10% used in the primary screen. From the secondary screening, 15 molecules with a median polish score ≥2 for all well were considered as hits. Together with the bioactive control, TSA, 15 of the potentially active compounds were selected for their significant effect on luciferase expression (Z′ value > 0.5) (Figure 1a). We purchased them and further investigated manually their specificity to TPE. For this purpose, we compared the effect of the drugs inC33A cells stably transfected with a eGFP reporter construct devoid of telomeric sequence and consequently randomly inserted (pCMV) and cells with terminal integration of eGFP (pCMV Telo)15,21 (Figure 1a). For further testing, we preferred the use of an eGFP reporter system since it is possible to discriminate between an increase expression in eGFP in cells either silenced or expressing the reporter at a low level. This is not feasible with luciferase, which is detected globally from whole cell lysate. Inhibition of TPE was confirmed for seven out of the 16 molecules tested (Figure 1b).
Figure 1.

Schematic representation of the screening procedure. (a) C33A cells carrying a telomeric firefly luciferase reporter gene were gown in the presence of hygromycin and transferred to 96 plates 24 h before addition of the drug. The 1,120 molecules of the Prestwick collection of chemicals were added at a final concentration of 25 µM per molecule as recommended. For each molecule, luminescence was measured after 24 h incubation in the presence of the drug from whole cell lysates. The level of luminescence was compared with the value obtained for trichostatin A, used as bioactive control or DMSO, used as bioinactive control. A total of 60 molecules were selected as anti-TPE agents and tested in a secondary automated screening. Finally, 15 molecules were retained with a potential anti-TPE effect and further investigated manually to identify molecules having a specific effect on a telomere-associated reporter gene (see (b)). Formulations of the two final hits shown are boxed. (b) C33A cells stably transfected with either the pCMV-GFP vector (pCMV) or the pCMV-Telo vector were treated for 24 h with a final concentration of 25 µM of the 16 drugs identified after the secondary screening. Data are mean values of three independent experiments ± SD. Statistical significance was determined using a Mann–Whitney test *P < 0.01. TPE, telomere position effect.
Importantly, two polyphenols belonging to the flavone family, acacetin and chrysin (Figures 1a and 2a), specifically and significantly affect TPE when we compared their effect on cells transfected with pCMV-Telo vs. pCMV (Figure 1b). The specific anti-TPE effect of both drugs was further confirmed using increasing drug concentrations within a range that is not toxic to the cells (12.5–50 μM, Supplementary Figure 2 online). A significant increase in eGFP expression was observed with 25 and 50 μM of each drug, a range of concentration previously reported in other systems.22,23 A concentration of 25 μM was used for further investigation.
Figure 2.

Acacetin and chrysin specifically alleviate TPE in human cells. (a) Molecular structure of acacetin and chrysin. (b) C33A and myoblast primary cells were treated with acacetin or chrysin and expression of FRG2 and GAS11 mRNA level was determined by RT-qPCR. (c) Based on structure-function relation, five additional molecules were tested manually for their specificity with regard to TPE. (d) C33A cells stably transfected with either the pCMV telo vector or the pCMV vector were incubated for 24 and 72 h in the presence of the drug resuspended in DMSO or DMSO alone and eGFP expression was measured by flow cytometry. The histogram display the ratio of intensity between cells treated with the different drugs and cells incubated with DMSO. No significant difference was observed between cells stably transfected wit the pCMV telo vector compared with cells transfected with the pCMV vector indicated that, on the contrary to acacetin and chrysin, none of the five molecules shown in panel (c) alleviate TPE. TPE, telomere position effect.
Acacetin and chrysin upregulate the expression of natural subtelomeric genes
To further validate the role of acacetin and chrysin in TPE, we analyzed, by quantitative RT-PCR, the expression of two natural subtelomeric genes whose expression was previously reported to be regulated by telomeres: GAS11 located at the 16q subtelomere24 and FRG2 located at the 4q subtelomere and suspected to play a role in facio-scapulo-humeral dystrophy, a genetic disease involving TPE.25,26 Experiments were conducted in the transformed C33A cell line and primary human myoblast cells. For both genes, we observed an increase of their mRNA level relative to β-actin after treatment with chrysin or acacetin, in contrast to control cells treated with DMSO (Figure 2b). These results indicate that acacetin and chrysin increase expression of natural subtelomeric genes, which is in agreement with their effects on TPE reporter genes. They also suggest that these compounds can alleviate TPE in human primary cells.
The anti-TPE effects of acacetin and chrysin are dependent upon their chemical structure
Phenolic compounds or polyphenols is a large group of compounds comprising at least 8,000 known structures. In order to investigate the correlation between chemical structure and anti-TPE function, additional structurally related compounds have been tested. None of the five congeners related to acacetin and chrysin showed the same specificity toward TPE (Figure 2c,d). For luteolin, there is trend toward TPE alleviating at 72 hours that might reflect a weak specific effect on TPE. However, we were unable to pursue the kinetics with this drug since cell proliferation was greatly affected at later time points. Therefore, the effects on telomeres are strictly dependent on the compounds chemical structure, ruling out a general effect of polyphenolic compounds on telomere chromatin.
Acacetin and chrysin uncap telomeres
In C33A cells, treatment with acacetin and chrysin showing TPE alleviation are accompanied neither with a massive cell death or cell cycle alterations after 6 days of treatment (Supplementary Figure 3a online) nor with mean telomere length shortening after 4 days of treatment, i.e., well after the 24 hours of treatment sufficient to inhibit TPE (Supplementary Figure 3b online). These results indicate that TPE alleviation by acacetin and chrysin is not caused by growth arrest or telomere shortening.
Since reversal of TPE is not associated with telomere shortening, we further investigated the pathway by which the drugs modulate telomeric silencing. To this aim, we treated the C33A cells as well as the primary fibroblast line IMR90 with acacetin or chrysin and assayed the formation of telomere damage induced foci by immunostaining of both TRF1 and the DNA damage checkpoint protein 53 BP1. Drug addition leads to telomere deprotection (Figure 3a,b) and also significantly increases the frequency of telomeric abnormalities observed in metaphase spreads (Figure 3c) with a significant increase of sister telomere fusions and the presence of multiple telomeric signals, an aberrant telomere structure believed to reflect defective telomere replication.27,28,29 Impressively, these defects in telomere protection are observed as soon as 24-h post-treatment in both cell lines suggesting a direct effect on telomere integrity and not the secondary consequence of increased senescence after multiple rounds of impaired replication as we might expect for the primary IMR90 cells. In agreement with the telomere deprotection triggered by acacetin and chrysin, both drugs trigger premature growth arrest at the G1/S transition of the primary IMR90 cells (Supplementary Figure 4 online). These results show that acacetin and chrysin, in addition to inhibiting TPE, profoundly affect the capacity of telomere to protect chromosome ends.
Figure 3.
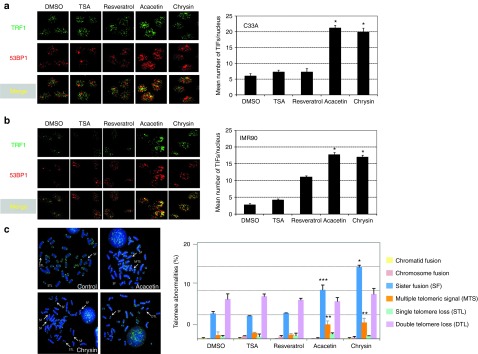
Treatment with acacetin and chrysin increases telomere dysfunction. Representative images of confocal sections of 53BP1 foci (red) and TRF1 (green) in nuclei of C33A (a) and IMR 90 (b) cells treated with of 25 µM of the different drugs or mock-treated with DMSO. Merging shows the colocalization between 53BP1 and TRF1 foci (TIFs). Histograms indicates the quantification of TIFs per nucleus after incubation of either C33A or IMR90 cells for 24 hours in the presence of the drug and represent the mean value of three independent experiments. Significant differences between DMSO and drug-treated cells are indicated with asterisks (*P < 0.001, **P < 0.002). P values were calculated using a t test. (c) Representative images of the main telomeric aberrations encountered in cells treated for 24 hours with 25 µM of the different drugs and quantification as the percentage of events per chromosome. Thirty metaphases were counted for each condition. TIF, telomere damage induced foci.
Telomere deprotection is not sufficient to alleviate TPE
Since acacetin and chrysin both alleviate TPE and uncap telomeres, we asked whether telomere deprotection is sufficient to alleviate TPE. To this purpose, we depleted either TRF2 or POT1 expression in C33A cells and measured telomere damage induced foci rate and eGFP level in these cells. As expected, depletion in TRF2 or POT1 leads to an increase in telomere damage induced foci formation (Figure 4a–c). Although not significantly, telomere proximal eGFP expression decreases in cells depleted for TRF2 or POT1 with a more pronounced effect for POT1 (Figure 4d). This shows that the increased expression of the GFP-reporter gene observed for acacetin and chrysin can not be merely attributed to telomere deprotection and shelterin inhibition, suggesting an unprecedented mechanism of action linking telomere protection to TPE in human cells.
Figure 4.
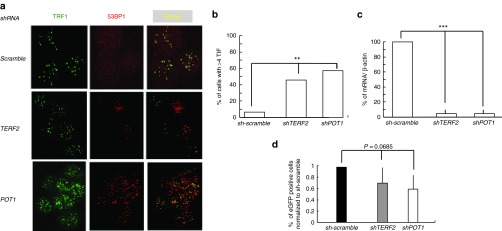
Telomere dysfunction induced by shelterin alteration does not alleviate TPE. (a) Representative images of confocal sections of TRF1 (green) and 53BP1 foci (red) in nuclei of C33A cells transduced with shRNA directed either against TERF2 or POT1 transcripts or against control (scramble). Merging shows the colocalization between 53BP1 and TRF1 foci named TIFs. (b) Histograms display the quantification of TIFs per nucleus and represent the mean value of three independent experiments with the SD shown as error bars. (c) Expression of TERF2 or POT1 mRNA in cells transfected with shRNA lentivectors. (d) Percentage of eGFP-positive cells measured by flow cytometry analysis. Results are the average of three independent experiments (n = 3, error bars ± SD). P value corresponds to the comparison of shTERF2 or shPOT1 and sh-scramble (F-test). TIF, telomere damage induced foci; TPE, telomere position effect.
Discussion
In order to identify new molecules able to modulate telomere structure, we used a radically new readout procedure based on a gene expression reporter constructs sensitive to TPE. As a proof of principle to this approach, we successfully identified two new telomere drugs among the 1,120 molecules of the Prestwick collection of chemicals. Specifically, we found that the treatment of human cells by chrysin and acacetin, two compounds belonging to the flavonoid family, alleviates TPE and induces a DNA damage response at telomeres both in cancer and primary cells. Moreover, we showed that these compounds upregulate the expression of GAS11 and FRG2, two genes naturally found at subtelomeric regions and known to be regulated by telomeres. Both molecules display a closely related backbone differing only by the presence of an O-methylated residue (O-CH3) (Figure 2b). Moreover, other assayed polyphenols did not show any effect on TPE. Taken together, these results suggest that the specificity for telomeres that we observed in our screening depends on the structure of the molecules and is not on a general property of polyphenolic compounds.
Cells treated by the two flavonoids exhibit several types of telomere dysfunctions: activation of the DNA damage response pathway indicating telomere deprotection, and increased rate of sister telomere fusion and multiple telomeric signals, indicative of telomere injuries during the S and G2 phases. Since telomere deprotection triggered by inhibition of TRF2 or POT1 does not alleviate TPE, we conclude that chrysin and acacetin deprotect telomeres in a way that specifically alters TPE.
Acacetin and chrysin are found in food, traditional herbal medicine or proposed as dietary supplement for their vascular relaxant and antioxidant activities.30 They are used either as inhibitors of glutathione reductase and inhibitor of topoisomerase I (acacetin)31 or as an antifungal and inhibitor of 5-lipoxygenase (chrysin).32 They have never been described as targeting telomeres. Since they are also regarded as core chemicals for cancer prevention by inhibiting cell proliferation in vitro33 as well as angiogenesis and tumor growth in vivo,34 our results raise the interesting possibility that their telomere uncapping and anti-TPE activities contribute to their anti-cancer effects. However, their potent telomere uncapping and growth inhibition effects on primary IMR90 cells may impede their use in clinics at least at the doses used here. Further studies are necessary to evaluate whether these compounds can exhibit differential telomere and growth effects between cancer and normal cells. Overall, chrysin and acacetin emerge as valuable model molecules to design multitarget drugs against cancer progression, combining antiangiogenic, antitopoisomerase, antioxidant, and antitelomere properties, which might be optimized by the “SOSA Approach” used to design the Prestwick chemical library.19
In conclusion, this study validates the use of human TPE-based cell assays to screen for new categories of drugs targeting telomeres. This opens new avenues for the identification of antitelomere molecules, which may have a high clinical value.
Materials and methods
Cell culture. The human primary myoblasts were obtained from a fetal biopsy after collagenase/dispase dissociation (Broucqsault et al., in preparation) and grown in HamF10 15% SVF, 1% penicillin/streptomycin, and fibroblast growth factor 10 ng/ml. Experiments were performed using cells between the fifth and seventh passage. The human epithelial cervix adenocarcinoma C33-A cells were maintained in Dulbecco's Modified Eagle's Medium with l-alanyl-l-glutamine (GlutaMAXTMI), d-glucose and sodium pyruvate (Invitrogen). The human fetal lung fibroblasts IMR90 cell line was maintained in MEM alpha modification with l-glutamine and without nucleosides (PAA) supplemented with nonessential amino acids (PAA, Pasching, Austria). For both C33A and IMR90 media were supplemented with 10% FBS (Life Technologies, Paisley, UK) and 1% penicillin–streptomycin, 10,000 units/ml (Invitrogen). All cell lines were grown at 37 °C, 5% CO2, in a humidified atmosphere.
Constructs, transfections, and flow cytometry. The pCMV and pCMVTelo plasmids carrying an eGFP reporter gene are described in.15 For automated screening, eGFP was replaced by the luciferase sequence obtained from the pGL3 vector (Promega, Madison, WI), inserted downstream of the CMV promoter. Cloning details are available upon request. The conditions of transfection of the linearized vectors with a modified calcium phosphate method15 were optimized in order to obtain a single integration per cell. Three days post-transfection, cells were treated with Hygromycin B (Roche Diagnostics, Meylan, France) at a final concentration of 400 µg/ml and kept under permanent selection. In these conditions, nearly 90% of hygromycin-resistant cells transfected with a linearized telomeric fragment harbor the reporter gene at a telomeric location.15,21,35 Harvested cells were analyzed using a FACScan flow cytometer and data were processed using the CellQuestPro software (Becton-Dickinson, Le Pont de Claix, France). The percentage of eGFP-positive cells was determined using nontransfected cells as the baseline for autofluorescence. For luciferase, reporter expression was determined by luminescence using the ONE-Glo™ Luciferase Assay System Kit (Promega).
Screening. The screening facility was composed of a TECAN GENESIS 200 workstation completed with a nine-stacker Carousel with barcod reader, a Te-shake 2-position plate shaker and a 6-position Dark Incubator (TECAN), a Kendro 42-position Cytomat 2C cell incubator (THERMO), a TECAN PowerWasher 384 with a 96-well wash head, and a FLUOstar Optima plate reader (BMG Lab technologies, Ortenberg, Germany). The platform is controlled with the TECAN Gemini 4.00 and FACTS 4.81 softwares. Luminescence was measured with the FLUOStar Optima Plate reader. After cell treatment, raw data were exported to an excel file and compared with bioactive and bioinactive controls. The statistical significance of each well in the 96-well plate assay was assessed by calculating the Z′ factor using DMSO as the bioinactive control and TSA as the bioactive one for anti-TPE activity. This factor takes into account the data variation associated with the signal measurement and reveals the overall quality of the assay. The Z′ factor was calculated using the following equation:18
 |
C+ is the bioactive control (TSA), C− is the bioinactive control (DMSO); µ, the mean of fluorescence intensity and σ, the SD. A DMSO/TSA ratio of 2.7 and a Z′ factor of 0.71 were obtained from 30 wells indicating that the assay is robust and suitable for automated screening since the Z′ factor value is above 0.5.
Drug treatment. Cells were split one day before drug addition at a density of 8,000 cells/well for 96-well plate. At day 1, media were removed and drugs were added into fresh media at a concentration of 25 µM, a concentration is in the range of concentration used in cell-based screens. TSA was obtained from SIGMA, other drugs were purchased from Prestwick Chemicals. For time course assays, cells were maintained in the presence of the drugs.
Total RNA extraction. Total RNA was extracted from cell lines using the RNeasy kit (Qiagen, Germantown, MD) and treated with DNaseI following the manufacturer's instructions. After extraction, the integrity of total RNA was examined on a 1.2% agarose gel containing 1 µg/ml ethidium bromide. RNA concentration was measured at 260 nm, purity of the total RNA was determined by the 260 nm/280 nm ratio with expected values between 1.8 and 2 and RNA samples were stored at −20 °C until use.
DNA extraction. Total DNA was extracted using the DNeasy Blood & Tissue kit (Qiagen). DNA concentration was measured at 260 nm, purity of the total DNA extracted was determined by the 260 nm/280 nm ratio with expected values between 1.8 and 2 and DNA samples were stored at 4 °C until use.
Southern blot. Genomic DNA was digested with HinfI/RsaI enzymes and separated by electrophoresis on a 0.7% agarose gel for 23 hours at 60 V. The presence of DNA and the efficiency of digestion were verified by UV in the presence of ethidium bromide. Prior to transfer, the gel was depurinated (0.25 mol/l HCl), denatured (0.5 mol/l NaOH/1 mol/l NaCl), and neutralized (1 mol/l ammonium acetate). DNA was transferred onto a Hybond XL Nylon membrane (GE Healthcare, Velizy-Villacoublay, France) in SSC 20×. The membrane was then neutralized in 250 mmol/l phosphate buffer pH 7.2 at 50 °C, prehybridized for 1 hour at 50 °C (6× SSC, 0.5× SDS, 1% milk) and hybridized overnight at 50 °C (6× SSC, 0.1× SDS, 1% milk) in the presence of a dCTP α32P labeled DNA probe (random priming). After hybridization, membranes were washed at increasing stringency and exposed to a phosphoimager intensifying screen (Molecular Dynamics). Signal intensities were analyzed with the Image Gauge software. The probe was a 700 nucleotides fragment of tandem telomeric (T2AG3) repeats.
Immuno-fluorescence detection and telomere induced foci analysis. Cells were grown on slides and treated with molecules for different time points, then fixed for 20 min in the presence of methanol at −20 °C, or by addition of 4% formaldehyde at room temperature for 10–15 min, incubated for 1 h with the blocking buffer (0.8× PBS, 50 mmol/l NaCl, 0.5% Triton X100, 3% milk), followed by an 1 h incubation at 37 °C with the primary antibody (mouse monoclonal anti-TRF1 (ABCAM-ab10579) and rabbit polyclonal to 53BP1 (NOVUS BIOLOGICALS: NB 100–305) in blocking buffer. Cells were then washed with 0.8× PBS, 50 mmol/l NaCl, 1.5% milk, and incubated in the presence of a donkey polyclonal antimouse ALEXA488 antibody (A21202; Molecular Probes) and donkey polyclonal antirabbit ALEXA555 antibody (A-31572; Molecular Probes). After washing with 0.8× PBS, 50 mmol/l NaCl, 0.1% Trition X-100, slides were counterstained with DAPI and mounted in Vectashield (Vector Laboratories, Burlingame, CA).
Metaphases preparation. Approximately 18 h after platting, colcemid was added to the medium at a final concentration of 60 ng/ml (KaryoMAX, Invitrogen) for 30 min. Cells were harvested and resuspended in 75 mmol/l KCl for 15 min and the methanol:acetic acid (3:1) fixative was added. After centrifugation, cells were resuspended in fixative by vortexing and left overnight at 4 °C. Cells were centrifuged and an aliquot was spread on slides. Dried slides were stored at −20 °C until use.
Fluorescence in situ hybridization with a FITC-conjugated PNA probe (5′ CCCTAA 3′)3. Slides were fixed for 2 min at RT in 4% paraformaldehyde, followed by dehydration through ethanol series (50, 75, and 100%, 5 min each). Fluorochrome-coupled PNA probe (AATCCC) was then applied to the slides (0.3 ng PNA probe/µl, dissolved in 70% formamide, 10 mmol/l Tris pH 7.2, 1% blocking reagent (Roche Biochemicals)). A denaturation at 80 °C was applied for 3 min, followed by a 2 h hybridization at room temperature. Washes were the following: 15 min at RT in 70% formamide, 10 mmol/l Tris pH 7.2 and 15 min in 50 mmol/l Tris pH 7.5, 150 mmol/l NaCl, 0.05% Tween-20. Finally, slides were counterstained with DAPI and mounted in Vectashield (Vector Laboratories).
Lentiviral production, infection and gene silence by shRNA. Human Embryonic Kidney 293 phoenix cells were grown in DMEM with 10% fetal calf serum. VSVg-pseudotyped self inactivating (SIN) lentiviruses, expressing shCtrl vector or shTRF2 (Sigma, NM_005652.2-1320s) or shPOT1(Sigma, NM_015450.1-1934s), were produced by transient transfection of Human Embryonic Kidney 293 Phenix cell lines with two other packaging plasmids, p8.91 and pVSVg, by calcium phosphate precipitation. 48 and 72 h post-transfection, the infectious supernatant was collected and applied to the target cells. Cells were collected 1 week after infection for further experiments and knock-downefficiency of the targeted genes was determined by RT-qPCR.
Primers used for RT-qPCR:
TERF2 : 5′- GACCTTCCAGCAGAAGATGCT-3′ and 5′- GTTGGAGGATTCCGTAGCTG-3′. POT1: 5′-TGGGTATTGTACCCCTCCAA and 5′-GATGAAGCATTCCAACCACGG-3′. FRG2: 5′-TGCCAAGACACAGCAGGGAACT and 5′-CCCGAGACCTATGCCGCTTGC. Sequence of primers for GAS11 can be found in.24
Statistics. All quantitative results represent the mean ± SD. For automated screening, the statistical significance of each well in the 96-well plate assay (primary screening) was assessed by using the trim polish procedure with polish set to 10%. In the secondary screening, median polish (50%) was used in order to increase test stringency. To determine overall survival, we used a log-rank (Mantel-Cox) test. For the percentage of GFP-positive cells, the significance of differences between the means was determined using the Mann–Whitney two-tailed test. A P value of <0.05 was considered as statistically significant. For the telomere damage induced foci assays, statistics were calculated using a t test. To compare variances, a F-test was used.
SUPPLEMENTARY MATERIAL Figure S1. Screening of molecules affecting human TPE. Figure S2. Dose response assay. Figure S3. Effect of acacetin and chrysin on cell cycle and telomere length. Figure S4. Acacetin and chrysin induce premature growth arrest of IM90 cells.
Acknowledgments
We acknowledge the facilities of IFR 128 (microscopy (PLATIM) and Flow Cytometry) and IRCAN (microscopy (PICMI) and cytometry (CYTOMED)). We also thank the ChemAxon company (http://www.chemaxon.com) which has allowed the academic TAMIS software team (S. Wieczorek, S. Aci, C. Charavay and S. Roy) and C. Barette to freely use the MarvinView package, a helpful Graphic User Interface for viewing structure files and query results. A.B was the recipient of a fellowship from Association Française contre les Myopathies (AFM) and Fondation pour la Recherche Médicale (FRM). NB is the recipient of a fellowship from Association Française contre les Myopathies (AFM). This work was supported by grants from AFM (to EG and FM), the Ligue Nationale contre le Cancer (to EG, équipe labelisée) and the National Natural Science Foundation of China (Grant Numbers: 81000875, 81270433) (to JY).
Supplementary Material
Screening of molecules affecting human TPE.
Dose response assay.
Effect of acacetin and chrysin on cell cycle and telomere length.
Acacetin and chrysin induce premature growth arrest of IM90 cells.
References
- Blackburn EH, Greider CW, Szostak JW. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med. 2006;12:1133–1138. doi: 10.1038/nm1006-1133. [DOI] [PubMed] [Google Scholar]
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- Giraud-Panis MJ, Pisano S, Poulet A, Le Du MH, Gilson E. Structural identity of telomeric complexes. FEBS Lett. 2010;584:3785–3799. doi: 10.1016/j.febslet.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Blackburn EH. The end of the (DNA) line. Nat Struct Biol. 2000;7:847–850. doi: 10.1038/79594. [DOI] [PubMed] [Google Scholar]
- Buseman CM, Wright WE, Shay JW. Is telomerase a viable target in cancer. Mutat Res. 2012;730:90–97. doi: 10.1016/j.mrfmmm.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Chan SS, Chang S. Telomere dysfunction and tumour suppression: the senescence connection. Nat Rev Cancer. 2008;8:450–458. doi: 10.1038/nrc2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet. 2001;28:155–159. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- Ségal-Bendirdjian E, Gilson E. Telomeres and telomerase: from basic research to clinical applications. Biochimie. 2008;90:1–4. doi: 10.1016/j.biochi.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Ye J, Wu Y, Gilson E. Dynamics of telomeric chromatin at the crossroads of aging and cancer. Essays Biochem. 2010;48:147–164. doi: 10.1042/bse0480147. [DOI] [PubMed] [Google Scholar]
- Biroccio A, Rizzo A, Elli R, Koering CE, Belleville A, Benassi B, et al. TRF2 inhibition triggers apoptosis and reduces tumourigenicity of human melanoma cells. Eur J Cancer. 2006;42:1881–1888. doi: 10.1016/j.ejca.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Salvati E, Leonetti C, Rizzo A, Scarsella M, Mottolese M, Galati R, et al. Telomere damage induced by the G-quadruplex ligand RHPS4 has an antitumor effect. J Clin Invest. 2007;117:3236–3247. doi: 10.1172/JCI32461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonet T, Zaragosi LE, Philippe C, Lebrigand K, Schouteden C, Augereau A, et al. The human TTAGGG repeat factors 1 and 2 bind to a subset of interstitial telomeric sequences and satellite repeats. Cell Res. 2011;21:1028–1038. doi: 10.1038/cr.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Xiong Y, Kim H, He Q, Li Y, Chen R, et al. Human telomeric proteins occupy selective interstitial sites. Cell Res. 2011;21:1013–1027. doi: 10.1038/cr.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Zou Y, Shay JW, Wright WE. Telomere position effect in human cells. Science. 2001;292:2075–2077. doi: 10.1126/science.1062329. [DOI] [PubMed] [Google Scholar]
- Koering CE, Pollice A, Zibella MP, Bauwens S, Puisieux A, Brunori M, et al. Human telomeric position effect is determined by chromosomal context and telomeric chromatin integrity. EMBO Rep. 2002;3:1055–1061. doi: 10.1093/embo-reports/kvf215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottaviani A, Gilson E, Magdinier F. Telomeric position effect: from the yeast paradigm to human pathologies. Biochimie. 2008;90:93–107. doi: 10.1016/j.biochi.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Pedram M, Sprung CN, Gao Q, Lo AW, Reynolds GE, Murnane JP. Telomere position effect and silencing of transgenes near telomeres in the mouse. Mol Cell Biol. 2006;26:1865–1878. doi: 10.1128/MCB.26.5.1865-1878.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Wermuth CG. Selective optimization of side activities: another way for drug discovery. J Med Chem. 2004;47:1303–1314. doi: 10.1021/jm030480f. [DOI] [PubMed] [Google Scholar]
- Malo N, Hanley JA, Carlile G, Liu J, Pelletier J, Thomas D, et al. Experimental design and statistical methods for improved hit detection in high-throughput screening. J Biomol Screen. 2010;15:990–1000. doi: 10.1177/1087057110377497. [DOI] [PubMed] [Google Scholar]
- Ottaviani A, Rival-Gervier S, Boussouar A, Foerster AM, Rondier D, Sacconi S, et al. The D4Z4 macrosatellite repeat acts as a CTCF and A-type lamins-dependent insulator in facio-scapulo-humeral dystrophy. PLoS Genet. 2009;5:e1000394. doi: 10.1371/journal.pgen.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan MH, Lai CS, Wang YJ, Ho CT. Acacetin suppressed LPS-induced up-expression of iNOS and COX-2 in murine macrophages and TPA-induced tumor promotion in mice. Biochem Pharmacol. 2006;72:1293–1303. doi: 10.1016/j.bcp.2006.07.039. [DOI] [PubMed] [Google Scholar]
- Wu HJ, Sun HY, Wu W, Zhang YH, Qin GW, Li GR. Properties and molecular determinants of the natural flavone acacetin for blocking hKv4.3 channels. PLoS ONE. 2013;8:e57864. doi: 10.1371/journal.pone.0057864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Y, Xu JF, Li Y, Chavez L, Riethman HC, Lansdorp PM, et al. Telomere length and the expression of natural telomeric genes in human fibroblasts. Hum Mol Genet. 2003;12:1329–1336. doi: 10.1093/hmg/ddg139. [DOI] [PubMed] [Google Scholar]
- Gabellini D, Green MR, Tupler R. Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell. 2002;110:339–348. doi: 10.1016/s0092-8674(02)00826-7. [DOI] [PubMed] [Google Scholar]
- Rijkers T, Deidda G, van Koningsbruggen S, van Geel M, Lemmers RJ, van Deutekom JC, et al. FRG2, an FSHD candidate gene, is transcriptionally upregulated in differentiating primary myoblast cultures of FSHD patients. J Med Genet. 2004;41:826–836. doi: 10.1136/jmg.2004.019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez P, Thanasoula M, Muñoz P, Liao C, Tejera A, McNees C, et al. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;23:2060–2075. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Lenain C, Bauwens S, Rizzo A, Saint-Léger A, Poulet A, et al. TRF2 and apollo cooperate with topoisomerase 2alpha to protect human telomeres from replicative damage. Cell. 2010;142:230–242. doi: 10.1016/j.cell.2010.05.032. [DOI] [PubMed] [Google Scholar]
- Woodman OL, Chan ECh. Vascular and anti-oxidant actions of flavonols and flavones. Clin Exp Pharmacol Physiol. 2004;31:786–790. doi: 10.1111/j.1440-1681.2004.04072.x. [DOI] [PubMed] [Google Scholar]
- Boege F, Straub T, Kehr A, Boesenberg C, Christiansen K, Andersen A, et al. Selected novel flavones inhibit the DNA binding or the DNA religation step of eukaryotic topoisomerase I. J Biol Chem. 1996;271:2262–2270. doi: 10.1074/jbc.271.4.2262. [DOI] [PubMed] [Google Scholar]
- Bajgai SP, Prachyawarakorn V, Mahidol C, Ruchirawat S, Kittakoop P. Hybrid flavan-chalcones, aromatase and lipoxygenase inhibitors, from Desmos cochinchinensis. Phytochemistry. 2011;72:2062–2067. doi: 10.1016/j.phytochem.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Jaganathan SK, Mandal M. Antiproliferative effects of honey and of its polyphenols: a review. J Biomed Biotechnol. 2009;2009:830616. doi: 10.1155/2009/830616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LZ, Jing Y, Jiang LL, Jiang XE, Jiang Y, Rojanasakul Y, et al. Acacetin inhibits VEGF expression, tumor angiogenesis and growth through AKT/HIF-1a pathway. Biochem Biophys Res Commun. 2011;413:299–305. doi: 10.1016/j.bbrc.2011.08.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottaviani A, Schluth-Bolard C, Rival-Gervier S, Boussouar A, Rondier D, Foerster AM, et al. Identification of a perinuclear positioning element in human subtelomeres that requires A-type lamins and CTCF. EMBO J. 2009;28:2428–2436. doi: 10.1038/emboj.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Screening of molecules affecting human TPE.
Dose response assay.
Effect of acacetin and chrysin on cell cycle and telomere length.
Acacetin and chrysin induce premature growth arrest of IM90 cells.