

Abstract
MicroRNAs are small noncoding RNAs that have critical roles in regulating a number of cellular functions through transcriptional silencing. They have been implicated as oncogenes and tumor suppressor genes (oncomirs) in several human neoplasms. We used an integrated genomics and functional screening strategy to identify potential oncomirs in the pediatric neoplasm neuroblastoma. We first identified microRNAs that map within chromosomal regions that we and others have defined as frequently deleted (1p36, 3p22, and 11q23-24) or gained (17q23) in high-risk neuroblastoma. We then transiently transfected microRNA precursor mimics or inhibitors into a panel of six neuroblastoma cell lines that we characterized for these genomic aberrations. The majority of transfections showed no phenotypic effect, but the miR-34a (1p36) and miR-34c (11q23) mimics showed dramatic growth inhibition in cell lines with 1p36 hemizygous deletion. In contrast, there was no growth inhibition by these mimics in cell lines without 1p36 deletions. Quantitative reverse transcription-PCR showed a perfect correlation of absent miR-34a expression in cell lines with a 1p36 aberration and phenotypic effect after mimetic add-back. Expression of miR-34a was also decreased in primary tumors (n = 54) with 1p36 deletion (P = 0.009), but no mutations were discovered in resequencing of the miR-34a locus in 30 neuroblastoma cell lines. Flow cytometric time series analyses showed that the likely mechanism of miR-34a growth inhibition is through cell cycle arrest followed by apoptosis. BCL2 and MYCN were identified as miR-34a targets and likely mediators of the tumor suppressor phenotypic effect. These data support miR-34a as a tumor suppressor gene in human neuroblastoma.
Introduction
Neuroblastoma is an embryonal tumor of the postganglionic sympathetic nervous system with a median age of diagnosis of 17 months. It is an important childhood disease and remains responsible for 15% of pediatric cancer mortalities (1). Genomic aberrations that correlate strongly with disease aggressiveness have been well characterized, and include high-level amplification of the neural-specific MYC homologue MYCN, unbalanced gain of 17q23-qter, and hemizygous deletion of 1p36 and 11q23-q24 (1-4). Other than MYCN, the genes being targeted by these prognostically relevant chromosomal aberrations are not known.
MicroRNAs are endogenously expressed small noncoding RNAs that are involved in posttranscriptional gene regulation (5). MicroRNA-encoded genes are transcribed by PolII into primiRNAs. They are then processed by Drosha into stem-loop structures called pre-miRNAs and transported out of the nucleus. The enzyme Dicer cleaves a portion of the stem-loop into a 20- to 25-nucleotide double-stranded molecule. One of the strands is incorporated into the RNA-induced silencing complex, which then recognizes partially complementary sequences in the 3′-untranslated region of target mRNAs. The protein product of the target mRNA is down-regulated, either through translation inhibition or mRNA destabilization/sequestration. Recently, microRNAs have been implicated as oncogenes and tumor suppressor genes in several cancers and tend to map to fragile sites within the cancer genome (6-8). As reviewed by Kent and Mendell, experimentally validated tumor suppressors include miR-15a, 16-1, 143, 145, let-7, and 17-5p and oncogenes include miR-155, 17-5p, 21, 372, 18a, 19a-b, 20a, and 92 (7). Recently, miR-34a-c have been shown to be regulated by the tumor suppressor p53 (9-13). Because regulation of microRNA expression also seems critical for normal embryonal development (14), we hypothesized that the loss or gain of microRNAs in the pediatric embryonal tumor neuroblastoma could contribute to the acquisition of an aggressive phenotype that is seen in approximately half of all cases.
Results and Discussion
We identified eight microRNAs that map within chromosomal regions frequently deleted (1p36, 3p22 and 11q23) or gained (17q23) in high-risk neuroblastoma (Table 1; refs. 15, 16). The published common regions of deletion or gain previously listed were queried using the University of California Santa Cruz Genome Browser web site3 Build 36.1 and revealed eight miRNAs. They include miR-34a, miR-200, miR-429, miR-551, miR-34b, miR-34c, miR-26a, and miR-21. We transiently transfected these miRNA precursor mimics or inhibitors into a panel of six neuroblastoma cell lines (SK-NAS, IMR-5, NLF, SY5Y, NBL-S, and SK-N-DZ) that we characterized for these genomic aberrations (16) and monitored their growth (Fig. 1). All but one of the mimics (miR-34b) was able to inhibit the growth of at least one cell line (Table 1). Only miR-34a (1p36.23) and miR-34c (11q23) mimic overexpression caused growth inhibition in several cell lines, as defined in the analysis section as CImax miRNA/CImax ≤ 60%. Interestingly, miR-34a growth inhibition correlated with the 1p36 deletion status (Table 1; Fig. 2). The cell line panel was expanded to include two additional cell lines with (NB16 and LAN-5) and without (NGP and SK-N-SH) a 1p36 deletion. Again, there was a tight genotype/phenotype correlation as all cells with a 1p36 deletion were susceptible to growth inhibition and those without a 1p36 deletion were not inhibited by miR-34a mimic add-back (Fig. 1). NGP is the one apparent discrepancy, but this line has a complicated 1p36 duplication event (17). In contrast, miR-34b had a modest effect on the growth of the cells, despite very similar sequence similarities of the miR-34 family members and comparable posttransfection miR-34b levels by reverse transcription-PCR (RT-PCR; Supplementary Fig. S1). In order to investigate whether miR-34a also decreased tumorigenicity, soft agar assays were done in two 1p36-deleted cell lines. There was a 48% (109 versus 57 colonies) and 32% (92 versus 62 colonies) decrease in colony formation in miR-34a–transfected lines compared with control, in NLF and SK-N-AS, respectively (Fig. 3).
Table 1. Summary of the Functional MicroRNA Screen.
| Genomic Status | Growth Inhibitory Effect | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1p36 | 11q23 | 3p22 | 17q | ||||||||||
| cell line | 1p36 | 3p | 11q | 17q | MYCN | 34A | 200 | 429 | 551 | 34B | 34C | 26A | 21 |
| SKNAS | Y | Y | Y | Y | N | Y | Y | N | N | N | Y | N | N |
| NLF | Y | N | N | Y | Y | Y | N | N | Y | N | Y | Y | N |
| IMR5 | Y | Y | Y | Y | Y | Y | Y | Y | N | N | Y | N | N |
| NB16 | Y | Y | Y | Y | N | Y | N | Y | |||||
| LAN5 | Y | N | N | Y | Y | Y | |||||||
| SY5Y | N | N | N | Y | N | N | Y | N | N | N | N | N | N |
| NBLS | N | N | Y | Y | N | N | N | N | N | N | N | Y | Y |
| SKNDZ | N | N | Y | Y | Y | N | N | N | N | N | N | Y | N |
| SKNSH | N | N | N | Y | N | N | |||||||
| NGP | dup | Y | Y | Y | Y | Y | N | N | |||||
NOTE: miR-34a and miR-34c emerge as candidate tumor suppressor genes from a microRNA screen. The left half of the table summarizes the deletion (1p36, 3p22, and 11q23), gain (17q23), or amplification (MYCN) status of the cell lines used in the panel. The right half of the table summarizes the growth-inhibitory effect of the mimic or inhibitor. “Y” indicates that the growth was inhibited; “N” indicates it was not inhibited. A yellow shaded box indicates concordance between the growth effect and the genomic status for a proposed tumor suppressor gene/oncogene and a pink box indicates discordance. Gray shading for cell lines in the secondary panel indicates that the experiment was not done.
Figure 1.
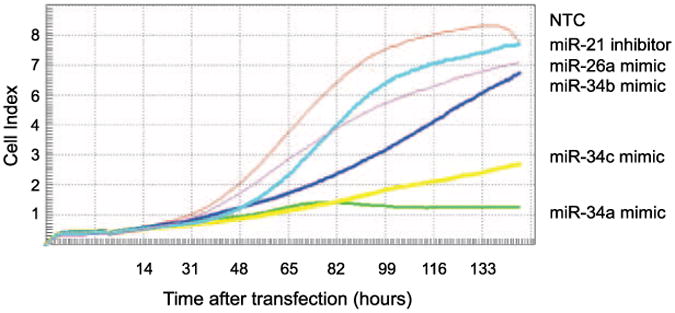
Representative example of the micro-RNA screen results as measured by the RT-CES microelectronic cell sensor system in the cell line SK-N-AS. The nontargeting control microRNA (NTC) produces a growth curve with its maximal cell index of 8.3. miR-34a and miR-34c overexpression have growth-inhibitory effects, whereas miR-34b, miR-26a, and miR-21 inhibition do not. Cell index is a measurement derived from electrical impedance of cells interacting with a microelectrode on the bottom of the 96-well plate.
Figure 2.
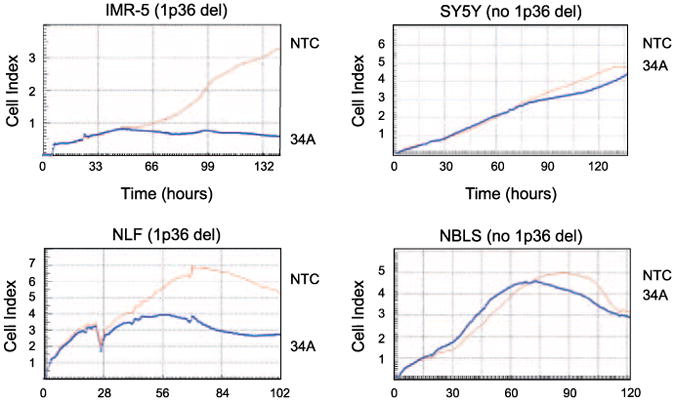
miR-34a inhibits cell lines with a genomic miR-34a deletion on 1p36. RT-CES growth curves show that forced miR-34a overexpression inhibits the growth of neuroblastoma cell lines with a 1p36 deletion (IMR5 and NLF), but does not affect those without a 1p36 deletion (SY-5Y and NBLS). NTC, miRNA nontargeting control; 34A, miR-34a mimic.
Figure 3.

miR-34a overexpression decreases colony formation in soft agar. A. A bar graph compares the number of colonies grown in soft agar from 1p36 deleted cell lines NLF and SK-N-AS transfected with a nontargeting (NTC) microRNA control versus a miR-34a (34A) mimic. Columns, averages from triplicate agar plates; bars, SD; P values calculated by t testing. B. Digital photomicrographs of colonies on agar plates. Magnification, ×10.
We then analyzed miR-34a and miR-34c expression by RT-PCR in the cell line panel used in the screen, and 54 primary neuroblastoma tumors that represent the spectrum of the disease and for which RNA expression levels and genome-wide copy number aberrations were already characterized (15, 16, 18). There was low to absent expression of miR-34a RNA in the cell lines with miR-34a allelic deletion, and high expression in those lines without a miR-34a deletion (Fig. 4A, Table 2). In addition, there was lower miR-34a expression in primary tumors with a hemizygous deletion of the miR-34a locus compared with those without a 1p36 deletion (P = 0.009). miR-34c expression was low or absent in the majority of primary tumors, regardless of 11q23 deletion status, but was expressed abundantly in fetal brain (data not shown). Therefore, given the profound phenotype with mimic overexpression and correlation between endogenous microRNA expression levels and 1p36 deletion status, we prioritized miR-34a for further evaluation.
Figure 4.

A. RT-PCR of endogenous miR-34a levels and endogenous control 5S RNA in the primary cell line screen panel showing absent miR-34a in SK-N-AS, IMR-5, NLF, and NGP. B. Box plot comparing miR-34a expression levels in primary tumor samples with and without a deletion in the region of miR-34a demonstrating that there is a statistically significant decrease in miR-34a expression levels in primary tumors with a 1p36 deletion.
Table 2. miR-34a Genomic Deletion Status Correlates with miR-34a RNA Expression Levels and the Growth-inhibitory Effect of miR-34a Overexpression.
| Cell line | MYCN | miR-34a deletion | miR-34a level | mimic effect |
|---|---|---|---|---|
| SKNAS | N | Y | 0.00 | Y |
| IMR5 | Y | Y | 0.05 | Y |
| NLF | Y | Y | 0.06 | Y |
| LAN5 | Y | Y | 0.70 | Y |
| NB16 | N | Y | 1.40 | Y |
| NGP | N | N* | 0.04 | Y |
| SY5Y | N | N | 20.00 | N |
| NBLS | N | N | 6.60 | N |
| SKNSH | N | N | 7.90 | N |
| SKNDZ | N | N | 3.93 | N |
NOTE: The designation “Y” in the appropriate columns means MYCN-amplified, miR-34a – deleted, or growth-inhibited by miR-34a overexpression. miR-34a level is quantitative measurement of endogenous miR-34a levels measured by quantitative RT-PCR normalized to the control RNU43.
Allele duplicated.
miR-34a is an intergenic noncoding RNA that maps to 1p36.23 (11). The family is highly conserved from Caenorhabditis elegans to Homo sapiens(19). miR-34a was initially identified in retinoblastoma and mouse brain (19, 20), and has reduced expression in several cancer cell lines and primary tumors (9, 10, 21). In neuroblastoma, miR-34a levels have been shown to be low or absent in neuroblastoma cell lines and primary tumors (22), and this report supports these findings in a larger panel. This is the first report, however, to show that there is a correlation between miRNA deletion status, expression level, and growth inhibition with miR-34a overexpression.
Direct resequencing of the mature miR-34a and miR-34c genomic loci in 30 neuroblastoma cell lines showed no sequence variations (data not shown). Other potential mechanisms for silencing the retained miR-34a allele include epigenetic alterations such as hypermethylation or histone acetylation of the miR-34a promoter. The miR-34 family promoter regions each map within predicted CpG islands (11). miR-34a expression has been shown to increase 8-fold after histone deacetylase inhibition, but does not increase after demethylation in a bladder cancer cell line (23). Another mechanism of silencing could be through the disruption of a regulatory sequence. Several recent reports have shown that there is a p53 binding site upstream of miR-34a that, when mutated, decreases the transcription of miR-34a, and when reexpressed, induces genes that regulate apoptosis and cell cycle progression (10-12). Although p53 mutations are common in many adult solid tumors, they are rare in de novo neuroblastoma (24). It has yet to be determined whether there are mutations in this p53 binding site in neuroblastoma that could account for decreased miR-34a expression.
Because miRNAs regulate many aspects of cellular function, we examined the effect of miR-34a overexpression on the cell cycle. In a panel of two high (SY-5Y and NBL-S) and four low (SK-N-AS, IMR-5, NLF and NGP) endogenous miR-34a expressors, we saw initiation of G1 arrest by 48 hours after miRNA mimic transfection only in low endogenous expressors (Fig. 5A-C, Supplementary Fig. S2). There was a decrease in phosphorylated RB in the lines with miR-34a mimic add-back (Fig. 5D). By PI staining alone, few cells were in the sub-G0 fraction after mimic transfection (average 2%, all time points; range, 0.4-7%). Because a sub-G0 peak is only detected by PI and flow cytometry late in apoptosis when the DNA has already fragmented, we assayed for apoptosis using Annexin V. At 48 hours after transfection, there was no increase in Annexin V positivity (3%) in the nontargeting control versus the miR-34a mimic–treated cells (Supplementary Fig. S3). At 72 hours, however, the miR-34a mimic–treated cells were 16% Annexin V–positive. Therefore, we propose that miR-34a–mediated growth inhibition occurs via cell cycle arrest and subsequent programmed cell death.
Figure 5.

miR-34a overexpression causes inhibition of the cell cycle and apoptosis in cell lines with a low endogenous miR-34a. A. Summary of the cell cycle distribution at 48 h after transfection of the nontargeting control microRNA mimic (NTC, open columns) and miR-34a mimic (shaded columns) shows an increase in the G0/G1 peak and decrease in the S and G2-M peaks suggestive of cell cycle inhibition in SK-N-AS, IMR-5, and NLF only. Columns, mean percentage of cells gated; bars, SD. B. Representative cell cycle histogram from NLF at 48 h after transfection. C. Fluorescence-activated cell sorting scatter plot of the nontargeting control mimic microRNA and miR-34a mimic–transfected NLF cells at 72 h show early apoptosis in the miR-34a – transfected cell line (16% vs. 0.81%). Y-axis, PI staining; X-axis, Annexin V staining. Bottom right, Annexin V–positive yet PI-negative cells, indicative of early apoptosis. Top right, double-positive cells suggestive of late apoptosis. D. A Western blot of proteins assayed 24 to 96 h after miR-34a transfection in NLF cells show that there is a decrease in phosphorylated Rb, consistent with cell cycle inhibition.
BCL2 plays an important role in the pathogenesis of many human cancers through its ability to inhibit apoptosis and has been previously shown to play a role in tumorigenesis through dysregulation of microRNAs (6). BCL2 is expressed in neuroblastoma cell lines and primary tumors (25). It is a computationally predicted target of miR-34a in each of the four microRNA prediction target databases we queried [Miranda, Pictar, TargetScan (5-way), and DIANA-microT4] with one to six predicted miR-34a–binding sites in BCL2, and has also been shown to be a direct target of miR-34a (9). In order to determine whether or not BCL2 knockdown would phenocopy forced miR-34a expression, we did a small interfering RNA (siRNA) knockdown of BCL2 in the 1p36-deleted low endogenous miR-34a–expressing cell lines SK-N-AS, NLF, and IMR-5. As shown in Fig. 6A and Supplementary Fig. S4, knockdown of BCL2 protein levels resulted in growth inhibition similar to forced miR-34a overexpression. In addition, miR-34a mimic overexpression resulted in decreased BCL2 mRNA and protein by 48 hours (Fig. 6B; Supplementary Fig. S4). This suggests that in some neuroblastoma cell lines, the effect of miR-34a–mediated BCL2 protein inhibition may also be in part mediated by an effect on BCL2 mRNA.
Figure 6.
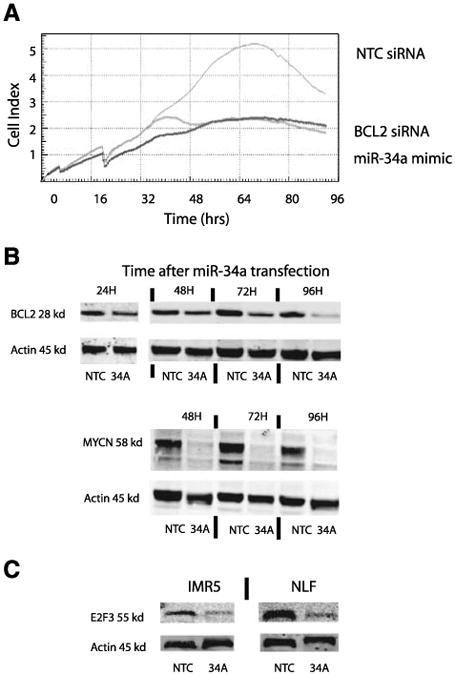
Inhibition of BCL2 may mediate some of the growth-inhibitory effects of miR-34a overexpression. A. Knockdown of BCL2 by siRNA phenocopies miR-34a growth inhibition of NLF cells as measured by the RT-CES microelectronic cell sensor system. B. A time course of miR-34a overexpression in NLF shows that BCL2 and MYCN protein expression decreases over time. NTC, nontargeting control–treated cell line; 34A, the miR-34a mimic–treated cell line. C. miR-34a transfection down-regulates E2F3 protein expression in NLF and IMR-5 cell lines by 48 h.
We were able to confirm that the computationally predicted miR-34a target neuroblastoma oncogene MYCN also had decreased protein expression after miR-34a transfection (Fig. 4B; ref. 26). Although Welch et al. (22) were unable to document direct miR-34a targeting of MYCN, Wei et al. show MYCN to be a direct target of miR-34a in MYCN-amplified neuroblastoma cell lines (26). Regardless of whether direct or indirect, the result of abolished high MYCN protein levels after miR-34a transfection is profound, and likely to mediate some of the growth inhibition seen with miR-34a overexpression. siRNA knockdown of MYCN in neuroblastoma has been shown to result in cell cycle arrest, decreased CDK6 levels, and decreased Rb phosphorylation (27, 28). Therefore, decreased MYCN protein levels could contribute to our finding of cell cycle arrest, apoptosis, and decreased Rb phosphorylation. However, recent studies highlight that miRNAs influence entire networks and pathways, and their effect is not likely due to a single targeted mRNA. Gene expression arrays analysis after miR-34 induction (9, 11) highlight this fact by demonstrating significant down-regulation of multiple genes involved in cell cycle control with validation of direct effect of the miR-34 computationally predicted targets E2F3/5, CDK4/6, and cyclin E. There are likely many other direct and indirect targets that cause miR-34a cell cycle arrest and apoptosis in neuroblastoma. Welch et al. showed that miR-34a causes down-regulation of E2F3 (22). We confirmed this finding in our miR-34a–deleted cell lines NLF and IMR5 (Fig. 6C) and are using both genomic and proteomic approaches to identify other targets in our tumor model.
It has been known for over two decades that somatically acquired deletion of chromosome band 1p36 is associated with an aggressive neuroblastoma phenotype, but identification of the genes targeted by these deletions has proven elusive. Recently, there is evidence that TP73(29), CHD5(30, 31), KIF1B(32), CAMTA1(30), and CASTOR(33) might be 1p36 neuroblastoma suppressor genes, but mutations in these protein encoding genes are extremely rare. Our data suggest that miR-34a is a strong candidate for a neuroblastoma suppressor gene as we showed a very tight correlation with 1p allelic status, miR-34a expression, and functional consequence of forced miR-34a expression. Like other examples of candidate 1p36 suppressor genes, mutations of the retained allele are not a common mechanism towards achieving biallelic inactivation. Additional work is necessary to understand the mechanism for loss of miR-34a and miR-34c expression in neuroblastoma, but it is now clear that functional inactivation of microRNAs are very likely involved in the evolution of high-risk neuroblastomas. Ongoing work should define whether or not microRNAs are also involved in neuroblastoma initiation, and/or if precise definition of microRNA targets might lead to novel therapeutic strategies for patients with high-risk neuroblastoma.
Materials and Methods
Cell Culture
Neuroblastoma cell lines SK-N-AS, IMR-5, NLF, LA-N-5, NB16, NGP, SY5Y, NBL-S, SK-N-SH, and SK-N-DZ were obtained from the CHOP neuroblastoma cell line bank and cultured as described (15). In order to verify cell line integrity, all lines were routinely genotyped (AmpFLSTR identifier kit; Applied Biosystems), and Mycoplasma tested.
Functional miRNA Screen
A total of 1 × 10 to 3 × 10 neuroblastoma cells were plated in triplicate overnight in antibiotic-free complete medium in the 96-well RT-CES microelectronic cell sensor system (ACEA; ref. 34). The cells were then transiently transfected with 200 μL containing 10 nmol/L of a mature microRNA mimic or inhibitor, nontargeting negative control, and positive control (PLK siRNA) using 0.1% v/v Dharmafect I, according to the manufacturer's protocol (Dharmacon). All miRNA mimics, inhibitors, and siRNAs were from Dharmacon. In brief, 35 μL of 0.2 Amol/L miRNA mimic and 35 μL of serum-free medium were combined with 0.7 μL of Dharmafect I in 70 μL of serum-free media and incubated for 20 min at room temperature, and then 560 μL of antibiotic-free complete medium was added. The culture medium was gently removed from the plated cells and replaced by 200 μL of fresh medium containing the microRNA mimic or inhibitor. Cell growth was monitored continuously and recorded as a cell index every 30 min for a minimum of 96 h. The “cell index” is derived from the change in electrical impedance as the living cells interact with the biocompatible microelectrode surface in the microplate well effectively measuring cell number, shape, and adherence (34). TaqMan MicroRNA RT-PCR assays (ABI) were done for the miR-34a, miR-34b, and miR-34c mimics in SK-N-AS and for the miR-34a mimic in all cell lines in the panel to verify mimic overexpression (described below).
Soft Agar Tumorigenicity Assay
miR-34a–deleted cell lines NLF and SK-N-AS were transfected with a 10 nmol/L miR-34a mimic and nontargeting control mimic as described above. After 12 h, cells were harvested and 5,000 cells were plated in 0.35% soft agar in triplicate (35). After 16 days of growth, viable colonies were stained with 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium blue liquid substrate (Sigma) overnight and then photographed with a Nikon 4500 CoolPix Digital Camera mounted on a Nikon SMZ dissection microscope. Jpg digital images were opened in Adobe Photoshop 6.0, magnified 100%, and colonies that were greater than or equal in size to a 5-pixel standard were marked and counted. Results are displayed as an average of the triplicates with SDs, with P values obtained by paired t test. Representative digital photomicrographs are 10× magnification.
MicroRNA RT-PCR
Total RNA was isolated according to Ambion's mirVana microRNA extraction protocol. The mature miR-34a, miR-34b, miR-34c, and control RNU43 were measured by TaqMan microRNA RT-PCR on the ABI 7900 Real-time PCR System according to the manufacturer's protocol (ABI). Expression values were quantified with corresponding standard curves. Cell line results are from two independent experiments done in triplicate, and primary tumor samples were assayed as a single experiment in triplicate. End point RT-PCR of miR-34a, miR-34b, miR-34c, and 5S were done on 50 ng of cell line total RNA using the mirVana RT-PCR miRNA Detection Kit (Ambion). Products were separated and visualized on a 4% agarose gel.
Flow Cytometry
A total of 1 × 10 cultured cells were transiently transfected as described above with the 10 nmol/L miR-34a mimic or nontargeting control for 24, 48, 72, and/or 96 h. For DNA content analysis, harvested cells were fixed overnight in ethanol, stained with PI with RNase (BD PharMingen) and analyzed on a FACSCalibur flow cytometer. For apoptosis analysis, harvested cells were incubated with an Annexin V-FITC antibody and PI, with appropriate controls (BD Phar-Mingen) and analyzed on a Becton Dickinson analytic FACSCalibur flow cytometer. The 48-h time points were done in triplicate and the other time points were single experiments.
SiRNA Knockdown of BCL2
A total of 1 × 104 SK-N-AS, NLF, and IMR-5 cells were plated in triplicate in the RT-CES microelectronic cell sensor system (ACEA) or a parallel 96-well plate for mRNA knockdown analysis. Twenty hours later, nontargeting control and BCL2 siRNA knockdown was done with 10 nmol/L of ONTARGETplus SMART POOL siRNA (Dharmacon) as described above for the microRNA mimic transfection. Impedance was monitored continuously and recorded as a cell index every 30 min for a minimum of 96 h. Three independent experiments were done in triplicate.
BCL2 RT-PCR
Forty-eight hours after siRNA or miRNA transfection, total RNA was extracted from the cells that had been plated in a 96-well plates using the Qiagen mini extraction kit, with DNase treatment. Two hundred nanograms of total RNA was oligo-dT primed and reverse transcribed using Superscript II reverse transcriptase (Invitrogen). BCL2, HPRT, TBP, and cyclophilin B expression levels were measured by quantitative RT-PCR using TaqMan gene expression assays (ABI), quantified on corresponding standard curves, and normalized to the geometric mean of the three housekeeping genes (18). Two independent experiments were done in triplicate.
Immunoblotting
Forty micrograms of neuroblastoma cell lysates collected at 24, 48, 72, and/or 96 h after miR-34a mimic transfection were separated on an 8% to 12% NuPage Gel with MES buffer (Invitrogen) and transferred to a polyvinylidene difluoride membrane. Primary antibody dilutions included 1:500 BCL2 (sc-7382) and 1:500 E2F3 (sc-28308; Santa Cruz Biotechnology), 1:500 phosphorylated RB1 (Cell Signaling), 1:300 MYCN (BD PharMingen), and 1:1,000 h-actin (Cell Signaling). Membranes were incubated in primary antibody in Odyssey Blocking buffer (Li-Cor) 1:1 v/v with PBS overnight at 4 °C. The membranes were washed in 0.1% PBS-T and then incubated for 1 h at room temperature with 1:1,000 fluorescently labeled secondary antibody. After washing, fluorescently labeled secondary antibodies were visualized and quantified on the Odyssey Infrared Imaging Center (Li-Cor).
Genomic Sequencing
PCR products encoding the pre-miRNA sequences were amplified from genomic DNA from 30 cell lines with the following primers (pre–miR-34a forward, 5′cctccccacatttccttctt; reverse, 5′ggcatctctcgcttcatctt; pre–miR-34b forward, 5′cagctacgcgtgttgtgc; reverse, 5′caggcatcttctctcgaagg; and pre–miR-34c forward, 5′aaagcaaaatccctggaggt; reverse, 5′gatgcacaggcagctcatt), purified and then bidirectly sequenced on a ABI 3730 DNA Analyzer using the same primers as for PCR amplification. DNA chromatograms were visually inspected for potential sequence variants.
Analysis and Statistical Methods
In the cell line functional screen, microRNAs were defined as growth-inhibitory if the ratio of the mimic-treated cell index to the nontargeting control cell index was ≤60% at the time when the nontargeting control reached its maximal cell index.
For miR-34a expression comparisons, cell line and primary tumor samples were defined as having a genomic deletion in the region of miR-34a as analyzed on a customized bacterial artificial chromosome–based microarray platform (16), compared by paired t testing and displayed as box plots.
Supplementary Material
Acknowledgments
Grant support: NIH grants R01-CA87847 (J.M. Maris), R01-CA39771 (G.M. Brodeur), U10-CA98543 (Children's Oncology Group), the Alex's Lemonade Stand Foundation (J.M. Maris), Super Jake Foundation (K.A. Cole), NIH T32 training grant (K.A. Cole), the Audrey Evans Endowed Chair (G.M. Brodeur), and the Abramson Family Cancer Research Institute (J.M. Maris).
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–20. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 2.Bown N, Cotterill S, Lastowska M, et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N Engl J Med. 1999;340:1954–61. doi: 10.1056/NEJM199906243402504. [DOI] [PubMed] [Google Scholar]
- 3.Attiyeh EF, London WB, Mosse YP, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med. 2005;353:2243–53. doi: 10.1056/NEJMoa052399. [DOI] [PubMed] [Google Scholar]
- 4.Michels E, Vandesompele J, De Preter K, et al. ArrayCGH-based classification of neuroblastoma into genomic subgroups. Genes Chromosomes Cancer. 2007;46:1098–108. doi: 10.1002/gcc.20496. [DOI] [PubMed] [Google Scholar]
- 5.Engels BM, Hutvagner G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene. 2006;25:6163–9. doi: 10.1038/sj.onc.1209909. [DOI] [PubMed] [Google Scholar]
- 6.Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 7.Kent OA, Mendell JT. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene. 2006;25:6188–96. doi: 10.1038/sj.onc.1209913. [DOI] [PubMed] [Google Scholar]
- 8.Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bommer GT, Gerin I, Feng Y, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 10.Chang TC, Wentzel EA, Kent OA, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–52. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raver-Shapira N, Marciano E, Meiri E, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–43. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 13.Tarasov V, Jung P, Verdoodt B, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G(1)-arrest. Cell Cycle. 2007;6 doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- 14.Plasterk RH. Micro RNAs in animal development. Cell. 2006;124:877–81. doi: 10.1016/j.cell.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 15.Mosse YP, Greshock J, Margolin A, et al. High-resolution detection and mapping of genomic DNA alterations in neuroblastoma. Genes Chromosomes Cancer. 2005;43:390–403. doi: 10.1002/gcc.20198. [DOI] [PubMed] [Google Scholar]
- 16.Mosse YP, Diskin SJ, Wasserman N, et al. Neuroblastomas have distinct genomic DNA profiles that predict clinical phenotype and regional gene expression. Genes Chromosomes Cancer. 2007;46:936–49. doi: 10.1002/gcc.20477. [DOI] [PubMed] [Google Scholar]
- 17.Amler LC, Bauer A, Corvi R, et al. Identification and characterization of novel genes located at the t(1;15)(p36.2;q24) translocation breakpoint in the neuroblastoma cell line NGP. Genomics. 2000;64:195–202. doi: 10.1006/geno.1999.6097. [DOI] [PubMed] [Google Scholar]
- 18.Wang Q, Diskin S, Rappaport E, et al. Integrative genomics identifies distinct molecular classes of neuroblastoma and shows that multiple genes are targeted by regional alterations in DNA copy number. Cancer Res. 2006;66:6050–62. doi: 10.1158/0008-5472.CAN-05-4618. [DOI] [PubMed] [Google Scholar]
- 19.Dostie J, Mourelatos Z, Yang M, Sharma A, Dreyfuss G. Numerous microRNPs in neuronal cells containing novel microRNAs. RNA. 2003;9:180–6. doi: 10.1261/rna.2141503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–9. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 21.Gaur A, Jewell DA, Liang Y, et al. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 2007;67:2456–68. doi: 10.1158/0008-5472.CAN-06-2698. [DOI] [PubMed] [Google Scholar]
- 22.Welch C, Chen Y, Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007;26:5017–22. doi: 10.1038/sj.onc.1210293. [DOI] [PubMed] [Google Scholar]
- 23.Saito Y, Liang G, Egger G, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–43. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 24.Carr J, Bell E, Pearson AD, et al. Increased frequency of aberrations in the p53/MDM2/p14(ARF) pathway in neuroblastoma cell lines established at relapse. Cancer Res. 2006;66:2138–45. doi: 10.1158/0008-5472.CAN-05-2623. [DOI] [PubMed] [Google Scholar]
- 25.Reed JC, Meister L, Tanaka S, et al. Differential expression of bcl2 protooncogene in neuroblastoma and other human tumor cell lines of neural origin. Cancer Res. 1991;51:6529–38. [PubMed] [Google Scholar]
- 26.Wei JS, Song Y, Chen Q, Durinck S, Yu L, Veenstra T, Khan J. miR-34a is a potential tumor suppressor gene on 1p36 in neuroblastoma; American Association for Cancer Research Annual Meeting; Los Angeles CA. April 14-18, 2007. [Google Scholar]
- 27.Bell E, Premkumar R, Carr J, et al. The role of MYCN in the failure of MYCN amplified neuroblastoma cell lines to G1 arrest after DNA damage. Cell Cycle. 2006;5:2639–47. doi: 10.4161/cc.5.22.3443. [DOI] [PubMed] [Google Scholar]
- 28.Woo CW, Tan F, Cassano H, Lee J, Lee KC, Thiele CJ. Use of RNA interference to elucidate the effect of MYCN on cell cycle in neuroblastoma. Pediatr Blood Cancer. 2008;50:208–12. doi: 10.1002/pbc.21195. [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa T, Takahashi M, Ozaki T, et al. Negative autoregulation of p73 and p53 by DNp73 in regulating differentiation and survival of human neuroblastoma cells. Cancer Lett. 2003;197:105–9. doi: 10.1016/s0304-3835(03)00090-9. [DOI] [PubMed] [Google Scholar]
- 30.Okawa ER, Gotoh T, Manne J, et al. Expression and sequence analysis of candidates for the 1p36.31 tumor suppressor gene deleted in neuroblastomas. Oncogene. 2008;27:803–10. doi: 10.1038/sj.onc.1210675. [DOI] [PubMed] [Google Scholar]
- 31.Bagchi A, Papazoglu C, Wu Y, et al. CHD5 is a tumor suppressor at human 1p36. Cell. 2007;128:459–75. doi: 10.1016/j.cell.2006.11.052. [DOI] [PubMed] [Google Scholar]
- 32.Caren H, Ejeskar K, Fransson S, et al. A cluster of genes located in 1p36 are down-regulated in neuroblastomas with poor prognosis, but not due to CpG island methylation. Mol Cancer. 2005;4:10. doi: 10.1186/1476-4598-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Z, Yang X, Tan F, Cullion K, Thiele CJ. Molecular cloning and characterization of human Castor, a novel human gene upregulated during cell differentiation. Biochem Biophys Res Commun. 2006;344:834–44. doi: 10.1016/j.bbrc.2006.03.207. [DOI] [PubMed] [Google Scholar]
- 34.Kirstein SL, Atienza JM, Xi B, et al. Live cell quality control and utility of real-time cell electronic sensing for assay development. Assay Drug Dev Technol. 2006;4:545–53. doi: 10.1089/adt.2006.4.545. [DOI] [PubMed] [Google Scholar]
- 35.To C, Seiden I, Liu N, Wigle D, Tsao MS. High expression of Met/hepatocyte growth factor receptor suppresses tumorigenicity in NCI-H1264 lung carcinoma cells. Exp Cell Res. 2002;273:45–53. doi: 10.1006/excr.2001.5433. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.