

ABSTRACT
Clathrin-mediated endocytosis (CME) is conserved among eukaryotes and has been extensively analyzed at a molecular level. Here, we present an analysis of CME in the human fungal pathogen Candida albicans that shows the same modular structure as those in other fungi and mammalian cells. Intriguingly, C. albicans is perfectly viable in the absence of Arp2/3, an essential component of CME in other systems. In C. albicans, Arp2/3 function remains essential for CME as all 15 proteins tested that participate in CME, including clathrin, lose their characteristic dynamics observed in wild-type (WT) cells. However, since arp2/3 cells are still able to endocytose lipids and fluid-phase markers, but not the Ste2 and Mup1 plasma membrane proteins, there must be an alternate clathrin-independent pathway we term Arp2/3-independent endocytosis (AIE). Characterization of AIE shows that endocytosis in arp2 mutants relies on actin cables and other Arp2/3-independent actin structures, as inhibition of actin functions prevented cargo uptake in arp2/3 mutants. Transmission electron microscopy (TEM) showed that arp2/3 mutants still formed invaginating tubules, cell structures whose proper functions are believed to heavily rely on Arp2/3. Finally, Prk1 and Sjl2, two proteins involved in patch disassembly during CME, were not correctly localized to sites of endocytosis in arp2 mutants, implying a role of Arp2/3 in CME patch disassembly. Overall, C. albicans contains an alternative endocytic pathway (AIE) that relies on actin cable function to permit clathrin-independent endocytosis (CIE) and provides a system to further explore alternate endocytic routes that likely exist in fungal species.
IMPORTANCE
There is a well-established process of endocytosis that is generally used by eukaryotic cells termed clathrin-mediated endocytosis (CME). Although the details are somewhat different between lower and higher eukaryotes, CME appears to be the dominant endocytic process in all eukaryotes. While fungi such as Saccharomyces cerevisiae have proven excellent models for dissecting the molecular details of endocytosis, loss of CME is so detrimental that it has been difficult to study alternate pathways functioning in its absence. Although the fungal pathogen Candida albicans has a CME pathway that functions similarly to that of S. cerevisiae, inactivation of this pathway does not compromise growth of yeast-form C. albicans. In these cells, lipids and fluid-phase molecules are still endocytosed in an actin-dependent manner, but membrane proteins are not. Thus, C. albicans provides a powerful model for the analysis of CME-independent endocytosis in lower eukaryotes.
Introduction
Endocytosis is a fundamental mechanism cells employ both to capture macromolecules from the extracellular environment and to selectively recycle and internalize membrane lipids and membrane-bound proteins (1, 2). One of the first described endocytic pathways is clathrin-mediated endocytosis (CME) (3). Today, CME is well characterized and has been identified as evolutionarily conserved from humans to fungi. Molecular mechanisms governing CME have been elucidated in great detail with the help of live-cell, multicolor fluorescent microscopy techniques. Studies in Saccharomyces cerevisiae as well as in mammalian cells have shaped the picture of CME as a highly coordinated process in which more than 60 proteins cooperate to drive vesicle internalization (4). In mammalian cells, these proteins are grouped into 7 functional modules, while S. cerevisiae has 5 such functional modules (5–7). The 5 modules in S. cerevisiae are (i) the early module, (ii) the coat module, (iii) the Wiskott-Aldrich syndrome protein/myosin (Myo/WASP) module, (iv) the actin module, and (v) the scission (or amphiphysin) module (4, 8–12).
During CME in yeast, Syp1 of the early module is among the first proteins to arrive at the nascent endocytic site (12, 13). The elements of the coat module arrive next—first clathrin followed by other coat components like Sla2 (a homologue of mammalian HIP1-R) and the EH-domain proteins End3 and Pan1 (14, 15). Collectively, the functions of clathrin, End3, Pan1, and Sla2 as part of the early and coat modules include, respectively, initial membrane bending and linking coat components to the actin cytoskeleton. The Myo/WASP module arrives subsequently; this includes Vrp1 (the yeast WIP homologue) and several Arp2/3 complex activators, like Las17 (the yeast WASP homologue) and type I myosins (Myo3 and Myo5). The actin module is recruited next and includes in part actin, Arp2/3, Cap1 and Cap2 (two capping proteins), the fimbrin Sac6 (an actin-bundling protein), Abp1 (an actin binding protein), two kinases (Prk1 and Ark1) that regulate endocytic proteins, and the phosphatidylinositol-4,5-bisphosphate phosphatase Sjl2 (5, 16, 17). Together, the components of the actin module orchestrate the completion of an endocytic event (in concert with the scission module). Specifically, the function of the Arp2/3-mediated branched actin network is proposed to provide the force that together with type I myosin motor activity extends vesicle invagination (15, 18). As the nascent endocytic vesicle invaginates, facilitated by the dynamin-like protein Vps1, two amphiphysins (Rvs161 and Rvs167) of the scission module are recruited and help release the vesicle, which then traffics along actin filaments to ultimately fuse with endosomes (19–22).
In addition to our detailed understanding of CME, recent studies have started to examine alternate pathways that do not rely on clathrin. Examples of such clathrin-independent endocytosis (CIE) are found in Caenorhabditis elegans, Drosophila melanogaster, and mammalian cells (23–25). The functions of CIE include endocytosis of membrane and fluids, which can reach up to 70% of the endocytic capacity of a cell (26–28). In fungi, however, it was not clear whether CIE exists, as CME was long thought to be the sole endocytic route (29). However, one recent report has described a Rho1-dependent pathway in the yeast Saccharomyces cerevisiae that seems to be clathrin independent (30). Conveniently, the fungal pathogen Candida albicans has the potential to functionally and unambiguously separate different endocytic pathways. In C. albicans, robust endocytosis occurs in the absence of one key player that drives CME, the Arp2/3 complex (31). Thus, this molecularly manipulable dimorphic fungus can allow the study of different endocytic processes; such alternate endocytic pathways may be important in a human fungal pathogen that needs to recycle damaged plasma membrane and cell wall components to cope with the plasma membrane and cell-wall-targeting drugs used to treat patients in hospitals.
In this study, we investigate Arp2/3-independent endocytosis (AIE) and CME in C. albicans. We first establish that in C. albicans, CME is driven by an equivalent modular design, with individual proteins showing functional similarities compared to other fungal systems. We then provide evidence that AIE does not rely on any of the functional modules involved in CME, including clathrin. Furthermore, AIE differs from CME in that two plasma membrane (PM) proteins are not endocytosed, suggesting a cargo-specific role of AIE versus CME. We further provide evidence that actin cables are important for endocytosis during AIE. Finally, using electron microscopy (EM) to visualize invaginating tubules, our data show that these structures still form in the absence of Arp2/3, raising the possibility that AIE could rely on Arp2/3-independent invaginating tubule formation to take up cargo. Collectively, our data identify novel features of a CIE pathway (AIE) in C. albicans and lay the groundwork to further elucidate how endocytosis works without Arp2/3.
RESULTS
Analysis of CME in C. albicans.
We initially investigated whether CME works in a modular design in C. albicans similar to that in S. cerevisiae. To do this, we recorded the dynamics of green fluorescent protein (GFP)-tagged proteins at every stage of the CME pathway using high-speed spinning-disk confocal or total internal reflection fluorescence (TIRF) microscopy (see Movie S1 in the supplemental material). We then applied several forms of quantitative motion analysis to assess CME; we measured the mean square displacement (MSD) of GFP signals over time, calculated the percentage of patches that left the origin; determined the average lateral track displacement within the plane of the plasma membrane (from now on referred to as “average track displacement length”), and established the average track duration time (Fig. 1 and 2). Syp1 represents one of the earliest components of CME in yeast. Analysis in C. albicans similarly showed that when Arp2/3 was present, Syp1 of the early module had an average track displacement length of 0.08 µm (standard deviation [SD], ±0.05 µm) and an average track duration of 101 s (SD, ±60 s) and that 86% of Syp1 signals left the origin.
FIG 1 .

CME is heavily affected without Arp2/3. Dynamics of GFP-tagged components at every stage of CME were recorded in live cells by spinning-disk confocal microscopy and quantitatively analyzed. Recorded displacement data of individual patches (n > 20) were aligned at the start of their lifetime and then averaged. MSD plots for WT cells were truncated at the time when 50% of spots have disappeared, corresponding to the median lifetime. For mutant cells, MSD plots represent the first 70 s of recording. Components of the early (A), coat (B), Myo/WASP (C), actin (D), and scission (E) module show dynamic behavior in WT cells. In arp2 cells, early, coat, and Myo/WASP components lost dynamics and remained static at the plasma membrane, while some actin module as well as scission module components retained partial dynamics in arp2 mutants. Prk1 and Sjl2 did not localize to any clear patch structures, and the dynamics could not be quantitatively analyzed in arp2 cells. The results for WT cells are in blue, and those for arp2 cells are in red. MSD plots correspond to means ± SEM. See Movies S1 and S2 in the supplemental material.
FIG 2 .

Arp2/3 is required for productive functioning of CME. Live-cell imaging data were analyzed for three additional quantitative elements to demonstrate that CME becomes nonproductive without Arp2/3. (A) The percentage of patches (n > 20) leaving the origin was defined in two ways; patches that disappeared (applied for the early, coat, Myo/WASP, and scission modules) and patches that moved more than 100 nm from the sites of first detection (applied for the actin module). In a few cases, Pan1 and End3 patches disappeared in arp2 cells, which likely correspond to patches moving out of the focal plane. (B) Track displacement length measures the distance between first and last patch detection, in arp2 cells corresponding to a maximum of 128 s. (C) Track duration refers to how long a patch was observed over a 2-min period. All graphs are expressed as means ± SD.
The coat components analyzed (Sla2, Pan1, and End3) showed an initial stationary phase followed by a mobile phase when patches moved away from the membrane. The average track durations varied and ranged from 66 ± 22 s (Sla2) and 63 ± 30 s (End3) to 36 ± 11 s (Pan1), while the average track displacement length and the percentage of patches leaving the origin were similar for all coat components, with displacement lengths of 0.20 ± 0.12 µm (Pan1), 0.22 ± 0.12 µm (End3), and 0.32 ± 0.18 µm (Sla2) and at least 97% of patches leaving the origin. The average track duration for Myo/WASP components varied between 13 ± 5 s (Myo5 and Vrp1) and 31 ± 23 s (Wal1), with all components showing similar average track displacement lengths, which were 0.07 ± 0.03 µm (Vrp1), 0.07 ± 0.05 µm (Myo5), and 0.08 ± 0.05 µm (Wal1), and with 100% of patches leaving the origin.
The actin module components showed average track durations and average track displacement lengths for Cap2, Cap1, Sac6, and Abp1 of 11 ± 4, 13 ± 5, 16 ± 6, and 20 ± 8 s, and 0.38 ± 0.19, 0.41 ± 0.27, 0.45 ± 0.30, and 0.49 ± 0.24 µm, respectively. All observed patches left the origin. Sjl2 and Prk1 had average track durations of 10 ± 6 and 15 ± 6 s, respectively. Average track displacements were 0.38 ± 0.26 µm for Sjl2 and 0.41 ± 0.23 µm for Prk1, and all Sjl2 and Prk1 spots left the origin. Finally, Rvs167 of the scission module showed an average track duration of 5 ± 2 s and average displacement length of 0.15 ± 0.10 µm, with 100% of observed Rvs167 signals disappearing from the origin.
Overall, the analysis showed that, in the presence of Arp2/3, CME components display similar dynamic characteristics and a modular design as observed in other fungal and mammalian species, thus emphasizing the conservation of CME among eukaryotes.
Endocytosis can occur in the absence of Arp2/3.
We have previously demonstrated that arp2/3 mutants are viable in C. albicans (31). Here, we used these deletion mutants to study Arp2/3-independent endocytosis (AIE). We began by testing whether CME components are still functional without Arp2/3. For Syp1, the average track displacement length was 0.04 ± 0.03 µm, the average track duration was >120 s (128 s was the maximum length of observation), and no Syp1 signals disappeared (see Fig. 1 and 2; see Movie S1 in the supplemental material). Components of the coat module and the Myo/WASP module correctly localized to the plasma membrane but remained static and did not cycle off the membrane, demonstrating that losing Arp2/3 complex function has a profound impact on these early modules.
Imaging of actin patches in arp2 cells was more difficult as the same patch signal tended to drift in and out of the focal plane. Consequently, it was unclear whether patches internalized or whether they showed increased lateral movement along the membrane. We therefore analyzed patch behavior of actin module components in arp2 mutants by recording 20 z stacks over time. This demonstrated that more than 70% of patches move at least 100 nm from where they were first detected, with an average displacement length of 0.27 ± 0.27 µm (Cap2), 0.20 ± 0.17 µm (Cap1), 0.43 ± 0.45 µm (Sac6), and 0.29 ± 0.34 µm (Abp1). Importantly, almost all patches in arp2 mutants were detected during the entire length of observation (128 s) and did not appear to internalize, which is in stark contrast to WT cells, where nearly 100% of patch signals internalized (Fig. 2; see Movie S1 in the supplemental material). The number of patches was reduced in arp2 cells more than 4-fold with usually 20 or less Cap1, Cap2, Sac6 or Abp1 GFP signals present. These signals frequently appeared as actin filament-like structures, suggesting that the signals may be remnants of nonfunctional patches, thus supporting the view that Arp2/3 is instrumental for the proper movement and turnover of the tested actin patch module components. Similar actin filament structures have previously been observed in various CME-deficient S. cerevisiae strains (18, 32, 30).
Analyzing patch dynamics for Prk1, Rvs161, and Sjl2 was not possible in arp2 mutants as Prk1 and Rvs161 were not clearly localized to any structures and Sjl2 appeared associated with filamentous-like structures (see Movie S1 in the supplemental material; data for Rvs161 not shown). Finally, only 14.6% of Rvs167 spots disappeared from the origin, with the remaining 85.4% of signals staying stationary at the membrane. The few spots (14.6%) that disappeared in arp2 cells showed WT-like tracking durations, but when all Rvs167 signals were averaged, the track duration and track displacement length for Rvs167 were >111 ± 27 s and 0.08 ± 0.08 µm in arp2 cells, respectively, indicating that the scission module is heavily affected.
Remarkably, when imaging GFP coupled to clathrin light chain (Clc1) in the absence of Arp2/3, we found that clathrin existed in two populations: one remained static at the membrane and in the vast majority of cases (≥95%, n ≥ 200) was detected during the entire time of observation (128 s), while the second clathrin population appeared intracellular and retained dynamics similar to those of the WT cells. In order to further establish whether clathrin is involved in AIE, we observed Clc1 by TIRF microscopy in arp2 cells. Time-lapse movies over 6 min (see Movie S2 in the supplemental material) confirmed the spinning-disk microscopy observations above that membrane-bound clathrin remains static at the membrane in arp2 cells and does not internalize, suggesting that the clathrin light chain is dispensable for AIE at the plasma membrane.
Overall, our detailed analysis of CME in the absence of Arp2/3 demonstrated that the CME pathway is dramatically affected, with most components, including clathrin, completely losing their functionality at sites of endocytosis. This suggests that any cargo uptake in AIE must follow a clathrin-independent endocytic pathway in C. albicans.
Ste2 and Mup1 PM proteins are not endocytosed through AIE in C. albicans.
In mammalian cells, different CIE pathways are cargo specific. In C. albicans, AIE allows endocytosis of lipids and fluids (31), but it was unclear whether AIE is capable of internalizing PM proteins. To test this, we used the GFP-labeled pheromone receptor Ste2 as a marker for receptor-mediated endocytosis. While the GFP-signal disappeared from the membrane in WT cells after 40 min of pheromone induction, the signal remained stable at the membrane in the absence of Arp2/3 (Fig. 3A). When we used a second inducible marker for PM protein endocytosis, the methionine permease Mup1, we found the same results: no GFP signal was detected at the plasma membrane in WT cells after addition of methionine, but signals persisted at the membrane in the absence of Arp2/3 (Fig. 3B). To rule out that the vacuolar background staining observed in the Ste2 and Mup1 assays was not camouflaging minor uptake capacities in the absence of Arp2/3, we developed pHluorin (34) for use in C. albicans. pHluorin is a pH-sensitive GFP variant that is detected at cytosolic pH but not at the lower pH in the vacuole (35). Mup1-pHluorin disappeared completely in WT cells after 60 min of incubation, while Mup1-pHluorin was still detected at the plasma membrane in cells lacking Arp2/3, even after extended overnight incubation periods (see below).
FIG 3 .
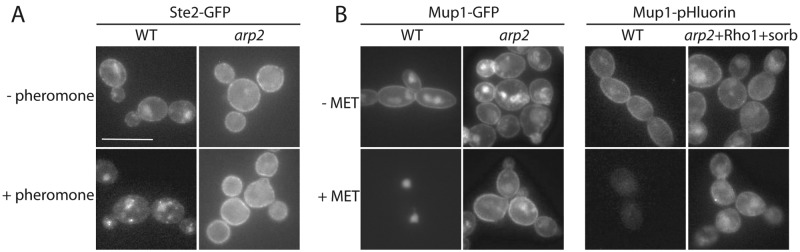
Plasma membrane proteins (Ste2 and Mup1) are not endocytosed without Arp2/3. (A) Internalization of the Ste2 pheromone receptor was assayed after 40-min incubation in the presence of α-factor pheromone. This results in dominant vacuolar accumulation of Ste2-GFP in WT cells with some signals visible as vesicles trafficking en route to the vacuole. In arp2 cells, no strong vacuolar signal or moving vesicles were observed during time-lapse movies (not shown). (B) Mup1 internalization was stimulated by the addition of methionine. Mup1-GFP signal disappeared in WT cells at the plasma membrane, but it was still visible in arp2 cells (right). When Mup1 was coupled to the pH-sensitive GFP variant, pHluorin, the Mup1-pHluorin signal almost completely disappeared in WT cells upon addition of methionine, while Mup1-pHluorin was still observed in arp2 cells that carried a CaRho1 overexpression, treated with sorbitol, and incubated overnight (left). Bar, 10 µm.
Because a recent study suggested that relieving turgor pressure by providing osmotic support reduces the requirement for actin during endocytosis in S. cerevisiae (36), we grew the Arp2/3-deficient cells in the presence of 1.5 M sorbitol to test whether this can suppress defects in PM protein endocytosis. As well, based on a report showing that increased Rho1 activity suppresses PM protein endocytosis in S. cerevisiae adaptor mutants (ENT1, ENT2, Yap1801, and Yap1802) (30), we hypothesized that overexpression of the functional homolog of S. cerevisiae Rho1p in C. albicans (CaRho1p) might allow Mup1 endocytosis in Arp2/3 mutants (37). While quantitative PCR (qPCR) analysis showed that we could overexpress the C. albicans RH01 (CaRHO1) message 8.5-fold in arp2 cells, no difference in substrate uptake was observed in the presence of either this CaRho1 overexpression, of osmotic support, or of a combination of the two in terms of Mup1-pHluorin endocytosis in arp2 mutants, even after extended overnight incubations (Fig. 3B). These results show that not every cargo (e.g., Ste2 and Mup1 PM proteins) is endocytosed through AIE in C. albicans.
Actin cable dynamics are not blocked in the absence of Arp2/3.
Studies in fungal cells suggest that CIE relies on actin cables (30). Therefore, to investigate whether actin cables and other Arp2/3-independent actin structures play a role during AIE in C. albicans, we first addressed whether actin dynamics are affected during AIE in C. albicans. In order to visualize the actin cytoskeleton in live cells, we expressed LIFEACT-GFP (38) in WT cells, which labeled actin patches and cables, while in arp2/3 mutants, no actin patches were observed. In general, two populations of actin cables were observed in WT and arp2 cells: long cables that elongated and depolymerized and shorter, fast-moving cables that did not appear to grow or shrink. The first actin cable population was observed in small and midsize budded cells, originating at the bud neck and elongating toward the mother cell along the polarity axis. Cable depolymerization proceeded in the same direction, starting from the bud neck toward the mother cell. The rates of actin polymerization for long actin cables were similar in the WT (0.20 ± 0.07 µm/s) and arp2 mutants (0.22 ± 0.04 µm/s, n > 10 in each case). In addition to these long cables that usually span from the bud neck to the distal side of the cell, a second population of actin cables was observed that were typically short, originating from different places and moving almost twice as fast as long cables polymerized; 0.47 ± 0.13 µm/s for WT and 0.49 ± 0.12 µm/s for arp2 cells (n > 10) (Fig. 4; see Movie S3 in the supplemental material). These observations corroborate previous findings of actin filament dynamics in WT cells of S. cerevisiae (39).
FIG 4 .

Actin filament dynamics are not affected without Arp2/3 function. LIFEACT-GFP was used to visualize actin dynamics and showed that the rate of actin filament elongation was not affected in arp2 cells. Two populations of actin filaments observed in WT and arp2 cells are illustrated by arrowheads (first image in the time series) and arrows (subsequent images). See Movie S3 in the supplemental material.
Together, our data demonstrate that actin cable dynamics are not dramatically affected in the absence of Arp2/3. However, we did observe that actin cables appeared less oriented and often disorganized, with cables frequently aligning perpendicularly to the polarity axis in arp2 cells (not shown). Overall, this analysis suggests that actin cables retain function in the absence of the Arp2/3 complex during AIE in C. albicans.
Cargo uptake is actin dependent during AIE.
To investigate directly whether actin structures are involved in lipid endocytosis (FM4-64 uptake) during AIE, we blocked actin dynamics with cytochalasin A (CA). Because CA completely abolished all fluorescent signals in arp2 mutants carrying LIFEACT-GFP, while WT cells were more resistant and showed nonmotile actin patches at the membrane (Fig. 5A), we performed the following experiments with arp2 cells only. We blocked actin dynamics and then added FM4-64, washed all nonadherent dye away, and chased dye internalization while CA (or dimethyl sulfoxide [DMSO] for control cultures) was continuously present, before blocking endocytosis. This procedure showed that, whereas 95% of control cells treated with DMSO had FM4-64 internalization and showed a clear vacuolar staining, none of the CA-treated cells showed dye uptake. In most cases, the dye appeared evenly distributed at the plasma membrane, and only occasionally did we observe the dye as concentrated punctae at the periphery. Such dye accumulation at the plasma membrane likely indicates that cargo loading into vesicles is not affected in arp2/3 mutants, which is consistent with previous time-lapse observations (31). To rule out that cells were dead after CA treatment and to show that the effect of CA is reversible, we performed two control experiments. First, arp2 cells were assessed for viability after treatment with CA and found to be as viable as when treated with DMSO (Fig. 5B, bottom). Second, CA-treated cells were washed and recovery of actin dynamics was assessed at 4, 8, and 24 h, showing that at 4 and 8 h, only a few cells (2% and 11%, respectively) showed actin dynamics and that at 24 h the majority of cells (>90%) had regained actin dynamics. Together, these data demonstrate that arp2 mutants endocytose FM4-64 in an actin-dependent manner, suggesting that cargo uptake is an active process that depends on actin cable filaments and possibly other Arp2/3-independent structures during AIE.
FIG 5 .

FM4-64 endocytosis is actin dependent in arp2 mutants. (A, top) Schematic representation of the experimental setup. Cells were treated with 10 µg/ml cytochalasin A (CA) or DMSO for 3 min, at which time LIFEACT-GFP signals were assessed. Only arp2 cells completely lost LIFEACT-GFP signals (left). In a second experiment, arp2 cells were treated with 10 µg/ml CA for 3 min, FM4-64 was added at 20 µg/ml for 5 min, unbound dye was washed away and cells were chased for another 20 min to allow dye internalization. During this 30-min procedure, either DMSO or CA was present in the corresponding solutions. Endocytosis was blocked after 30 min by adding sodium azide and sodium fluoride, and arp2 cells were assessed for endocytic capacity (right, n > 150). (B) Two control experiments demonstrating that treatment of arp2 cells for 30 min with CA does not significantly reduce viability (left), and blocking actin dynamics with CA is reversible with cells recovering almost completely after 24 h of washing CA away (right). The bottom represents the schematic setup of the experiment in panel B. All experiments were done with arp2 mutants, expect the left figure in part A.
Arp2/3 mutants show invaginating tubules.
Arp2/3 nucleation has been suggested to provide the force to drive the invagination and scission process during CME (15, 18). To test whether invaginating tubules still form in the absence of Arp2/3 in C. albicans, we examined invaginating structures using transmission electron microscopy (TEM). Invaginating tubules were on average 121 and 117 nm long in WT and ARP2 revertant cells, respectively (Fig. 6A), standing in good agreement with studies in S. cerevisiae (40). Surprisingly, in arp2/3 mutants invaginating structures still formed but were on average longer than those in WT cells, ranging from 179 to 202 nm in arp2 and arp2/arp3 mutants, respectively. In addition, invaginating structures appeared uniformly tubular in control cells with maximum diameters of 100 nm, while in mutant cells, some invagination-like structures appeared close to the membrane, with diameters exceeding 100 nm (Fig. 6B). Together, the data suggest that invaginating structures originating from the plasma membrane still form in the absence of Arp2/3 and might play a role during AIE (see Discussion).
FIG 6 .

Invaginating tubules form independently of Arp2/3. (A) Transmission electron microscopy (TEM) showed that on average invaginating tubules are longer in arp2/3 mutants compared to control strains. Statistical significance was assessed using Student’s t test. (B) Invagination diameters at the membrane do not exceed 100 nm in control cells, while in arp2/3 mutants invagination diameters were larger than 100 nm in up to 28% (arp2) or 24% (arp2/arp3) of cases. (C) Examples of invaginating tubules. Bar, 100 nm.
DISCUSSION
The discovery of clathrin-mediated endocytosis (CME) more than 4 decades ago has provided the first fundamental paradigm of how cells traffic extracellular material and cell membrane components into the cell (3, 23). CME is a highly regulated process and conserved from yeast to mammals (5, 6). Recently, several endocytotic pathways that do not rely on clathrin have emerged, for instance, in Caenorhabditis elegans, Drosophila melanogaster, and in mammalian cells (23, 24, 41). However, in fungi CME was long thought to be the sole route of endocytosis, a view that has only recently started to change (29, 30).
Here, we examined Arp2/3-independent endocytosis (AIE) in the human pathogen C. albicans and compared it to CME. We demonstrate that (i) the molecular mechanisms of CME in WT C. albicans cells are similar to those in other eukaryotic systems, supporting a high level of evolutionary conservation of CME; (ii) that none of the 15 proteins tested, including clathrin, that participate in CME appear to contribute to AIE since they do not show proper functional characteristics, therefore highlighting how fundamentally differently these pathways operate in this fungal species; and (iii) that AIE is dependent on Arp2/3-independent actin structures, most likely actin cables.
Arp2/3 function during CME.
The involvement of the Arp2/3 complex in CME has been well documented and includes, for example, organization of different components of the CME machinery (42–45). Much of this current knowledge has been obtained through mutational analysis coupled to live-cell microscopy in either S. cerevisiae or Schizosaccharomyces pombe, two model organisms that are inviable in the absence of the Arp2/3 complex. In contrast, Arp2/3 is not essential for survival in C. albicans and arp2/3 mutants of this fungus are capable of lipid- and fluid-phase endocytosis, albeit at slightly reduced rates (31). Using this resource, our data show that every stage of the CME pathway tested becomes inactive without Arp2/3 (Fig. 1 and 2; see Movie S1 in the supplemental material).
Among the most dramatic effects of losing Arp2/3 is that some CME components (Prk1, Sjl2, and Rvs161) were not localized to any clear structures (see Movie S1 in the supplemental material; data for Rvs161 not shown). This confirms that the Arp2/3 complex is necessary to correctly localize different components of the CME machinery to sites of endocytosis (43–45), which in the case of Prk1 could directly depend on Arp2 and Arc40, two physical interaction partners of the Arp2/3 complex (46, 47). Besides this known function, our data revealed another previously unappreciated role of Arp2/3: disassembly of CME modules. Without Arp2/3, Prk1 and Sjl2 are not correctly localized to sites of endocytosis, but they are necessary in WT cells for separate actin patch disassembly mechanisms during CME (44, 48–51). Therefore, Arp2/3 appears to contribute to actin patch disassembly by facilitating recruitment of Prk1 and Sjl2 to sites of endocytosis. The lack of proper localization of various disassembly factors would also provide an explanation why components of the early, coat, and Myo/WASP module are trapped at the plasma membrane in arp2 mutants. Taken together, besides establishing the conserved nature of CME in WT cells in C. albicans, our data suggest that Arp2/3 contributes to different patch disassembly processes during CME.
Given the abundant scientific evidence that links Arp2/3 to the invagination and scission processes during CME (15, 18), we were surprised to see invaginating structures in arp2/3 mutants (Fig. 6). Of note, invaginating structures have previously been observed in several S. cerevisiae mutants that lack components at critical stages of the CME machinery (lipid-phase segregation or the vesicle scission machinery); both sjl1 sjl2 and vps1 mutants in S. cerevisiae show membrane invaginations (21, 52). Our TEM data clearly link Arp2/3 to membrane invaginations; invaginations are longer and have wider necks close to the membrane in arp2/3 mutants. Future studies are required to delineate the precise function of the invaginations in arp2/3 mutants and whether such structures represent CME-independent routes of endocytosis.
AIE in C. albicans.
One surprising observation was that clathrin light chain was divided into two distinct populations in arp2/3 mutants, as observed by time-lapse microscopy: one population lost its dynamic behavior and remained static at sites of endocytosis at the plasma membrane, while the second population retained some intracellular dynamics (see Movies S1 and S2 in the supplemental material). The observation that plasma membrane-bound clathrin did not turn around, together with the fact that clathrin light chain did not appear to cycle between the membrane-bound and intracellular locations in arp2 cells, suggests that endocytosis in the absence of Arp2/3 is clathrin-independent. The mobile, intracellular clathrin population is likely involved in intracellular trafficking at the trans-Golgi network (TGN). It remains to be established, however, whether these intracellular clathrin signals are fully functional: for instance, whether budding of clathrin-coated vesicles (CCV) without Arp2/3 occurs, as a previous report in S. cerevisiae has shown that Arp2/3 accumulates at TGN-derived CCVs (53). The evidence presented here indicates that some aspects of the overall clathrin population in arp2/3 mutants do play essential roles and hence are Arp2/3 independent, as deletion of the clathrin heavy and light chains in C. albicans is lethal (see Fig. S1 in the supplemental material).
Why does membrane-bound clathrin require Arp2/3 to carry out receptor uptake? It is possible that receptors are not internalized during AIE because clathrin is missing the Arp2/3-driven force generation to endocytose such bulky cargo, while fluid and lipids are less bulky and accordingly do not require the combination of a structured clathrin cage, the Arp2/3 force, as well as other components of CME to endocytose. How cargo passes the plasma membrane during AIE without the apparent help of clathrin and how the dye (FM4-64) is trafficked to the vacuole are interesting questions that remain to be investigated. Time-lapse movies of FM4-64 uptake suggest that vesicles are not involved (31), or if they are, they are too faint or small to be detected by current imaging techniques.
Clathrin-independent endocytic pathways in fungal cells.
In mammalian cells, different endocytotic pathways exist that do not require clathrin. Examples of such clathrin-independent pathways include caveolae and caveolin endocytosis, macropinocytosis, flotillin-dependent endocytosis, Arf6-associated uptake as well as RhoA-dependent and Cdc42-dependent uptake routes (26). In fungal cells, the only described clathrin-independent pathway is mediated by Rho1, a small GTPase from the Rho/Rac subfamily, in S. cerevisiae (30). Both Rho1-dependent endocytosis in S. cerevisiae and AIE in C. albicans appear to rely on clathrin-independent endocytic strategies, so one can wonder whether AIE uses similar mechanisms as the Rho1-mediated pathway (Fig. 7). We started to address this question by testing PM protein endocytosis in arp2 mutants after overexpressing CaRho1, the functional homolog of S. cerevisiae Rho1p (38). When CaRho1 was overexpressed in C. albicans, PM protein (Mup1 and Ste2) endocytosis did not occur, even after addition of osmotic support, a condition that has previously been shown to reduce the requirement of actin during endocytosis (36). Concluding that Rho1-mediated endocytosis and AIE are two distinct pathways seems premature. It remains a possibility that CaRho1 does not have the same effector, Bni1, in C. albicans, as in S. cerevisiae, where Bni1 is a direct effector of Rho1. Support of this comes from the S. pombe homolog of Bni1 in yeast, For3, which is an effector for Cdc42 and Rho3 but not Rho1 (54).
FIG 7 .

Model of clathrin-mediated endocytosis (CME) and clathrin-independent endocytosis (CIE) in fungal cells. (A) CME in Candida albicans, Saccharomyces cerevisiae, and Schizosaccharomyces pombe work in a similarly modular way. First, the early module arrives at sites of endocytosis, followed by proteins of the coat, then the Myo/WASP, the actin, and finally the amphyphysin module. After vesicle scission, the cargo-loaded vesicle traffics via endosomes to the vacuole. (B) Currently, two CIE pathways in fungal cells have been described: Rho1-dependent CIE in S. cerevisiae and Arp2/3-independent CIE in C. albicans. Both routes seem not to rely on protein modules of the CME pathway, and yet cargo such as the membrane-impermeable dye FM4-64 still accumulate in the vacuole over time. Rho1-CIE relies on Rho1 and Bni1, while both Rho1-dependent and Arp2/3-independent CIE rely on Arp2/3-independent actin structures. If and how cargo is loaded into vesicles and how cargo crosses the membrane and traffics to the vacuole are unknown both in Rho1-CIE and Arp2/3-independent CIE. Whether Rho1-CIE and Arp2/3-independent endocytosis are one or two distinct ways for CIE remains to be shown. See text for details.
Independent of whether Rho1 and AIE share the same or distinct pathways, our data support the idea that actin cables are involved in AIE; the chemical disruption of actin cables did not allow any endocytosis in arp2/3 mutants (Fig. 5). We therefore suggest that Arp2/3-unrelated actin functions could compensate for endocytotic uptake in arp2/3 mutants, an idea that is supported by microarray experiments that showed that in arp2/3 mutants, several actin-related genes were overexpressed (31).
Similar to mammalian systems, several CIE pathways might exist in fungal species. Clearly, the details of CIE in fungal organisms warrant further investigation, particularly whether CIE pathways play similar roles in fungi as they do in mammalian cells, where CIE routes can contribute the majority of membrane and fluid fraction taken up into the cell (up to 70%) and are involved in repairing plasma membrane, cellular polarization, as well as modulation of intercellular signaling (26–28). Taking advantage of C. albicans to functionally differentiate AIE from other endocytic pathways, this pathogenic fungus provides an ideal starting point to expand our knowledge of how endocytosis works, particularly in the context of how damaged plasma membrane or cell wall components are recycled while being exposed to clinically relevant plasma membrane/cell wall-targeting drugs. Advances in our understanding of endocytosis might one day help to combat fungal drug resistance and ultimately lead to improved therapy options for patients.
MATERIALS AND METHODS
Strains and media.
All strains are derived from SN148 (55) and are listed in Table S1 in the supplemental material together with primers used in this study, which are listed in Table S2 in the supplemental material. Strain construction followed standard transformation protocols, using homologous recombination for genomic marker integration, and selection on synthetic media (see below) that contained the necessary auxotrophic supplements, as described previously (56). Correct marker insertion was verified by diagnostic PCR (57). In order to have sufficient auxotrophic markers available for subsequent experiments, we first constructed strain CaEE341, which served as the arp2 deletion background throughout this study. CaEE341 was created by deleting the first ARP2 allele as described (31), using LEU2 from Candida maltose as a marker (58). The second ARP2 allele was deleted with the nourseothricin-based approach, using pSFS2A as described previously (56). Briefly, 485 bp of upstream homologous sequence to ARP2 was amplified with primers oEE166 and oEE382 and cloned in pEE33 (31), between the KpnI/XhoI sites. This resulted in plasmid pEE59, which was digested with KpnI and SacII prior to transformation.
To tag proteins with GFP, 120-mer oligonucleotides were used to amplify CaGFPγ (59) cassettes and inserted at the 3′ end of the open reading frame (ORF), thus not interfering with the endogenous promoters. For all GFP-tagged strains that we used for quantitative motion analysis, the second allele was disrupted, thus leaving only the GFP-tagged version in the genome. These GFP-tagged strains did not show any significant defects in terms of growth rate or morphology, and all GFP-tagged strains created in the WT background formed hyphae, indicating that the GFP constructs did not significantly interfere with functionality (see Table S1 in the supplemental material).
In order to fuse STE2 to GFP, we used plasmid pADH-STE2-GFP (60). STE2 pheromone receptor internalization was carried out by adding 30 µg/ml of a synthetic 13-amino-acid version of the α-factor mating pheromone dissolved in 50% methanol (1 mg/ml). Cells were imaged after 40 min of pheromone addition.
Strains were routinely grown in standard YPD medium (1% yeast extract, 2% peptone, 2% dextrose, supplemented with 50 mg/ml uridine), synthetic complete (SC) medium (2% dextrose, 6.7% yeast nitrogen base without amino acids, 1.5% of a complete amino acid mix, supplemented with 50 mg/ml of each uridine, histidine, leucine, and arginine), or SC−met medium, which is identical to SC medium, except that SC−met medium lacked methionine and was adjusted to pH 5.5. Mup1 internalization was induced by adding 20 µg/ml of methionine to cells grown overnight in SC−met medium. Because arp2 cells grew poorly in SC−met medium, we incubated arp2 mutants carrying Mup1-GFP or Mup1-pHluorin in YPD overnight and then diluted cells into fresh SC or SC−met medium before imaging. Sorbitol was added for 45 min in strain CaEE720. For experiments involving cytochalasin A (CA), the drug was dissolved to 1 mg/ml in DMSO and stored at −20°C. Cells were grown in SC medium for all experiments involving CA. The FM4-64 uptake was done as previously described in SC medium (31), except that endocytosis was blocked by the addition of sodium azide and sodium fluoride (final concentration, 10 mM).
Plasmids.
The LIFEACT-GFP plasmid (pEE113) was constructed by amplifying the CaGFPγ sequence (59) with primers oEE653 and oEE663 and cloned between PstI and SpeI sites in pFA-GFP-URA3 (58). Primer oEE653 contains the first 51 amino acids from S. cerevisiae ABP140, which is sufficient for actin binding (39). Primers oEE653 and oEE663 also contain 100 bp of homology to up- and downstream regions of ACT1, which are used to insert pEE113 at the actin locus. Prior to transformation, pEE113 was digested with PstI and SpeI. Expression of LIFEACT-GFP did not appear to affect C. albicans physiology as hyphal formation was not influenced in WT cells and the sensitivity to cytochalasin A (CA), a drug that binds barbed ends of actin filaments (61, 62), was the same as that in the control cells (data not shown).
The pH-sensitive GFP variant pHluorin was created based on the superecliptic pHluorin sequence (63). In order to do this, the QuikChange multisite-directed mutagenesis kit (Agilent Technologies) was used to exchange the following sites in CaGFPγ (59): S147D, N149Q, S202F, Q204T, A206T, Q80R, T167I, S175G, using primers oEE622 and oEE623 and oEE680, oEE681, and oEE682. This resulted in plasmid pCaEE114.
Overexpression of RHO1 was achieved using the actin promoter from pACT1 (64) fused to the Rho1 coding sequence, resulting in plasmid pACT1-RHO1, where the Rho1 sequence was amplified with primers oEE705 and oEE723 and inserted between the HindIII and XmaI sites. pACT1-RHO1 was digested with StuI and inserted at the RPS1 locus. Rho1 overexpression was confirmed by real-time quantitative PCR as described previously (65), using as a reference the coding sequence for constitutively expressed C. albicans snoRNAs (primers pAsCaSnoR46F1 and pAsCaSnoR46R1) (64). All plasmids created in this study were verified by sequencing.
Fluorescence microscopy.
Cells were grown to the log phase in SC or SC−met medium, gently concentrated, and applied to precleaned slides, which were covered with coverslips. Slide cleaning was done as described previously (66). Only yeast-phase cells were used for microscopy. Cells were grown at 30°C for wide-field microscopy and at room temperature for confocal microscopy. For wide-field microscopy, an inverted Leica DMIRE2 microscope with a 100× immersion oil objective and a 10× projection lens was used. Images were acquired using OpenLab (version 5.5.0). For confocal microscopy, image data were collected using a customized WaveFX spinning-disk confocal microscope (Quorum Technologies) built on a Leica DM6000 inverted body, with a 100-by-1.46 NA Plan-Apochromat objective, piezo stage (ASI), and DAQ control board, laser merge module (Spectral), 493- and 561-nm solid-state lasers (Coherent), and a water-cooled bt-EMCCD camera with photon counting enabled (Hamamatsu). Metamorph (version 7.7.3.0.) was used for image acquisition. Data were collected in two ways—either (i) in one medial focal plane with a 50- to 200-ms exposure time at intervals of 50 ms to 1 s for a duration of 2 to 6 min or (ii) as stacks (200-nm z steps in 20 focal planes) with a capture time of ~2.5 s/stack for 2 to 6 min. Metamorph’s stream function for time and stack was used when acquiring data as stacks, which resulted in negligibly small intervals between stacks.
TIRF microscopy was done using a Zeiss Axiovert Z1 microscope chassis, 100-by-1.45 NA Plan-Apochromat objective lens, and the Zeiss TIRF III slider. Diode-pumped solid-state lasers (Cobolt Jive, Cobolt Calypso) were coupled to fiber optic cables in free space and introduced into the Zeiss slider. Images were recorded using an Andor iXon+ DV-897 EMCCD camera and MetaMorph software.
Image analysis.
Quantitative motion analysis of patch dynamics was done using Imaris ×64 (version 7.2.3) developed by Bitplane AG. Between 20 and 120 spots were quantitatively analyzed for every component of the clathrin-mediated pathway. Briefly, the spot detection algorithm in Imaris was used to measure spot dynamics over time by setting the estimated diameter to 0.35 µm with the background subtraction selected. The quality attribute in the filter type setting was manually used to set the signal threshold in order to distinguish background from patch signals. The autoregressive motion algorithm was used with parameters set between 0.5 and 0.1 µm for maximum distance (“Max Distance”), and the “Max Gap Size” option was set between 1 and 3. All tracks created this way were visually inspected and had to satisfy the following criteria: (i) the patch was initially present at the cell cortex, (ii) the patch was clearly distinguishable from other patches (i.e., did not merge with another patch), and (iii) the entire lifetime from patch appearance to disappearance was recorded. The last criterion did not apply to arp2 cells, as patches often did not disappear. Statistics in the form of mean square displacement (MSD), track duration, and track displacement length were exported into Excel, which was used to create graphs. MSD plots for WT cells were truncated when 50% of the patches had disappeared, thus representing the median lifetime, or in the case of arp2 cells, MSD plots represent the first 70 s of analysis. “Cells leaving origin” was defined as signals that either moved more than 100 nm from where they were first detected (applied for the actin module) or signals that disappeared (applied for the early, coat, Myo/WASP, and scission modules).
Electron microscopy.
Samples used for transmission electron microcopy (TEM) were processed according to standard techniques as described previously (60). Briefly, samples were fixed with 3% EM-grade glutaraldehyde in 1× sodium cacodylate buffer (pH 7.4) at room temperature. After fixation, samples were washed in buffer and resuspended in an aqueous 4% permanganate solution, washed, stained with uranyl acetate, dehydrated through a graded series of ethyl alcohol, and embedded in Epon resin. Ultrathin sections of 80 nm were cut with a Reichert-Jung (Heidelberg, Germany) UltracutE ultramicrotome, placed on Formvar-coated slot copper grids, and viewed with a FEI Tecnai12 BioTwinG2 electron microscope (Eindhoven, The Netherlands). Digital images were acquired with an AMT XR-60 CCD digital camera system. The TEM analyses were carried out at Stony Brook University at the Microscopy Imaging Center.
SUPPLEMENTAL MATERIAL
Strains used in this study.
Oligonucleotides used in this study.
Clathrin is essential for survival in C. albicans. Conditional mutants of clathrin heavy chain (chc1/tetO-CHC1) or light chain (clc1/tetO-CLC1) were created using a tetracycline-regulatable cassette (tetO) (67). Cells were grown for 4 days on SC medium, which allows growth, or on SC medium containing 100 µg/ml of tetracycline, which turns off the respective promoter. Growth of clc1/tetO-CLC1 was severely affected on repressing conditions and no single colony of chc1/tetO-CHC1 was observed. Two independently derived isolates were tested for clc1/tetO-CLC1. Download
Summary of dynamic behavior of GFP-tagged components in CME. Images were acquired by spinning disk confocal microscopy for at least 2 min in one single medial focal plane. Components in the pathway are grouped into modules. Download
Clathrin remains static at the plasma membrane in arp2 cells. WT and arp2 cells carrying Clc1-GFP were imaged by TIRF microscopy for 6 min. In WT cells, clathrin frequently appears and disappears from the membrane, while in arp2 cells, all clathrin signal remained static at the membrane. Download
Actin dynamics in the absence of Arp2/3. LIFEACT coupled to GFP was used to visualize actin dynamics in living cells. Spinning disk confocal images acquired over 4 min in one single medial focal plane showed two populations of actin filaments in WT and arp2 cells. See Fig. 4 and the description in the text for details. Download
ACKNOWLEDGMENTS
We thank Judith Lacoste (McGill CIAN Facility) for assisting with confocal microscopy, Aleksandrs Spurmanis and Claire Brown (McGill Imaging Facility) for advice on image analysis, and Cory Glowinski (Bitplane AG) for advice on using Imaris. We also thank Susanne Bechstedt, Michal Wieczorek, and Gary Brouhard (McGill) for assisting with TIRF microscopy and Andrea Walther and Jürgen Wendland (Carlsberg Laboratory) for sharing reagents.
This work was supported by CIHR grants MOP42516 to M.W. and MOP64404 to J.V. and NIH grant RO1AI047837 to J.B.K. E.E. was supported by a CIHR Systems Biology Scholarship and the Carpenter Fellowship. E.N. was supported through a CDMC CREATE fellowship.
Footnotes
Citation Epp E, Nazarova E, Regan H, Douglas LM, Konopka JB, Vogel J, Whiteway M. 2013. Clathrin- and Arp2/3-independent endocytosis in the fungal pathogen Candida albicans. mBio 4(5):e00476-13. doi:10.1128/mBio.00476-13.
REFERENCES
- 1. Doherty GJ, McMahon HT. 2009. Mechanisms of endocytosis. Annu. Rev. Biochem. 78:857–902 [DOI] [PubMed] [Google Scholar]
- 2. Kirchhausen T. 2009. Imaging endocytic clathrin structures in living cells. Trends Cell Biol. 19:596–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pearse BM. 1976. Clathrin: a unique protein associated with intracellular transfer of membrane by coated vesicles. Proc. Natl. Acad. Sci. U. S. A. 73:1255–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kaksonen M, Toret CP, Drubin DG. 2006. Harnessing actin dynamics for clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 7:404–414 [DOI] [PubMed] [Google Scholar]
- 5. Kaksonen M, Toret CP, Drubin DG. 2005. A modular design for the clathrin- and actin-mediated endocytosis machinery. Cell 123:305–320 [DOI] [PubMed] [Google Scholar]
- 6. Taylor MJ, Perrais D, Merrifield CJ. 2011. A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PLOS Biol. 9:e1000604. 10.1371/journal.pbio.1000604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Doyon JB, Zeitler B, Cheng J, Cheng AT, Cherone JM, Santiago Y, Lee AH, Vo TD, Doyon Y, Miller JC, Paschon DE, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Drubin DG. 2011. Rapid and efficient clathrin-mediated endocytosis revealed in genome-edited mammalian cells. Nat. Cell Biol. 13:331–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Galletta BJ, Cooper JA. 2009. Actin and endocytosis: mechanisms and phylogeny. Curr. Opin. Cell Biol. 21:20–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Galletta BJ, Mooren OL, Cooper JA. 2010. Actin dynamics and endocytosis in yeast and mammals. Curr. Opin. Biotechnol. 21:604–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robertson AS, Smythe E, Ayscough KR. 2009. Functions of actin in endocytosis. Cell. Mol. Life Sci. 66:2049–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Girao H, Geli MI, Idrissi FZ. 2008. Actin in the endocytic pathway: from yeast to mammals. FEBS Lett. 582:2112–2119 [DOI] [PubMed] [Google Scholar]
- 12. Stimpson HE, Toret CP, Cheng AT, Pauly BS, Drubin DG. 2009. Early-arriving Syp1p and Ede1p function in endocytic site placement and formation in budding yeast. Mol. Biol. Cell 20:4640–4651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boettner DR, D'Agostino JL, Torres OT, Daugherty-Clarke K, Uygur A, Reider A, Wendland B, Lemmon SK, Goode BL. 2009. The F-BAR protein Syp1 negatively regulates WASp-Arp2/3 complex activity during endocytic patch formation. Curr. Biol. 19:1979–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Newpher TM, Smith RP, Lemmon V, Lemmon SK. 2005. In vivo dynamics of clathrin and its adaptor-dependent recruitment to the actin-based endocytic machinery in yeast. Dev. Cell 9:87–98 [DOI] [PubMed] [Google Scholar]
- 15. Kaksonen M, Sun Y, Drubin DG. 2003. A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell 115:475–487 [DOI] [PubMed] [Google Scholar]
- 16. Tonikian R, Xin X, Toret CP, Gfeller D, Landgraf C, Panni S, Paoluzi S, Castagnoli L, Currell B, Seshagiri S, Yu H, Winsor B, Vidal M, Gerstein MB, Bader GD, Volkmer R, Cesareni G, Drubin DG, Kim PM, Sidhu SS, Boone C. 2009. Bayesian modeling of the yeast SH3 domain interactome predicts spatiotemporal dynamics of endocytosis proteins. PLOS Biol. 7:e1000218. 10.1371/journal.pbio.1000218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Toret CP, Lee L, Sekiya-Kawasaki M, Drubin DG. 2008. Multiple pathways regulate endocytic coat disassembly in Saccharomyces cerevisiae for optimal downstream trafficking. Traffic 9:848–859 [DOI] [PubMed] [Google Scholar]
- 18. Sun Y, Martin AC, Drubin DG. 2006. Endocytic internalization in budding yeast requires coordinated actin nucleation and myosin motor activity. Dev. Cell 11:33–46 [DOI] [PubMed] [Google Scholar]
- 19. Huckaba TM, Gay AC, Pantalena LF, Yang HC, Pon LA. 2004. Live cell imaging of the assembly, disassembly, and actin cable-dependent movement of endosomes and actin patches in the budding yeast, Saccharomyces cerevisiae. J. Cell Biol. 167:519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Toshima JY, Toshima J, Kaksonen M, Martin AC, King DS, Drubin DG. 2006. Spatial dynamics of receptor-mediated endocytic trafficking in budding yeast revealed by using fluorescent alpha-factor derivatives. Proc. Natl. Acad. Sci. U. S. A. 103:5793–5798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smaczynska-de Rooij II, Allwood AG, Aghamohammadzadeh S, Hettema EH, Goldenberg MW, Ayscough KR. 2010. A role for the dynamin-like protein Vps1 during endocytosis in yeast. J. Cell Sci. 123:3496–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Douglas LM, Martin SW, Konopka JB. 2009. BAR domain proteins Rvs161 and Rvs167 contribute to Candida albicans endocytosis, morphogenesis, and virulence. Infect. Immun. 77:4150–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mayor S, Pagano RE. 2007. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 8:603–612 [DOI] [PubMed] [Google Scholar]
- 24. Glodowski DR, Chen CC, Schaefer H, Grant BD, Rongo C. 2007. RAB-10 regulates glutamate receptor recycling in a cholesterol-dependent endocytosis pathway. Mol. Biol. Cell 18:4387–4396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Windler SL, Bilder D. 2010. Endocytic internalization routes required for delta/notch signaling. Curr. Biol. 20:538–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sandvig K, Pust S, Skotland T, van Deurs B. 2011. Clathrin-independent endocytosis: mechanisms and function. Curr. Opin. Cell Biol. 23:413–420 [DOI] [PubMed] [Google Scholar]
- 27. Howes MT, Kirkham M, Riches J, Cortese K, Walser PJ, Simpson F, Hill MM, Jones A, Lundmark R, Lindsay MR, Hernandez-Deviez DJ, Hadzic G, McCluskey A, Bashir R, Liu L, Pilch P, McMahon H, Robinson PJ, Hancock JF, Mayor S, Parton RG. 2010. Clathrin-independent carriers form a high capacity endocytic sorting system at the leading edge of migrating cells. J. Cell Biol. 190:675–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Howes MT, Mayor S, Parton RG. 2010. Molecules, mechanisms, and cellular roles of clathrin-independent endocytosis. Curr. Opin. Cell Biol. 22:519–527 [DOI] [PubMed] [Google Scholar]
- 29. Prosser DC, Wendland B. 2012. Conserved roles for yeast Rho1 and mammalian RhoA GTPases in clathrin-independent endocytosis. Small GTPases 3:229–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prosser DC, Drivas TG, Maldonado-Báez L, Wendland B. 2011. Existence of a novel clathrin-independent endocytic pathway in yeast that depends on Rho1 and formin. J. Cell Biol. 195:657–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Epp E, Walther A, Lépine G, Leon Z, Mullick A, Raymond M, Wendland J, Whiteway M. 2010. Forward genetics in Candida albicans that reveals the Arp2/3 complex is required for hyphal formation, but not endocytosis. Mol. Microbiol. 75:1182–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Newpher TM, Lemmon SK. 2006. Clathrin is important for normal actin dynamics and progression of Sla2p-containing patches during endocytosis in yeast. Traffic 7:574–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reference deleted. [Google Scholar]
- 34. Miesenböck G, De Angelis DA, Rothman JE. 1998. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394:192–195 [DOI] [PubMed] [Google Scholar]
- 35. Prosser DC, Whitworth K, Wendland B. 2010. Quantitative analysis of endocytosis with cytoplasmic pHluorin chimeras. Traffic 11:1141–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aghamohammadzadeh S, Ayscough KR. 2009. Differential requirements for actin during yeast and mammalian endocytosis. Nat. Cell Biol. 11:1039–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kondoh O, Tachibana Y, Ohya Y, Arisawa M, Watanabe T. 1997. Cloning of the RHO1 gene from Candida albicans and its regulation of beta-1,3-glucan synthesis. J. Bacteriol. 179:7734–7741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. 2008. Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5:605–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang HC, Pon LA. 2002. Actin cable dynamics in budding yeast. Proc. Natl. Acad. Sci. U. S. A. 99:751–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Idrissi FZ, Grötsch H, Fernández-Golbano IM, Presciatto-Baschong C, Riezman H, Geli MI. 2008. Distinct acto/myosin-I structures associate with endocytic profiles at the plasma membrane. J. Cell Biol. 180:1219–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Windler SL, Bilder D. 2010. Endocytic internalization routes required for delta/notch signaling. Curr. Biol. 20:538–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martin AC, Welch MD, Drubin DG. 2006. Arp2/3 ATP hydrolysis-catalysed branch dissociation is critical for endocytic force generation. Nat. Cell Biol. 8:826–833 [DOI] [PubMed] [Google Scholar]
- 43. Fazi B, Cope MJ, Douangamath A, Ferracuti S, Schirwitz K, Zucconi A, Drubin DG, Wilmanns M, Cesareni G, Castagnoli L. 2002. Unusual binding properties of the SH3 domain of the yeast actin-binding protein Abp1: structural and functional analysis. J. Biol. Chem. 277:5290–5898 [DOI] [PubMed] [Google Scholar]
- 44. Stefan CJ, Padilla SM, Audhya A, Emr SD. 2005. The phosphoinositide phosphatase Sjl2 is recruited to cortical actin patches in the control of vesicle formation and fission during endocytosis. Mol. Cell. Biol. 25:2910–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cope MJ, Yang S, Shang C, Drubin DG. 1999. Novel protein kinases Ark1p and Prk1p associate with and regulate the cortical actin cytoskeleton in budding yeast. J. Cell Biol. 144:1203–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jin M, Cai M. 2008. A novel function of Arp2p in mediating Prk1p-specific regulation of actin and endocytosis in yeast. Mol. Biol. Cell 19:297–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tarassov K, Messier V, Landry CR, Radinovic S, Serna Molina MM, Shames I, Malitskaya Y, Vogel J, Bussey H, Michnick SW. 2008. An in vivo map of the yeast protein interactome. Science 320:1465–1470 [DOI] [PubMed] [Google Scholar]
- 48. Zeng G, Yu X, Cai M. 2001. Regulation of yeast actin cytoskeleton-regulatory complex Pan1p/Sla1p/End3p by serine/threonine kinase Prk1p. Mol. Biol. Cell 12:3759–3772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zeng G, Cai M. 1999. Regulation of the actin cytoskeleton organization in yeast by a novel serine/threonine kinase Prk1p. J. Cell Biol. 144:71–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Toshima J, Toshima JY, Martin AC, Drubin DG. 2005. Phosphoregulation of Arp2/3-dependent actin assembly during receptor-mediated endocytosis. Nat. Cell Biol. 7:246–254 [DOI] [PubMed] [Google Scholar]
- 51. Sun Y, Carroll S, Kaksonen M, Toshima JY, Drubin DG. 2007. PtdIns(4,5)P2 turnover is required for multiple stages during clathrin- and actin-dependent endocytic internalization. J. Cell Biol. 177:355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Singer-Krüger B, Nemoto Y, Daniell L, Ferro-Novick S, De Camilli P. 1998. Synaptojanin family members are implicated in endocytic membrane traffic in yeast. J. Cell Sci. 111:3347–3356 [DOI] [PubMed] [Google Scholar]
- 53. Carreno S, Engqvist-Goldstein AE, Zhang CX, McDonald KL, Drubin DG. 2004. Actin dynamics coupled to clathrin-coated vesicle formation at the trans-Golgi network. J. Cell Biol. 165:781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nakano K, Imai J, Arai R, Toh-E A, Matsui Y, Mabuchi I. 2002. The small GTPase Rho3 and the diaphanous/formin For3 function in polarized cell growth in fission yeast. J. Cell Sci. 115:4629–4639 [DOI] [PubMed] [Google Scholar]
- 55. Noble SM, Johnson AD. 2005. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot. Cell 4:298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Epp E, Vanier G, Harcus D, Lee AY, Jansen G, Hallett M, Sheppard DC, Thomas DY, Munro CA, Mullick A, Whiteway M. 2010. Reverse genetics in Candida albicans predicts ARF cycling is essential for drug resistance and virulence. PLOS Pathog. 6:e1000753. 10.1371/journal.ppat.1000753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Walther A, Wendland J. 2008. PCR-based gene targeting in Candida albicans. Nat. Protoc. 3:1414–1421 [DOI] [PubMed] [Google Scholar]
- 58. Schaub Y, Dünkler A, Walther A, Wendland J. 2006. New pFA-cassettes for PCR-based gene manipulation in Candida albicans. J. Basic Microbiol. 46:416–429 [DOI] [PubMed] [Google Scholar]
- 59. Zhang C, Konopka JB. 2010. A photostable green fluorescent protein variant for analysis of protein localization in Candida albicans. Eukaryot. Cell 9:224–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Alvarez FJ, Douglas LM, Rosebrock A, Konopka JB. 2008. The Sur7 protein regulates plasma membrane organization and prevents intracellular cell wall growth in Candida albicans. Mol. Biol. Cell 19:5214–5225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cooper JA. 1987. Effects of cytochalasin and phalloidin on actin. J. Cell Biol. 105:1473–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Akashi T, Kanbe T, Tanaka K. 1994. The role of the cytoskeleton in the polarized growth of the germ tube in Candida albicans. Microbiology 140:271–280 [DOI] [PubMed] [Google Scholar]
- 63. Ng M, Roorda RD, Lima SQ, Zemelman BV, Morcillo P, Miesenböck G. 2002. Transmission of olfactory information between three populations of neurons in the antennal lobe of the fly. Neuron 36:463–474 [DOI] [PubMed] [Google Scholar]
- 64. Nicholls S, Straffon M, Enjalbert B, Nantel A, Macaskill S, Whiteway M, Brown AJ. 2004. Msn2- and Msn4-like transcription factors play no obvious roles in the stress responses of the fungal pathogen Candida albicans. Eukaryot. Cell 3:1111–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sellam A, Hogues H, Askew C, Tebbji F, van Het Hoog M, Lavoie H, Kumamoto CA, Whiteway M, Nantel A. 2010. Experimental annotation of the human pathogen Candida albicans coding and noncoding transcribed regions using high-resolution tiling arrays. Genome Biol. 11:R71. 10.1186/gb-2010-11-7-r71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rauch A, Nazarova E, Vogel J. 2010. Analysis of microtubules in budding yeast. Methods Cell Biol. 97:277–306 [DOI] [PubMed] [Google Scholar]
- 67. Roemer T, Jiang B, Davison J, Ketela T, Veillette K, Breton A, Tandia F, Linteau A, Sillaots S, Marta C, Martel N, Veronneau S, Lemieux S, Kauffman S, Becker J, Storms R, Boone C, Bussey H. 2003. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol. Microbiol. 50:167–181 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Strains used in this study.
Oligonucleotides used in this study.
Clathrin is essential for survival in C. albicans. Conditional mutants of clathrin heavy chain (chc1/tetO-CHC1) or light chain (clc1/tetO-CLC1) were created using a tetracycline-regulatable cassette (tetO) (67). Cells were grown for 4 days on SC medium, which allows growth, or on SC medium containing 100 µg/ml of tetracycline, which turns off the respective promoter. Growth of clc1/tetO-CLC1 was severely affected on repressing conditions and no single colony of chc1/tetO-CHC1 was observed. Two independently derived isolates were tested for clc1/tetO-CLC1. Download
Summary of dynamic behavior of GFP-tagged components in CME. Images were acquired by spinning disk confocal microscopy for at least 2 min in one single medial focal plane. Components in the pathway are grouped into modules. Download
Clathrin remains static at the plasma membrane in arp2 cells. WT and arp2 cells carrying Clc1-GFP were imaged by TIRF microscopy for 6 min. In WT cells, clathrin frequently appears and disappears from the membrane, while in arp2 cells, all clathrin signal remained static at the membrane. Download
Actin dynamics in the absence of Arp2/3. LIFEACT coupled to GFP was used to visualize actin dynamics in living cells. Spinning disk confocal images acquired over 4 min in one single medial focal plane showed two populations of actin filaments in WT and arp2 cells. See Fig. 4 and the description in the text for details. Download