

ABSTRACT
Overexpression of the Staphylococcus aureus multidrug efflux pump MepA confers resistance to a wide variety of antimicrobials. mepA expression is controlled by MarR family member MepR, which represses mepA and autorepresses its own production. Mutations in mepR are a primary cause of mepA overexpression in clinical isolates of multidrug-resistant S. aureus. Here, we report crystal structures of three multidrug-resistant MepR variants, which contain the single-amino-acid substitution A103V, F27L, or Q18P, and wild-type MepR in its DNA-bound conformation. Although each mutation impairs MepR function by decreasing its DNA binding affinity, none is located in the DNA binding domain. Rather, all are found in the linker region connecting the dimerization and DNA binding domains. Specifically, the A103V substitution impinges on F27, which resolves potential steric clashes via displacement of the DNA binding winged-helix-turn-helix motifs that lead to a 27-fold reduction in DNA binding affinity. The F27L substitution forces F104 into an alternative rotamer, which kinks helix 5, thereby interfering with the positioning of the DNA binding domains and decreasing mepR operator affinity by 35-fold. The Q18P mutation affects the MepR structure and function most significantly by either creating kinks in the middle of helix 1 or completely unfolding its C terminus. In addition, helix 5 of Q18P is either bent or completely dissected into two smaller helices. Consequently, DNA binding is diminished by 2,000-fold. Our structural studies reveal heretofore-unobserved allosteric mechanisms that affect repressor function of a MarR family member and result in multidrug-resistant Staphylococcus aureus.
IMPORTANCE
Staphylococcus aureus is a major health threat to immunocompromised patients. S. aureus multidrug-resistant variants that overexpress the multidrug efflux pump mepA emerge frequently due to point mutations in MarR family member MepR, the mepA transcription repressor. Significantly, the majority of MepR mutations identified in these S. aureus clinical isolates are found not in the DNA binding domain but rather in a linker region, connecting the dimerization and DNA binding domains. The location of these mutants underscores the critical importance of a properly functioning allosteric mechanism that regulates MepR function. Understanding the dysregulation of such allosteric MepR mutants underlies this study. The high-resolution structures of three such allosteric MepR mutants reveal unpredictable conformational consequences, all of which preclude cognate DNA binding, while biochemical studies emphasize their debilitating effects on DNA binding affinity. Hence, mutations in the linker region of MepR and their structural consequences are key generators of multidrug-resistant Staphylococcus aureus.
Introduction
Staphylococcus aureus is a leading pathogen causing nosocomial infections. This Gram-positive bacterium is responsible for moderately severe infections of the lower respiratory tract, skin, bones, and joints; surgical site infections; nosocomial bacteremia; and cardiovascular infection (1). S. aureus may also cause other dramatic forms of infection, such as necrotizing fasciitis and necrotizing pneumonia (2).
Bacteria employ a wide variety of means to negate the toxic effects of antimicrobial therapeutics, which include modification of the drug and drug target, increased hindrance of drug entry into the cell, and drug efflux from the cell (3–7). Polyspecific ligand efflux is a major factor rendering bacteria simultaneously insusceptible to a wide array of toxic substances. Such expulsion is achieved through the function of multidrug resistance (MDR) efflux pumps, transmembrane proteins capable of binding structurally and chemically dissimilar antimicrobials and transporting them out of the cell by utilizing the energy of the proton motive force, a Na+ ion gradient, or ATP hydrolysis (8, 9). MDR efflux pumps belong to one of five different protein families: the ATP binding cassette (ABC), major facilitator superfamily (MFS), resistance-nodulation-division (RND), small multidrug resistance (SMR), and multidrug and toxin extrusion (MATE) transporters (10, 11). The MATE family of MDR pumps is the least abundant, with only one protein, MepA, encoded by the S. aureus chromosome (12). MepA has been demonstrated to confer resistance to various dyes, including ethidium, pentamidine, bis-indoles, the biocides cetrimide and chlorhexidine, and antibiotics, such as tigecycline (12–16).
The expression of mepA is regulated by the transcription factor MepR, which is a repressor belonging to the MarR family of transcription regulators (12, 14, 17). MepR binds cognate DNA as a dimer. Each MepR protomer consists of a winged-helix-turn-helix (wHTH) DNA binding domain and a dimerization domain. MepR represses mepA transcription by binding to two inverted repeats located in the operator region of the mepA promoter that spans the −35 and −10 hexamers. MepR also represses its own transcription by binding to a single inverted repeat in the mepR operator region, which encompasses the −10 hexamer and transcription start site. Both mepA and mepR sites contain a GTTAG signature sequence, which is required for high-affinity MepR-DNA interaction (14, 18). Derepression occurs upon exposure of the MepR-DNA complex to positively charged, lipophilic antimicrobials that are also substrates of MepA. Binding of these cytotoxic compounds by MepR is proposed to lead to conformational changes in the protein, which are incompatible with high-affinity DNA association (12, 14, 18).
Multidrug-resistant strains of S. aureus overexpressing mepA have been discovered in clinical isolates and can be selected in the laboratory (19). Sequencing data demonstrated that mepA overexpression is due frequently to the defective repressor function of MepR that results from either truncation or substitution mutations. The most common single mutation, identified in 12 out of 22 mepA-overexpressing clinical strains with mutations in the mepR gene, is the rather conservative alanine-to-valine mutation at position 103 (A103V). Residue A103 is located in helix 5 of MepR in the linker region connecting the wHTH DNA binding domain to the dimerization domain. Two additional mutations, Q18P and F27L, were identified and are located in the C-terminal part of helix 1, which also belongs to the linker region. Interestingly, Q18P resulted in the most dramatic overexpression of mepA, 198-fold, which is even greater than that observed for mepR truncations and consistent with the complete loss of the repressor function of MepR. Although the initial functional consequences of these MepR mutations have been assessed previously using electrophoretic mobility shift assay (EMSA) and in vitro transcription analysis (18, 19), the mechanisms underlying their loss of function have been elusive in great part due to the lack of germane structural data.
Here, we provide those data and describe the crystal structures of MepR mutants A103V, F27L, and Q18P, as well as the structure of wild-type (WT) MepR in a conformation compatible with cognate DNA binding. Comparison of the structure of each mutated protein with that of our new WT MepR structure reveals heretofore-unseen allosteric mechanisms, which result in the abrogation of the repressor function of each mutant. Additionally, we carried out isothermal titration calorimetry (ITC) studies on the binding of the mepR operator site by WT MepR and the MepR mutants to understand the thermodynamic changes that are effected by the substitutions, which result in multidrug-resistant strains.
RESULTS
Crystal structure of MepR in a DNA binding conformation.
The crystal structure of a new WT MepR was obtained as a part of our effort to crystallize a MepR-DNA complex. Although excess mepR operator DNA was present, the asymmetric unit contained only a MepR dimer. We designate this new apoMepR protein WT MepR. The structure was refined to 3.45-Å resolution (Rwork = 22.48%/Rfree = 26.43%) and exhibited excellent stereochemistry, with 97.8% of residues in the favored region and 2.2% in the allowed region of the Ramachandran plot. Each protomer of MepR is composed of six α helices and two β strands (Fig. 1A and C). The dimerization domain is composed of residues 1 to 14 of helix 1, 111 to 119 of helix 5, and 120 to 139 of helix 6 from both MepR subunits. Helix 2 (residues 31 to 44), helix 3 (residues 48 to 58), helix 4 (the recognition helix; residues 60 to 76), and the wing (residues 77 to 94) form the DNA binding domain. The linker region, which includes the C-terminal part of helix 1 (residues 15 to 30) and the N-terminal part of helix 5 (residues 95 to 110), connects the wHTH and dimerization domains (Fig. 1A). Inspection of the dimer of the current WT MepR structure and that reported previously by Kumaraswami et al. (18) (Protein Data Bank [PDB] accession code 3eco) indicated a large conformational difference, and the superimposition of the two MepR dimers results in the strikingly large root mean square deviation (RMSD) of 6.7 Å. In contrast, structural alignment of only their dimerization domains produces an RMSD of 0.72 Å. Similarly, no marked changes occur in the structure of the core of DNA binding domains, which superimpose with an RMSD of 0.5 to 0.9 Å with, as expected, the wings producing the largest deviations. The largest structural changes associated with the new WT MepR conformation are found in the linker region.
FIG 1 .

(A) Structure of wild-type MepR in a DNA-binding-compatible conformation; the mutation sites are shown as red spheres, and corresponding residues are labeled. (B) Structural alignment of MepR and S. enterica SlyA bound to DNA. (C) Cartoon representation of the WT MepR-DNA complex model with the secondary structure elements of one MepR subunit labeled; the distance between the recognition helices is indicated by a double-headed black arrow. (D) Structural alignment of MepR in a DNA-binding-compatible form with the wild-type apoMepR reported previously by Kumaraswami et al. (18): only the dimerization domains, including residues 1 to 16 and 116 to 139 of both protomers, were superimposed.
The conformation of the present WT MepR dimer and, particularly, the position and orientation of the recognition helices (where the center-to-center distance between the recognition helices using the Cα atoms of the 2-fold-related L68 residues is 36.1 Å) would allow ready docking of the MepR dimer to B-form DNA (the distance between two consecutive major grooves is 34 Å) (Fig. 1C). Again, this conformation is in sharp contrast to that observed in the previously reported structure of MepR (18) (PDB accession code 3eco), where the center-to-center distance between the DNA binding recognition helices is 43 Å. Further analysis suggests that to assume a B-DNA binding conformation, each wHTH domain of WT MepR must rotate 40° from its location in the MepR 3eco structure (Fig. 1D). To create a model of a WT MepR-cognate DNA complex, we superimposed the WT MepR structure onto the structure of MarR family member SlyA from the SlyA-DNA complex (PDB accession code 3q5f) (20). The RMSD of the structural alignment of WT MepR with DNA-bound SlyA is relatively high, 3.2 Å, but with the major differences originating from the position of MepR wings (Fig. 1B). Inspection of the crystal packing of the WT MepR structure reveals that its wings contribute significantly to crystallization contacts (data not shown), and thus, their position deviates from the optimal DNA binding conformation seen in the SlyA-DNA complex. In addition, the distance between the centers of recognition helices of WT MepR is slightly larger than that in SlyA bound to its cognate DNA (36.1 Å in MepR versus 35.3 Å in SlyA). This difference we also attributed to crystal packing. Despite these deviations, the overall conformation of the WT MepR protein is very similar to conformation of DNA-bound SlyA (Fig. 1B) and to other reported MarR family members bound to cognate DNA with the exception of DNA-bound ST1710 (21–25) (see Fig. S1 in the supplemental material). Inspection of the MepR recognition helix and comparison of it to other complexes of DNA-bound MarR family proteins suggest that MepR residues T60, T63, S65, N66, and R69 might recognize the MepR binding site by making direct hydrogen bond or water-mediated contacts with bases of the major groove. Interestingly, residue P61 of SlyA establishes van der Waals contacts with the TTA box of its TTAGC signature sequence. This proline residue is conserved in MepR (P62), and the identical TTA box is present in the GTTAG signature sequence of the mepR operator binding site. Even though the MepR P62A mutant was found to bind mepR and mepA operator DNA with wild-type-like affinity (19), the rigid prolyl side chain of P62 may still be involved in DNA specificity by its selection against particular DNA sequences, rather than in favor of one. Residue R87 is conserved among MarR family members (see Fig. S2) and, in most available MarR family member-DNA complexes, makes hydrogen bonds to the O2 atom of thymines of AT-rich minor grooves (20–22, 24, 25). A similar interaction is anticipated in the MepR-mepR and MepR-mepA operator complexes. Thus, our model of the MepR-DNA complex affords a reasonable representation of the WT MepR-DNA complex and, more importantly, allows us to compare the structures of the three nonfunctional MepR mutants to the DNA-bound form of MepR (Fig. 1C).
A103V alters the optimal positioning of the wHTH motifs of MepR.
The substitution of valine for residue A103 was found in more than 50% of the clinically isolated S. aureus strains that overexpressed mepA in conjunction with a MepR substitution mutation. Although this mutation is relatively conservative, it results in at least a 10-fold-weaker repressor activity (19). Residue 103 is located in a region of MepR that connects its DNA binding and dimerization domains and is likely to play an important role in the conformational flexibility of the wHTH motif (Fig. 1A). Such plasticity is necessary to achieve an optimal orientation of this DNA binding motif for interaction with cognate DNA. Sequence alignment of MepR with Pseudomonas aeruginosa MexR, Escherichia coli MarR, Bacillus subtilis OhrR, and Salmonella enterica SlyA shows that A103 is not conserved among various members of the MarR family (see Fig. S2 in the supplemental material). Moreover, the equivalent positions in other proteins are occupied with large charged residues. A recent study by Schindler et al. (19) using an electrophoretic mobility shift assay (EMSA) demonstrated that the impaired repressor function of the A103V mutant is due to diminished affinity for cognate DNA. To characterize quantitatively how the A103V change affected DNA binding activity of MepR, we used isothermal titration calorimetry (ITC). WT MepR and MepR(A103V) were titrated into the mepR operator DNA. Analysis of the resulting thermograms demonstrates that upon mutation the affinity toward DNA drops more than 27-fold, from 37 nM to 1 µM (Fig. 2A).
FIG 2 .

The mechanism of the defective repressor function of MepR(A103V). (A) Titrations of mepR operator DNA with WT MepR, MepR(A103V), and MepR(A103S). The solid lines represent theoretical fits to the experimental data (closed squares); fitting parameters are provided in Table 1. (B) Structural alignment of wild-type apoMepR (PDB accession code 3eco) with MepR(A103V); only the dimerization domains were superimposed, as in Fig. 1D. (C) Local structural alterations due to the A103V substitution; the wHTH motifs of MepR(A103V) and the WT MepR were superimposed by aligning the C-terminal portion of helix 1 and the N-terminal portion of helix 5 of the mutant with corresponding regions of the WT MepR. (D) Displacement of the helices of the wHTH motif of MepR(A103V).
To determine the structural mechanism of repressor function inactivation by the A103V mutation, we crystallized the MepR(A103V) protein. The crystals diffracted to 1.6-Å resolution, and the structure was solved by molecular replacement using the apoMepR dimer (PDB accession code 3eco) as the search model. The final model was refined to Rwork/Rfree = 18.95%/21.74% and exhibited excellent stereochemistry, with 99.7% of residues in the most favored region and 0.3% in the additionally allowed region of the Ramachandran plot. Interestingly, MepR(A103V) crystallized in the same space group as the previously solved wild-type apoMepR protein (see Table S1 in the supplemental material) (18). The superimposition of the dimers of these structures produces an RMSD of 1.9 Å, with the major differences accumulated in the DNA binding lobes (Fig. 2B). Structural alignment of the DNA binding domains of MepR(A103V) and DNA-binding compatible WT MepR reveals the underlying cause of the weaker DNA binding of the mutated protein (Fig. 2C and D). The replacement of alanine at position 103 with valine, which is bulkier and more hydrophobic and possesses branched γ-methyl groups, creates steric repulsion with residue F27, which is located on the C-terminal part of helix 1. As a result, the N terminus of helix 5, which includes position 103, is displaced toward the core of the DNA binding domain and forces residue F104 to change its conformation by rotating the phenyl ring toward helix 1. This rotameric change of the F104 side chain would lead to an even larger steric clash between the N terminus of helix 5 and C terminus of helix 1 (Fig. 2C). To avoid such a clash, the N-terminal part of helix 5 moves toward helix 2, pushing it toward helix 3, which then drives toward helix 4, the recognition helix of the DNA binding domain. As a consequence of the translocation of helix 3, helix 4 rotates ~20° clockwise toward the sugar-phosphate DNA backbone (Fig. 2D). The outcome of this series of rearrangements of the wHTH motif is the displacement of the recognition helix from an orientation that is suitable for interaction with the DNA major groove. The fact that this change occurs in both subunits augments this conformational effect and provides a structural rationale for how such a relatively small change in a somewhat solvent-exposed residue results in an almost 30-fold drop in DNA affinity. That β branching at position 103 is critical to this allosteric loss of DNA binding affinity is bolstered by our ITC studies on MepR(A103S), which binds DNA as tightly as does wild-type MepR (Fig. 2A). Serine, which is a smaller, more hydrophilic amino acid than valine and critically is not β branched, is able to assume a conformation which is sterically compatible with residue F27 of WT MepR.
TABLE 1 .
Thermodynamic parameters of MepR binding to the mepR operator sitea
| MepR variant | n | Kd, nM | ΔH, kcal/mol | ΔS, cal/(mol ⋅ degree) |
|---|---|---|---|---|
| WT | 1.92 | 36.6 | 8.73 | 63.3 |
| A103V | 2.01 | 1,000.0 | 9.16 | 58.2 |
| A103S | 2.15 | 42.6 | 6.65 | 56.0 |
| F27L | 2.08 | 1,250.0 | 9.07 | 57.4 |
| F27L/F104A | 2.09 | 32.3 | 8.57 | 63.0 |
| Q18P | 2.00 | 61,350.0 | 20.50 | 87.9 |
| Q18A | 2.16 | 34.1 | 9.10 | 64.7 |
n, stoichiometry of binding (n = 2 assumes that two protomers of MepR, which form a dimer, bind one duplex DNA); Kd, dissociation constant of the MepR-DNA complex; ΔH, enthalpy change of the reaction; ΔS, entropy change of the reaction.
F27L affects DNA binding by bending helix 5.
Residue F27 is located at the C terminus of helix 1 (Fig. 1A). Similar to residue A103, this position belongs to the linker region that connects the DNA binding and dimerization domains, and like MepR(A103V), MepR(F27L) has been identified in a multidrug-resistant S. aureus clinical isolate that overexpresses the MepA multidrug transporter. Intriguingly, a sequence alignment of MepR and other members of the MarR family reveals that leucine is highly conserved at position 27 (see Fig. S2 in the supplemental material). In order to understand the effects of the F27L change, we first characterized the mepR operator DNA binding of MepR(F27L). The data show a 35-fold reduction in DNA binding affinity: the Kd (dissociation constant) for MepR(F27L) is 1.3 µM and the Kd for WT MepR is 37 nM (Fig. 3A).
FIG 3 .

The effect of the F27L substitution on the linker region of MepR. (A) Titrations of mepR operator DNA with MepR(F27L) and MepR(F27L/F104A). The solid lines represent theoretical fits to the experimental data (closed squares); fitting parameters are provided in Table 1. (B) The structure of native MepR(F27L) superimposed on apoMepR (PDB accession code 3eco) and the structure of selenomethionine-substituted MepR(F27L). Helix 5, bent in both MepR(F27L) structures, is indicated by arrows; the distance between the recognition helices of the selenomethionine-substituted MepR(F27L) is 58 Å and indicated by a double-headed black arrow. (C) A structural alignment of the DNA binding domains of MepR-DNA model and native MepR(F27L). The area surrounding the mutation is magnified to demonstrate the details of the structural rearrangements in helix 5 caused by the F27L substitution.
To elucidate the structural basis of such a marked decrease in DNA binding affinity by this seemingly conservative and conserved substitution, we crystallized MepR(F27L). Two different crystal forms were obtained. One form diffracted to 1.8-Å resolution and took space group P21. The structure was solved by molecular replacement using the structure of apoMepR (PDB accession code 3eco) as the search model. The asymmetric unit contained one MepR dimer. The model was refined to Rwork/Rfree = 17.0%/20.3%, respectively; 100% of residues were in the favored region of the Ramachandran plot, 98.6% of which were in the most favored region, and 1.4% were in the additionally allowed region. The dimer of MepR(F27L) superimposes poorly on the WT 3eco structure (RMSD, ~1.8 Å), again with the most prominent mismatch in the orientation of DNA binding lobes (Fig. 3B). The second crystal form took the rhombohedral space group R32. We were unable to solve the structure using molecular replacement and therefore expressed, purified, and crystallized selenomethionine-substituted MepR(F27L). The crystals of SeMet MepR(F27L) diffracted to 2.37-Å resolution. The structure was solved using single-wavelength anomalous dispersion (SAD) and density modification. The model was refined to an Rwork of 23.1% and Rfree of 25.6% and revealed good stereochemistry with 99.3% of residues in the favored region of the Ramachandran plot (98.5% were in the most favored region and 0.8% were in an additionally allowed region). The asymmetric unit contained one protomer of MepR with the biological dimer created by crystallographic symmetry. The structure of the SeMet-substituted MepR(F27L) dimer revealed a novel conformation, with the wHTH motifs spread a striking 58 Å (center-to-center distance of recognition helices 4 and 4′) (Fig. 3B).
When the structures of WT MepR and MepR(F27L) are compared, the most striking difference is the presence of a kink in helix 5 at position 106 in both MepR(F27L) subunits (Fig. 3B and C). A similar structural change occurs in selenomethionine-substituted MepR(F27L) (Fig. 3B). Closer investigation of this region reveals that upon replacement of F27 with leucine, one of the δ methyl groups of the latter residue would clash with residue F104. In order to avoid unfavorable steric interaction with L27, the side chain of F104 rotates ~90°. However, without additional changes, the new rotamer of the F104 would clash with the residue F108 and the entire C-terminal portion of helix 5. The conflict is resolved by creating a kink in helix 5 (Fig. 3C). As a result, the N-terminal segment of helix 5 along with the DNA binding domain rotates approximately 45° from the wild-type MepR conformation. Alignment of Mep(F27L) onto our model of the DNA-bound WT MepR (Fig. 3C) indicates that the kink in helix 5 creates a substantial structural obstacle for the productive interaction of the second wHTH motif with the neighboring major groove of DNA and thereby provides an explanation for the resulting nanomolar-to-micromolar drop in mepR operator binding affinity.
Support for our supposition that the clash between the δ methyl group of a leucine at residue 27 and the phenyl ring side chain of F104 caused the allosteric loss of the DNA binding function of MepR(F27L) was provided by the creation of the MepR double mutant, MepR(F27L/F104A). This additional substitution removes the steric clash between L27 and residue 104 and thus precludes the need to kink helix 5. Indeed, the ITC data demonstrated that the MepR(F27L/F104A) double mutant completely eliminated the negative effects of the F27L single change and bound the mepR operator site with an affinity (Kd = 32 nM) equal to that of wild-type MepR binding (Fig. 3A).
Q18P severely distorts the linker region of MepR in multiple ways.
The mutant strain that contained the Q18P substitution in MepR was isolated in in vitro selection experiments and exhibited mepA overexpression levels comparable to or even greater than those of truncated MepR variants, suggesting severe structural and functional defects instigated by the mutation (19). Residue Q18 is located in the middle of helix 1 and, as found for residues F27 and A103, contributes to the linker region connecting the dimerization and DNA binding domains (Fig. 1A). Interestingly, prolines are found in the linker regions of other MarR family members, including S. enterica SlyA, where prolines are located in the C-terminal portion of helix 1 and at the N-terminal part of helix 5, and in B. subtilis OhrR, where prolines are found at the C terminus of helix 1 and in the middle of helix 5. In both proteins, these residues, as expected, create kinks, which appear to assist the wHTH domains in adopting an orientation for optimal DNA binding. To quantitate the effect of the Q18P substitution on the DNA binding ability of MepR(Q18P), ITC experiments were done (Fig. 4A). Introduction of a proline at position 18 results in an approximately 2,000-fold decrease in DNA binding affinity: the dissociation constant increases from 37 nM for WT MepR to ~61 µM for MepR(Q18P). Such a dramatic drop in DNA binding affinity would account for the essentially complete loss of the repressor function of this mutant. To ensure that the loss of affinity was not the result even in part of the loss of any Gln18-DNA interaction, the MepR(Q18A) protein was created and its DNA binding affinity was tested (see Fig. S3 in the supplemental material). MepR(Q18A) binds DNA with wild-type affinity (Kd = 34 nM), proving that the disruption of any putative Gln18-DNA interaction does not affect the stability of the protein-DNA complex.
FIG 4 .

The effect of the Q18P substitution on the structure and function of MepR. (A) Titrations of mepR operator DNA with MepR(Q18P). The solid lines represent theoretical fits to the experimental data (closed squares); fitting parameters are provided in Table 1. (B) The overlay of the four MepR(Q18P) dimers located in the asymmetric unit after alignment of their dimerization domains. Only one monomer of each dimer is shown in color. P18 is shown as appropriately colored spheres. The insets show the effects of Q18P mutation on helix 1 and helix 5, which are indicated by black arrows. (C to E) The alignment of the WT MepR-DNA model (WT MepR is shown in red) with each of the four MepR(Q18P) conformational variants (shown in blue, green, orange, and magenta). The structures were aligned as in panel B. (F) The locations of the recognition helices of the four MepR(Q18P) conformational variants and the WT MepR protein as they would be found in consecutive major grooves of B-DNA. The color code is the same as in panels C to E. A more detailed description of the structural distortions of MepR helices 1 and 5 effected by the Q18P mutation is provided in Text S2 and Fig. S4 in the supplemental material.
To provide a more detailed mechanism of the loss of the repressor activity of MepR(Q18P), we solved the crystal structure of this mutant. The protein crystallized in the monoclinic space group P21 with 4 dimers in the asymmetric unit. The structure was refined to Rwork/Rfree = 21.1%/26.8%. The final models exhibit good stereochemistry, with 98.2% of residues in the most favored region of the Ramachandran plot and 1.7% in the additionally allowed region. The four dimers exhibit striking conformational heterogeneity (Fig. 4B). Pairwise alignment of all four MepR(Q18P) dimers in the asymmetric unit produces RMSDs in the range of 4 to 6 Å, with the exception of one pair, which superimposes somewhat better, with an RMSD equal to 1.85 Å. Structural alignment of each MepR(Q18P) dimer and the DNA-binding-compatible WT MepR also reveals large deviations, with their RMSDs ranging from 3.8 to 6.1 Å.
Despite the severe distortions in the overall quaternary conformation, the structures of the DNA binding and dimerization domains of each MepR(Q18P) protomer are essentially identical to those of wild-type MepR. The most dramatic structural changes that the Q18P mutation imposes are found in the linker region. In two dimers (orange and blue, Fig. 4B, insets), the effect of Q18P mutation manifests itself as a kink in the middle of helix 1, which is required to accommodate the prolyl ring, and a consequential 45° rotation of the C-terminal part of the helix. As a result of this rearrangement, a second kink is created in helix 5 at either position 108 or 112, and the N-terminal portion of helix 5 is rotated by ~45°. In the two other conformational variants of the MepR(Q18P) dimer (green and magenta, Fig. 4B, insets), the glutamine-to-proline mutation leads to a similar kink at position 18 of helix 1 as described above or remarkably results in the complete unfolding of the C terminus of helix 1. In addition, helix 5 undergoes a yet more significant structural rearrangement whereby the central part of the helix, spanning residues 106 to 110, unfolds and thereby breaks helix 5 into two smaller helices. Such dissection of helix 5 of these two dimers of the MepR(Q18P) mutant is identical to that of helix 5 of the MarR family member OhrR from Xanthomonas campestris (26). In OhrR, the presence of organic hydroperoxides triggers the formation of the disulfide bond between cysteine residues in helix 1 and helix 5, which leads to the disruption of helix 5 and its subdivision into two smaller helices. Additionally, OhrR helix 6 and its 2-fold mate, helix 6′, swap places to allow reconfiguration of the dimer interface, which in turn alters the location of the wHTH and results in the inability to bind cognate DNA. Interestingly, OhrR is preconfigured for the partition of helix 5 due to the presence of residue P122 in the unfolding region. Whether the splitting of helix 5 in MepR(Q18P), and potentially in WT MepR, bears any functional significance, as in OhrR, and is not simply the outcome of the Q18P substitution will require further structural investigations into the mechanisms of drug induction of mepA and mepR transcription by MepR.
As expected, the alignment of the structures of the MepR(Q18P) protein with that of the DNA-binding-compatible WT MepR indicates that none of the independent Q18P dimers would bind the mepR or mepA operator site (Fig. 4C to E). The mutation-induced distortions in helices 1 and 5 cause reconfigurations of MepR dimers, which lead to an increased center-to-center distance between recognition helices, numerous potential clashes with the sugar-phosphate backbone of DNA, and large rotations of the wHTH motifs. None of the recognition helices of the four MepR(Q18P) variants are configured to dock into consecutive major grooves (Fig. 4F).
Altogether, the Q18P substitution not only introduces severe distortion at the place of its actual location on helix 1 but also triggers dramatic structural changes in neighboring helix 5. Further, the flexibility of the linker connecting the DNA binding and dimerization domains is altered to the extent that none of the wHTH motifs is able to assume readily a proper DNA binding conformation, leading to the observed 2,000-fold decrease in binding affinity.
DISCUSSION
Bacterial multidrug resistance is a major threat in the hospital environment, particularly to patients with weakened immune systems. The most common causative agents of nosocomial infections are S. aureus, P. aeruginosa, and E. coli. These bacteria protect themselves from a variety of antimicrobial drugs by expressing multidrug efflux pumps, for example, MepA in S. aureus or the tripartite transporters MexAB-OprM in P. aeruginosa (7) and AcrAB-TolC in E. coli (5), which pump a variety of chemically and structurally dissimilar toxins from the cell. Often, multidrug resistance occurs due to mutations in the transcription factors regulating the expression of efflux pump genes. In P. aeruginosa, clinical isolates exhibiting increased levels of transcription of the mexAB operon originate from a variety of mutations in the MexR repressor, which, like MepR, is a member of the MarR family of transcription regulators (27–30). The bulk of these mutations mapped to the DNA binding domain, including the G58E, R70W, R83H, and L95F changes, suggesting that they either directly interfere with MexR-DNA interactions or affect the correct folding of the wHTH motif. Similar results were obtained by mutational analysis of MarR, a transcription repressor of the marRAB operon that is responsible for the emergence of resistance to multiple antibiotics in E. coli (31). Analyses of S. aureus clinical isolates overexpressing the MepA efflux pump as well as in vitro selection of multidrug-resistant strains also revealed defective function of its transcriptional repressor, MepR. Premature truncations of MepR that cause uncontrolled mepA expression were often found, as well as mutations that affect the folding of the DNA binding domain, such as G97E and G97W (18, 19). Interestingly, and unlike what has been observed for other MarR family members, a large fraction of inactivating substitutions, including A103V, F27L, and Q18P, were located in the linker region of MepR. The crystal structures of these mutants presented in the current study in conjunction with ITC data highlight the importance of the linker region for the allosteric regulation of specific DNA recognition by MepR and likely other multidrug binding MarR family members.
Our ITC studies on MepR(A103V) and MepR(F27L) revealed that these proteins were still able to bind the mepR operator site, although with substantially weakened affinity (~1 µM). Importantly, the decreased DNA binding of these mutants is caused not by any structural defect or change in the wHTH motifs but rather by the poorer ability of the mutant MepR dimers to assume a mepR operator-specific DNA binding conformation. Therefore, binding of the MepR(A103V) and MepR(F27L) mutants to cognate DNA can be divided into two steps: (i) the structural rearrangement of the mutant MepR necessary to adopt the conformation of the wild-type protein and (ii) binding to mepR or mepA operator DNA. Since the model of our WT MepR-DNA complex suggests that neither residue A103 nor residue F27 interacts with nucleic acid directly, the second event in the scheme should be identical to the binding of the wild-type MepR. Taking into account these assumptions, the change in the Gibbs free energy of DNA binding by a mutant MepR (ΔGmut) is equal to the sum of ΔGconf, the unfavorable term representing the free energy change associated with any structural rearrangements required to achieve the WT MepR-like DNA binding conformation, and ΔGWT, the favorable Gibbs free energy change of WT MepR binding to DNA:
| (1) |
Considering that ΔG = −RTlnK and rearranging equation 1, we can calculate ΔGconf:
| (2) |
where Kmut and KWT are the equilibrium association constants for mutant and wild-type MepR, respectively. Solving for ΔGconf reveals ΔGconf[MepR(F27L)] = +8.82 kJ/mol and ΔGconf[MepR(A103V)] = +8.17 kJ/mol.
Application of the same logic and approach to describe the conformational cost of the Q18P substitution to assume a structure that can bind the mepR operator sequence reveals a Gibbs free energy term, ΔGconf[MepR(Q18P)], of +18.39 kJ/mol, which is significantly larger and even more unfavorable than that for the A103V and F27L mutants. The result of this calculation is not surprising considering the amount of structural “damage” that Q18P does to MepR (Fig. 4), and regardless of whether or not this calculation is fully correct, our structural and biochemical data show indisputably that the Q18P change has a much more dramatic effect than either the F27L or the A103V substitution. Clearly, the affinity of MepR(Q18P) for the mepR operator site is extremely weak. Whether or not MepR(Q18P) is even able to take a mepR operator sequence-specific binding conformation as opposed to a nonspecific DNA binding conformation will require additional studies.
The loss of DNA binding affinity and repressor function of MepR(F27L) is particularly interesting as it reveals insight into a possible multidrug binding induction pathway of MepR. Upon association with a germane ligand, MepR dissociates from its cognate DNA binding sites. However, the exact mechanism is unknown because no structures of any MepR-drug complex have been solved. Examination of the structures of MarR-effector/inducer complexes (32, 33) suggests that the drug binding pocket is located between helix 3 of the wHTH motif and the linker region. Structural comparison of the ligand-bound MarR transcriptional regulators PcaV bound to protocatechuate (PDB accession code 4fht), MarR bound to kanamycin (PDB accession code 4emo), MTH313 (MarR) bound to salicylate (PDB accession code 3bpx), and MepR(F27L) and the DNA binding conformation of WT MepR suggests a potential key step in the mechanism for ligand-induced conformational changes in MepR that cause DNA dissociation (Fig. 5). All described MepR ligands, including ethidium, 4′,6-diamidino-2-phenylindole (DAPI), and dequalinium, are significantly larger than salicylate and protocatechuate and are comparable in size to kanamycin. If the location of the ligand binding site of MepR is similar to those of PcaV, MarR, and MTH313, the binding of these MepR ligands would result in a steric clash with residue F108 and the C-terminal portion of helix 5 in the same manner as that in which the molecule of kanamycin from the MarR-kanamycin complex is clashing with helix 5 of MepR (Fig. 5). This unfavorable steric interaction could be eliminated by breaking helix 5, as observed in MepR(F27L), and the consequential rearrangement of the wHTH motifs that ultimately lead to DNA release. Thus, the alternative rotamer of MepR residue F104 observed in the MepR(F27L) structure is likely key to this process and is emulating the effect of MepR induction by an effector molecule. The importance of residue F104 and its large side chain is underscored by the restoration of DNA binding to wild-type affinity upon its mutation to alanine in the presence of the F27L change (Fig. 3A).
FIG 5 .
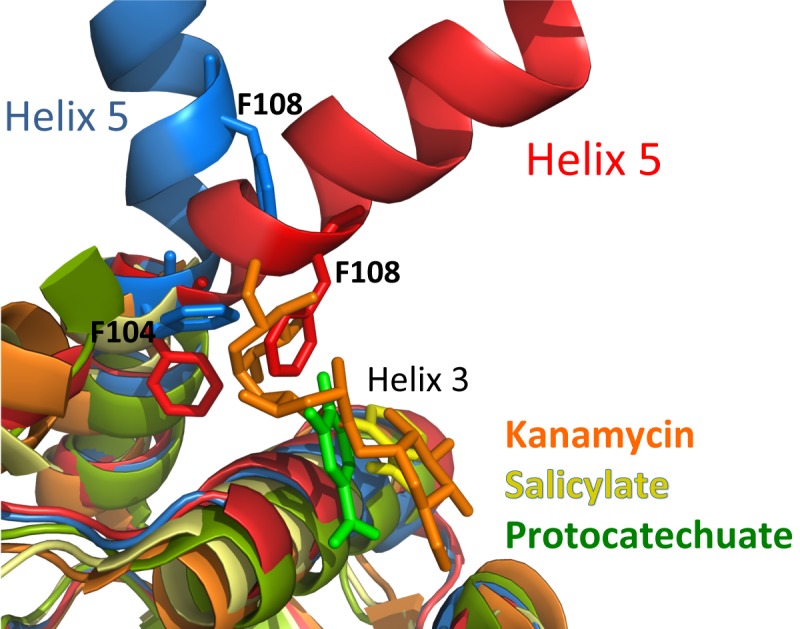
Structural alignment of selected MarR family members in their ligand-bound forms, MepR(F27L) (blue), and WT MepR in its DNA-binding conformation (red). The S. aureus MarR-kanamycin complex (4EM0) is shown in orange, the Streptomyces coelicolor PcaV-protocatechuate (4FHT) complex is shown in green, and the Methanobacterium thermautotrophicum MTH313-salicylate complex (3BPX) is shown in yellow. For clarity, large portions of helices 1 and 5 were removed, except for helix 5 in WT MepR and MepR(F27L).
In the current work, we describe the thermodynamic characterization of three MepR mutants, Q18P, F27L, and A103V, that have been identified in multidrug-resistant strains of S. aureus and present the structural mechanisms underpinning their inability to repress the mepR and mepA genes. We also present the crystal structure of wild-type MepR in its DNA binding conformation and provide a model of the MepR-DNA complex. Although the model does not provide atomic details of MepR-DNA interactions, the general architecture of the complex allows the ready visualization of how these three MepR mutations altered the ability of this repressor to bind cognate DNA. Intriguingly, none of these mutations affects DNA binding directly but rather elicits multidrug resistance through allosteric mechanisms. We demonstrate that these mutations impart their deleterious effects either by transmitting negative structural perturbations from the mutation site to the winged-helix-turn-helix motif, as in MepR(A103V), or by altering the conformational mobility of the DNA binding domains of MepR(F27L) and MepR(Q18P). Independent of the exact allosteric mechanism taken, in all cases the optimal orientation of the recognition helices and wings for interaction with the major and minor grooves of B-DNA is precluded or requires additional free energy to obtain. Interestingly, the allosteric regulation of DNA binding is observed for another MarR-type regulator, RovA, which controls the expression of virulence factors in Yersinia species (24). In RovA, the C terminus of helix 5 undergoes unfolding upon an increase in temperature to 37°C, which is associated with the environmental transition of this pathogen in its colonization of and persistence in its human host. The resulting structural changes are transmitted from the dimerization domain to the DNA binding domain and trigger the dissociation of this transcription factor and upregulation of the expression of multiple virulence genes. Combined with our studies on MepR, it is becoming clearer that helix 5 of MarR family members is one of the key components of allosteric regulation by inducing agents.
MATERIALS AND METHODS
Cloning, mutagenesis, and protein expression and purification.
Most experiments were conducted using recombinant MepR containing a noncleavable C-terminal hexahistidine tag. The plasmid used for expression has been described previously (14). Protein expression was achieved using strain C41(DE3) (Lucigen) or BL21(DE3) One Shot (Invitrogen), and the corresponding growth protocol was described by Kumaraswami et al. (18). Protein purification was conducted according to the protocol of Kumaraswami et al. with slight modification (see Text S1 in the supplemental material). In addition to the above-described protein, we also purified MepR in which the hexahistidine tag was removed. The mepR gene was cloned into plasmid pMCSG7 using the standard protocol for ligation-independent cloning (34). The protein expressed from pMCSG7mepR contains an N-terminal hexahistidine tag followed by a tobacco etch virus (TEV) protease cleavage site. After removal of the tag, the N-terminal methionine of MepR is replaced by an asparagine and an N-terminal serine is added. The protein was expressed and purified as described above with the following exception: before applying the gel filtration chromatography step, the hexahistidine tag was removed by digesting the protein overnight with several milligrams of TEV protease at room temperature (22°C). All mutagenesis was carried out using standard protocols and the QuikChange site-directed mutagenesis kit (Agilent Technologies). Selenomethionine-substituted MepR was expressed using the methionine inhibitory pathway protocol, described previously (35), and purified as described for native MepR.
Crystallization, data collection, and structure solution.
The details of protein crystallization, X-ray diffraction data collection, and structure solution are provided in Text S1 in the supplemental material. Selected data collection and refinement statistics of all crystals are provided in Table S1. All figures were created using the PyMOL molecular graphics system, version 1.5.0.4, Schrödinger, LLC.
Isothermal titration calorimetry.
All reactions were carried out at 25°C using a VP-ITC calorimeter (Microcal). Data were collected and analyzed using the manufacturer-supplied software package, Origin 7.0 (OriginLab Corp.). Each ITC run included an initial 4-µl injection followed by 30 10-µl injections of the MepR solution into the sample cell containing the DNA (total volume = 1.45 ml). The DNA used in each experiment was the mepR operator sequence modified slightly to obtain the palindrome 5′ TAT TTA GTT AGA TAT CTA ACT AAA TA 3′. DNA was purchased from IDT and annealed by heating at 95°C for 5 min followed by slow cooling to room temperature on the bench. The buffer used in all ITC experiments was 20 mM Tris HCl (pH 7.5) (room temperature), 150 mM NaCl, and 5 mM MgCl2. The following concentrations were used in experiments: WT MepR, 400 µM protein and 24 µM DNA; MepR(A103V), 413 µM protein and 14 µM DNA; MepR(A103S), 270 µM protein and 15 µM DNA; MepR(F27L), 280 µM protein and 15 µM DNA; MepR(F27L/F104A), 405 µM protein and 17 µM DNA; MepR(Q18P), 200 µM protein and 10 µM DNA; MepR(Q18A), 170 µM protein and 10 µM DNA.
SUPPLEMENTAL MATERIAL
Materials and methods. Download
Structural distortions in the MepR α1 and α5 helices effected by the Q18P mutation. Download
Selected statistics of data collection, structure solution, and refinement.
The alignment of MepR in its DNA binding conformation with other members of the MarR family that have been crystallized with their cognate DNA. (A) The alignment of MepR with SlyA (3Q5F), MosR (4FX4), RovA (4AIJ), OhrR (1Z9C), and SCO3205 (3ZPL) demonstrates similar architectures of the protein-DNA complexes, with the recognition helices binding to consecutive major grooves and the wings interacting with the flanking minor grooves. For clarity, only the DNA from the MepR-DNA model is shown. (B) The comparison of MepR to ST1710 reveals significant conformational differences, especially pronounced in DNA binding domains. Download
Sequence alignment of S. aureus MepR, P. aeruginosa MexR, E. coli MarR, B. subtilis OhrR, and S. enterica SlyA. The boxes indicate the mutation sites of MepR: Q18, F27, and A103. The sequence alignment was created using ClustalW and shaded using BoxShade 3.2. Download
The role of MepR residue Q18 in DNA binding. (A) The MepR-DNA complex model showing potential interactions between Q18 (red spheres) and DNA. (B) ITC experiment of mepR operator DNA binding to MepR(Q18A). The solid lines represent theoretical fits to the experimental data (closed squares); fitting parameters are provided in Table 1. Download
Alignment of MepR in its DNA binding conformation with MepR(Q18P). Only one subunit of the dimer is shown. Solid arrows point toward residues or structural elements of interest. Dashed arrows demonstrate the displacements of wHTH motifs of the Q18P mutant from its WT MepR location. P18 is shown in spheres. Key secondary structural elements and residues are labeled. Download
ACKNOWLEDGMENTS
We thank the beamline scientists at APS 22-BM and 22-ID for their assistance with X-ray diffraction data collection.
This work was supported in part by funds from Duke University Medical School and VA Biomedical Laboratory Research and Development grant IO1BX000465.
Footnotes
Citation Birukou I, Tonthat NK, Seo SM, Schindler BD, Kaatz GW, Brennan RG. 2013. The molecular mechanisms of allosteric mutations impairing MepR repressor function in multidrug-resistant strains of Staphylococcus aureus. mBio 4(5):e00528-13. doi:10.1128/mBio.00528-13.
REFERENCES
- 1. Bereket W, Hemalatha K, Getenet B, Wondwossen T, Solomon A, Zeynudin A, Kannan S. 2012. Update on bacterial nosocomial infections. Eur. Rev. Med. Pharmacol. Sci. 16:1039–1044 [PubMed] [Google Scholar]
- 2. Otto M. 2012. MRSA virulence and spread. Cell. Microbiol. 14:1513–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nikaido H. 1994. Prevention of drug access to bacterial targets: permeability barriers and active efflux. Science 264:382–388 [DOI] [PubMed] [Google Scholar]
- 4. Nikaido H. 2009. Multidrug resistance in bacteria. Annu. Rev. Biochem. 78:119–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seeger MA, Diederichs K, Eicher T, Brandstätter L, Schiefner A, Verrey F, Pos KM. 2008. The AcrB efflux pump: conformational cycling and peristalsis lead to multidrug resistance. Curr. Drug Targets 9:729–749 [DOI] [PubMed] [Google Scholar]
- 6. Higgins CF. 2007. Multiple molecular mechanisms for multidrug resistance transporters. Nature 446:749–757 [DOI] [PubMed] [Google Scholar]
- 7. Lister PD, Wolter DJ, Hanson ND. 2009. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22:582–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borges-Walmsley MI, McKeegan KS, Walmsley AR. 2003. Structure and function of efflux pumps that confer resistance to drugs. Biochem. J. 376:313–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Köhler T, Pechère JC, Plésiat P. 1999. Bacterial antibiotic efflux systems of medical importance. Cell. Mol. Life Sci. 56:771–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lynch AS. 2006. Efflux systems in bacterial pathogens: an opportunity for therapeutic intervention? An industry view. Biochem. Pharmacol. 71:949–956 [DOI] [PubMed] [Google Scholar]
- 11. Paulsen IT, Brown MH, Skurray RA. 1996. Proton-dependent multidrug efflux systems. Microbiol. Rev. 60:575–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaatz GW, McAleese F, Seo SM. 2005. Multidrug resistance in Staphylococcus aureus due to overexpression of a novel multidrug and toxin extrusion (MATE) transport protein. Antimicrob. Agents Chemother. 49:1857–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DeMarco CE, Cushing LA, Frempong-Manso E, Seo SM, Jaravaza TA, Kaatz GW. 2007. Efflux-related resistance to norfloxacin, dyes, and biocides in bloodstream isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 51:3235–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kaatz GW, DeMarco CE, Seo SM. 2006. MepR, a repressor of the Staphylococcus aureus MATE family multidrug efflux pump MepA, is a substrate-responsive regulatory protein. Antimicrob. Agents Chemother. 50:1276–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McAleese F, Petersen P, Ruzin A, Dunman PM, Murphy E, Projan SJ, Bradford PA. 2005. A novel MATE family efflux pump contributes to the reduced susceptibility of laboratory-derived Staphylococcus aureus mutants to tigecycline. Antimicrob. Agents Chemother. 49:1865–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Opperman TJ, Williams JD, Houseweart C, Panchal RG, Bavari S, Peet NP, Moir DT, Bowlin TL. 2010. Efflux-mediated bis-indole resistance in Staphylococcus aureus reveals differential substrate specificities for MepA and MepR. Bioorg. Med. Chem. 18:2123–2130 [DOI] [PubMed] [Google Scholar]
- 17. Wilkinson SP, Grove A. 2006. Ligand-responsive transcriptional regulation by members of the MarR family of winged helix proteins. Curr. Issues Mol. Biol. 8:51–62 [PubMed] [Google Scholar]
- 18. Kumaraswami M, Schuman JT, Seo SM, Kaatz GW, Brennan RG. 2009. Structural and biochemical characterization of MepR, a multidrug binding transcription regulator of the Staphylococcus aureus multidrug efflux pump MepA. Nucleic Acids Res. 37:1211–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schindler BD, Seo SM, Jacinto PL, Kumaraswami M, Birukou I, Brennan RG, Kaatz GW. 2013. Functional consequences of substitution mutations in MepR, a repressor of the Staphylococcus aureus mepA multidrug efflux pump gene. J. Bacteriol. 195:3651–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dolan KT, Duguid EM, He C. 2011. Crystal structures of SlyA protein, a master virulence regulator of Salmonella, in free and DNA-bound states. J. Biol. Chem. 286:22178–22185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brugarolas P, Movahedzadeh F, Wang Y, Zhang N, Bartek IL, Gao YN, Voskuil MI, Franzblau SG, He C. 2012. The oxidation-sensing regulator (MosR) is a new redox-dependent transcription factor in Mycobacterium tuberculosis. J. Biol. Chem. 287:37703–37712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hong M, Fuangthong M, Helmann JD, Brennan RG. 2005. Structure of an OhrR-OhrA operator complex reveals the DNA binding mechanism of the MarR family. Mol. Cell 20:131–141 [DOI] [PubMed] [Google Scholar]
- 23. Kumarevel T, Tanaka T, Umehara T, Yokoyama S. 2009. ST1710-DNA complex crystal structure reveals the DNA binding mechanism of the MarR family of regulators. Nucleic Acids Res. 37:4723–4735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Quade N, Mendonca C, Herbst K, Heroven AK, Ritter C, Heinz DW, Dersch P. 2012. Structural basis for intrinsic thermosensing by the master virulence regulator RovA of Yersinia. J. Biol. Chem. 287:35796–35803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stevenson CE, Assaad A, Chandra G, Le TB, Greive SJ, Bibb MJ, Lawson DM. 7 June 2013, posting date Investigation of DNA sequence recognition by a streptomycete MarR family transcriptional regulator through surface plasmon resonance and X-ray crystallography. Nucleic Acids Res.[Epub ahead of print.] 10.1093/nar/gkt523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Newberry KJ, Fuangthong M, Panmanee W, Mongkolsuk S, Brennan RG. 2007. Structural mechanism of organic hydroperoxide induction of the transcription regulator OhrR. Mol. Cell 28:652–664 [DOI] [PubMed] [Google Scholar]
- 27. Andrésen C, Jalal S, Aili D, Wang Y, Islam S, Jarl A, Liedberg B, Wretlind B, Mårtensson LG, Sunnerhagen M. 2010. Critical biophysical properties in the Pseudomonas aeruginosa efflux gene regulator MexR are targeted by mutations conferring multidrug resistance. Protein Sci. 19:680–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Higgins PG, Fluit AC, Milatovic D, Verhoef J, Schmitz FJ. 2003. Mutations in GyrA, ParC, MexR and NfxB in clinical isolates of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 21:409–413 [DOI] [PubMed] [Google Scholar]
- 29. Llanes C, Hocquet D, Vogne C, Benali-Baitich D, Neuwirth C, Plésiat P. 2004. Clinical strains of Pseudomonas aeruginosa overproducing MexAB-OprM and MexXY efflux pumps simultaneously. Antimicrob. Agents Chemother. 48:1797–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saito K, Akama H, Yoshihara E, Nakae T. 2003. Mutations affecting DNA-binding activity of the MexR repressor of mexR-mexA-mexB-oprM operon expression. J. Bacteriol. 185:6195–6198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alekshun MN, Kim YS, Levy SB. 2000. Mutational analysis of MarR, the negative regulator of marRAB expression in Escherichia coli, suggests the presence of two regions required for DNA binding. Mol. Microbiol. 35:1394–1404 [DOI] [PubMed] [Google Scholar]
- 32. Davis JR, Brown BL, Page R, Sello JK. 2013. Study of PcaV from Streptomyces coelicolor yields new insights into ligand-responsive MarR family transcription factors. Nucleic Acids Res. 41:3888–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saridakis V, Shahinas D, Xu X, Christendat D. 2008. Structural insight on the mechanism of regulation of the MarR family of proteins: high-resolution crystal structure of a transcriptional repressor from Methanobacterium thermoautotrophicum. J. Mol. Biol. 377:655–667 [DOI] [PubMed] [Google Scholar]
- 34. Eschenfeldt WH, Lucy S, Millard CS, Joachimiak A, Mark ID. 2009. A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol. Biol. 498:105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Doublié S. 1997. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 276:523–530 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and methods. Download
Structural distortions in the MepR α1 and α5 helices effected by the Q18P mutation. Download
Selected statistics of data collection, structure solution, and refinement.
The alignment of MepR in its DNA binding conformation with other members of the MarR family that have been crystallized with their cognate DNA. (A) The alignment of MepR with SlyA (3Q5F), MosR (4FX4), RovA (4AIJ), OhrR (1Z9C), and SCO3205 (3ZPL) demonstrates similar architectures of the protein-DNA complexes, with the recognition helices binding to consecutive major grooves and the wings interacting with the flanking minor grooves. For clarity, only the DNA from the MepR-DNA model is shown. (B) The comparison of MepR to ST1710 reveals significant conformational differences, especially pronounced in DNA binding domains. Download
Sequence alignment of S. aureus MepR, P. aeruginosa MexR, E. coli MarR, B. subtilis OhrR, and S. enterica SlyA. The boxes indicate the mutation sites of MepR: Q18, F27, and A103. The sequence alignment was created using ClustalW and shaded using BoxShade 3.2. Download
The role of MepR residue Q18 in DNA binding. (A) The MepR-DNA complex model showing potential interactions between Q18 (red spheres) and DNA. (B) ITC experiment of mepR operator DNA binding to MepR(Q18A). The solid lines represent theoretical fits to the experimental data (closed squares); fitting parameters are provided in Table 1. Download
Alignment of MepR in its DNA binding conformation with MepR(Q18P). Only one subunit of the dimer is shown. Solid arrows point toward residues or structural elements of interest. Dashed arrows demonstrate the displacements of wHTH motifs of the Q18P mutant from its WT MepR location. P18 is shown in spheres. Key secondary structural elements and residues are labeled. Download