

Abstract
Protein kinase A (PKA) is a prototype of multidomain signaling proteins functioning as allosteric conformational switches. Allosteric transitions have been the subject of extensive structural and dynamic investigations focusing mainly on folded domains. However, the current understanding of the allosteric role of partially unstructured linkers flanking globular domains is limited. Here, we show that a dynamic linker in the regulatory subunit (R) of PKA serves not only as a passive covalent thread, but also as an active allosteric element that controls activation of the kinase subunit (C) by tuning the inhibitory preequilibrium of a minimally populated intermediate (apo R). Apo R samples both C-binding competent (inactive) and incompetent (active) conformations within a nearly degenerate free-energy landscape and such degeneracy maximally amplifies the response to weak (∼2RT), but conformation-selective interactions elicited by the linker. Specifically, the R linker that in the R:C complex docks in the active site of C in apo R preferentially interacts with the C-binding incompetent state of the adjacent cAMP-binding domain (CBD). These unanticipated findings imply that the formation of the intermolecular R:C inhibitory interface occurs at the expense of destabilizing the intramolecular linker/CBD interactions in R. A direct implication of this model, which was not predictable solely based on protein structure, is that the disruption of a linker/CBD salt bridge in the R:C complex unexpectedly leads to increased affinity of R for C. The linker includes therefore sites of R:C complex frustration and frustration-relieving mutations enhance the kinase inhibitory potency of R without compromising its specificity.
Keywords: allostery, cAMP, dynamics, NMR, intrinsically disordered proteins
Regulation of signaling systems often relies on multidomain proteins, which function as conformational switches controlled by allosteric effectors (1–13). The structural and dynamic changes experienced by folded domains during effector-dependent allosteric transitions have been extensively investigated (1–21). However, the current understanding of the allosteric role of partially unstructured linkers is at best scant. Although the role of covalent linkage in colocalization of protein domains is well known, it has recently been hypothesized that linkers, although generally quite flexible, have evolved to serve not simply as passive covalent threads connecting one domain to the next (i.e., “beads-on-a-string” model), but also as active components of functionally relevant allosteric networks (22–25). To test this hypothesis, here we have investigated the allosteric role of a critical linker in the regulatory subunit (R) of protein kinase A (PKA). This linker bridges the inhibitory site (IS) and a critical cAMP-binding domain (CBD) of R [i.e., RIα (119-244) or CBD domain A (CBD-A), ref. 26, Fig. 1A]. The R construct spanning the IS, the linker, and CBD-A [i.e., RIα (91-244) or RA in short, Fig. 1A] exhibits affinities for the catalytic subunit of PKA (C) and for cAMP as well as structures that are similar to those observed for longer RIα constructs (26–29). RA therefore inhibits C in a cAMP-dependent manner (27–29) and constitutes a functional regulatory unit. In addition, a similar R construct is also found fused to the RET oncogene kinase (30).
Fig. 1.

(A) Domain organization of the RIα subunit of PKA. (B–D) Structures of RIα (91-244) of PKA bound to either the C-subunit (H state, ref. 31, red) (C) or cAMP (B state, ref. 32, green) (D). (B) Superimposition of the H and B conformations. (E) Surface representation of the RIα (91-244):C holoenzyme structure with the C subunit shown in gray.
The structures of RA have been solved both in complex with C and cAMP (Fig. 1 B–E), providing the first picture of an R:C complex and revealing that CBD-A is composed of a structurally invariant β-subdomain and a more dynamic α-subdomain (27, 28, 31, 32). The α-subdomain tertiary structure changes dramatically depending on whether RA is bound to C or to cAMP (Fig. 1 B–D). When bound to C, CBD-A adopts a conformation denoted as “H” (inhibited Holoenzyme or inactive, analogous to “T” in the original Monod-Wyman-Changeux (MWC) model, ref. 4) state (Fig. 1 C and E) (31). In the H state, the inhibitory region docks in the active site of the kinase, and the α-subdomain of CBD-A provides additional contacts with C that result in an extended R:C interface, covering ∼3,000 Å2 (Fig. 1 C and E) (31). When bound to cAMP, CBD-A adopts a conformation denoted as “B” (active cAMP-Bound, analogous to “R” in the original MWC model, ref. 4) state (Fig. 1 B and D) (32). In apo RA, the H and B states coexist in a dynamic equilibrium (28), but their C vs. cAMP selectivity patterns are markedly different due to the H vs. B differences at the level of the α-subdomain. The C subunit exhibits higher affinity for the H- rather than the B state, whereas cAMP binds the B state more tightly than the H state, i.e., H and B represent the inactive and active forms of RA, respectively. As a result of the B selectivity of cAMP, cAMP binding to apo RA lowers the population of the H state, causing an effective weakening of the R:C interface and leading to the activation of PKA.
Whereas the crystal structures of the H and B states of RA (28, 31, 32) have been pivotal in understanding the cAMP-dependent activation of PKA, the allosteric role of the flexible RA linker [i.e., RIα (99-118)] remains elusive due to lack of electron density and to crystal packing for several linker residues (28, 32, 33). Does the RA linker control the cAMP-dependent activation of PKA? If so, how does cAMP affect the RA linker? Here, we address these questions using solution NMR and fluorescence and we propose a model showing that the RA linker, although dynamic and only partially structured, tunes the H vs. B equilibrium of apo RA. Using a thermodynamic linkage model tested through kinase bioassays, we also show that the position of the apo RA equilibrium is a key determinant of the cAMP-dependent activation of PKA, although apo R is only transiently populated in vivo. In addition, the covariance analysis of NMR chemical shifts reveals that the RA linker is an active element of the cAMP-dependent allosteric networks controlling kinase function. Our results illustrate potentially general principles explaining how flexible linkers exploit weak but conformation-selective interactions to control enzymatic activity and to be in turn controlled by allosteric effectors.
Results
The Impact of the Linker on the Globular cAMP-Binding Domain of RA Is Comparable to That of cAMP-Binding.
To probe the relevance of the RIα (91-118) linker, we compared the effects of linker deletion and of cAMP-binding. For this purpose, we measured both the apo RIα (91-244) vs. apo RIα (119-244) chemical shift differences (Fig. 2A, red bars) and the apo RIα (119-244) vs. cAMP-bound RIα (119-244) chemical shift map (Fig. 2A, black bars). Fig. 2A shows that both linker deletion and cAMP binding cause pervasive perturbations spanning the whole CBD-A [i.e., RIα (119-244)]. Although the extensive impact of cAMP binding is fully consistent with the long-range conformational changes expected for the H-to-B transition (Fig. 1B), the marked ppm variations caused by the linker deletion were somewhat unexpected because the linker is known to be only minimally structured based on prior hydrogen-deuterium exchange MS data (34). The lack of well-defined secondary structure in the linker is confirmed by the probability profiles derived from the secondary NMR chemical shifts (Fig. S1 A and B). Furthermore, the linker appears mostly flexible in the picosecond–nanosecond timescale, as indicated by the proton nitrogen nuclear overhauser effect data (Fig. S1C). Given that the linker is only partially structured and flexible, it is likely that it interacts only weakly with CBD-A.
Fig. 2.

RIα (91-118) linker shifts the B vs. H equilibrium of apo CBD-A toward the B state. (A) apo RIα (119-244) vs. apo RIα (91-244) (red) and apo vs. cAMP-bound RIα (119-244) (black) chemical shift differences. (B) B-state molar fraction (xB) vs. ΔG/RT (black) computed for a two-state equilibrium, where ΔG = GH − GB. The red curve is the first derivative of xB vs. ΔG/RT. (C) Representative H–N correlation peaks for apo, cAMP-, and C-bound RIα (91-244). (D) As C with C-bound RIα (91-244) replaced by apo RIα (119-244). Arrows indicate the effect of linker addition on the H vs. B equilibrium. (E) Correlation between the percent of B state in CBD-A and ΔLΔUFGoH2O, which quantifies the strength of the linker–CBD interactions (Table S1). (F) Scheme for the linker-induced shift of the CBD-A H vs. B equilibrium.
To reconcile the weakness of the linker/CBD-A interactions with the pervasiveness of the linker effects on CBD-A (Fig. 2A), we propose a hypothesis based on two key notions. First, the linker/CBD-A interactions, although weak, are state-selective, i.e., the linker preferentially interacts with either the B or the H state of CBD-A. Second, the B and H free energies of apo RA are nearly degenerate (i.e., GB ∼ GH). The energetic degeneracy of the B and H states maximizes the sensitivity of the relative B vs. H populations to weak state-selective interactions, as predicted by the theory of molecular sensors (35) and as illustrated for a simple two-state thermodynamic model in Fig. 2B. Fig. 2B shows that, when starting from a situation approaching free-energy degeneracy (ΔB,HG = 0 kcal/mol) with equal populations for both states (xH = xB = 0.5), a state-selective interaction equivalent to as little as RT [R = gas constant, T = temperature (K)] (∼0.6 kcal/mol) results in a population change of ∼50%. Weak but state-selective interactions of the linker have therefore the potential to cause global changes similar to those observed in Fig. 2A, provided that the B and H free energies in apo RA do not excessively deviate from near degeneracy (Fig. 2B, gray zone). To test this hypothesis we investigated further apo RA (36).
The B and H States of apo CBD-A Are Nearly Degenerate and Thus the B and H Populations Are Highly Sensitive to Weak State-Selective Interactions Such as Those Between the Linker and CBD-A.
To assess the relative B vs. H populations of apo RA, we compared the HN correlation spectra of apo RA to those of the same construct bound to either C or cAMP, which approximate the H and B states, respectively (Fig. 2C). For several residues that are remote from the C- and cAMP-binding interfaces (Fig. S2 C and D), the apo, C-, and cAMP-bound cross-peaks exhibit a clearly linear pattern with the apo state observed in the middle of the two bound states (Fig. 2C). This linear pattern is conserved in longer R constructs (Fig. S2 A and B) and is indicative of fast chemical exchange (37) between the B and H states in apo RA. The central position of the apo cross-peaks (Fig. 2C) points to comparable B and H populations, i.e., near degeneracy of the B and H states. This finding provides initial support of our hypothesis about the sensitivity of CBD-A to weak state-selective interactions.
To test if the linker is able to provide state-selective interactions and consequently to modify the state populations, we compared the hetero-nuclear single quantum coherence (HSQC) spectra of apo and cAMP-bound RIα (91-244) with that of apo RIα (119-244), without the linker segment (Fig. 2D). Again a linear pattern was observed, with the peaks of apo RIα (119-244) shifted away from those of cAMP-bound RIα (91-244), which approximates the B state (Fig. 2D). This observation suggests that the deletion of the (91-118) linker causes a consistent decrease in the population of the B state, pointing to B-selective linker/CBD-A interactions. To independently confirm this result and quantify the strength of the linker/CBD-A interactions, we measured the free energy of unfolding of both RIα (119-244) and RIα (91-244), each in the apo, cAMP- and Rp-cAMPS–bound states (Fig. S3). The Rp-cAMPS analog is a well-known reverse-agonist of PKA that is selective for the H state of CBD-A and therefore it is useful to promote the stabilization of the inactive H state (Fig. S2E) (28).
The Linker Preferentially Interacts with the B- Rather than the H-State of CBD-A.
The urea-unfolding profiles in Fig. S3 fully confirm the chemical shift-based results of Fig. 2D, indicating that the linker selectively interacts with the B- as opposed to the H-state of CBD-A. The urea-induced unfolding of RA was monitored through intrinsic fluorescence and, because all of the Trp residues present in the RA constructs (i.e., W188 and W222) are confined to CBD-A (38), the fluorescence data of Fig. S3 report primarily on the unfolding of the globular CBD-A. Fig. S3A illustrates that in the presence of excess cAMP, whereby RA populates mainly the B state, the deletion of the (91-118) linker destabilizes CBD-A by about 1.5 kcal/mol (Table S1). However, when RA is bound to the Rp-cAMPS reverse-agonist and populates primarily the H state, the removal of the (91-118) linker does not result in any significant destabilization of CBD-A (Fig. S3B and Table S1), indicating that the interactions of the linker with the H state of CBD-A are negligible compared with those between the linker and the B state of CBD-A.
As a further control for the B vs. H selectivity in the linker/CBD-A interactions, we also examined the urea-induced unfolding of apo RA with state populations that are intermediate between those of the cAMP- and Rp-cAMPS-bound states (Fig. 2C and Fig. S3C). As expected, the variation in the free energy of unfolding caused by the linker deletion in apo RA is in between the corresponding values observed in the presence of cAMP and Rp-cAMPS (Fig. 2E and Table S1), further confirming the B vs. H selectivity of the linker/CBD-A interactions. Such state-selective interactions contribute a change in the B vs. H free-energy difference of ∼1.4 kcal/mol (Fig. 2E and Table S1), which is slightly larger than 2RT ∼ 1.2 kcal/mol (Fig. 2B) and corresponds to an approximately one-order-of-magnitude variation in the B vs. H equilibrium constant (Fig. 2F). Further proof of the B vs. H preferential interaction of the linker is provided by paramagnetic relaxation enhancements (PREs) (Fig. S4 D–F). The B-selective linker/CBD-A interactions lead to two major implications discussed below, i.e. (i) the RA linker is an active element of the cAMP-dependent allosteric networks of RA and (ii) the RA linker tunes both kinase inhibition and activation.
The cAMP-Dependent Allosteric Networks of RA Extend Beyond the Globular Domain to Sites Within the Linker Region.
Because both cAMP and the linker interact preferentially with the B- as opposed to the H-state of CBD-A (Fig. 2F), we anticipate that the allosteric networks controlled by cAMP reach sites in the linker region as well. To test this prediction, we implemented for RA the chemical shift covariance analysis (CHESCA), which has been shown to be effective in mapping effector-dependent networks in other allosteric systems based on a small library of agonist and antagonists (39). This library includes cAMP, the reverse-agonist Rp-cAMPS, the partial agonist 2′-OMe-cAMP, and the agonist Sp-cAMPS (27, 40). Based on the residue-specific chemical shift changes caused by these ligands (Fig. S4 A and B), we computed a residue vs. residue correlation matrix (Fig. 3A). Fig. 3A clearly indicates that several correlations occur between the flexible linker region and the globular CBD-A (red highlight), providing initial support of our hypothesis about the coupling between cAMP and the linker. A further confirmation of this hypothesis is provided by agglomerative clustering (AC), which reveals that the largest residue cluster derived from the correlation matrix of Fig. 3A spans both CBD-A and several sites within the linker region (Fig. 3 A and B).
Fig. 3.

cAMP-dependent allosteric network of R reaches the linker region. (A) Chemical shift correlation matrix of RIα (91-244). Black dots identify residue pairs with absolute correlation coefficients ≥0.98. Grid lines connect residues in the largest agglomerative cluster (cluster 1). The linker region is highlighted in red. Key CBD-A/linker correlations are boxed by red lines. (B) Cluster 1 dendrogram. Residues linked through the dendrogram are labeled. Linker residues are in red. (C and D) Representative interresidue chemical shift correlations involving the linker region.
The AC residue cluster spanning both the linker and CBD-A (Fig. 3 A and B) underlies the cAMP-dependent activation of PKA, because the linear chemical shift correlations defining such network exhibit a point distribution that correlates well with PKA function: the active (i.e., cAMP and Sp-cAMPS) and inactive (i.e., apo and Rp-cAMPS) forms of RA are well separated (Fig. 3 C and D), with the partial agonist 2′-OMe-cAMP found in between these two sets (Fig. 3 C and D). Because the PKA activation profiles of these cAMP analogs were originally measured for the full-length R subunit (40), the CHESCA results (Fig. 3 C and D) support the relevance of the RA construct used in these NMR analyses. Note that the AC cluster underlying the cAMP-dependent activation of PKA includes the RA residues used in Fig. 2C (i.e., L221 and G159), confirming the suitability of these residues to monitor the B vs. H equilibrium independently of potentially obscuring binding effects. The results from the AC analysis are also more formally and more generally confirmed by the singular value decomposition (SVD) (Fig. S4C and Table S2) of the chemical shift matrix (SI Text) (39).
Overall, the CHESCA of RA (Fig. 3 and Fig. S4) corroborates our hypothesis that, although flexible, the RA linker is not simply a passive covalent thread between the inhibitory sites and CBD-A, but an active element of the functional allosteric network of RA. In particular, Fig. 3A reveals that several residues in the 112-116 linker region define a contiguous segment involved in extensive correlations with multiple CBD-A residues, suggesting that at least some of these residues may contribute to the preferential B vs. H interactions of the linker with CBD-A. This prediction was tested using two representative and previously undescribed point mutations targeting the 112-116 site, i.e., R113A and I116A. These mutants are expected to cause a loss of B-selective interactions and therefore to result in a shift of the apo RA equilibrium away from the B state. Such expected equilibrium shift was observed in the HSQC spectra of both mutants, showing that the peaks of apo R113A-RA and of apo I116A-RA shift away from those measured for cAMP-bound wild type (wt)-RA (Fig. S5E).
The B-state selective linker/CBD-A interactions mediated by R113 appear significant enough to result in B vs. H differential solvent exposure, as probed by H/H exchange (Fig. S5 A–D). The H/H exchange spectra were acquired for both the cAMP- and the Rp-cAMPS-bound forms of RA, which promote the stabilization of the B and H states of CBD-A, respectively (Fig. S5 A–D). The comparative cAMP vs. Rp-cAMPS H/H analysis reveals that residues 111-114 are more exposed in the Rp-cAMPS-bound form than in the presence of cAMP (Fig. S5 B and D, dotted box), confirming that R113 and the adjacent residues define a key site for the B-selective interaction of the linker with CBD-A. In agreement with these findings, the side chain of R113 forms multiple salt bridges with CBD-A residues (i.e., D146 and D149) in several crystal structures solved in the presence of cAMP or the Sp-cAMPS agonist (Fig. S6 E–G) (28, 33).
The salt bridges between R113 and D146/D149 detected in the crystal structures of Fig. S6 E–G are most likely preserved also in solution for the B state of RA, as confirmed by the H/H data of Fig. S5 A–D showing that several residues in the 141-150 region are more exposed in the Rp-cAMPS–bound form than in the presence of cAMP, similarly to what is observed for R113. However, it would have been challenging to establish the B-state selectivity of the linker/CBD-A contacts solely based on the crystal structures, because R113 is affected by crystal packing (28, 33) and R113 interacts with CBD-A also in the RA:C complex (Fig. S6 C and D) (31). In addition, the linker/CBD-A interface is significantly more extended in the RA:C complex (Fig. 1C) than in the RA:cAMP complex (Fig. 1D). In the former the linker becomes more structured by docking into the active site of C (Fig. 1E), whereas in the latter no electron density was observed for residues 91-111 of the linker (Fig. 1D). However, R113 mediates B-selective interactions, which are likely to be a feature shared by all PKA R-subunit isoforms because this residue is conserved (Fig. S6A). Such state-selective linker/CBD-A interactions loci are critical to tune the position of the B vs. H equilibrium in apo RA, as schematically illustrated in Fig. 4A.
Fig. 4.

Linker controls both inhibition and activation of PKA by modulating the B vs. H equilibrium of apo CBD-A. (A) Thermodynamic model of cAMP-dependent activation of PKA. The linker (red) is dynamic and samples conformations in which it is fully solvent exposed (“linker out” and conformations in which it transiently approaches the well-folded CBD-A (“linker in”), preferentially interacting with the B state of CBD-A (gray) and shifting the overall H vs. B apo equilibrium toward its midpoint (i.e., L = [H]apo/[B]apo ∼ 1). The H and B conformations exhibit high affinity for C and cAMP, respectively (thick lines), but weaker cross-interactions occur (thin dotted lines) (Table S3). The α- and β-subdomains of CBD-A are shown schematically in both the inactive (H) and active (B) conformations. (B) PKA activation by cAMP for wt RIα (91-244) (black circles) and the I116A (red circles) and R113A (blue circles) mutants, which weaken the B-selective interaction of the linker with CBD-A. Data were fit by activation curves (solid lines) computed based on the thermodynamic model in A (SI Text). (C) PKA inhibition by wt (black) and R113A (blue) RIα (91-244).
The RA Linker Tunes both Kinase Inhibition and Activation.
To evaluate how the B vs. H equilibrium of apo RA affects the cAMP-dependent activation of PKA, we developed a thermodynamic linkage model (Fig. 4A), which takes into account not only the selectivity of C and cAMP for the H and B states, respectively, but also the possibility of weaker cross-interactions (i.e., C:B state and cAMP:H-state, Table S3 and Eq. S1–S10). This model reproduces well the experimental profile for the cAMP-dependent activation of C inhibited by wt RA (Fig. 4B) and predicts that, although the apo form of the PKA R subunit is only transiently and minimally populated in vivo, the position of the B vs. H equilibrium of apo R (i.e., L = [H]apo/[B]apo) is a key determinant of PKA inhibition and activation. As L increases, the average affinity of R for C (Ka,R:C) and the half maximal effective concentration for activation (EC50) for the cAMP-dependent activation of PKA are both predicted to increase (Eq. S5 and Fig. S5F). These predictions were experimentally tested using the R113A mutant, which leads to increased L values, as proved by NMR (Fig. S5E). As expected based on the NMR data, the R113A mutation in the linker leads to several-fold increased EC50 and Ka,R:C values relative to wt (Fig. 4 B and C and Fig. S5F), further confirming the thermodynamic model of Fig. 4A and the role of the linker in tuning both kinase inhibition and activation.
Discussion
Our results directly support the notion that the dynamic RA linker serves the function of tuning the inhibition and the activation of PKA through weak but state-selective interactions, such as those mediated by R113 and I116, whose impact is amplified by the free-energy near degeneracy of apo RA. Considering that both residues are remote from the R:C and R:cAMP interfaces (Fig. S6 B and I), the effect of these sites on PKA activation is remarkable. Also unexpected was the enhanced stability of the RA:C holoenzyme resulting from the R113A mutation (Fig. 4C), despite the loss of the salt bridges mediated by the R113 guanidinium in RA:C (Fig. S6 C and D). These linker/H-state contacts are not affected by crystal packing and the apparently counterintuitive enhancement in the affinity of RA for C caused by the R113A RA mutation is explained considering that the salt bridges formed by R113 in C-bound wt RA (H state; Fig. S6 C and D) are weaker than those generated by R113 in the B state of apo wt RA (Fig. 5A and Fig. S6 E–G). Consequently, the R113A mutation causes only a marginal increase in the free energy of the RA:C complex, but a more significant increase in the free energy of apo RA, which samples the B state as well (Fig. 5). The overall net effect of the R113A mutation in RA is to increase the magnitude of the binding free energy for the RA:C complex (Fig. 5), i.e., R113A RA is a more potent kinase inhibitor than wt RA, as observed in Fig. 4C.
Fig. 5.
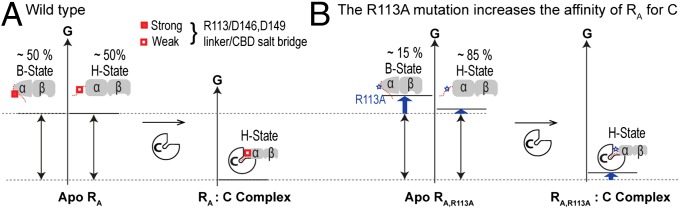
Effect of the R113A RA linker mutation on the binding free energy of the RA:C kinase-inhibitory complex. (A) Free-energy changes occurring upon formation of the wt RA:C complex. The C subunit selects primarily for the H state of apo wt RA. (B) As in A but for the R113A RA:C complex. In apo R113A RA, the H state is more populated than in apo wt RA, because R113 forms stable salt bridges in the B- but not in the H state of apo wt RA. For clarity, the weak cross-interactions between C and the B state of apo RA are not shown.
The enhanced stability of the R113A RA:C complex vs. the wt RA:C complex (Figs. 4C and 5) suggests that the interactions mediated by the R113 guanidinium in the wt RA:C complex (Fig. S6 C and D) are frustrated, although the contacts between the R113 guanidinium and the H state of CBD-A are partially stabilized by the C subunit as the IS at the N terminus of the linker is recruited into the active site of the kinase (Fig. 1 C and E). The local structural frustration (41) originating from the RA linker within the inhibitory RA:C complex is absent in cAMP-bound RA, because cAMP selects for the B state of CBD-A. Therefore, we conclude that structurally frustrated linkers have evolved to assist small molecules, such as cAMP, in effectively disrupting large (∼3,000 Å2) protein–protein interfaces, such as that in the RA:C complex.
In conclusion, we have shown that the dynamic linker of the functional RA construct of PKA (Fig. 1A) controls the position of the active vs. inactive conformational equilibrium of the apo cAMP-binding domain A (CBD-A). The active vs. inactive near degeneracy of the apo CBD-A amplifies the functional impact of weak but state-selective linker/CBD-A interactions, e.g., salt bridges between R113 in the linker and D146/D149 in CBD-A, which are preferentially formed in the B state (active) rather than the H state (inactive) of CBD-A. These state-selective linker/CBD-A interactions result in an effective coupling between the linker and CBD-A. We have shown both theoretically and experimentally that such linker/CBD-A coupling leads to two previously unanticipated consequences. First, the cAMP-dependent allosteric networks of RA unexpectedly extend well beyond the folded CBD-A domain to the more flexible linker. Second, the effect of the linker on the minimally populated apo RA intermediate is critical to tune not only the cAMP sensitivity of kinase activation (EC50), but also kinase inhibition half maximal inhibitory concentration (IC50). This is because the RA:C inhibitory interface, which selects for the H-state of CBD-A, is formed at the expense of locally destabilizing the intramolecular linker/CBD-A interactions; i.e., the linker includes discrete sites of local structural frustration in the RA:C inhibitory complex. Such frustration has evolved to assist the disruption of large protein–protein interfaces by small allosteric ligands and, when relieved by deletion mutations (e.g., R113A), leads to an R construct with enhanced kinase inhibitory potency without affecting the R:C interface. Overall, our results imply that not all multidomain proteins are adequately described by a beads-on-a-string model because selected linkers provide an additional level of functional regulation that goes beyond the colocalization of different structural modules, suggesting that multidomain systems are not only the product of gene fusion but of a higher degree of evolution. In addition, although the results presented here apply to the monomeric deletion RIα constructs used, they provide a necessary first step toward the full assessment of the role of the linker interactions in longer RIα constructs and in the quaternary structure of PKA.
Materials and Methods
Details for protein expression and purification, NMR data acquisition for the RIα (91-244), holoenzyme RIα (91-244):C complex, and for RIα (91-379), thermodynamic modeling and measurements of PKA inhibition, and cAMP-dependent activation are provided in SI Text. RIα (119-244) was expressed and purified according to previously published protocols (42–44).
All spectra were recorded at a temperature of 306 K on a Bruker AV 700 spectrometer equipped with TCI cryoprobe. All spectra were processed with NMRpipe (45) using linear prediction, unless otherwise specified, and a resolution-enhancing 60° shifted sine-squared bell window function. The NMR spectra were analyzed with Sparky (46) using Gaussian line-fitting. Assignments were obtained either through triple-resonance 3D experiments [i.e., HNCO, HNCA, HN(CO)CA, CBCA(CO)NH, and HNCACB] (47) and/or through spectral comparisons, if no ambiguities were present. HN NOE data were acquired as previously reported (48, 49) and processed without linear prediction. A 10-s recycle delay was used to include a 5-s proton saturation period. Data were collected in 10 sets of saturated and unsaturated interleaved spectra. All spectra were coadded before processing using NMRpipe, resulting in a total of 40 scans per serial file for each NOE experiment (with and without 1H saturation). The steady-state NOE values were computed as the ratio of the intensities in saturated to unsaturated spectra. The error on the NOE values were gauged based on the SD between fit heights in replicate spectra. Some of the least sensitive NMR experiments were acquired using the RIα (96-244) construct, which is more soluble than RIα (91-244) but preserves very similar spectra, i.e., 94% of RIα (96-244) HSQC peaks are overlapped with those of RIα (91-244). The secondary structure probabilities were determined using the secondary chemical shifts via the PECAN software (50). Combined chemical shift changes in Fig. 2A were measured as √(Δδ1H2+0.22Δδ15N2). In Fig. 2E, the percent of B state was assessed using four residues that sense the position of the H vs. B equilibrium (i.e., V115, G178, G223, and L221) and assuming that the RIα (91-244):C and RIα (91-244):cAMP complexes represent the H and B states, respectively.
Uniformly 15N-labeled RIα (91-244) was concentrated to 10 μM in 50 mM 3-(N-morpholino)propanesulfonic acid (MOPS) (pH 7), 100 mM NaCl, 10 mM MgCl2, 5 mM DTT, 0.02 mM 15N-acetylglycine, 0.02% sodium azide, and 5% 2H2O. Because the apo state and the four ligand-bound states (i.e., Rp-cAMPS-, Sp-cAMPS-, cAMP-, and 2′-OMe-cAMP-bound states) were used as the five perturbation sets for CHESCA, stock solutions of Rp-cAMPS, Sp-cAMPS, 2′-OMe-cAMP (BIOLOG), and cAMP (Sigma) were prepared using the same buffer as for the protein solution. Starting from the apo samples, stock solutions of the cyclic nucleotide ligands (25 mM) were titrated into the protein solution to full saturation (the final concentrations of ligands were 3 mM). Sensitivity-enhanced 15N-1H HSQC spectra with 256 (t1) and 1024 (t2) complex points and spectral widths of 31.82 and 14.06 ppm for the 15N and 1H dimensions, respectively, were recorded with 128 scans and a recycle delay of 1.70 s. The 1H and 15N carrier frequencies were set at the water resonance and in the middle of the amide region, respectively. The combined 15N and 1H chemical shifts (δ1H + 0.2 δ15N) from the HSQC spectra of apo and Rp-cAMPS-, Sp-cAMPS-, cAMP-, and 2′-OMe-cAMP-bound RIα (91-244) referenced to 15N-acetylglycine were compiled into a chemical shift matrix, which was then analyzed using AC and SVD, according to previously published protocols (39). All nodes in the dendrogram of Fig. 3B correspond to correlation coefficients with absolute values ≥0.98.
Supplementary Material
Acknowledgments
We thank Dr. F. Fogolari, Dr. D. Blumenthal, Dr. A. Kornev, Dr. M. Keshwani, L. Matthews, B. VanSchouwen, and M. Elgamal for helpful discussions, and the Canadian Institute of Health Research (G.M.) and the National Sciences and Engineering Research Council (G.M.) for financial support. We are also grateful to Prof. Brian E. McCarry (1946-2013) for his contributions to the NMR Facilities of McMaster University used for this work.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312644110/-/DCSupplemental.
References
- 1.Kuriyan J, Eisenberg D. The origin of protein interactions and allostery in colocalization. Nature. 2007;450(7172):983–990. doi: 10.1038/nature06524. [DOI] [PubMed] [Google Scholar]
- 2.Ruschak AM, Kay LE. Proteasome allostery as a population shift between interchanging conformers. Proc Natl Acad Sci USA. 2012;109(50):E3454–E3462. doi: 10.1073/pnas.1213640109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vendruscolo M. Protein regulation: The statistical theory of allostery. Nat Chem Biol. 2011;7(7):411–412. doi: 10.1038/nchembio.603. [DOI] [PubMed] [Google Scholar]
- 4.Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308(5727):1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 5.Gardino AK, et al. Transient non-native hydrogen bonds promote activation of a signaling protein. Cell. 2009;139(6):1109–1118. doi: 10.1016/j.cell.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kern D, Zuiderweg ER. The role of dynamics in allosteric regulation. Curr Opin Struct Biol. 2003;13(6):748–757. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 7.Aghazadeh B, Lowry WE, Huang XY, Rosen MK. Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell. 2000;102(5):625–633. doi: 10.1016/s0092-8674(00)00085-4. [DOI] [PubMed] [Google Scholar]
- 8.Long D, Brüschweiler R. Atomistic kinetic model for population shift and allostery in biomolecules. J Am Chem Soc. 2011;133(46):18999–19005. doi: 10.1021/ja208813t. [DOI] [PubMed] [Google Scholar]
- 9.Smock RG, Gierasch LM. Sending signals dynamically. Science. 2009;324(5924):198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tzeng SR, Kalodimos CG. Dynamic activation of an allosteric regulatory protein. Nature. 2009;462(7271):368–372. doi: 10.1038/nature08560. [DOI] [PubMed] [Google Scholar]
- 11.Boehr DD, Nussinov R, Wright PE. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol. 2009;5(11):789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Religa TL, Sprangers R, Kay LE. Dynamic regulation of archaeal proteasome gate opening as studied by TROSY NMR. Science. 2010;328(5974):98–102. doi: 10.1126/science.1184991. [DOI] [PubMed] [Google Scholar]
- 13.Hayashi I, Wilde A, Mal TK, Ikura M. Structural basis for the activation of microtubule assembly by the EB1 and p150Glued complex. Mol Cell. 2005;19(4):449–460. doi: 10.1016/j.molcel.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 14.Berman HM, et al. The cAMP binding domain: An ancient signaling module. Proc Natl Acad Sci USA. 2005;102(1):45–50. doi: 10.1073/pnas.0408579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13(9):831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manley G, Loria JP. NMR insights into protein allostery. Arch Biochem Biophys. 2012;519(2):223–231. doi: 10.1016/j.abb.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masterson LR, et al. Dynamics connect substrate recognition to catalysis in protein kinase A. Nat Chem Biol. 2010;6(11):821–828. doi: 10.1038/nchembio.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishiyama N, et al. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell. 2010;141(1):117–128. doi: 10.1016/j.cell.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 19.Tzeng SR, Kalodimos CG. Protein activity regulation by conformational entropy. Nature. 2012;488(7410):236–240. doi: 10.1038/nature11271. [DOI] [PubMed] [Google Scholar]
- 20.Dhulesia A, Gsponer J, Vendruscolo M. Mapping of two networks of residues that exhibit structural and dynamical changes upon binding in a PDZ domain protein. J Am Chem Soc. 2008;130(28):8931–8939. doi: 10.1021/ja0752080. [DOI] [PubMed] [Google Scholar]
- 21.Marlow MS, Dogan J, Frederick KK, Valentine KG, Wand AJ. The role of conformational entropy in molecular recognition by calmodulin. Nat Chem Biol. 2010;6(5):352–358. doi: 10.1038/nchembio.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarkar P, Saleh T, Tzeng SR, Birge RB, Kalodimos CG. Structural basis for regulation of the Crk signaling protein by a proline switch. Nat Chem Biol. 2011;7(1):51–57. doi: 10.1038/nchembio.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmad A, et al. Heat shock protein 70 kDa chaperone/DnaJ cochaperone complex employs an unusual dynamic interface. Proc Natl Acad Sci USA. 2011;108(47):18966–18971. doi: 10.1073/pnas.1111220108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma B, Tsai CJ, Haliloğlu T, Nussinov R. Dynamic allostery: Linkers are not merely flexible. Structure. 2011;19(7):907–917. doi: 10.1016/j.str.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhuravleva A, Gierasch LM. Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc Natl Acad Sci USA. 2011;108(17):6987–6992. doi: 10.1073/pnas.1014448108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kornev AP, Taylor SS, Ten Eyck LF. A generalized allosteric mechanism for cis-regulated cyclic nucleotide binding domains. PLOS Comput Biol. 2008;4(4):e1000056. doi: 10.1371/journal.pcbi.1000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anand GS, et al. Cyclic AMP- and (Rp)-cAMPS-induced conformational changes in a complex of the catalytic and regulatory (RIα) subunits of cyclic AMP-dependent protein kinase. Mol Cell Proteomics. 2010;9(10):2225–2237. doi: 10.1074/mcp.M900388-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Badireddy S, et al. (2011) Cyclic AMP analog blocks kinase activation by stabilizing inactive conformation: Conformational selection highlights a new concept in allosteric inhibitor design. Mol Cell Proteomics 10:M110.004390. [DOI] [PMC free article] [PubMed]
- 29.Huang LJ, Taylor SS. Dissecting cAMP binding domain A in the RIalpha subunit of cAMP-dependent protein kinase. Distinct subsites for recognition of cAMP and the catalytic subunit. J Biol Chem. 1998;273(41):26739–26746. doi: 10.1074/jbc.273.41.26739. [DOI] [PubMed] [Google Scholar]
- 30.Bongarzone I, et al. Molecular characterization of a thyroid tumor-specific transforming sequence formed by the fusion of ret tyrosine kinase and the regulatory subunit RI α of cyclic AMP-dependent protein kinase A. Mol Cell Biol. 1993;13(1):358–366. doi: 10.1128/mcb.13.1.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim C, Xuong NH, Taylor SS. Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science. 2005;307(5710):690–696. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- 32.Su Y, et al. Regulatory subunit of protein kinase A: Structure of deletion mutant with cAMP binding domains. Science. 1995;269(5225):807–813. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- 33.Wu J, Jones JM, Nguyen-Huu X, Ten Eyck LF, Taylor SS. Crystal structures of RIalpha subunit of cyclic adenosine 5′-monophosphate (cAMP)-dependent protein kinase complexed with (Rp)-adenosine 3′,5′-cyclic monophosphothioate and (Sp)-adenosine 3′,5′-cyclic monophosphothioate, the phosphothioate analogues of cAMP. Biochemistry. 2004;43(21):6620–6629. doi: 10.1021/bi0302503. [DOI] [PubMed] [Google Scholar]
- 34.Hamuro Y, et al. Mapping intersubunit interactions of the regulatory subunit (RIalpha) in the type I holoenzyme of protein kinase A by amide hydrogen/deuterium exchange mass spectrometry (DXMS) J Mol Biol. 2004;340(5):1185–1196. doi: 10.1016/j.jmb.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 35.Vallée-Bélisle A, Ricci F, Plaxco KW. Thermodynamic basis for the optimization of binding-induced biomolecular switches and structure-switching biosensors. Proc Natl Acad Sci USA. 2009;106(33):13802–13807. doi: 10.1073/pnas.0904005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das R, Abu-Abed M, Melacini G. Mapping allostery through equilibrium perturbation NMR spectroscopy. J Am Chem Soc. 2006;128(26):8406–8407. doi: 10.1021/ja060046d. [DOI] [PubMed] [Google Scholar]
- 37.Palmer AG., 3rd NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104(8):3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 38.Leon DA, Canaves JM, Taylor SS. Probing the multidomain structure of the type I regulatory subunit of cAMP-dependent protein kinase using mutational analysis: Role and environment of endogenous tryptophans. Biochemistry. 2000;39(19):5662–5671. doi: 10.1021/bi992819z. [DOI] [PubMed] [Google Scholar]
- 39.Selvaratnam R, Chowdhury S, VanSchouwen B, Melacini G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proc Natl Acad Sci USA. 2011;108(15):6133–6138. doi: 10.1073/pnas.1017311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Christensen AE, et al. cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J Biol Chem. 2003;278(37):35394–35402. doi: 10.1074/jbc.M302179200. [DOI] [PubMed] [Google Scholar]
- 41.Ferreiro DU, Hegler JA, Komives EA, Wolynes PG. On the role of frustration in the energy landscapes of allosteric proteins. Proc Natl Acad Sci USA. 2011;108(9):3499–3503. doi: 10.1073/pnas.1018980108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Das R, Melacini G. A model for agonism and antagonism in an ancient and ubiquitous cAMP-binding domain. J Biol Chem. 2007;282(1):581–593. doi: 10.1074/jbc.M607706200. [DOI] [PubMed] [Google Scholar]
- 43.Das R, et al. cAMP activation of PKA defines an ancient signaling mechanism. Proc Natl Acad Sci USA. 2007;104(1):93–98. doi: 10.1073/pnas.0609033103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Das R, et al. Dynamically driven ligand selectivity in cyclic nucleotide binding domains. J Biol Chem. 2009;284(35):23682–23696. doi: 10.1074/jbc.M109.011700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 46. Goddard TD, Kneller DG (2006) Sparky 3 (Univ California, San Francisco)
- 47.Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Magn Reson Spectrosc. 1999;34:93–158. [Google Scholar]
- 48.Renner C, Schleicher M, Moroder L, Holak TA. Practical aspects of the 2D 15N-[1h]-NOE experiment. J Biomol NMR. 2002;23(1):23–33. doi: 10.1023/a:1015385910220. [DOI] [PubMed] [Google Scholar]
- 49.Das R, et al. Entropy-driven cAMP-dependent allosteric control of inhibitory interactions in exchange proteins directly activated by cAMP. J Biol Chem. 2008;283(28):19691–19703. doi: 10.1074/jbc.M802164200. [DOI] [PubMed] [Google Scholar]
- 50.Eghbalnia HR, Bahrami A, Wang L, Assadi A, Markley JL. Probabilistic identification of spin systems and their assignments including coil-helix inference as output (PISTACHIO) J Biomol NMR. 2005;32(3):219–233. doi: 10.1007/s10858-005-7944-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.