

Abstract
Background
In Bacillus mycoides, as well as in other members of the B. cereus group, the tubulin-like protein of the division septum FtsZ is encoded by the distal gene of the cluster division and cell wall (dcw). Along the cluster the genes coding for structural proteins of the division apparatus are intermingled with those coding for enzymes of peptidoglycan biosynthesis, raising the possibility that genes with this different function might be coexpressed. Transcription of ftsZ in two model bacteria had been reported to differ: in B. subtilis, the ftsZ gene was found transcribed as a bigenic mRNA in the AZ operon; in E. coli, the transcripts of ftsZ were monogenic, expressed by specific promoters. Here we analyzed the size and the initiation sites of RNAs transcribed from ftsZ and from other cluster genes in two B. mycoides strains, DX and SIN, characterized by colonies of different chirality and density, to explore the correlation of the different morphotypes with transcription of the dcw genes.
Results
In both strains, during vegetative growth, the ftsZ-specific RNAs were composed mainly of ftsZ, ftsA-ftsZ and ftsQ-ftsA-ftsZ transcripts. A low number of RNA molecules included the sequences of the upstream murG and murB genes, which are involved in peptidoglycan synthesis. No cotranscription was detected between ftsZ and the downstream genes of the SpoIIG cluster. The monogenic ftsZ RNA was found in both strains, with the main initiation site located inside the ftsA coding sequence. To confirm the promoter property of the site, a B. mycoides construct carrying the ftsA region in front of the shortened ftsZ gene was inserted into the AmyE locus of B. subtilis 168. The promoter site in the ftsA region was recognized in the heterologous cellular context and expressed as in B. mycoides.
Conclusions
The DX and SIN strains of B. mycoides display very similar RNA transcription specificity. The ftsZ messenger RNA can be found either as an independent transcript or expressed together with ftsA and ftsQ and, in low amounts, with genes that are specific to peptidoglycan biosynthesis.
Keywords: B. mycoides, dcw cluster, ftsZ, Initiation of transcription
Background
Bacillus mycoides, a Gram positive soil rod bacillus of the B. cereus species-group [1], is characterized by hyphal colonies with cells connected at the poles in long filaments. These filaments converge into bundles that mainly curve clock- or counter-clockwise in two kinds of bacilli, both of which were attributed to B. mycoides[2].
We have previously isolated [3] examples of the two types from the environment and followed the process of colony formation on agar of two strains, i.e. DX with the right-curving colony branches and SIN with the left-curving colony branches. The initial cell filaments formed after seeding the cells on agar already showed the strain-specific turn direction and the colonies in the advanced growth stage appeared with a different overall density. The differences prompted a genetic characterization of the strains beyond the identical metabolic properties detected by monitoring 50 enzymatic reactions using the API50CH test. Genomic similarity of DX and SIN was thus checked by examining the region of the dcw (division cell wall) cluster, composed of a group of fundamental genes coding for several proteins of the division apparatus and for enzymes of peptidoglycan biosynthesis [3]. The distribution in the cells of the sites of new peptidoglycan synthesis, which was also analyzed in these strains, was found to be very similar [4]. A very limited number of DX and SIN nucleotides differs along the dcw region. This points to a close evolutionary relationship between the two strains as well as between the members of the B. cereus group. Comparative genome analysis of a large number of bacilli attributed to the group recently led to the proposal that they should be classified as a single species [1].
Here we extended sequencing to additional genes of the cluster and, in order to better characterize these different strains, we examined the RNAs expressed in vegetative cells. In particular, we focused on the specific transcripts of the genes coding for two proteins, FtsZ and FtsA, which are the building blocks of the Z ring assembly for septum formation during cell division. Among the various bacilli, the expression of these two genes was examined only in B. subtilis[5,6]. Both papers reported that ftsA and ftsZ form an operon, transcribed as a bigenic ftsA-ftsZ RNA. In the Northern blot shown by Gholamhoseinian et al. [5], the ftsZ probe binds to a band with the length of a single-gene transcript, but it was not investigated further because it was considered as a degradation product. We found instead that in both B. mycoides strains, in addition to polycistronic transcripts, ftsZ is transcribed as the single-gene RNA, independently of ftsA.
Results and discussion
Northern blot analysis of transcripts
In B. mycoides, ftsA and ftsZ occupy the 3’ end of the dcw cluster, separated by 39 bp of non-coding DNA. Transcripts of these two genes were sized in Northern blots of SIN and DX vegetative RNA (Figure 1).
Figure 1.
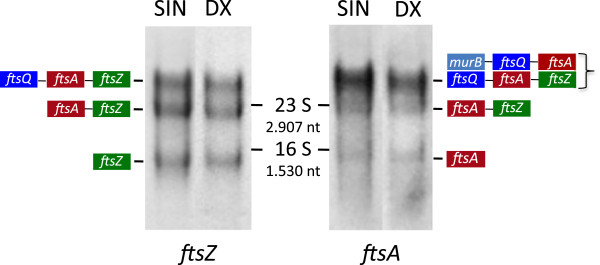
Northern blot analysis of RNA from exponentially growing B. mycoides SIN and DX. SIN and DX total RNA was electrophoresed in formaldehyde-agarose and blotted. The same filter was hybridized first to ftsZ and, after stripping, to ftsA DNA probes. The position of ribosomal 23S (2907 bases) and 16S (1530 bases) RNA on the filter is indicated. FtsZ and ftsA RNAs in the band below 16S rRNA are monogenic transcripts. The band below the position of the 23 S rRNA contains the ftsA-ftsZ bigenic transcripts. The transcripts of the genes ftsQ-ftsA-ftsZ are within the uppermost bands together with the transcripts murB-ftsQ-ftsA, detected only by the ftsA probe.
The ftsZ DNA probe detected three main RNA components in SIN and DX: the shortest one, found just below the position of the 16S B. mycoides ribosomal RNA (1530 nucleotides), was the size of the monogenic transcript, since the ftsZ coding region spans 1155 nucleotides from the starting ATG to the termination triplet; the second hybridization band, below the 23S ribosomal RNA (2907 nucleotides), harbored transcripts with the length of ftsA plus ftsZ; the third band, in the upper part of the gel, carried RNAs corresponding to the size of three genes, thus ftsQ-ftsA-ftsZ since ftsZ is not cotranscribed with downstream genes.
The ftsA probe, hybridized to the same filters, revealed three ftsA-specific RNA bands. The fastest one migrated slightly less than the monogenic ftsZ RNA band, which is in keeping with the 144 bp longer coding sequence of the ftsA gene; the second ftsA-specific band colocalized with the ftsZ bicistronic transcripts; the third band in the uppermost position was broader and more intense than the other two bands, indicating that ftsA was particularly abundant in long transcripts, mostly ftsQ-ftsA-ftsZ RNA. The intensity of the uppermost band is higher when probed with ftsA than when probed with ftsZ, indicating that a fraction of the transcripts does not contain ftsZ but carries the RNA of the murB gene, located upstream of ftsQ (Figure 1, schematics).
These results show that the bulk of the ftsA and ftsZ-specific RNAs were in molecules spanning one, two and three gene units, though the low level of detection and molecular weight definition of the Northern blots required further analysis.
Primer extension analysis of ftsZ, ftsA and ftsQ RNA
In order to map the initiation sites of the observed RNAs, the vegetative SIN and DX RNAs were analyzed by Primer Extension (PE) (Figure 2). FtsZ transcripts were hybridized to primer ZB (Table 1), annealing to RNA at nucleotide position +103 relative to the A of the first ATG codon of the ftsZ open reading frame (+1). Two cDNA bands, elongated by reverse transcriptase (RT) starting from this primer, stopped at positions −14 and −140 (Figure 2A and Additional file 1). The −140 cDNA, which mapped inside the coding sequence of the preceding gene ftsA, was more abundant than the one at −14. The fact that the −14 position lies in the spacer region between ftsA and ftsZ, at the upper end of the ribosome binding site (RBS), suggests that this RNA may originate from a longer RNA, such as the one at −140, protected from degradation by ribosomes bound to the RBS.
Figure 2.

Determination of ftsZ, ftsA and ftsQ RNA 5’ ends by primer extension (PE) in B. mycoides SIN (S) and DX (D). 5’ 32P-labeled primers were hybridized to total RNA, extended by reverse transcriptase and the cDNAs separated by 6% urea-PAGE electrophoresis. The numbers on the right side of the autoradiograms indicate the position of the cDNA 3’ ends relative to the ORF first nucleotide (+1). The thick lateral bar indicates the approximate position in the gel of the next upstream gene. A) PE from primer ZB annealing to ftsZ RNA at +103; B) PE from primer Arev annealing to ftsA RNA at +80; C) PE from primer Qrev annealing to ftsQ RNA at +52 (Table 1). Here two different SIN cDNA preparations were loaded on the gel. A schematic view of the major cDNA products is shown in the inset. M = MW marker 32P-labeled DNAs. GATC = 35S-dATP labeled M13mp18 ladder.
Table 1.
Primers used in this study
| Primer name | Primer sequence | gene | (a) |
|---|---|---|---|
|
Northern probes | |||
| Zfor |
AAAGTWATCGGTGTCGGCGGWGGC |
ftsZ |
+43 |
| Zrev |
CAGAAATACCTTGAACCCCTTGGCG |
ftsZ |
+595 |
| Ain |
GAACAGCAATGAAATATATGTTG |
ftsA |
+3 |
| N2R |
ACCGTCTACAATGAACTGTC |
ftsA |
+411 |
|
Primer Extension | |||
| prex |
GCCCAAACCGCACTCGCAC |
ftsW |
+95 |
| Wrev |
AATCCATTCTCTGTACCAATG |
murG |
+125 |
| Rip2 |
GTTGCTTAGYAGCCAGTTTC |
murG |
+1030 |
| Qrev |
TCTTTARCTTTGGTACACGATC |
ftsQ |
+52 |
| Arev |
TCATTAACCATTTCACCAATGATG |
ftsA |
+80 |
| N2R |
ACCGTCTACAATGAACTGTC |
ftsA |
+411 |
| ZB |
CACCGTGTTCAATCATACGG |
ftsZ |
+103 |
| ZD |
ACAACCAAACAACGTCGGCG |
spoIIGA |
+74 |
| ZDbis |
CCTAACACAAGCCTCCATC |
spoIIGA |
+158 |
| BigD |
CCCAAATGCTGTATACACAATAAGTAACGAG |
spoIIGA |
+273 |
|
RT-PCR | |||
| Zfin |
CTTTTATCGTCTACGACGGTTAC |
ftsZ |
+1158 |
| Zin |
CATGTTAGAGTTTGATACTACTC |
ftsZ |
−1 |
| Ain |
GAACAGCAATGAAATATATGTTG |
ftsA |
+3 |
| Afin |
CCCATAAATAACGGAATGCACG |
ftsA |
+1297 |
| Qin |
CGTACATGAARAAYAGTAARG |
ftsQ |
−5 |
| Mbin |
GAGATTGTCTATGGAACAATTAG |
murB |
−10 |
| MGin |
ACAGCTGAAACNCTTATTCGTG |
murG |
+964 |
| Fw |
CATCAGCACCGTATCGRATG |
ftsW |
+601 |
|
Mini-ftsZ |
|
|
(b) |
| Hind5 |
GACAAGCTTATATTGGTGTTCGTGAG |
ftsA |
+1056 |
| Eco5 |
GGCGAATTCGCTAATTGATCTTGAG |
ftsZ |
+39 |
| Eco3 |
CACGAATTCAAAACAACGTGAAGTTAAG |
ftsZ |
+1035 |
| Bam3 |
GGCGGATCCAAAAAGGAGCATGAAAGCTC |
spacer |
+28 |
| Amy5 | GCCGCGATTTCCAATGAGG | pJPR1 | +245 |
(a) Position of the primer 5’ nucleotide on the corresponding gene numbering beginning from the first codon of the gene (+1).
(b) Position on the gene of the first complementary primer base after the added restriction site evidenced bold.
cDNA bands were also detected in a gel position close to the 1650 bp MW marker, thus mapping within the spacer region between ftsA and the upstream gene ftsQ. Additional bands were visible in the upper part of the sequencing gels, where compression does not allow size definition. These data indicate that ftsZ is transcribed as a monogenic RNA and a bigenic ftsA-ftsZ RNA, thereby confirming the Northern blot data.
Initiation sites of ftsA-specific RNAs were analyzed by PE from primer Arev (+ 80 in ftsA, Table 1). Three minor cDNAs mapped at −9, -57 and −77 and a major one at −222 from the first nucleotide of the ftsA ORF, all of them within the 400 bp spacer region between ftsQ and ftsA (Figure 2B and Additional file 1). The major −222 RNA transcript resembles the vegetative P3 transcript of B. subtilis initiating at −285 from the ftsA ORF [6]. The −222 start site is preceded by the same modules for sigmaA recognition as the B. subtilis promoter, mapped within the sbp gene that separates ftsQ from ftsA in B. subtilis. In B. mycoides, there is no open reading frame in the Q-A spacer region, but only similarity to B. subtilis sbp in short dispersed sequences.
Figure 2C shows the ftsQ-specific cDNAs extended from primer Qrev (+52, Table 1). cDNA elongation stopped at nucleotide −30 both in SIN and DX. The two strains differed in this location insofar as a cDNA band was present at −27/28 in DX alone and one at −53 in SIN alone. In this region, one base difference between the two strains changes the stability of a stem composed of two inverted repeats of 11 nucleotides. Several cDNA ends, which were either strain-specific or common to both strains, were visible within the upstream murB gene sequences. The RNA initiation sites located upstream of murB indicate the cotranscription of ftsQ with murB and probably with murG, though gel compression prevents a precise length determination of the cDNAs.
RT-PCR analysis of dcw transcripts
The high MW transcripts were instead highlighted by RT-PCR analysis (Figure 3). Using B. mycoides RNAs controlled for the absence of DNA, cDNA was synthesized from the Zfin primer which is complementary to the 3’end of ftsZ. PCR amplifications of the cDNA were then produced using this downstream primer and descending primers from each of the sequenced B. mycoides dcw genes (Table 1). The longest amplification product (lane B of the agarose gel) indicated the existence of RNA transcribed from 5 genes, murG, murB, ftsQ, ftsA and ftsZ. The PCR did not detect molecules including ftsW/spoVE sequences (lane A).
Figure 3.

RT-PCR analysis of RNA transcripts from the dcw genes in B. mycoides. Purified vegetative RNA of B. mycoides DX was reverse transcribed from primers complementary to the 3’ end of ftsZ (Zfin) and to the 3’ end of ftsA (Afin). The control cDNAs (lanes -) were without RT in the reaction. cDNAs were PCR amplified using Zfin (A-F) and Afin (G-H) as downstream primers. Upstream primers were specific for each gene (Table 1). Multigene ftsZ RNAs included murG and murB, though not ftsW transcripts.
The cDNA prepared using the primer Afin, complementary to the end of the ftsA gene, was also amplified using Afin as the downstream primer and upstream primers specific for murB and for ftsQ (Figure 3, lanes G, H). Although a simple PCR does not provide a precise quantification, the murB-ftsQ-ftsA RNA and the ftsQ-ftsA RNA are better represented than the RNA ftsQ-ftsA-ftsZ, which is in accordance with the Northern blot data.
The continuous coverage by RNA transcripts of the dcw cluster from murG to ftsZ has recently been reported in another member of the B. cereus group, the B. anthracis Ames ancestor, in the study of the whole genome transcriptome. The shotgun sequencing of cDNA (RNA-Seq) obtained from RNA transcribed under various growth conditions provided a map of transcription start sites and operon structure in the B. anthracis genome; in this study the ftsZ gene was found to be cooperonic with ftsA, ftsQ, murB and murG. [7].
Heterologous expression of a ftsZ minigene
Monogenic transcripts of the ftsZ gene, guided by at least three promoters located within the ftsA coding region, have been described in E. coli[8]. In the Gram positive model bacillus, B. subtilis, the ftsZ RNA was only considered as a part of the bigenic transcript of the AZ operon, directed by the activity of three promoters of different growth phase specificity located upstream of ftsA[5,6]. In our assays, Northern blots and PE data indicated transcription of ftsZ as a single gene; thus we decided to search for a bona fide promoter upstream of the RNA start sites seen in the experiments.
When determined by the primer extension technique, the real initiation point of a messenger RNA can sometimes be uncertain owing to RNA processing or to premature termination of the reverse transcriptase at secondary structures of the RNA. Our hypothesis was that if a specific promoter drove transcription of the ftsZ monogenic RNA, this mechanism could work in a similar cellular context. We thus chose to insert the B. mycoides DNA region harboring the putative −140 and −14 ftsZ initiation sites at the chromosomal amyE locus of B. subtilis. The −140 site is within the 3’ coding region of ftsA and the −14 site in the spacer region between ftsA and ftsZ (Additional file 1).
We created a shortened B. mycoides DX ftsZ gene, missing the central coding region, to make it easily distinguishable from the endogenous B. subtilis gene. The minigene was preceded by the 286 bp region containing the −140 and the −14 putative initiation sites and followed by 28 bp of the 3’ non-coding region after the ftsZ termination codon. The construct was inserted at the B. subtilis str.168 amyE locus after cloning into the pJPR1 integrative vector (amyE:: Pxylcat[9]). Plasmid pJPR1 carries the 5’ and 3’ regions of the B. subtilis amyE gene for integration of the recombinant sequences into the chromosome by a double cross-over. The sequences inserted into the plasmid cloning site and eventually integrated at the amyE site become controlled by the strong promoter Pxyl, which is induced by xylose but is normally blocked by a tight repressor (Figure 4B).
Figure 4.

Initiation of mini-ftsZ RNA transcripts in B. subtilis. The B. mycoides mini-ftsZ DNA construct was cloned into pJPR1 and inserted at the AmyE site of B. subtilis 168 (see methods). Transcripts of the construct were detected in total B. subtilis RNA by primer extension from the labeled primer Amy5 (Table 1) specific to the amyE 5’ region located 245 nt downstream of the inserted construct. A) Autoradiogram of PE. Lanes1 and 2: transcripts originating from the Pxyl promoter, induced by 5% xylose for 18 and 3 hours. Lane 3: the faint transcripts of the ftsZ minigene present in the non-induced B. subtilis recombinant strain are indicated by asterisks and map at −140 and −10 from the first nucleotide of the minigene ftsZ ORF as in B. mycoides. These bands are not present in the control B. subtilis strain (lane 4). B) schematic view of the construct in pJPR1. C) Schematic representation of the cDNAs indicated by asterisks in A. The red circle marks the position of the terminator structure 3’ to the B. mycoides ftsZ ORF. M = MW marker DNA. GATC = M13MP18 sequence ladder.
RNAs transcribed in the recombinant and wild type B. subtilis strains were analyzed by primer extension (Figure 4A), with the labeled primer Amy5 (Table 1) annealed to RNA of the 5’ AmyE region 245 nucleotides downstream of the minigene construct.
In addition to the aspecific bands present in both lanes, two faint but clear cDNA bands were detected in the recombinant (Figure 4A, lane 3) though not in the control B. subtilis (Figure 4A, lane 4). These bands are magnified in the lateral view. The longer cDNA (575 bp) maps at the nucleotide located at −140 bp from the starting ATG of the inserted mini-ftsZ, which is the same initiation site as that found for the RNA transcribed in B. mycoides. The second cDNA (465 bp) maps located in the short spacer region between ftsA and ftsZ containing the −14 site. The data show that the heterologous region is recognized by the B. subtilis transcription machinery as containing promoter elements and is hence transcribed as in the original context. As for the −14 RNA that starts at the RBS preceding the ftsZ ATG, it is still difficult to establish whether this shorter RNA is a maturation product of the longer RNA or an independent transcript.
When the pxyl promoter was induced by xylose for 18 hr (lane 1) and 3 hr (lane 2), strong cDNA bands were produced. The most intense band at position 255 is composed of a stop of the RT at the termination sequence located at the end of the B. mycoides mini-ftsZ. However, the RT also bypasses the terminator hairpin-loop structure and extends the cDNA up to the vector promoter site, forming the top band, which is about 800 bases in length. The lower bands are due to cDNA terminations in the vector sequences between the Amy5 primer and the minigene.
Termination sequences
Transcription termination in E. coli is helped by specific proteins such as Rho [10], while Rho independent termination sites, in the form of RNA hairpins followed by a polyU stretch [11], are commonly found in Gram positive bacilli. The close parenthood of B. mycoides with the B. cereus group members prompted us to make use of the prediction program of Transcription Terminators, developed for Firmicutes, at the TransTerm-HP site [12]. The presumed termination sequences considered were those relative to B. weihenstephanensis[13], the annotated genome with the highest similarity to the DX isolate. Only 34 nucleotide differences are present between DX and B. weihenstephanensis in the 10.731 bp dcw region we analyzed, while the number of nucleotide variations in the same DNA region is more than ten times greater comparing DX with other B. cereus group members. An additional element pointing to the close similarity of the two strains is the identity in length and in sequence of the very variable spacer region that separates the dcw cluster from the SpoIIG operon.
The TransTerm-HP site had revealed several hairpin-loop structures in B. weihenstephanensis, with the specific characteristics of a terminator, in the spacer DNAs located between the gene coding regions. Figure 5 shows the location in the dcw and SpoIIG clusters of these putative terminators. The DNA sequences that form the structures are shown below the drawing. They are 100% identical in DX and in B. weihenstephanensis. Six out of seven are assigned a 100% confidence score by the algorithm of the program, and the seventh, between sigmaE and sigmaG, has an 89% score. The SIN termination structures are not identical, but maintain the characteristic of terminators with one or a few different nucleotides, the same level of diversity existing for instance between the terminators of B. weihenstephanensis and those of B. anthracis Ames.
Figure 5.
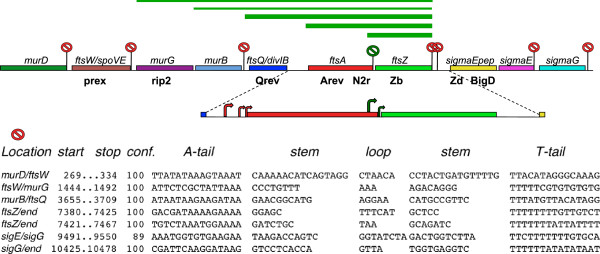
Transcriptional terminators within the B. mycoides dcw and spoIIG gene clusters. Red labels mark the position of the putative terminators. The DX termination sequences displayed are 100% identical to those predicted at the TransTerm-HP site for B. weihenstephanensis KBAB4 (Accession NC_010184, from coordinates 3780796 to 3790953). The green label between ftsA and ftsZ indicates a hairpin structure not recognized there as a potential terminator. The three large green bars over the genes represent the main ftsZ-specific RNAs and the green thin bars the minor ones. The primers used to detect RNA 5’ ends by primer extension are indicated below the genes. The curved arrows in the enlarged region show the main ftsA and ftsZ RNA start sites.
The short 39 bp DNA region between ftsA and ftsZ can also be folded into a hairpin structure with a calculated stability of −7.8 ΔG, though it is not recognized as a potential terminator by the TransTerm-HP site and is tagged with a different color in the figure. Downstream of the dcw cluster, in the group composed of three genes, SpoIIA-sigmaE processing peptidase, prosigmaE and sigmaG, putative termination sequences are located between prosigmaE and sigmaG and after sigma G, at the end of the group.
The putative terminators are located at the boundary between genes of different specificity, which code either for enzymes of peptidoglycan biosynthesis or for structural proteins of the division septum, meaning that terminators are found between the mur/fts genes and not between the mur/mur or fts/fts genes. Two consecutive terminator hairpins close the dcw cluster immediately after the ftsZ gene.
In B. anthracis, another member of the B. cereus group, the genome-wide coverage of DNA by RNA transcripts has been analyzed at the single nucleotide level [7]. The high-throughput sequencing of total RNA (RNA-Seq), in various growth conditions, provided a map of transcript start sites and operon structure throughout the genome. Discontinuity of RNA transcripts in B. anthracis, along the dcw region shown in Figure 5 was observed in tracts corresponding to those harboring putative termination sites, with one exception: no interruption in RNA coverage was found at any point between murB and ftsQ, and no RNA initiation sites were consequently reported in this intergenic region [14].
We did instead find cDNAs terminating in this location in B. mycoides. Our experimental data, obtained by PE and RT-PCR, are thus in keeping with the results reported in the literature, since we found transcripts made up of five genes: murG, murB, ftsQ, ftsA and ftsZ. Moreover, the Northern blot showed ftsZ and ftsA RNA in the form of monogenic mRNAs, as well as of ftsA-ftsZ, ftsQ-ftsA-ftsZ and murB-ftsQ-ftsA RNAs.
The spoIIG operon
The B. mycoides dcw cluster is closely followed by three genes expressed by the same DNA strand, forming a group homologous to the spoIIG operon that has been extensively characterized in B. subtilis[15-17]. The first gene, spoIIGA, encodes the protease required to activate the product of the second gene, pro-sigmaE, synthesized as an inactive precursor with an N-terminal prosequence. In B. subtilis, the region located between the dcw and the spoIIG clusters carries the high molecular weight bpr gene, a bacillopeptidase. In SIN and DX, the region between these clusters is short, non-coding and of different length (respectively 260 and 415 bp), and is identical along 70 nucleotides after ftsZ and 145 nucleotides before SpoIIGA. Only B. weihenstephanensis, in the B. cereus group, harbors a 415 bp spacer 100% identical to that of the DX strain, which points to the phylogenetic linkage of these two bacilli.
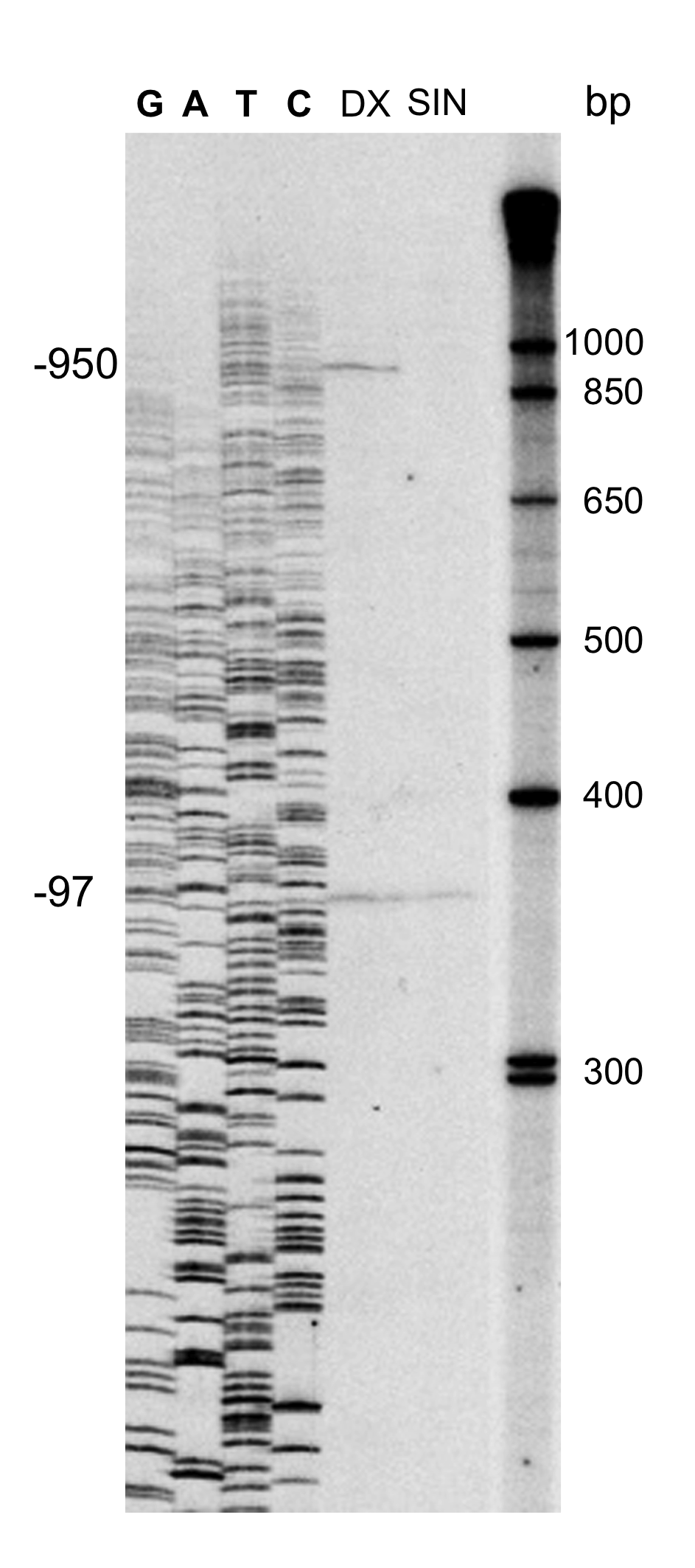
As the vicinity of the two clusters dcw and spoIIG might have a functional meaning, we searched for transcripts linking their genes. RT was performed with the BigD oligonucleotide (Table 1), which anneals at +273 relative to the first in frame ATG of the sigmaE processing peptidase (SpoIIGA). The primer was elongated up to −97 bp upstream of the spoIIGA ATG, in the spacer region that is identical in the B. mycoides DX and SIN strains. In the DX strain only, a higher band mapped inside the 3’ coding region of ftsZ. No elongation products included the complete ftsZ gene, thereby excluding a co-transcription of genes belonging to the two clusters (Additional file 2).
Conclusions
Here we show that the organization and transcription of the dcw genes in the B. mycoides DX and SIN strains is not dissimilar, if we exclude minor variations that are most likely irrelevant to colony shape. Although only bicistronic transcripts were reported in B. subtilis, the novel finding is that ftsZ RNA is expressed as a single-gene transcript in the vegetative cells of these Gram positive bacilli. Multigenic ftsZ transcripts are also present, connecting the division genes to the upstream genes encoding enzymes of peptidoglycan biosynthesis. No common transcript was instead found between ftsZ and the downstream genes of the SpoIIG cluster.
Methods
Strains
B. mycoides DX and SIN are sporogenic bacilli of the soil isolated from the environment and maintained in the lab [3].
RNA extraction
Exponentially growing cells were sedimented by centrifugation and blocked on ice in cell stop solution (5% phenol in ethanol). Cells were pelleted, suspended in 50% phenol in LETS buffer (10 mM Tris–HCl pH 8.0, 10 mM EDTA, 1% SDS, 10 mM DTT) and mechanically broken by vortexing with glass beads. After sedimentation at maximum speed in Eppendorf centrifuge, the supernatant was extracted twice with pH 4.3 phenol, then twice with chloroform and precipitated with 2M ammonium acetate final concentration and 1 volume isopropanol. The pellet was washed with 80% ethanol, air-dried and resuspended in RNAse-free water. DNA was digested with DNAseI (Amplification Grade, Invitrogen), according to the manufacturer’s indications.
Analysis of RNA
Northern blots: electrophoresis of RNA in agarose-formaldehyde gels, blot and hybridization were conducted as described in Sambrook et al. [18]. The ftsZ specific DNA probe (551 bp) was obtained by PCR amplification of B. mycoides SIN DNA with primers Zfor and Zrev and the ftsA probe (408 bp) with primers Ain and N2R (Table 1). Amplified DNAs were labelled using a nick-translation kit (Boehringer).
For primer extension analysis, oligonucleotide primers were labeled at the 5’ end using T4 polynucleotide kinase and [γ-32P]ATP according to standard protocols [18]. 4 pmol of labeled oligonucleotides and 10 μg RNA were coprecipitated, suspended in 30 μl formamide buffer (10 mM PIPES pH 6.4, 0.1 M NaCl 0.1 mM EDTA 80% formamide) and incubated for 3 hrs at 30°C for annealing. Samples were diluted with 5 volumes water and precipitated with 0.25 M NaCl and ethanol. After 80% ethanol washing, the samples were dried in the air, suspended in 20 μl Super Script II Reverse Transcriptase buffer (Invitrogen) plus 10 mM DTT, 0.5 mM dNTP and 20 units RT and incubated at 42°C for 90 min. The enzyme was inactivated by heating at 70°C for 10 min and the RNA complementary to the cDNA digested away with RNAse H. Samples were precipitated with 0.25 M NaCl and ethanol, sedimented, washed with ethanol, dried and resuspended in 4 μl formamide-dye for electrophoresis on 6% acrylamide sequencing gels [18].
RT-PCR
Two cDNAs were prepared using RNA purified from DNA (see above), one using the primer Zfin, which is complementary to the end of ftsZ, and a second using the primer Afin, which is complementary to the 3’ region of ftsA. The RNA was coprecipitated with the primers, suspended in formamide buffer, annealed and reverse transcribed as described above. The Zfin cDNA was amplified with Zfin as downstream primer and with Ain, Qin, Mbin, MGin and FW as upstream primers. The Afin cDNA was also amplified with Mbin and Qin. These primers are shown in Table 1. Control cDNA preparations were also prepared, omitting Reverse Transcriptase, to monitor possible residual DNA, and amplified. The PCR conditions were: 52°C for annealing and 7.5 min for elongation.
Construction and expression of the ftsZ minigene
The DX region from coordinates 5947 to 6271 (GenBank AY129555), containing 246 bp of the 3’ ftsA coding region, the 39 bp ftsA/ftsZ spacer and the initial 39 bp of ftsZ, was amplified with primers which added an upstream HindIII site and a downstream EcoRI site (primers Hind5 and Eco5, see Table 1) and restricted at those sites. The DNA region containing the final 121 bp of the ftsZ ORF and 28 bp after the termination codon (coordinates 7267 to 7415) was amplified with the primers Eco3 and Bam3 (Table 1) that carry EcoRI and BamHI sites, respectively, and was restricted. Plasmid pJPR1 [9] (‘amyE cat PxylamyE’ bla, a gift from J. Rawlings) was digested with HindIII and BamHI in the polylinker region, ligated to the prepared DNA fragments and transformed into E. coli Hb101. The correct recombinant plasmid was chosen by sequencing and used to transform competent B. subtilis 168. The ftsZ minigene became integrated at the amyE site as a result of a double crossing-over event between the 5’ and 3’ amyE regions carried upstream and downstream of the cloning site in pJPR1. Integration was controlled by sequencing. RNA transcribed from the minigene in the recombinant B. subtilis 168 was detected by primer extension with primer Amy5 (Table 1) annealing to the 5’ region of the amyE locus, 245 nucleotides downstream of the inserted minigene. Induction of the pxyl promoter by 5% xylose in TS was for 18 h and 3 h.
Termination sequences
The putative B. mycoides termination sequences were detected on the basis of their identity to those predicted for B. weihenstephanensis at the TransTerm-HP site (http://transterm.cbcb.umd.edu.). The region of the B. weihenstephanensis KBAB4 genome considered was from coordinates 3780796 to 3790953 (Accession NC_010184), containing the genes of the dcw cluster from murD to ftsZ and the following spoIIG operon.
Sequence data
Sequences of the B. mycoides SIN and DX partial dcw clusters are deposited as GenBank AY129554 (SIN) and AY129555 (DX).
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TS participated in the design of the study and carried out the experiments. LT and GC added new data and confirmed previous data. CDF participated in the design and coordination of the study. EB conceived the study, organized the sequence data and drafted the manuscript. All the authors read and approved the final manuscript.
Supplementary Material
Putative initiation sites of ftsQ, ftsA and ftsZ RNA as determined by primer extension. The gene sequences are those of the B. mycoides DX strain (accession AY12555.2). The DNA complementary to the PE primers is highlighted in turquoise, as are the nucleotides of RNA start. Initiation and termination codons of the ORFs are in red. The hexamers corresponding to consensus TATA-box promoter motifs (17) and the ribosome binding sites are underlined.
Determination of SpoIIGA RNA 5’ ends by Primer Extension. The three genes of the SpoIIG cluster are encoded downstream of the dcw cluster, by the same DNA strand. The distance between the two clusters is 415 bp in DX and 260 bp in SIN. Primer extension started from primer BigD at position +273 of the first gene of the cluster, SpoIIGA. The DX and SIN cDNAs (two lanes each) were both elongated to position −97 upstream of the SpoIIGA first codon ATG, in the spacer region that is identical in both strains. A second cDNA termination, present only in DX, mapped within the 3’ end of the ftsZ coding region at −950.
{kind=link}
Contributor Information
Tiziana Santini, Email: tiziana.santini@uniroma1.it.
Luana Turchi, Email: luana.turchi@uniroma1.it.
Giulia Ceccarelli, Email: giulia.ceccarelli85@gmail.com.
Carmen Di Franco, Email: carmen.difranco@uniroma1.it.
Elena Beccari, Email: elena.beccari@uniroma1.it.
Acknowledgements
This work was supported by the Italian Space Agency with ASI contract n° 1/R/290/02 and ASI-MoMa project 2006–2009 to EB. Institutional funds for EB came from the CNR Istituto di Biologia e Patologia Molecolari IBPM. Science Faculty funds from the Sapienza University of Rome supported CDF. We thank Giuseppe Pisaneschi for his valuable technical assistance.
References
- Schmidt TR, Scott EJ II, Dyer DW. Whole-genome phylogenies of the family Bacillaceae and expansion of the sigma factor gene family in the Bacillus cereus species-group. BMC Genomics. 2011;12:430. doi: 10.1186/1471-2164-12-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gause GF. Some physiological properties of dextral and of sinistral forms in Bacillus mycoides flügge. Biol Bull Woods Hole MA. 1939;76:448–465. doi: 10.2307/1537751. [DOI] [Google Scholar]
- Di Franco C, Beccari E, Santini T, Pisaneschi G, Tecce G. Colony shape as a genetic trait in the pattern-forming Bacillus mycoides. BMC Microbiol. 2002;2(33):1–15. doi: 10.1186/1471-2180-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchi L, Santini T, Beccari E, Di Franco C. Localization of new peptidoglycan at poles in Bacillus mycoides, a member of the Bacillus cereus group. Arch Microbiol. 2012;194(10):887–892. doi: 10.1007/s00203-012-0830-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholamhoseinian A, Shen Z, Wu J-J, Piggot P. Regulation of transcription of the cell division gene ftsA during sporulation of Bacillus subtilis. J Bacteriol. 1992;174(14):4647–4656. doi: 10.1128/jb.174.14.4647-4656.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzy-Treboul G, Karmazyn-Campelli C, Stragier P. Developmental regulation of transcription of the Bacillus subtilis ftsAZ operon. J Mol Biol. 1992;224:967–979. doi: 10.1016/0022-2836(92)90463-T. [DOI] [PubMed] [Google Scholar]
- Passalacqua KD, Varadarajan A, Ondov BD, Okou DT, Zwick ME, Bergman NH. Structure and complexity of a bacterial transcriptome. J Bacteriol. 2009;191(10):3203–3211. doi: 10.1128/JB.00122-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flardh K, Garrido T, Vicente M. Contribution of individual promoters in the ddlB-ftsZ region to the transcription of the essential cell-division gene ftsZ in Escherichia coli. Mol Microbiol. 1997;24(5):927–936. doi: 10.1046/j.1365-2958.1997.4001762.x. [DOI] [PubMed] [Google Scholar]
- Jones LJ, Carballido-Lopez R, Errington J. Control of cell shape in bacteria: helical, actin-like filaments in Bacillus subtilis. Cell. 2001;104:913–922. doi: 10.1016/S0092-8674(01)00287-2. [DOI] [PubMed] [Google Scholar]
- Hollands K, Proshkin S, Sklyarova S, Epshtein V, Mironov A, Nudler E, Groisman EA. Riboswitch control of Rho-dependent transcription termination. Proc Natl Acad Sci USA. 2012;109(14):5376–5381. doi: 10.1073/pnas.1112211109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KS, von Hippel PH. Transcriptional termination at intrinsic terminators: the role of the RNA hairpin. Proc Natl Acad Sci USA. 1995;92:8793–8797. doi: 10.1073/pnas.92.19.8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsford CL, Ayanbule K, Salzberg SL. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007;8:R22. doi: 10.1186/gb-2007-8-2-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner S, Mayr R, Francis KP, Prüss BM, Kaplan T, Wiessner-Gunkel E, Stewart GS, Scherer S. Bacillus weihenstephanensis sp. nov. is a new psychrotolerant species of the Bacillus cereus group. Int J Syst Bacteriol. 1998;48(Pt 4):1373–1382. doi: 10.1099/00207713-48-4-1373. [DOI] [PubMed] [Google Scholar]
- Martin J, Zhu W, Passalacqua KD, Bergman N, Borodovsky M. Bacillus anthracis genome organization in light of whole transcriptome sequencing. BMC Bioinformatics. 2010;11(Suppl 3):S10. doi: 10.1186/1471-2105-11-S3-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satola S, Kirchman PA, Moran CP Jr. SpoOA binds to a promoter used by σA RNA polymerase during sporulation in Bacillus subtilis. Proc Natl Acad Sci USA. 1991;88:4533–4537. doi: 10.1073/pnas.88.10.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington J. Bacillus subtilis sporulation: regulation of gene expression and control of morphogenesis. Microbiol Rev. 1993;57(1):1–33. doi: 10.1128/mr.57.1.1-33.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Moran CP Jr. Promoter activation by repositioning of RNA polymerase. J Bacteriol. 2008;190:3110–3117. doi: 10.1128/JB.00096-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press C.S.H; 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Putative initiation sites of ftsQ, ftsA and ftsZ RNA as determined by primer extension. The gene sequences are those of the B. mycoides DX strain (accession AY12555.2). The DNA complementary to the PE primers is highlighted in turquoise, as are the nucleotides of RNA start. Initiation and termination codons of the ORFs are in red. The hexamers corresponding to consensus TATA-box promoter motifs (17) and the ribosome binding sites are underlined.
Determination of SpoIIGA RNA 5’ ends by Primer Extension. The three genes of the SpoIIG cluster are encoded downstream of the dcw cluster, by the same DNA strand. The distance between the two clusters is 415 bp in DX and 260 bp in SIN. Primer extension started from primer BigD at position +273 of the first gene of the cluster, SpoIIGA. The DX and SIN cDNAs (two lanes each) were both elongated to position −97 upstream of the SpoIIGA first codon ATG, in the spacer region that is identical in both strains. A second cDNA termination, present only in DX, mapped within the 3’ end of the ftsZ coding region at −950.