

Abstract
The vascular adventitia acts as a biological processing center for the retrieval, integration, storage, and release of key regulators of vessel wall function. It is the most complex compartment of the vessel wall and is comprised of a variety of cells including fibroblasts, immunomodulatory cells (dendritic and macrophages), progenitor cells, vasa vasorum endothelial cells and pericytes, and adrenergic nerves. In response to vascular stress or injury, resident adventitial cells are often the first to be activated and re-programmed to then influence tone and structure of the vessel wall, to initiate and perpetuate chronic vascular inflammation, and to act to stimulate expansion of the vasa vasorum, which can act as a conduit for continued inflammatory and progenitor cell delivery to the vessel wall.
This review presents the current evidence demonstrating that the adventitia acts as a key regulator of vascular wall function and structure from the “outside-in.”
Keywords: vasa vasorum, inflammation, pulmonary hypertension, fibroblast, progenitor cell
The Adventitia: Complex and Dynamic Compartment of the Vessel Wall
The arterial wall is a heterogeneous three-layered structure composed of an intima, media and adventitia. Each layer exhibits specific histologic, biochemical and functional characteristics and, as such, each contributes in unique ways to maintaining vascular homeostasis and to regulating the vascular response to stress or injury.
Whereas endothelial and smooth muscle cells, the principal cellular constituents of the intima and media respectively, have received much attention from vascular biologists, the adventitia in general, and the principal cells contained therein, have been largely overlooked. This is intriguing because the connective tissue stroma is an important structural component of all tissues in vertebrate animals. The adventitial stroma is considered to consist of an ECM scaffold containing fibroblasts, blood and lymphatic vessels, nerves, progenitor and immune cells, making the adventitia the most complex and heterogeneous compartment of the vessel wall (Fig. 1). Adventitial cells are capable of sensing and directing responses to a wide array of stimuli through reciprocal communication with other adventitial cells as well as with cells of the neighboring tissues (Fig. 2). An increasing volume of experimental data supports the idea that the vascular adventitia acts as a biological processing center for the retrieval, integration, storage, and release of key regulators of vessel wall function. Indeed, the adventitial compartment can act as the principal “injury-sensing tissue” of the vessel wall. In response to hormonal, inflammatory, and environmental stresses such as hypoxia/ischemia or vascular distention, resident adventitial cells (fibroblasts, immune and progenitor cells) are often the first vascular wall cells to exhibit evidence of “activation” by increases in cell proliferation, upregulation of expression of contractile, extracellular matrix, matricellular, and adhesion proteins, as well as by the secretion of chemokines, cytokines, growth, and angiogenic factors capable of directly and potently affecting resident vascular wall cell phenotype, and regulating vasa vasorum expansion (1, 2). Furthermore, under conditions of elevated blood pressure, the adventitia becomes the predominant wall component due to its pronounced stiffening behavior (3). Thus, the adventitia is capable of regulating vascular structure and function from the “outside–in.”
Figure 1. Complex cellular composition of the vascular adventitia.
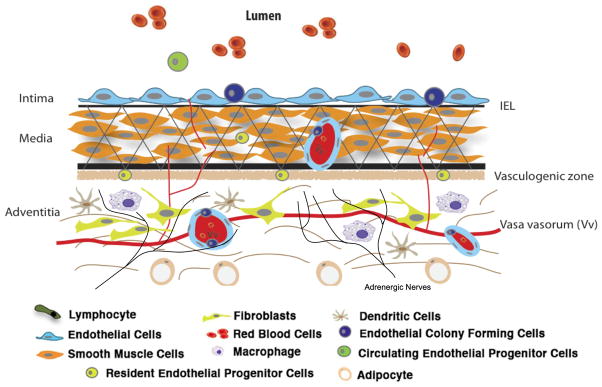
Unlike the normal intima and media, which are composed of endothelial and smooth muscle cells respectively, the normal adventitia is comprised of a wide variety of cell types, including fibroblasts, resident progenitor cells, immunomodulatory cells (dendritic cells, macrophages, T-lymphocytes), vasa vasorum endothelial cells and adrenergic nerves.
Figure 2. Fibroblasts play a central role in the control of vascular function.

Fibroblasts produce and organize elements of the extracellular matrix (ECM), and also degrade structural elements of the ECM; they secrete a complex mixture of growth factors, cytokines, chemokines; they communicate with neural cells, with cells of hematopoietic origin (dendritic cells, macrophages, T-lymphocytes), with SMC, endothelial and epithelial cells; importantly, this communication is reciprocal (5). (Adapted from: Sorrell et al., “Fibroblasts: a diverse population at the center of it all.”).
Adventitial Fibroblast - a “Sentinel Cell”
The most abundant cell type in vascular adventitia is the fibroblast. Unfortunately, a reliable and specific marker for the fibroblast has yet to be found. All currently utilized markers to identify the fibroblast, including vimentin, FSP-1, discoidin-domain receptor2, and prolyl-4-hydroxylase are potentially problematic as they are also expressed in other cell types and are not present in all fibroblasts. Therefore, to identify fibroblasts, investigators have to rely on the lack of markers for other cell lineages (non-lymphoid, non-endothelium, and non-epithelium), along with morphologic, functional, and biochemical characteristics.
Stromal fibroblasts are believed to arise from at least three distinct cellular origins: primary mesenchyme, local epithelial-mesenchymal transition (EMT), and bone marrow-derived precursors (4, 5). The numerous sources/origins of fibroblasts likely contribute to the now well-accepted notion of fibroblast heterogeneity. Fibroblasts exhibit organ and tissue site-specific gene expression patterns, which are as divergent as the gene expression patterns observed among distinct lineages of white blood cells (5, 6). Moreover, fibroblast heterogeneity within specific tissues, including the lung and pulmonary artery adventitia, has been reported (5, 7, 8). The existence within the lung and lung vasculature of fibroblast subsets with an increased propensity to contribute to fibro-proliferative responses is supported by studies demonstrating that fibroblasts from a fibrogenic milieu functionally differ from those in normal tissues (8–10). In the lung, the most extensively characterized marker used to differentiate specific fibroblast subpopulations, is the glycosyl phosphatidyl inositol (GPI)-anchored protein Thy-1. Thy-1-negative and Thy-1-positive mouse lung fibroblasts differ morphologically and have different secretory profiles (11). Thy-1(−) cells within the lung appear to be a more consistent fibrogenic subtype, exhibit greater proliferative responses to PDGF-AA and CTGF, secrete twice as much latent TGF-β as Thy-1(+) cells, and express five times more α-SM-actin, a characteristic of the myofibroblast phenotype. Recent data show that Thy-1(−/−) mice exhibit more severe lung fibrosis (increased collagen accumulation) and increased Smad 2/3 phosphorylation (indicating higher levels of active TGF-β) in response to intratracheal bleomycin than do Thy-1(+) control mice (9). Interestingly, the myofibroblasts in fibroblastic foci in lung tissues from individuals with IPF/UIP are Thy-1 (−), despite the fact that the majority of fibroblasts from normal lungs are Thy-1(+) (9).
Adventitial fibroblasts (AF) were originally thought to simply provide mechanical strength to tissues by producing extracellular matrix (ECM), which forms a supporting framework. However, one of the most consistent findings in experimental models of systemic vascular injury and hypertension is early and often dramatic adventitial remodeling (1, 2), wherein AF has been suggested to be a “sentinel cell” in the vessel wall responding to various stimuli as the first vascular wall cells to exhibit evidence of “activation”. The AF has been suggested to be the most appropriate cell for “sensing” hypertensive states (12). Adventitial remodeling in systemic vasculature has often being characterized by increases in AF proliferation, which precede and exceed endothelial and SMC proliferation (13, 14).
Similar findings have emerged from experimental studies in the pulmonary circulation. Early and often dramatic evidence of adventitial remodeling is documented in hypoxia-induced pulmonary hypertension, with AF undergoing the earliest and most significant increases in proliferation of all vascular wall cell types (14, 15). Less pronounced but still significant adventitial changes are noted in high-flow and monocrotaline models of pulmonary hypertension, as well as in idiopathic forms of human pulmonary hypertension (16, 17).
Significant “imprinted” phenotypic differences have been reported in pulmonary AF derived from pulmonary hypertensive versus control animals, including far greater proliferative responses to pro-mitogenic stimuli including hypoxia, changes in the signaling pathways that elicit proliferation (18), and a marked pro-inflammatory phenotype characterized by increased production of chemokines, cytokines, matricellular proteins, and adhesion molecules (19, 20). Das et al. have described significant changes in the functional role of the atypical PKCζ isozyme, such that it acted as a pro-proliferative kinase for AF from chronically hypoxic pulmonary hypertensive animals as opposed to its anti-proliferative actions in fibroblasts from normoxic controls (18). These observations raise the possibility that chronic hypoxia leads to the emergence and expansion of AFs that have lost their ability to limit stimulus-induced proliferation. Whether chronic hypoxia, or any stimulus for that matter, modulates intracellular signaling patterns to change fibroblast phenotype and there is a recruitment and expansion of a unique AF subset remain to be elucidated. Emerging evidence suggests that the stable changes in the proliferative and inflammatory phenotype of AF from the pulmonary hypertensive vessel wall are due to increases in HDAC activity and thus may be considered to have undergone epigenetic modification (20, 21).
Signaling Mechanisms Regulating Fibroblast Responses to Stress
The molecular basis of the distinct environmental “stimulus-sensing” capability of the AF remains incompletely understood. Further, there appear to be vascular bed specific properties of AF, with some suggesting that responses of pulmonary artery fibroblasts are distinct from those in systemic arteries (22). As noted, pulmonary AF exhibit the earliest and most sustained proliferative response to hypoxic exposure. Several signaling pathways have been suggested as important regulators of hypoxia-induced proliferation of AFs, including activation of Gαi and Gαq family members, perhaps in a ligand-independent fashion, with subsequent stimulation of PKC and mitogen-activated protein (MAP) kinase family members (18, 23), as well as activation of PI3K, and synergistic interaction with Akt, mTOR, and p70 ribosomal protein S6 kinase (24, 25). Noteworthy, for yet unknown reasons, hypoxia fails to initiate similar proliferative responses in pulmonary arterial SMC when tested under identical ex vivo conditions (22, 23). Hypoxia activates a diverse array of transcription factors (26), however, the degree and nature of the global transcriptional response to hypoxia is likely both cell-type and cell-state specific. Furthermore, the diverse nature of hypoxia-induced cellular responses was reported in hypoxic human pulmonary AF, as for example, differential HIF-1α/HIF-2α dependency, where proliferative responses were found to be solely HIF2α-dependent, whereas the migratory responses were controlled by both HIF-1α and HIF-2α (25, 27). Among other transcription factors involved in hypoxia-induced AF proliferative responses to hypoxia, Egr-1 has been reported as an important contributor to the pathogenesis of pulmonary vascular remodeling (28, 29).
Dysregulation of replication repressor signals can also contribute to increased hypoxic proliferative responses in AF, as growth-limiting signaling pathways including PKCζ and mitogen activated protein (MAP) kinase phosphatase-1 (MKP-1) have been shown to be important repressors of proliferative signals in hypoxia-exposed AF (30), and emergence of fibroblast-like cells lacking anti-replication activity of PKCζ was reported in pulmonary adventitia of calves exposed to chronic hypoxia (18).
Hypoxia or other stressors may also affect AF proliferation by inducing the secretion of various autocrine/paracrine factors including ATP (discussed in “Extracellular Purines” below), angiotensin II (25), angiotensin-converting enzyme (ACE) (31), PDGF, and others.
Differentiation of Adventitial Fibroblasts into Myofibroblasts
Activation of fibroblasts by a variety of stimuli can result in their differentiation toward a myofibroblast phenotype, a process shown to be critical to a variety of fibrotic diseases including those in the lung (32). It is now recognized that a variety of other cells including epithelial, endothelial, and resident and circulating progenitor cells can also differentiate into myofibroblasts (Fig. 3). No matter their origin, myofibroblasts express α-SM-actin, the most frequently used marker for myofibroblast identification, which allows monitoring of this cell type during experimental and clinical conditions but clearly does not allow for identification of its origin. (32). Clearly α-SM-actin fails to distinguish between myofibroblasts and SMCs in situations of mixed populations. Moreover, SMCs lose expression of contractile proteins used as markers of their differentiated phenotype, when acquiring a synthetic phenotype in vivo or after being placed in culture. At present no single cytoskeletal protein allows reliable discrimination between myofibroblast and SMC. This becomes particularly important when one considers the potential role of myofibroblast in contributing to vascular pathology.
Figure 3.

Numerous cell types give rise to cells expressing a myofibroblast phenotype.
Early and dramatic increases in the appearance of α-SM-actin expressing myofibroblasts in the adventitia are observed in hypoxia-induced pulmonary hypertension, as well as in numerous other vasculopathies (33). Myofibroblasts are implicated as key participants in tissue remodeling because of their ability to perform multiple physiologic functions in response to change in the local environment, including production of collagen and other extracellular matrix proteins (elastin, fibronectin), as well as matricellular proteins including tenascin-C and osteopontin) (19, 34), production of a variety of growth factors, cytokines, and ROS that exert paracrine effects on medial SMC (discussed below). Myofibroblasts exhibit significant contractile capabilities with slow onset and sustained contraction in response to a variety of agonists, and their responses to vasodilatory stimuli differ from those of SMC, thus potentially contributing to the abnormalities of vasorelaxation observed in the setting of chronic pulmonary hypertension. Collectively, myofibroblast accumulation can directly contribute to changes in the tone and structure of the vessel wall under pathophysiologic conditions (10, 35). Further, the myofibroblast is capable of migrating from the adventitia to the media or even the intima, thus contributing to vascular pathologic remodeling (1, 36). Unfortunately, labeling and tracking myofibroblast movement in the pulmonary circulation is more difficult than in the systemic and direct proof of AF migration to the intima in pulmonary hypertension is lacking.
The differentiation of fibroblasts into myofibroblasts in the adventitia is regulated by a complex microenvironment consisting of growth factors, cytokines, adhesion molecules, and extracellular matrix molecules, including TGF-β, thrombin, endothelin, angiotensin-II, IL-6, and Fizz1 (10, 37–40). All these factors are upregulated by hypoxia and have been observed in the pulmonary artery adventitia of chronically hypoxic animals. Upregulation of these molecules is also observed in other lung vascular injury models including monocrotaline. Furthermore, hypoxia alone can stimulate myofibroblast differentiation and proliferation of pulmonary AF, yet these two distinct cellular responses to hypoxia are regulated by different intracellular signaling modules, such as proliferative responses utilize Gαi-initiated ERK1/2-dependent signaling, whereas hypoxia-induced α-SM-actin expression, in addition to Gαi-activation, utilizes JNK rather than ERK1/2 signaling (41). It should also be mentioned that not all fibroblasts differentiate into myofibroblasts under similar microenvironmental conditions.
Fibroblast–Matrix Interactions in Adventitia
The composition of the adventitial extracellular matrix (ECM) is principally regulated by fibroblasts. Major components of the adventitial ECM produced by fibroblasts are fibrillar collagens, with types I and III collagens as the most abundant (42). Under normal conditions fibroblasts remain in a quiescent, undifferentiated state, which is maintained by a homeostatic relationship between fibroblasts and this collagen enriched ECM. Activation of the fibroblast in response to stress or injury, leads to dramatic alterations in the production and relative composition of ECM proteins, which in turn have profound effects on vascular structure and function. In the progression of various vascular diseases, including restenosis, atherosclerosis, and pulmonary arterial hypertension, adventitial ECM composition is markedly altered. During the development of pulmonary hypertension, for example, marked increases in the production and accumulation of collagens and elastin in the adventitia have been documented, which is likely to affect stiffness of the vessel wall and have profound effects on flow dynamics and ultimately on right ventricular function (43, 44). Besides increased deposition of collagens, de novo accumulation and increased expression/deposition of cellular fibronectin (ED-A isoform), tenascin-C, and osteopontin in the adventitia have been reported in pulmonary hypertensive vessels (19, 45). Fibronectin, in particular its ED-A isoform, and tenascin-C (TN-C), and osteopontin have been reported to contribute to augmented proliferation of fibroblasts and potentially to their differentiation into myofibroblasts (19, 32, 46, 47). Deposition of fibronectin, TN-C, and osteopontin also coincides with the increased expression and activity of several matrix metalloproteinases (MMPs) responsible for degradation of the ECM components, including basement membrane collagen, interstitial collagen, fibronectin, and various proteoglycans. Indeed, upregulated expression of MMPs may be necessary for the fibroblast/myofibroblast to move through the adventitial matrix into the media and even intima (36). Proteolytic activities of MMPs are regulated by specific tissue inhibitors of metalloproteinases (TIMPs), the activity of which have been reported to be decreased in various vasculopathies, thus creating an environment conducive to cell migration (48). Excessive expression of MMPs and decreased expression of TIMPs would facilitate pathogenesis of tissue destructive processes, which have been observed in a wide variety of diseases including pulmonary hypertension (49). Targeting fibroblast-matrix interactions may provide novel therapeutic approaches in the prevention of vascular remodeling, as has been shown in animal models of monocrotaline- and hypoxia-induced pulmonary hypertension where inhibition of MMP activity attenuated vascular structural remodeling (48, 50).
Adventitia: Role in Vascular Inflammation (Macrophages, Dendritic cells, and Fibroblasts)
Vascular inflammation has traditionally been considered an “inside-out” response centered on leukocyte/monocyte recruitment to the intima of blood vessels. In this hypothesis, injured endothelium of blood vessels expresses surface adhesion molecules and inflammatory mediators that participate in monocyte homing and eventual transmigration into the intima and/or media. Growing experimental evidence, however, supports a new paradigm of an “outside-in” hypothesis, in which vascular inflammation is initiated and perpetuated in the adventitia and contributes to medial and intimal remodeling. In support of the “outside-in” hypothesis of adventitial regulation of inflammation are observations in a wide variety of vascular injuries that there is a rapid influx of leukocytes into the adventitial compartment (51). In experimental models of pulmonary hypertension, both chronic hypoxic exposure and monocrotaline treatment lead to the early appearance and persistence of inflammatory/progenitor cells in the adventitia of both large and small pulmonary arteries, followed by prominent vascular remodeling (52–55). A study in the hypoxic pulmonary circulation demonstrated a complex, time-dependent and pulmonary artery-specific upregulation of several cytokines/chemokines, their receptors, and adhesion molecules, which appear to be produced/expressed by AF and recruited monocytes, and are likely involved in the initiation and perpetuation of the inflammatory response in a “outside-in” fashion (45, 54, 56).
The adventitia can therefore be regarded as playing a critical role in initial steps of the pathogenesis of vascular inflammation and remodeling (57). The adventitia comprises canonical innate immune cells, specifically macrophages and dendritic cells (DCs), but also cells that have only recently been regarded capable of exerting immune functions, such as the AF (20). Macrophages and DCs are diffusely scattered in the pulmonary arteries of human and animals (45, 58). Increases in their numbers in the PA are observed in the setting of pulmonary hypertension, raising the possibility for a role in perpetuating inflammation, just as in the systemic circulation (45, 58). Additionally, in a variety of pathological conditions such as asthma (59), COPD (60), and pulmonary hypertension (61), DCs demonstrate a marked proclivity to reside in the adventitia, likely modulating inflammatory, immunological, and vascular cell proliferative processes. The presence of dendritic cells (DCs) in the lung has been known for over 25 years (62). The principal job of pulmonary DCs is to encounter putative self and non-self/environmental antigens, primarily at epithelial surfaces, and coordinate appropriate innate and acquired immunity responses Whether circulating or tissue-resident, DCs are normally immature until triggered by a multitude of signals, including ingestion of apoptotic cells, to express DC-SIGN, CD83 and additional markers of maturation (55, 63).
Macrophages, DCs, and AF are all equipped with the necessary machinery (e.g. toll-like receptors (TLRs), inflammasome components (NLRs)) to potently respond to a variety of exogenous and endogenous danger signals. Such activation results in generation of a host of cytokines, chemokines, as well as mediators of oxidative stress (ROS, NOS) and tissue remodeling (TIMPS, MMPs) by macrophages, DCs, and AF (20, 64, 65). Interestingly, the pattern of expression of TLRs appears to be vessel specific, which contributes to vessel-specific risk for inflammatory vasculopathies (66). Monocytes/macrophages and classical and plasmacytoid DCs originate from hematopoietic stem cells and are replaced continually from a common macrophage and DC precursor (67, 68). It has now become evident that certain tissue resident macrophages such as liver Kupffer cells, epidermal Langerhans cells, microglia, and pleural macrophages are able to proliferate and renew independently from the bone marrow. These cells represent a lineage of tissue macrophages derived from the yolk sac that are genetically distinct from bone marrow derived macrophages (68). Therefore, certain populations of tissue resident macrophages and DCs may be present in the adventitia and may constitute a distinct population with the ability to self renew and to exert distinct functional phenotypes. Functional plasticity and diversity is increasingly recognized to be an essential feature of the mononuclear phagocyte system and may be regarded a conditio sine qua non for the coordinated initiation, propagation and resolution of local immune responses (64, 69, 70). A diverse set of macrophage functional phenotypes has thus been demonstrated to be involved in a wide variety of pathologies; macrophages can promote or resolve fibrosis, promote insulin resistance and obesity, mediate in thermoregulation through generation of catecholamines, enable wound healing, promote and restrict T cell responses, regulate angiogenesis, promote or suppress tumor growth, fight pathogens, and control homeostasis in local immune networks such as the gut (64, 69–72).
Importantly, it is possible, based on the fact that no surface or functional marker definitively distinguish macrophages from DCs, that macrophages and DCs do not represent separate entities but rather two extremes of regulated functional activation states on a continuum of a yet unknown number of functional activities (73). Macrophages are capable of mounting strong pro-inflammatory cytokine responses (initiating innate immune responses) while DCs are strong antigen presenters and inducers of T cell responses (initiating adaptive immune response). However, as pointed out by D. Hume, each cell types can perform both functions in response to adequate stimulation (73).
Previously, fibroblast activation was regarded as relatively insignificant in regulating immune responses and immune interactions were attributed to lymphocytes, macrophages, and DCs. It is now becoming clear that many danger signals are not antigen-specific, and currently focus is shifting toward an expanded immune system, in which fibroblasts play an important role in innate immune responses. Importantly, there is convincing evidence that fibroblasts taken from diseased tissue, including the pulmonary artery adventitia, display a fundamentally different phenotype compared with fibroblasts taken from normal tissues at the same anatomical site (20, 74, 75).
As such, the activated AF appears to exert a functional plasticity reminiscent of that of macrophages, DCs in that they have been shown to express a combination of functional phenotypes including generation of pro-inflammatory cytokines and molecules necessary for antigen presentation and T-cell stimulation. This functional plasticity of the activated AF may therefore play a key role in initiating and propagating adventitial inflammation through generation of numerous cytokines and chemokines that create a microenvironment tailored to modulate the activation of tissue resident macrophages and DCs as well as promote recruitment of blood derived inflammatory monocytes (20, 76).
Therefore, in inflammatory responses in the adventitia of the PA, macrophage, DC, and AF activation can be fine-tuned over a large scale of functional phenotypes, including switching from pro-inflammatory to pro-fibrotic or pro-resolving functional phenotypes or complete deactivation. In the absence of a specific antigen, as could be the case in early stages of hypoxia induced or idiopathic PH, a functional phenotype characterized by generation of pro-inflammatory mediators may be displayed by macrophages, DCs, and fibroblasts. Over time, this phenotype may evolve with generation of pro-remodeling and pro-fibrogenic mediators by macrophages and fibroblasts. In contrast, in the presence of specific antigen(s), an antigen processing and T cell activating functional phenotype (DC phenotype) may prevail (64, 69–71, 76, 77).
Importantly, there is growing evidence that epigenetic marks may “lock” innate immune cells into a distinct functional phenotype, which may result in loss of functional plasticity and failure to respond to regulatory signals, such that AF retain a pro-inflammatory phenotype driving recruitment of inflammatory monocytes and DC precursors (20, 71, 77–80), (Fig. 4). The molecular basis for this persistently activated fibroblast phenotype at sites of chronic inflammation remains unclear, although findings suggest that NF-kB signaling pathway plays a critical role in perpetuating chronic persistent inflammatory responses (81, 82).
Figure 4. The constitutively activated “imprinted” phenotype of pulmonary artery AF is due, at least in part, to increased HDAC activity.
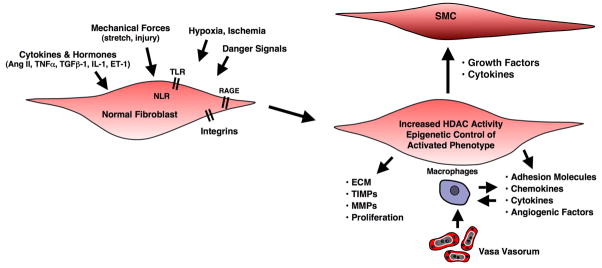
This fibroblast maintains a capacity to affect the phenotype of other adjacent cells including SMC, macrophages, and vasa vasorum endothelial cells.
Therefore, temporal-spatial dysregulation and/or failure in the normal “switch-off signal” in fibroblasts and/or macrophages, DCs may directly contribute to the persistence of a chronic inflammatory immune response.
The transition to a chronic inflammatory phenotype also requires changes in the adhesion molecule and chemokine receptor expression on fibroblasts and recruited hematopoietic cells respectively. Fibroblasts express and upregulate adhesion molecules including ICAM-1 and VCAM-1 which cause adhesion of leukocytes in response to a variety of stimuli. Secretion of cytokines, including TGF-b, by the activated fibroblast cause activation and upregulation of receptors such as CXCR4 on newly recruited hematopoietic cells, as well as secretion of SDF-1, the cognate ligand for CXCR4 (83). Thus, an environment is created in chronically inflamed tissues, whereby the adventitia acts as a “foster home” for leukocytes leading to their inappropriate/pathologic retention and survival (81), (Fig. 5).
Figure 5. Essential role of the adventitial fibroblast in initiating and perpetuating vascular inflammation and, consequently, vascular remodeling.

In response to hormonal, infectious, or environmental (hypoxia, hemodynamic stress, etc.) stimuli, the fibroblast is activated and secretes chemokines, cytokines and matricellular proteins involved in the recruitment of monocytes, lymphocytes and progenitor cells. With time, fibroblasts upregulate adhesion molecule expression, which promotes retention of leukocytes and progenitor cells within the adventitia. Some of the newly recruited cells can differentiate into fibroblasts and myofibroblasts, which perpetuate the cycle, thus leading to persistent inflammation and structural vascular remodeling. (PA = pulmonary artery).
Adventitial Vasa Vasorum
Vasa vasorum (VV) (in Latin “vessels of the vessels”) is a network of small blood vessels that provide large blood vessels (both arteries and veins > 0.5 mm in diameter), (84), nutrients and oxygen and may also serve to remove “waste products”.
In the systemic circulation, in a number of vasculopathies including atherosclerosis, type II diabetes, metabolic syndrome, restenosis and vasculitis, the adventitial VV undergoes marked expansion (54, 85–88). Expansion of the pulmonary artery VV is commonly observed in the setting of pulmonary artery obstruction, and patients with chronic thromboembolic obstruction of the pulmonary arteries demonstrate marked increases in the volume of adventitial VV, where the core of the non-resolving clots are recannulated by neovascular endothelialized structures originating from VV (89). Increased density of pulmonary adventitial VV has been described in patients with severe idiopathic pulmonary fibrosis and pulmonary hypertension and more recently in patients with iPAH (90). In animal models of experimental pulmonary hypertension marked expansion of the VV network in the adventitia and within the outer aspects of the media has been reported (56, 89, 91) (Fig. 6). Interestingly, it has been shown that blocking vasa vasorum angiogenesis with angiostatin reduces progression of advanced atherosclerosis (87). The mechanisms controlling expansion of the adventitial VV network are not well understood. There is some evidence suggesting that angiogenic responses are often associated with new nerve growth. The pulmonary vascular bed is innervated by the adrenergic nervous system residing in the adventitia and outer media of blood vessels, and norepinephrine has been shown to be one of the major signaling molecules released by these nerves (92). Increases in the numbers of adrenergic axons have been observed in the adventitial layer of even intra-acinar muscular arteries in at least some patients with pulmonary hypertension (88).
Figure 6. Angiogenic expansion of the vasa vasorum in the pulmonary artery adventitia of calves with severe hypoxia-induced pulmonary hypertension.

(A–E). Histopathology of large (A, C) and small (B, D) pulmonary arteries. Both histological, H&E (A, B) and immunofluorescent, PECAM-1/CD31 (C, D) staining’s demonstrate marked expansion of the vasa vasorum capillary network in adventitial, perivascular regions. Quantitative morphometric analyses demonstrated that the volume density (Vv) of vasa vasorum is significantly greater in neonatal calves with severe hypoxia-induced pulmonary hypertension compared with normoxic controls (E). (F, G), Angiogenic responses in the adventitia of a human patient with pulmonary fibrosis and associated pulmonary hypertension. Medium-sized pulmonary artery stained with CD31, demonstrating evidence for capillary network expansion in the perivascular area (medial/adventitial region, arrow) (F). CD31 immunohistochemical evidence of capillary proliferation (arrow) (G). Bars, 500 μm in A and C, 100 μm in B, D, F, and 25 μm in G.
An emerging hypothesis regarding VV expansion suggests that activation of fibroblasts plays a critical role, since in cancers of epithelial origin and in chronic inflammatory diseases like rheumatoid arthritis, stromal fibroblasts have been clearly implicated in the pathologic angiogenesis (93). Fibroblasts are frequently the first cell type to migrate to the wound site where they orchestrate reparative neovascularization, specifically to secrete cytokines and pro-angiogenic growth factors that regulate the formation of capillary-like networks by endothelial cells, and to provide a stabilizing force to newly formed vessels (94–96).
Fibroblasts are capable of producing many pro-angiogenic mediators, including VEGF, PDGF, endothelin, TGF-b, fibronectin, thrombin, S1000A4, known to be upregulated in the adventitia of chronically hypoxic hypertensive animals (45). This is in accordance with a number of previous studies, in which hypoxic conditions have been shown to induce pro-angiogenic phenotypes in a number of stromal cell types (97, 98). Interestingly, all of the aforementioned mediators have been found to be upregulated in the pulmonary arteries of human patients with various forms of pulmonary arterial hypertension (55, 99). Studies in the systemic circulation have also suggested that endothelin (ET-1), a factor well known to be upregulated in pulmonary hypertension, plays a critical role in coronary VV neovascularization in the setting of experimental hypercholesterolemia through local upregulation of VEGF (100). Similarly, Davie et al. have demonstrated that hypoxia-stimulated pulmonary AF exhibited pro-angiogenic properties and influenced the angiogenic phenotype of VVEC in matrigel in a process of cell-cell communication involving endothelin-1 (ET-1) (91). These data are consistent with the emerging concept that the endothelium of de novo forming microvessels receives and integrates pro-angiogenic signals from a number of non-endothelial cells, including fibroblasts (95, 96).
In the context of adventitial VV, it is worth mentioning pericytes, as recent studies are revealing their potentially important role in adventitial biology. Using a fibrin hydrogel culture system, Choi et al. showed that residing within “loose connective tissue” from various organs are mesenchymal stem cells that are precursors of precapillary pericytes (101). Goritz et al. showed that adventitial/perivascular pericytes can divide, migrate, and differentiate into scar forming stromal cells in the setting of spinal cord glial scar formation (102). The roles that pericytes play in the maintenance of vascular structures and in pathophysiological conditions of microvascular expansion are only now coming into full view, and the apparent plasticity of these cells bestows significant clinical importance to understand the underlying control mechanisms.
Extracellular Purines: Endogenous Mediators in Adventitia
Among the many factors that control vascular remodeling, extracellular purines and pyrimidines (ATP, ADP, AMP, adenosine, UTP and UDP) are the most ubiquitous, but paradoxically, among the least investigated endogenous signaling molecules. Concentrations of extracellular nucleotides are thought to be elevated in the local tissue microenvironment of various physiological and pathological conditions including hypoxia, inflammation, fluid shear stress, mechanical forces, osmotic shock, thrombosis, and sympathetic stimulation (103–106). Most of these environmental stimuli may play a role in controlling extracellular ATP levels in vascular wall adventitia (Fig. 7). The effects of extracellular nucleotides in target cells are mediated through P2Y (metabotropic) and P2X (ionotropic) purinergic receptors (61, 107). A number of studies support the idea that extracellular nucleotides are important regulators of vascular cell function and could contribute to the development of vascular disease (105, 108–110). Extracellular ATP has been implicated in the hyperplasia and hypertrophy of arterial walls in spontaneously hypertensive rats, in regulation of vascular permeability (111, 112), and in control of proliferation and migration of vascular SMCs and hematopoietic cells including monocytes (105, 110, 113–115). Importantly, in stimulating cell proliferation and migration, ATP acts synergistically with cytokines and integrins (105, 114–117) thereby supporting the physiological relevance of extracellular ATP under hypoxic and inflammatory conditions. Meantime, there is limited data on the role of extracellular purines in a regulation of angiogenic responses in microvascular endothelial cells and vasa vasorum endothelial cells in particular.
Figure 7. Sources of extracellular ATP in the pulmonary artery vascular wall.

ATP release and signaling is an integral part of hypoxia-induced response in vascular wall. ATP can be released as a result of combined action of hypoxia, inflammation, fluid shear stress (changes in blood flow), injury, mechanical forces (SMC contractility), and sympathetic neurotransmission (release from perivascular nerve together with noradrenalin and neuropeptides). Importantly, vasa vasorum endothelial cell have been identified as an abundant source of extracellular ATP in pulmonary artery adventitia (118).
Collectively, within adventitial microenvironment, extracellular ATP acts in a synergistic manner with multiple pro-inflammatory and growth-promoting factors in hypoxic conditions. (SMC = smooth muscle cells; blood cells = erythrocytes, platelets, monocytes).
Along with considerable progress in understanding a regulatory role of extracellular nucleotides in angiogenesis, their roles in hypoxia-induced adventitial remodeling and VV expansion are receiving attention. Studies with pulmonary AF demonstrated that hypoxia stimulates exogenous ATP release, which in turn acts as an autocrine/paracrine factor to stimulate fibroblast proliferation (105). Moreover, it was demonstrated that pulmonary adventitial VV endothelial cells (VVEC) release ATP in response to hypoxia, suggesting that VVEC represent a novel source of extracellular ATP in the pulmonary artery vessel wall, and that extracellular ATP may contribute to VV neovascularization (118). Furthermore, ATP induces pro-angiogenic responses in VVEC, including increased DNA synthesis, augmented migration and tube formation. These responses are mediated, in part, via activation of PI3K/mTOR, ERK1/2 signaling pathways and elevation of intracellular Ca2+ (119). Together, these observations support the idea that in addition to nerve and circulating blood cells, vascular cells themselves appear to be potent sources of ATP and other adenine nucleotides, and that local purinergic signaling networks initiated by hypoxic stress may alter endothelial cell phenotype and function. In addition, as all cell types in the vascular wall adventitia express purinergic receptors, extracellular nucleotides may act as intercellular signaling molecules, providing cell-to-cell communication by an autocrine/paracrine mechanism in the pulmonary vessel wall (Fig. 7). Interestingly, ATP-induced angiogenic responses have not been documented in pulmonary artery and aortic endothelial cells (119). Thus, it can be speculated that VVEC isolated from angiogenic active VV exhibit unique phenotypic characteristics, with a particular reliance on extracellular nucleotides produced by cells accumulating in the adventitial to stimulate growth.
Adventitial Reactive Oxygen Species (ROS) in Vascular Disease
Reactive oxygen species (ROS) generated in the pulmonary artery adventitia also contribute to “outside-in” effects on pulmonary vasoconstriction and pulmonary vascular remodeling (120–125). In the vascular adventitia, activated AFs produce ROS predominantly via NADPH oxidase, with both NOX2 and NOX4 identified as important fibroblast isoforms (120–122, 126, 127). Other important sources of vascular ROS include the mitochondria, uncoupled eNOS and xanthine oxidase (128–130). The production of ROS by the resident AF then promotes recruitment of inflammatory cells, which in turn generate more ROS through the inflammatory cell NADPH oxidase, greatly magnifying the impact of ROS (121). These different sources of ROS result in oxidant production in distinct cellular compartments, which determine the local redox state and the specific targets of ROS (120, 124, 131–133).
Superoxide generated in the adventitia can directly modulate selected targets or indirectly impact signaling pathways through its rapid spontaneous or enzymatic dismutation to hydrogen peroxide. These ROS modulate the phenotype of the pulmonary AF, including proliferation, migration, differentiation, and matrix production (124). ROS can function as signaling molecules in pulmonary vascular disease by targeting NF-kB, Nrf-2, MAPK, K+ channel regulation, and BMPR-2 signaling (120, 134–136). In addition, ROS can modulate multiple receptors and growth factors expressed by activated pulmonary AF such as ET-1, PDGF, TGF-b, FGF-2 and IGF-1 (20, 120).
ROS generated in the adventitia can function as paracrine molecules to modulate neighboring cells, contributing to vasoconstriction and vascular remodeling (122). Cascino et al. recently reported that adventitial-derived hydrogen peroxide impairs vascular relaxation by activating p38 MAPK in the medial smooth muscle cell (126). These data support other earlier reports in the systemic circulation demonstrating a paracrine effect of adventitial ROS on medial wall hypertrophy and vasoconstriction (121, 122, 125). Superoxide reacts rapidly with nitric oxide to decrease nitric oxide bioavailability and promote pulmonary vasoconstriction, even in the setting of adequate NO production. While ROS promote many pathologic processes, they are also important in adaptation to stress, which requires careful consideration when evaluating the impact of oxidative stress or designing approaches to block ROS production in the adventitia (125, 137–139).
The importance of adventitial ROS in regulating pulmonary vascular function is strongly supported by the high expression of the key antioxidant enzyme, extracellular superoxide dismutase (EC-SOD or SOD3) in the vascular adventitia (140). This is the dominant SOD isoform in the vasculature and is highly localized to the adventitia (133). Overexpression of SOD3 in the lung, which increases adventitial SOD3, protects against not only collagen deposition, but also medial and intimal wall remodeling in models of pulmonary hypertension (124, 141–143). In the systemic circulation, SOD3 overexpression has been shown to protect in various models associated with adventitial activation (144).
Collectively these studies support the key role for adventitial ROS production and adventitial oxidant/antioxidant imbalance in pulmonary vascular disease.
Adventitia: A Depot for Vascular Progenitor Cells
There is increasing experimental evidence demonstrating that the adventitial layer, in both the developing and adult blood vessels serves as a niche for stem and progenitor cells, including endothelial progenitor cells (EPCs), mesenchymal progenitor cells (MPC), smooth muscle and pericyte progenitors, hematopoietic stem cells (HSCs), mesenchymal stem cells (MSCs), and mesoangioblasts (145–149).
Zengin et al. described this progenitor cell niche in the adventitia, in the aorta of mice, as a specific “vasculogenic zone” that borders the outer media (146), and seems to co-localize with a zone of Sca-1+ cells (149). At present, the nature and origin of resident adventitial progenitor cells remain undetermined and controversial. One possibility to explain the presence of resident progenitor cells in the adventitia is that these cells are remnants of earlier developmental stages, and that mesenchymal precursors that remain located in the adventitia are capable of differentiating into different types of vascular cells under the appropriate circumstances. It is possible that there is a site-specific diversity of progenitor cell types, such that distinct progenitor cell subtypes reside in different vessels and subserve different roles in response to site-specific vascular injuries.
Recent in vitro experimental evidence suggests a wide spectrum of differentiation potential of adventitial progenitor cells, including SMCs (150), pericyte-like cells (151), CD34+ angiogenic progenitors and even macrophages (152). A recent study showed that rat aortic adventitia contained a distinct subset of immature immunocytes capable of proliferating, differentiating into macrophages and DCs, and stimulating angiogenesis (153).
Hoshino et al. isolated adventitial progenitor cells from human pulmonary arteries, expressing mesenchymal stem/progenitor cell (MPC) markers but negative for hematopoietic or endothelial markers that exhibited osteogenic, adipogenic or myogenic (myofibroblast) differentiation potentials (145). Progenitor cells isolated from “vasculogenic zone” were of a hematopoietic origin and when cultured in appropriate medium were capable to differentiate, toward endothelial, fibroblast/myofibroblast-like, or even immune cells, such as macrophages (146). Regulatory signals that control the phenotype specification of adventitial progenitors in response to local vessel wall environment are largely undetermined. Passman et al. reported that Sca-1+ cells, also characterized as CD34+, Flk-1+, CD140b+, but c-Kit−, localize to sonic hedgehog (Shh) signaling domain in the adventitia, and that Shh signaling plays an important role in the maintenance of resident SMC progenitors in the artery wall (149).
Adventitial progenitor cells may be involved in both physiologically beneficial and/or detrimental processes during homeostatic and pathogenic conditions. For example, since progenitor cells isolated from “vasculogenic zone” were shown to be capable of differentiating toward endothelial, fibroblast/myofibroblast-like, or even immune cell lineages, the authors speculated that these cells may serve as a source for postnatal vasculogenesis, contributing to tumor vascularization and local immune responses (146). A potentially different population of adventitial progenitor cells, expressing Sca-1, cKit, CD34 and Flk-1, and capable of differentiating into either smooth muscle or endothelial cells has also been described (150, 154). When these Sca-1+ cells, carrying the Lac-Z gene, were transferred to the adventitial side of vein grafts in ApoE-deficient mice, they migrated to the media and neointima, contributing to lesion formation/progression.
The adventitia may also serve as a repository for non-resident, circulating progenitor cells following vascular injury. Circulating progenitor cells have been implicated in the pathophysiology of a number of systemic and pulmonary vascular diseases (54, 155, 156). In sex-mismatched bone marrow transplant patients, Caplice et al. demonstrated that some α-SM-actin-positive cells in the neointima and in and around microvessels in adventitia originate from Y-chromosome+ cells administered at bone marrow transplantation (157). Others have demonstrated that circulating bone marrow-derived progenitor cells participate in the neovascularization of the adventitia and intima in atherosclerosis (158). We have previously demonstrated the appearance of cKit+ cells in pulmonary artery adventitia of chronically hypoxic calves in and around rapidly expanding adventitial vasa vasorum blood vessels (56) and similar findings have recently been reported in IPAH (90).
As discussed above, inflammation is an important component of systemic and pulmonary vascular diseases (54, 159). Contained within this inflammatory cell pool is a subset of monocytic mesenchymal precursors, termed fibrocytes, which are characterized by the dual expression of both leukocytic (CD45, CD11b, CD14) and mesenchymal (α-1-procollagen) markers (160, 161). Fibrocytes are rapidly recruited to sites of tissue injury and have been shown to differentiate into collagen-producing fibroblasts or even myofibroblasts (160–162). We have previously described a rapid and robust influx of fibrocytes into the adventitia of rats and calves with experimental hypoxia-induced pulmonary hypertension (163). When circulating monocytic cells were depleted in vivo via clodronate-liposomes, pulmonary vascular remodeling was markedly attenuated, thus suggesting that circulating fibrocyte recruitment is essential for hypoxic pulmonary vascular remodeling (163).
Collectively these observations support the idea that adventitia serves as a depot for progenitor cells, both of resident and circulating origins, that are critically involved in vascular repair in response to a wide variety of pathologic stimuli.
Being the most complex compartment of the vessel wall, the vascular adventitia can act as biological “central processing unit” that integrates key regulators of vessel wall function. The adventitia harbors a wide variety of cells with potent immunoregulatory function, such as fibroblasts, fibrocytes, macrophages and DCs, resident progenitor cells, but also vasa vasorum endothelial cells, and adrenergic nerves. In response to vascular stress or injury, these resident adventitial cells are often the first to be activated and subsequently re-programmed to modulate vascular tone and restructure the architecture of the vessel wall. The AF, being the most abundant cellular constituent of the adventitia, has been shown to be a critical regulator of vascular wall function. In response to vascular stresses including hypoxia, the AF undergoes epigenetically fixed functional changes which include proliferation, differentiation, production of extracellular matrix proteins, adhesion molecules, release of reactive oxygen species, growth factors and metalloproteinases (MMPs), which affect medial smooth muscle cell tone and growth directly. This activated AF phenotype is also characterized by generation of chemokines and cytokines that are capable of recruiting, retaining, and activating circulating and resident inflammatory and progenitor cells. Bidirectional interactions between activated fibroblasts and resident and recruited macrophages, DCs, and progenitor cells can thus create a microenvironment that is conducive to promoting a persistent and non-resolving inflammatory response, that includes expansion of the vasa vasorum (which acts as a conduit for further delivery of inflammatory/progenitor cells), with the end result of in irreversible functional and structural remodeling of the vessel wall.
Acknowledgments
Supported by:
NIH Grants: HL-084923-03 (Stenmark), HL-014985-36 (Stenmark), HL-086680 (Grayck) HL-086783 (Gerasimovskaya).
Contributor Information
Kurt R. Stenmark, Email: Kurt.Stenmark@ucdenver.edu.
Michael E. Yeager, Email: Michael.Yeager@ucdenver.edu.
Karim C. El Kasmi, Email: Karim.Elkasmi@ucdenver.edu.
Eva Nozik-Grayck, Email: Eva.Grayck@ucdenver.edu.
Evgenia V. Gerasimovskaya, Email: Evgenia.Gerasimovskaya@ucdenver.edu.
Min Li, Email: Min.Li@ucdenver.edu.
Suzette R. Riddle, Email: Suzette.Riddle@ucdenver.edu.
Maria G. Frid, Email: Maria.Frid@ucdenver.edu.
Literature Cited
- 1.Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, Ausoni S, Pauletto P. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: From innocent bystander to active participant. Circ Res. 2001;89:1111–1121. doi: 10.1161/hh2401.100844. [DOI] [PubMed] [Google Scholar]
- 2.Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M. Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda, Md. 2006;21:134–145. doi: 10.1152/physiol.00053.2005. [DOI] [PubMed] [Google Scholar]
- 3.Silver FH, Horvath I, Foran DJ. Viscoelasticity of the vessel wall: The role of collagen and elastic fibers. Crit Rev Biomed Eng. 2001;29:279–301. doi: 10.1615/critrevbiomedeng.v29.i3.10. [DOI] [PubMed] [Google Scholar]
- 4.Postlethwaite AE, Shigemitsu H, Kanangat S. Cellular origins of fibroblasts: Possible implications for organ fibrosis in systemic sclerosis. Current opinion in rheumatology. 2004;16:733–738. doi: 10.1097/01.bor.0000139310.77347.9c. [DOI] [PubMed] [Google Scholar]
- 5.Sorrell JM, Caplan AI. Fibroblasts-a diverse population at the center of it all. International review of cell and molecular biology. 2009;276:161–214. doi: 10.1016/S1937-6448(09)76004-6. [DOI] [PubMed] [Google Scholar]
- 6.Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, Brown PO. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99:12877–12882. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eyden B. Fibroblast phenotype plasticity: Relevance for understanding heterogeneity in “fibroblastic” tumors. Ultrastruct Pathol. 2004;28:307–319. doi: 10.1080/019131290882204. [DOI] [PubMed] [Google Scholar]
- 8.Jelaska A, Strehlow D, Korn JH. Fibroblast heterogeneity in physiological conditions and fibrotic disease. Springer Semin Immunopathol. 1999;21:385–395. [PubMed] [Google Scholar]
- 9.Hagood JS, Prabhakaran P, Kumbla P, Salazar L, MacEwen MW, Barker TH, Ortiz LA, Schoeb T, Siegal GP, Alexander CB, Pardo A, Selman M. Loss of fibroblast thy-1 expression correlates with lung fibrogenesis. Am J Pathol. 2005;167:365–379. doi: 10.1016/S0002-9440(10)62982-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi-Wen X, Chen Y, Denton CP, Eastwood M, Renzoni EA, Bou-Gharios G, Pearson JD, Dashwood M, du Bois RM, Black CM, Leask A, Abraham DJ. Endothelin-1 promotes myofibroblast induction through the eta receptor via a rac/phosphoinositide 3-kinase/akt-dependent pathway and is essential for the enhanced contractile phenotype of fibrotic fibroblasts. Mol Biol Cell. 2004;15:2707–2719. doi: 10.1091/mbc.E03-12-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barker TH, Hagood JS. Getting a grip on thy-1 signaling. Biochimica et biophysica acta. 2009;1793:921–923. doi: 10.1016/j.bbamcr.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGrath JC, Deighan C, Briones AM, Shafaroudi MM, McBride M, Adler J, Arribas SM, Vila E, Daly CJ. New aspects of vascular remodelling: The involvement of all vascular cell types. Exp Physiol. 2005;90:469–475. doi: 10.1113/expphysiol.2005.030130. [DOI] [PubMed] [Google Scholar]
- 13.Belknap JK, Orton EC, Ensley B, Tucker A, Stenmark KR. Hypoxia increases bromodeoxyuridine labeling indices in bovine neonatal pulmonary arteries. Am J Respir Cell Mol Biol. 1997;16:366–371. doi: 10.1165/ajrcmb.16.4.9115746. [DOI] [PubMed] [Google Scholar]
- 14.Meyrick B, Reid L. Hypoxia and incorporation of 3h-thymidine by cells of the rat pulmonary arteries and alveolar wall. Am J Pathol. 1979;96:51–70. [PMC free article] [PubMed] [Google Scholar]
- 15.Orton EC, LaRue SM, Ensley B, Stenmark K. Bromodeoxyuridine labeling and DNA content of pulmonary arterial medial cells from hypoxia-exposed and nonexposed healthy calves. Am J Vet Res. 1992;53:1925–1930. [PubMed] [Google Scholar]
- 16.Langleben D, Szarek JL, Coflesky JT, Jones RC, Reid LM, Evans JN. Altered artery mechanics and structure in monocrotaline pulmonary hypertension. J Appl Physiol. 1988;65:2326–2331. doi: 10.1152/jappl.1988.65.5.2326. [DOI] [PubMed] [Google Scholar]
- 17.Chazova I, Loyd JE, Zhdanov VS, Newman JH, Belenkov Y, Meyrick B. Pulmonary artery adventitial changes and venous involvement in primary pulmonary hypertension. Am J Pathol. 1995;146:389–397. [PMC free article] [PubMed] [Google Scholar]
- 18.Das M, Burns N, Wilson SJ, Zawada WM, Stenmark KR. Hypoxia exposure induces the emergence of fibroblasts lacking replication repressor signals of pkczeta in the pulmonary artery adventitia. Cardiovasc Res. 2008;78:440–448. doi: 10.1093/cvr/cvn014. [DOI] [PubMed] [Google Scholar]
- 19.Anwar A, Li M, Frid MG, Kumar B, Gerasimovskaya EV, Riddle SR, McKeon BA, Thukaram R, Meyrick BO, Fini MA, Stenmark KR. Osteopontin is an endogenous modulator of the constitutively activated phenotype of pulmonary adventitial fibroblasts in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2012 doi: 10.1152/ajplung.00050.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li M, Riddle SR, Frid MG, El Kasmi KC, McKinsey TA, Sokol RJ, Strassheim D, Meyrick B, Yeager ME, Flockton AR, McKeon BA, Lemon DD, Horn TR, Anwar A, Barajas C, Stenmark KR. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J Immunol. 2011;187:2711–2722. doi: 10.4049/jimmunol.1100479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao L, Chen C-N, Hajji N, Oliver E, Cotroneo E, Wharton J, Wang D, Li M, McKinsey TA, Stenmark KR, Wilkins MR. Histone deacetylation inhibition in pulmonary hypertension: Therapeutic potential of valproic acid (vpa) and suberoylanilide hydroxamic acid (saha) Circulation. 2012 doi: 10.1161/CIRCULATIONAHA.112.103176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welsh DJ, Scott PH, Peacock AJ. P38 map kinase isoform activity and cell cycle regulators in the proliferative response of pulmonary and systemic artery fibroblasts to acute hypoxia. Pulm Pharmacol Ther. 2005 doi: 10.1016/j.pupt.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 23.Das M, Bouchey DM, Moore MJ, Hopkins DC, Nemenoff RA, Stenmark KR. Hypoxia-induced proliferative response of vascular adventitial fibroblasts is dependent on g protein-mediated activation of mitogen-activated protein kinases. J Biol Chem. 2001;276:15631–15640. doi: 10.1074/jbc.M010690200. [DOI] [PubMed] [Google Scholar]
- 24.Gerasimovskaya EV, Tucker DA, Stenmark KR. Activation of phosphatidylinositol 3-kinase, akt, and mammalian target of rapamycin is necessary for hypoxia-induced pulmonary artery adventitial fibroblast proliferation. J Appl Physiol. 2005;98:722–731. doi: 10.1152/japplphysiol.00715.2004. [DOI] [PubMed] [Google Scholar]
- 25.Krick S, Hanze J, Eul B, Savai R, Seay U, Grimminger F, Lohmeyer J, Klepetko W, Seeger W, Rose F. Hypoxia-driven proliferation of human pulmonary artery fibroblasts: Crosstalk between hif-1alpha and an autocrine angiotensin system. Faseb J. 2005;19:857–859. doi: 10.1096/fj.04-2890fje. [DOI] [PubMed] [Google Scholar]
- 26.Cummins EP, Taylor CT. Hypoxia-responsive transcription factors. Pflugers Arch. 2005;450:363–371. doi: 10.1007/s00424-005-1413-7. [DOI] [PubMed] [Google Scholar]
- 27.Eul B, Rose F, Krick S, Savai R, Goyal P, Klepetko W, Grimminger F, Weissmann N, Seeger W, Hanze J. Impact of hif-1alpha and hif-2alpha on proliferation and migration of human pulmonary artery fibroblasts in hypoxia. Faseb J. 2006;20:163–165. doi: 10.1096/fj.05-4104fje. [DOI] [PubMed] [Google Scholar]
- 28.Yan SF, Lu J, Xu L, Zou YS, Tongers J, Kisiel W, Mackman N, Pinsky DJ, Stern DM. Pulmonary expression of early growth response-1: Biphasic time course and effect of oxygen concentration. J Appl Physiol. 2000;88:2303–2309. doi: 10.1152/jappl.2000.88.6.2303. [DOI] [PubMed] [Google Scholar]
- 29.Banks MF, Gerasimovskaya EV, Tucker DA, Frid MG, Carpenter TC, Stenmark KR. Egr-1 antisense oligonucleotides inhibit hypoxia-induced proliferation of pulmonary artery adventitial fibroblasts. J Appl Physiol. 2005;98:732–738. doi: 10.1152/japplphysiol.00821.2004. [DOI] [PubMed] [Google Scholar]
- 30.Short MD, Fox SM, Lam CF, Stenmark KR, Das M. Protein kinase czeta attenuates hypoxia-induced proliferation of fibroblasts by regulating map kinase phosphatase-1 expression. Mol Biol Cell. 2006;17:1995–2008. doi: 10.1091/mbc.E05-09-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morrell NW, Morris KG, Stenmark KR. Role of angiotensin-converting enzyme and angiotensin ii in development of hypoxic pulmonary hypertension. Am J Physiol. 1995;269:H1186–1194. doi: 10.1152/ajpheart.1995.269.4.H1186. [DOI] [PubMed] [Google Scholar]
- 32.Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005;13:7–12. doi: 10.1111/j.1067-1927.2005.130102.x. [DOI] [PubMed] [Google Scholar]
- 33.Wilcox JN, Okamoto EI, Nakahara KI, Vinten-Johansen J. Perivascular responses after angioplasty which may contribute to postangioplasty restenosis: A role for circulating myofibroblast precursors? Ann N Y Acad Sci. 2001;947:68–90. doi: 10.1111/j.1749-6632.2001.tb03931.x. dicussion 90–62. [DOI] [PubMed] [Google Scholar]
- 34.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- 35.Hinz B. Formation and function of the myofibroblast during tissue repair. The Journal of investigative dermatology. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 36.Li G, Chen SJ, Oparil S, Chen YF, Thompson JA. Direct in vivo evidence demonstrating neointimal migration of adventitial fibroblasts after balloon injury of rat carotid arteries. Circulation. 2000;101:1362–1365. doi: 10.1161/01.cir.101.12.1362. [DOI] [PubMed] [Google Scholar]
- 37.Malmstrom J, Lindberg H, Lindberg C, Bratt C, Wieslander E, Delander EL, Sarnstrand B, Burns JS, Mose-Larsen P, Fey S, Marko-Varga G. Transforming growth factor-beta 1 specifically induce proteins involved in the myofibroblast contractile apparatus. Mol Cell Proteomics. 2004;3:466–477. doi: 10.1074/mcp.M300108-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Bogatkevich GS, Tourkina E, Abrams CS, Harley RA, Silver RM, Ludwicka-Bradley A. Contractile activity and smooth muscle alpha-actin organization in thrombin-induced human lung myofibroblasts. Am J Physiol Lung Cell Mol Physiol. 2003;285:L334–343. doi: 10.1152/ajplung.00417.2002. [DOI] [PubMed] [Google Scholar]
- 39.Gallucci RM, Lee EG, Tomasek JJ. Il-6 modulates alpha-smooth muscle actin expression in dermal fibroblasts from il-6-deficient mice. The Journal of investigative dermatology. 2006;126:561–568. doi: 10.1038/sj.jid.5700109. [DOI] [PubMed] [Google Scholar]
- 40.Liu T, Dhanasekaran SM, Jin H, Hu B, Tomlins SA, Chinnaiyan AM, Phan SH. Fizz1 stimulation of myofibroblast differentiation. Am J Pathol. 2004;164:1315–1326. doi: 10.1016/S0002-9440(10)63218-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Short M, Nemenoff RA, Zawada WM, Stenmark KR, Das M. Hypoxia induces differentiation of pulmonary artery adventitial fibroblasts into myofibroblasts. Am J Physiol Cell Physiol. 2004;286:C416–425. doi: 10.1152/ajpcell.00169.2003. [DOI] [PubMed] [Google Scholar]
- 42.Prockop DJ, Kivirikko KI. Collagens: Molecular biology, diseases, and potentials for therapy. Annu Rev Biochem. 1995;64:403–434. doi: 10.1146/annurev.bi.64.070195.002155. [DOI] [PubMed] [Google Scholar]
- 43.Lammers S, Scott D, Hunter K, Tan W, Shandas R, Stenmark KR. Mechanics and function of the pulmonary vasculature: Implications for pulmonary vascular disease and right ventricular function. Compr Physiol. 2012;2:295–319. doi: 10.1002/cphy.c100070. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Chesler NC. Pulmonary vascular wall stiffness: An important contributor to the increased right ventricular afterload with pulmonary hypertension. Pulmonary circulation. 2011;1:212–223. doi: 10.4103/2045-8932.83453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burke DL, Frid MG, Kunrath CL, Karoor V, Anwar A, Wagner BD, Strassheim D, Stenmark KR. Sustained hypoxia promotes the development of a pulmonary artery-specific chronic inflammatory microenvironment. Am J Physiol Lung Cell Mol Physiol. 2009;297:L238–250. doi: 10.1152/ajplung.90591.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muro AF, Moretti FA, Moore BB, Yan M, Atrasz RG, Wilke CA, Flaherty KR, Martinez FJ, Tsui JL, Sheppard D, Baralle FE, Toews GB, White ES. An essential role for fibronectin extra type iii domain a in pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177:638–645. doi: 10.1164/rccm.200708-1291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tamaoki M, Imanaka-Yoshida K, Yokoyama K, Nishioka T, Inada H, Hiroe M, Sakakura T, Yoshida T. Tenascin-c regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am J Pathol. 2005;167:71–80. doi: 10.1016/S0002-9440(10)62954-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vieillard-Baron A, Frisdal E, Raffestin B, Baker AH, Eddahibi S, Adnot S, D’Ortho MP. Inhibition of matrix metalloproteinases by lung timp-1 gene transfer limits monocrotaline-induced pulmonary vascular remodeling in rats. Hum Gene Ther. 2003;14:861–869. doi: 10.1089/104303403765701150. [DOI] [PubMed] [Google Scholar]
- 49.Brauer PR. Mmps--role in cardiovascular development and disease. Front Biosci. 2006;11:447–478. doi: 10.2741/1810. [DOI] [PubMed] [Google Scholar]
- 50.Herget J, Novotna J, Bibova J, Povysilova V, Vankova M, Hampl V. Metalloproteinase inhibition by batimastat attenuates pulmonary hypertension in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 2003;285:L199–208. doi: 10.1152/ajplung.00167.2002. [DOI] [PubMed] [Google Scholar]
- 51.Okamoto E, Couse T, De Leon H, Vinten-Johansen J, Goodman RB, Scott NA, Wilcox JN. Perivascular inflammation after balloon angioplasty of porcine coronary arteries. Circulation. 2001;104:2228–2235. doi: 10.1161/hc4301.097195. [DOI] [PubMed] [Google Scholar]
- 52.Dorfmuller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358–363. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 53.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, Roedersheimer MT, van Rooijen N, Stenmark KR. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–669. doi: 10.2353/ajpath.2006.050599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stenmark KR, Davie NJ, Reeves JT, Frid MG. Hypoxia, leukocytes, and the pulmonary circulation. J Appl Physiol. 2005;98:715–721. doi: 10.1152/japplphysiol.00840.2004. [DOI] [PubMed] [Google Scholar]
- 55.Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S. Pathology of pulmonary hypertension. Clinics in chest medicine. 2007;28:23–42. vii. doi: 10.1016/j.ccm.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davie NJ, Crossno JT, Jr, Frid MG, Hofmeister SE, Reeves JT, Hyde DM, Carpenter TC, Brunetti JA, McNiece IK, Stenmark KR. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: Contribution of progenitor cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L668–678. doi: 10.1152/ajplung.00108.2003. [DOI] [PubMed] [Google Scholar]
- 57.Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007;75:640–648. doi: 10.1016/j.cardiores.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perros F, Dorfmuller P, Souza R, Durand-Gasselin I, Mussot S, Mazmanian M, Herve P, Emilie D, Simonneau G, Humbert M. Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur Respir J. 2007;29:462–468. doi: 10.1183/09031936.00094706. [DOI] [PubMed] [Google Scholar]
- 59.Lambrecht BN, Hammad H. Taking our breath away: Dendritic cells in the pathogenesis of asthma. Nat Rev Immunol. 2003;3:994–1003. doi: 10.1038/nri1249. [DOI] [PubMed] [Google Scholar]
- 60.Demedts IK, Bracke KR, Van Pottelberge G, Testelmans D, Verleden GM, Vermassen FE, Joos GF, Brusselle GG. Accumulation of dendritic cells and increased ccl20 levels in the airways of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175:998–1005. doi: 10.1164/rccm.200608-1113OC. [DOI] [PubMed] [Google Scholar]
- 61.Burnstock G. Purinergic signaling and vascular cell proliferation and death. Arterioscler Thromb Vasc Biol. 2002;22:364–373. doi: 10.1161/hq0302.105360. [DOI] [PubMed] [Google Scholar]
- 62.Sertl K, Takemura T, Tschachler E, Ferrans VJ, Kaliner MA, Shevach EM. Dendritic cells with antigen-presenting capability reside in airway epithelium, lung parenchyma, and visceral pleura. The Journal of experimental medicine. 1986;163:436–451. doi: 10.1084/jem.163.2.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annual review of immunology. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 64.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hume DA. Differentiation and heterogeneity in the mononuclear phagocyte system. Mucosal immunology. 2008;1:432–441. doi: 10.1038/mi.2008.36. [DOI] [PubMed] [Google Scholar]
- 66.Pryshchep O, Ma-Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel-specific toll-like receptor profiles in human medium and large arteries. Circulation. 2008;118:1276–1284. doi: 10.1161/CIRCULATIONAHA.108.789172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science (New York, NY. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F. A lineage of myeloid cells independent of myb and hematopoietic stem cells. Science (New York, NY. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 69.Gordon S, Mantovani A. Diversity and plasticity of mononuclear phagocytes. European journal of immunology. 2011;41:2470–2472. doi: 10.1002/eji.201141988. [DOI] [PubMed] [Google Scholar]
- 70.Sica A, Mantovani A. Macrophage plasticity and polarization: In vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Daley JM, Brancato SK, Thomay AA, Reichner JS, Albina JE. The phenotype of murine wound macrophages. Journal of leukocyte biology. 2010;87:59–67. doi: 10.1189/jlb.0409236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, Chawla A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011;480:104–108. doi: 10.1038/nature10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hume DA. Macrophages as apc and the dendritic cell myth. J Immunol. 2008;181:5829–5835. doi: 10.4049/jimmunol.181.9.5829. [DOI] [PubMed] [Google Scholar]
- 74.Brouty-Boye D, Pottin-Clemenceau C, Doucet C, Jasmin C, Azzarone B. Chemokines and cd40 expression in human fibroblasts. European journal of immunology. 2000;30:914–919. doi: 10.1002/1521-4141(200003)30:3<914::AID-IMMU914>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 75.Pap T, Muller-Ladner U, Gay RE, Gay S. Fibroblast biology. Role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis research. 2000;2:361–367. doi: 10.1186/ar113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stenmark KR, Frid MG, Yeager M, Li M, Riddle S, McKinsey T, El Kasmi KC. Targeting the adventitial microenvironment in pulmonary hypertension: A potential approach to therapy that considers epigenetic change. Pulmonary circulation. 2012;2:3–14. doi: 10.4103/2045-8932.94817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM, Carson WFt, Cavassani KA, Li X, Lukacs NW, Hogaboam CM, Dou Y, Kunkel SL. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. 2009;114:3244–3254. doi: 10.1182/blood-2009-04-217620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cavasin MA, Demos-Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N, Weiser-Evans MC, Harral J, Irwin DC, Anwar A, Yeager ME, Li M, Watson PA, Nemenoff RA, Buttrick PM, Stenmark KR, McKinsey TA. Selective class i histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res. 2012;110:739–748. doi: 10.1161/CIRCRESAHA.111.258426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, Paruchuri K, Mahabeleshwar GH, Dalmas E, Venteclef N, Flask CA, Kim J, Doreian BW, Lu KQ, Kaestner KH, Hamik A, Clement K, Jain MK. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest. 2011;121:2736–2749. doi: 10.1172/JCI45444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, Tsujimura T, Standley DM, Nakanishi K, Nakai K, Akira S. The jmjd3-irf4 axis regulates m2 macrophage polarization and host responses against helminth infection. Nature immunology. 2010;11:936–944. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 81.Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends in immunology. 2001;22:199–204. doi: 10.1016/s1471-4906(01)01863-4. [DOI] [PubMed] [Google Scholar]
- 82.Lo D, Feng L, Li L, Carson MJ, Crowley M, Pauza M, Nguyen A, Reilly CR. Integrating innate and adaptive immunity in the whole animal. Immunological reviews. 1999;169:225–239. doi: 10.1111/j.1600-065x.1999.tb01318.x. [DOI] [PubMed] [Google Scholar]
- 83.Buckley CD, Amft N, Bradfield PF, Pilling D, Ross E, Arenzana-Seisdedos F, Amara A, Curnow SJ, Lord JM, Scheel-Toellner D, Salmon M. Persistent induction of the chemokine receptor cxcr4 by tgf-beta 1 on synovial t cells contributes to their accumulation within the rheumatoid synovium. J Immunol. 2000;165:3423–3429. doi: 10.4049/jimmunol.165.6.3423. [DOI] [PubMed] [Google Scholar]
- 84.Wolinsky H, Glagov S. Nature of species differences in the medial distribution of aortic vasa vasorum in mammals. Circ Res. 1967;20:409–421. doi: 10.1161/01.res.20.4.409. [DOI] [PubMed] [Google Scholar]
- 85.Ritman EL, Lerman A. The dynamic vasa vasorum. Cardiovasc Res. 2007;75:649–658. doi: 10.1016/j.cardiores.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hayden MR, Tyagi SC. Vasa vasorum in plaque angiogenesis, metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: A malignant transformation. Cardiovasc Diabetol. 2004;3:1. doi: 10.1186/1475-2840-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moulton KS, Vakili K, Zurakowski D, Soliman M, Butterfield C, Sylvin E, Lo KM, Gillies S, Javaherian K, Folkman J. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci U S A. 2003;100:4736–4741. doi: 10.1073/pnas.0730843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Langheinrich AC, Kampschulte M, Buch T, Bohle RM. Vasa vasorum and atherosclerosis - quid novi? Thrombosis and haemostasis. 2007;97:873–879. [PubMed] [Google Scholar]
- 89.Mitzner W, Wagner EM. Vascular remodeling in the circulations of the lung. J Appl Physiol. 2004;97:1999–2004. doi: 10.1152/japplphysiol.00473.2004. [DOI] [PubMed] [Google Scholar]
- 90.Montani D, Perros F, Gambaryan N, Girerd B, Dorfmuller P, Price LC, Huertas A, Hammad H, Lambrecht B, Simonneau G, Launay JM, Cohen-Kaminsky S, Humbert M. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;184:116–123. doi: 10.1164/rccm.201006-0905OC. [DOI] [PubMed] [Google Scholar]
- 91.Davie NJ, Gerasimovskaya EV, Hofmeister SE, Richman AP, Jones PL, Reeves JT, Stenmark KR. Pulmonary artery adventitial fibroblasts cooperate with vasa vasorum endothelial cells to regulate vasa vasorum neovascularization: A process mediated by hypoxia and endothelin-1. Am J Pathol. 2006;168:1793–1807. doi: 10.2353/ajpath.2006.050754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kadowitz PJ, Knight DS, Hibbs RG, Ellison JP, Joiner PD, Brody MJ, Hyman AL. Influence of 5- and 6-hydroxydopamine on adrenergic transmission and nerve terminal morphology in the canine pulmonary vascular bed. Circ Res. 1976;39:191–199. doi: 10.1161/01.res.39.2.191. [DOI] [PubMed] [Google Scholar]
- 93.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature reviews. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 94.Martin TA, Harding KG, Jiang WG. Regulation of angiogenesis and endothelial cell motility by matrix-bound fibroblasts. Angiogenesis. 1999;3:69–76. doi: 10.1023/a:1009004212357. [DOI] [PubMed] [Google Scholar]
- 95.Nakatsu MN, Sainson RC, Aoto JN, Taylor KL, Aitkenhead M, Perez-del-Pulgar S, Carpenter PM, Hughes CC. Angiogenic sprouting and capillary lumen formation modeled by human umbilical vein endothelial cells (huvec) in fibrin gels: The role of fibroblasts and angiopoietin-1. Microvasc Res. 2003;66:102–112. doi: 10.1016/s0026-2862(03)00045-1. [DOI] [PubMed] [Google Scholar]
- 96.Velazquez OC, Snyder R, Liu ZJ, Fairman RM, Herlyn M. Fibroblast-dependent differentiation of human microvascular endothelial cells into capillary-like 3-dimensional networks. Faseb J. 2002;16:1316–1318. doi: 10.1096/fj.01-1011fje. [DOI] [PubMed] [Google Scholar]
- 97.Hlatky L, Tsionou C, Hahnfeldt P, Coleman CN. Mammary fibroblasts may influence breast tumor angiogenesis via hypoxia-induced vascular endothelial growth factor up-regulation and protein expression. Cancer Res. 1994;54:6083–6086. [PubMed] [Google Scholar]
- 98.Sharkey AM, Day K, McPherson A, Malik S, Licence D, Smith SK, Charnock-Jones DS. Vascular endothelial growth factor expression in human endometrium is regulated by hypoxia. J Clin Endocrinol Metab. 2000;85:402–409. doi: 10.1210/jcem.85.1.6229. [DOI] [PubMed] [Google Scholar]
- 99.Voelkel NF, Cool C. Pathology of pulmonary hypertension. Cardiol Clin. 2004;22:343–351. v. doi: 10.1016/j.ccl.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 100.Wilasrusmee C, Ondocin P, Bruch D, Shah G, Kittur S, Wilasrusmee S, Kittur DS. Amelioration of cyclosporin a effect on microvasculature by endothelin inhibitor. Surgery. 2003;134:384–389. doi: 10.1067/msy.2003.233. [DOI] [PubMed] [Google Scholar]
- 101.Choi MY, Kim HI, Yang YI, Kim JT, Jang SH, Park CM, Jang WH, Youn YC, Cheong SH, Choi CS, Kim DK, Lee SJ. The isolation and in situ identification of mscs residing in loose connective tissues using a niche-preserving organ culture system. Biomaterials. 2012;33:4469–4479. doi: 10.1016/j.biomaterials.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 102.Goritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisen J. A pericyte origin of spinal cord scar tissue. Science (New York, NY. 2011;333:238–242. doi: 10.1126/science.1203165. [DOI] [PubMed] [Google Scholar]
- 103.Bodin P, Burnstock G. Evidence that release of adenosine triphosphate from endothelial cells during increased shear stress is vesicular. J Cardiovasc Pharmacol. 2001;38:900–908. doi: 10.1097/00005344-200112000-00012. [DOI] [PubMed] [Google Scholar]
- 104.Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacology & therapeutics. 2006;112:358–404. doi: 10.1016/j.pharmthera.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 105.Gerasimovskaya EV, Ahmad S, White CW, Jones PL, Carpenter TC, Stenmark KR. Extracellular atp is an autocrine/paracrine regulator of hypoxia-induced adventitial fibroblast growth. Signaling through extracellular signal-regulated kinase-1/2 and the egr-1 transcription factor. J Biol Chem. 2002;277:44638–44650. doi: 10.1074/jbc.M203012200. [DOI] [PubMed] [Google Scholar]
- 106.Novak I. Atp as a signaling molecule: The exocrine focus. News in physiological sciences : an international journal of physiology produced jointly by the International Union of Physiological Sciences and the American Physiological Society. 2003;18:12–17. doi: 10.1152/nips.01409.2002. [DOI] [PubMed] [Google Scholar]
- 107.Burnstock G. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev. 2006;58:58–86. doi: 10.1124/pr.58.1.5. [DOI] [PubMed] [Google Scholar]
- 108.Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. International union of pharmacology lviii: Update on the p2y g protein-coupled nucleotide receptors: From molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Di Virgilio F, Solini A. P2 receptors: New potential players in atherosclerosis. British journal of pharmacology. 2002;135:831–842. doi: 10.1038/sj.bjp.0704524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Satterwhite CM, Farrelly AM, Bradley ME. Chemotactic, mitogenic, and angiogenic actions of utp on vascular endothelial cells. Am J Physiol. 1999;276:H1091–1097. doi: 10.1152/ajpheart.1999.276.3.H1091. [DOI] [PubMed] [Google Scholar]
- 111.Jacobson JR, Dudek SM, Singleton PA, Kolosova IA, Verin AD, Garcia JG. Endothelial cell barrier enhancement by atp is mediated by the small gtpase rac and cortactin. Am J Physiol Lung Cell Mol Physiol. 2006;291:L289–295. doi: 10.1152/ajplung.00343.2005. [DOI] [PubMed] [Google Scholar]
- 112.Kolosova IA, Mirzapoiazova T, Adyshev D, Usatyuk P, Romer LH, Jacobson JR, Natarajan V, Pearse DB, Garcia JG, Verin AD. Signaling pathways involved in adenosine triphosphate-induced endothelial cell barrier enhancement. Circ Res. 2005;97:115–124. doi: 10.1161/01.RES.0000175561.55761.69. [DOI] [PubMed] [Google Scholar]
- 113.Kaczmarek E, Erb L, Koziak K, Jarzyna R, Wink MR, Guckelberger O, Blusztajn JK, Trinkaus-Randall V, Weisman GA, Robson SC. Modulation of endothelial cell migration by extracellular nucleotides: Involvement of focal adhesion kinase and phosphatidylinositol 3-kinase-mediated pathways. Thrombosis and haemostasis. 2005;93:735–742. doi: 10.1267/THRO05040735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lemoli RM, Ferrari D, Fogli M, Rossi L, Pizzirani C, Forchap S, Chiozzi P, Vaselli D, Bertolini F, Foutz T, Aluigi M, Baccarani M, Di Virgilio F. Extracellular nucleotides are potent stimulators of human hematopoietic stem cells in vitro and in vivo. Blood. 2004;104:1662–1670. doi: 10.1182/blood-2004-03-0834. [DOI] [PubMed] [Google Scholar]
- 115.Rossi L, Manfredini R, Bertolini F, Ferrari D, Fogli M, Zini R, Salati S, Salvestrini V, Gulinelli S, Adinolfi E, Ferrari S, Di Virgilio F, Baccarani M, Lemoli RM. The extracellular nucleotide utp is a potent inducer of hematopoietic stem cell migration. Blood. 2007;109:533–542. doi: 10.1182/blood-2006-01-035634. [DOI] [PubMed] [Google Scholar]
- 116.Erlinge D. Extracellular atp: A growth factor for vascular smooth muscle cells. General pharmacology. 1998;31:1–8. doi: 10.1016/s0306-3623(97)00420-5. [DOI] [PubMed] [Google Scholar]
- 117.Huang N, Wang DJ, Heppel LA. Extracellular atp is a mitogen for 3t3, 3t6, and a431 cells and acts synergistically with other growth factors. Proc Natl Acad Sci U S A. 1989;86:7904–7908. doi: 10.1073/pnas.86.20.7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Woodward HN, Anwar A, Riddle S, Taraseviciene-Stewart L, Fragoso M, Stenmark KR, Gerasimovskaya EV. Pi3k, rho, and rock play a key role in hypoxia-induced atp release and atp-stimulated angiogenic responses in pulmonary artery vasa vasorum endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;297:L954–964. doi: 10.1152/ajplung.00038.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gerasimovskaya EV, Woodward HN, Tucker DA, Stenmark KR. Extracellular atp is a pro-angiogenic factor for pulmonary artery vasa vasorum endothelial cells. Angiogenesis. 2008;11:169–182. doi: 10.1007/s10456-007-9087-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Al Ghouleh I, Khoo NK, Knaus UG, Griendling KK, Touyz RM, Thannickal VJ, Barchowsky A, Nauseef WM, Kelley EE, Bauer PM, Darley-Usmar V, Shiva S, Cifuentes-Pagano E, Freeman BA, Gladwin MT, Pagano PJ. Oxidases and peroxidases in cardiovascular and lung disease: New concepts in reactive oxygen species signaling. Free radical biology & medicine. 2011;51:1271–1288. doi: 10.1016/j.freeradbiomed.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Csanyi G, Taylor WR, Pagano PJ. Nox and inflammation in the vascular adventitia. Free radical biology & medicine. 2009;47:1254–1266. doi: 10.1016/j.freeradbiomed.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Frazziano G, Champion HCMP, Pagano PJ. Nadph oxidase-derived ros and the regulation of pulmonary vessel tone. Am J Physiol Heart Circ Physiol. 2012 doi: 10.1152/ajpheart.00780.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Haurani MJ, Pagano PJ. Adventitial fibroblast reactive oxygen species as autacrine and paracrine mediators of remodeling: Bellwether for vascular disease? Cardiovasc Res. 2007;75:679–689. doi: 10.1016/j.cardiores.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 124.Nozik-Grayck E, Stenmark KR. Role of reactive oxygen species in chronic hypoxia-induced pulmonary hypertension and vascular remodeling. Adv Exp Med Biol. 2007;618:101–112. doi: 10.1007/978-0-387-75434-5_8. [DOI] [PubMed] [Google Scholar]
- 125.Tabima DM, Frizzell S, Gladwin MT. Reactive oxygen and nitrogen species in pulmonary hypertension. Free radical biology & medicine. 2012;52:1970–1986. doi: 10.1016/j.freeradbiomed.2012.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cascino T, Csanyi G, Al Ghouleh I, Montezano AC, Touyz RM, Haurani MJ, Pagano PJ. Adventitia-derived hydrogen peroxide impairs relaxation of the rat carotid artery via smooth muscle cell p38 mitogen-activated protein kinase. Antioxid Redox Signal. 2011;15:1507–1515. doi: 10.1089/ars.2010.3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rey FE, Pagano PJ. The reactive adventitia: Fibroblast oxidase in vascular function. Arterioscler Thromb Vasc Biol. 2002;22:1962–1971. doi: 10.1161/01.atv.0000043452.30772.18. [DOI] [PubMed] [Google Scholar]
- 128.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ros-hif-1alpha-kv1. 5 o2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294:H570–578. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 129.Jankov RP, Kantores C, Pan J, Belik J. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am J Physiol Lung Cell Mol Physiol. 2008;294:L233–245. doi: 10.1152/ajplung.00166.2007. [DOI] [PubMed] [Google Scholar]
- 130.Oishi PE, Wiseman DA, Sharma S, Kumar S, Hou Y, Datar SA, Azakie A, Johengen MJ, Harmon C, Fratz S, Fineman JR, Black SM. Progressive dysfunction of nitric oxide synthase in a lamb model of chronically increased pulmonary blood flow: A role for oxidative stress. Am J Physiol Lung Cell Mol Physiol. 2008;295:L756–766. doi: 10.1152/ajplung.00146.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, Karwande SV, Stringham JC, Bull DA, Gleich M, Kennedy TP, Hoidal JR. Transforming growth factor-beta1 induces nox4 nad(p)h oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L661–L673. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- 132.Takac I, Schroder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, Brandes RP. The e-loop is involved in hydrogen peroxide formation by the nadph oxidase nox4. J Biol Chem. 2011;286:13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ushio-Fukai M. Compartmentalization of redox signaling through nadph oxidase-derived ros. Antioxid Redox Signal. 2009;11:1289–1299. doi: 10.1089/ars.2008.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dennis KE, Aschner JL, Milatovic D, Schmidt JW, Aschner M, Kaplowitz MR, Zhang Y, Fike CD. Nadph oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol. 2009;297:L596–607. doi: 10.1152/ajplung.90568.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: The therapeutic potential of nrf2 activation. Molecular aspects of medicine. 2011;32:234–246. doi: 10.1016/j.mam.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 136.Lane KL, Talati M, Austin E, Hemnes AR, Johnson JA, Fessel JP, Blackwell T, Mernaugh RL, Robinson L, Fike C, Roberts LJ, 2nd, West J. Oxidative injury is a common consequence of bmpr2 mutations. Pulmonary circulation. 2011;1:72–83. doi: 10.4103/2045-8932.78107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, Shibata R, Sato K, Walsh K, Keaney JF., Jr Nadph oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation. 2011;124:731–740. doi: 10.1161/CIRCULATIONAHA.111.030775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, Anilkumar N, Ouattara A, Cave AC, Walker SJ, Grieve DJ, Charles RL, Eaton P, Brewer AC, Shah AM. Endothelial nox4 nadph oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol. 2011;31:1368–1376. doi: 10.1161/ATVBAHA.110.219238. [DOI] [PubMed] [Google Scholar]
- 139.Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP. Nox4 is a protective reactive oxygen species generating vascular nadph oxidase. Circ Res. 2012;110:1217–1225. doi: 10.1161/CIRCRESAHA.112.267054. [DOI] [PubMed] [Google Scholar]
- 140.Fukai T, Ushio-Fukai M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15:1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Van Rheen Z, Fattman C, Domarski S, Majka S, Klemm D, Stenmark KR, Nozik-Grayck E. Lung extracellular superoxide dismutase overexpression lessens bleomycin-induced pulmonary hypertension and vascular remodeling. Am J Respir Cell Mol Biol. 2011;44:500–508. doi: 10.1165/rcmb.2010-0065OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ahmed MN, Zhang Y, Codipilly C, Zaghloul N, Patel D, Wolin M, Miller EJ. Extracellular superoxide dismutase overexpression can reverse the course of hypoxia-induced pulmonary hypertension. Mol Med. 2012;18:38–46. doi: 10.2119/molmed.2011.00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xu D, Guo H, Xu X, Lu Z, Fassett J, Hu X, Xu Y, Tang Q, Hu D, Somani A, Geurts AM, Ostertag E, Bache RJ, Weir EK, Chen Y. Exacerbated pulmonary arterial hypertension and right ventricular hypertrophy in animals with loss of function of extracellular superoxide dismutase. Hypertension. 2011;58:303–309. doi: 10.1161/HYPERTENSIONAHA.110.166819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fleenor BS, Seals DR, Zigler ML, Sindler AL. Superoxide-lowering therapy with tempol reverses arterial dysfunction with aging in mice. Aging cell. 2012;11:269–276. doi: 10.1111/j.1474-9726.2011.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hoshino A, Chiba H, Nagai K, Ishii G, Ochiai A. Human vascular adventitial fibroblasts contain mesenchymal stem/progenitor cells. Biochemical and biophysical research communications. 2008;368:305–310. doi: 10.1016/j.bbrc.2008.01.090. [DOI] [PubMed] [Google Scholar]
- 146.Zengin E, Chalajour F, Gehling UM, Ito WD, Treede H, Lauke H, Weil J, Reichenspurner H, Kilic N, Ergun S. Vascular wall resident progenitor cells: A source for postnatal vasculogenesis. Development (Cambridge, England) 2006;133:1543–1551. doi: 10.1242/dev.02315. [DOI] [PubMed] [Google Scholar]
- 147.Campagnolo P, Cesselli D, Al Haj Zen A, Beltrami AP, Krankel N, Katare R, Angelini G, Emanueli C, Madeddu P. Human adult vena saphena contains perivascular progenitor cells endowed with clonogenic and proangiogenic potential. Circulation. 2010;121:1735–1745. doi: 10.1161/CIRCULATIONAHA.109.899252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cossu G, Bianco P. Mesoangioblasts--vascular progenitors for extravascular mesodermal tissues. Current opinion in genetics & development. 2003;13:537–542. doi: 10.1016/j.gde.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 149.Passman JN, Dong XR, Wu SP, Maguire CT, Hogan KA, Bautch VL, Majesky MW. A sonic hedgehog signaling domain in the arterial adventitia supports resident sca1+ smooth muscle progenitor cells. Proc Natl Acad Sci U S A. 2008;105:9349–9354. doi: 10.1073/pnas.0711382105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hu Y, Zhang Z, Torsney E, Afzal AR, Davison F, Metzler B, Xu Q. Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in apoe-deficient mice. J Clin Invest. 2004;113:1258–1265. doi: 10.1172/JCI19628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Howson KM, Aplin AC, Gelati M, Alessandri G, Parati EA, Nicosia RF. The postnatal rat aorta contains pericyte progenitor cells that form spheroidal colonies in suspension culture. Am J Physiol Cell Physiol. 2005;289:C1396–1407. doi: 10.1152/ajpcell.00168.2005. [DOI] [PubMed] [Google Scholar]
- 152.Pasquinelli G, Tazzari PL, Vaselli C, Foroni L, Buzzi M, Storci G, Alviano F, Ricci F, Bonafe M, Orrico C, Bagnara GP, Stella A, Conte R. Thoracic aortas from multiorgan donors are suitable for obtaining resident angiogenic mesenchymal stromal cells. Stem Cells. 2007;25:1627–1634. doi: 10.1634/stemcells.2006-0731. [DOI] [PubMed] [Google Scholar]
- 153.Zorzi P, Aplin AC, Smith KD, Nicosia RF. Technical advance: The rat aorta contains resident mononuclear phagocytes with proliferative capacity and proangiogenic properties. Journal of leukocyte biology. 2010;88:1051–1059. doi: 10.1189/jlb.0310178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Torsney E, Hu Y, Xu Q. Adventitial progenitor cells contribute to arteriosclerosis. Trends Cardiovasc Med. 2005;15:64–68. doi: 10.1016/j.tcm.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 155.Liu C, Nath KA, Katusic ZS, Caplice NM. Smooth muscle progenitor cells in vascular disease. Trends Cardiovasc Med. 2004;14:288–293. doi: 10.1016/j.tcm.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 156.Sata M, Saiura A, Kunisato A, Tojo A, Okada S, Tokuhisa T, Hirai H, Makuuchi M, Hirata Y, Nagai R. Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat Med. 2002;8:403–409. doi: 10.1038/nm0402-403. [DOI] [PubMed] [Google Scholar]
- 157.Caplice NM, Bunch TJ, Stalboerger PG, Wang S, Simper D, Miller DV, Russell SJ, Litzow MR, Edwards WD. Smooth muscle cells in human coronary atherosclerosis can originate from cells administered at marrow transplantation. Proc Natl Acad Sci U S A. 2003;100:4754–4759. doi: 10.1073/pnas.0730743100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hu Y, Davison F, Zhang Z, Xu Q. Endothelial replacement and angiogenesis in arteriosclerotic lesions of allografts are contributed by circulating progenitor cells. Circulation. 2003;108:3122–3127. doi: 10.1161/01.CIR.0000105722.96112.67. [DOI] [PubMed] [Google Scholar]
- 159.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 160.Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R. Circulating fibrocytes: Collagen-secreting cells of the peripheral blood. Int J Biochem Cell Biol. 2004;36:598–606. doi: 10.1016/j.biocel.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 161.Strieter RM, Keeley EC, Burdick MD, Mehrad B. The role of circulating mesenchymal progenitor cells, fibrocytes, in promoting pulmonary fibrosis. Transactions of the American Clinical and Climatological Association. 2009;120:49–59. [PMC free article] [PubMed] [Google Scholar]
- 162.Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. 2003;171:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 163.Frid M, Brunetti J, Burke D, Carpenter T, Davie N, Reeves J, Roedersheimer M, van Rooijen N, Stenmark K. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of monocyte/macrophage lineage. Am J Pathol. doi: 10.2353/ajpath.2006.050599. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]