

Background: DCCD is a classical inhibitor of F0F1 that modifies the proton binding site of the c subunit.
Results: Single-molecule analysis showed that a single modification in the multiple c subunit complex significantly inhibits F0F1.
Conclusion: Bound DCCD induces steric hindrance of the c subunit of the a subunit and hence blocks F0F1 rotation.
Significance: This is the first direct evidence to reveal the DCCD inhibition mechanism.
Keywords: ATP Synthase, Enzyme Inhibitors, Enzyme Kinetics, Enzyme Mechanisms, Enzyme Structure, F1F0 ATPase, Proton Transport, Single-molecule Biophysics
Abstract
N,N-Dicyclohexylcarbodiimide (DCCD) is a classical inhibitor of the F0F1-ATP synthase (F0F1), which covalently binds to the highly conserved carboxylic acid of the proteolipid subunit (c subunit) in F0. Although it is well known that DCCD modification of the c subunit blocks proton translocation in F0 and the coupled ATP hydrolysis activity of F1, how DCCD inhibits the rotary dynamics of F0F1 remains elusive. Here, we carried out single-molecule rotation assays to characterize the DCCD inhibition of Escherichia coli F0F1. Upon the injection of DCCD, rotations irreversibly terminated with first order reaction kinetics, suggesting that the incorporation of a single DCCD moiety is sufficient to block the rotary catalysis of the F0F1. Individual molecules terminated at different angles relative to the three catalytic angles of F1, suggesting that DCCD randomly reacts with one of the 10 c subunits. DCCD-inhibited F0F1 sometimes showed transient activation; molecules abruptly rotated and stopped after one revolution at the original termination angle, suggesting that hindrance by the DCCD moiety is released due to thermal fluctuation. To explore the mechanical activation of DCCD-inhibited molecules, we perturbed inhibited molecules using magnetic tweezers. The probability of transient activation increased upon a forward forcible rotation. Interestingly, during the termination F0F1, showed multiple positional shifts, which implies that F1 stochastically changes the angular position of its rotor upon a catalytic reaction. This effect could be caused by balancing the angular positions of the F1 and the F0 rotors, which are connected via elastic elements.
Introduction
F0F1-ATP synthase (F0F1) is a large multisubunit complex that catalyzes ATP synthesis from ADP and inorganic phosphate (Pi) by using the proton motive force that arises during oxidative phosphorylation and photosynthesis (1–4). F0F1 is composed of two structurally and functionally distinct parts: the F1 domain, which is responsible for catalysis, and the F0 domain, which conducts proton translocation across the membranes. Bacterial F0F1 has the simplest subunit composition: α3β3γδϵ for F1 and ab2c10–15 for F0. Although the number of c subunits can vary between species, it is 10 in Escherichia coli (5–8). Both F0 and F1 act as rotary molecular motors. F1 hydrolyzes ATP into ADP and inorganic phosphate (Pi) to rotate the γϵ complex against the surrounding α3β3 stator ring. Its catalytic sites reside on the three α-β interfaces. In contrast, F0 rotates the oligomer ring of the c subunit (c-ring) against the a subunit, and the γϵ complex binds to the c-ring to form the common rotary shaft. The stator components of F1 and F0 (the α3β3 ring and a subunit, respectively) are connected via the peripheral stalk, which is composed of the δ subunit and the b2 complex.
In physiological conditions, F0, when powered by the proton motive force, rotates the rotor complex (the γϵ-c10–15 complex) against the α3β3δ-ab2 stator complex, leading to a cycle of conformational changes in the α3β3 assembly and ATP synthesis from ADP and Pi. Conversely, when proton motive force diminishes, F1 hydrolyzes ATP, leading to reverse rotation of the rotor complex and causing F0 to pump protons in the reverse direction.
Single-molecule studies have extensively elucidated the chemomechanical coupling scheme and torque generation mechanism of F1 (5). The rotation consists of an elementary 120° step, which is consistent with the three-fold structural symmetry of F1 (9). Each 120° step is coupled with a single turnover of ATP hydrolysis and can be further resolved into discrete 80 and 40° substeps, which are triggered by ATP binding and ADP release or ATP-hydrolysis and Pi release, respectively (10–13). Although some uncertainties remain, a detailed coupling reaction scheme for F1 has been proposed (5).
In contrast to F1, the reaction scheme of the H+ translocation and the torque generation mechanism of F0 are poorly understood. One widely accepted working model is the two half-channels model (14). Here, the H+-conducting path of F0 is formed by two hemi-channels in the a subunit (15–17) and the H+-binding sites on the c-ring located on each of the individual c subunits. Under a proton motive force, H+ enters from the half-channel that faces the periplasm and reaches the H+-binding site of the c subunit. After one revolution of the c-ring, the bound H+ is transferred to the other half-channel that faces the cytoplasmic side and is finally released into the cytoplasmic space. Although structural details of the a-c interface are lacking, it is thought that torque is generated upon the H+-transferring steps between the a and c subunits. Thus, the postulated elementary angular step of the c-ring rotation is 360° divided by the number of c subunits, which translates into 36° for the bacterial c-ring (c10). Recent single-molecule studies have observed F0 rotation with multiple steps (up to 10) under ATP hydrolysis conditions, probably reflecting the structural symmetry of the c-ring (7, 8), whereas a three-step rotation was reported under ATP synthesis conditions (18).
The c subunit is composed of two transmembrane helices connected by a short loop on which the γϵ complex binds (19–22). The H+-binding site that forms the H+ translocation path is a highly conserved carboxyl residue (Asp-61 in E. coli) at the middle of the secondary helix located on the outer circumference of the c-ring. One NMR study showed that the pKa of the H+-binding carboxyl residue is higher than that of the carboxylate in aqueous condition, suggesting that the H+-binding carboxyl residue predominantly stays protonated in the membrane (23). Pogoryelov et al. (21, 24) proposed that when facing the exit half-channels, the H+-binding carboxyl residue orients out of the binding pocket and releases H+.
N,N-Dicyclohexylcarbodiimide (DCCD)2 is the gold standard inhibitor of F0F1 and has been widely used to study the corresponding coupling reaction. DCCD specifically reacts with the H+-binding carboxyl residue of the c subunit by forming a stable N-acyl urea (25) and blocks the coupling reaction of F0F1 (26, 27). Because DCCD reacts with carboxyl groups in the protonated state, carboxyl groups exposed in water solvent are much less reactive. Although F1 has a DCCD-reactive glutamate at the catalytic site, the glutamate residue is in a more aqueous condition than the H+-binding carboxyl residue of the c subunit. In addition, the glutamic acid of F1 requires a Mg2+-free solution to react with DCCD (28). Thus, reactivity of the glutamate residue of F1 is negligible. It was reported that the incorporation of a single DCCD molecule per F0 is sufficient to inhibit coupled ATPase activity (29). The crystal structure of the DCCD-modified c-ring (Protein Data Bank codes 2XQU, 2XQS, and 2XQT) showed that the cyclohexyl moieties are too large to enter the proton binding pocket of the c-subunit and protrude outwardly into the hydrophobic environment (24), implying that steric hindrance of the N-acyl urea with the a subunit blocks the rotation of the c-ring.
In single-molecule rotation assays, DCCD has been used to verify whether F0F1 retains the activity of the coupling reaction. Although pretreatment of the sample with DCCD decreases the probability of rotating particles (30), DCCD inhibition during rotation observation has not been attained. Therefore, it remains unclear how DCCD blocks the rotation. In the present study, we analyzed DCCD inhibition using the single-molecule rotation assay by introducing DCCD while observing rotating particles to characterize the rotary dynamics of DCCD-inhibited F0F1.
EXPERIMENTAL PROCEDURES
Preparation of F0F1-ATPase
Briefly, the 1.3 S subunit of Propionibacterium shermanii transcarboxylase, which contains a biotinylated domain, was fused to the N terminus of the β subunits for attaching the magnetic beads, and three histidine residues were introduced at the C terminus of the c subunits. Construction and purification of mutant E. coli F0F1-ATPase (EF0F1) for the rotational analysis were previously described by Iino et al. (31).
The E. coli strain RA1 (unc−/cyo−) was transformed with a F0F1 mutant plasmid and cultured in 1.2 liters of medium containing 30 μg/ml chloramphenicol for 16 h at 37 °C. Inverted membrane vesicles were prepared as follows. After the cell wall was digested with lysozyme treatment, the spheroplast was collected by centrifugation, resuspended, and broken down by sonication. The suspended mixture was centrifuged at 6,000 × g for 10 min at 4 °C to remove any cell debris. The supernatant containing the inverted membrane was transferred to a new tube and centrifuged at 300,000 × g for 20 min at 4 °C. The supernatant was discarded, and the pellet of membranes was resuspended in buffer A (100 mm HEPES-KOH (pH 7.5) and 50 mm KCl).
The purification of F0F1 was done as follows. The membrane suspension was solubilized with buffer B (20 mm HEPES-KOH (pH 7.5), 500 mm NaCl, 5 mm MgCl2, 200 μm ADP, 50 mm imidazole, 20% (v/v) glycerol, 1× protease inhibitor mixture, and 5 mm PAB, 0.8% (w/v) n-octyl-d-glucopyranoside (Sigma), and 2% (w/v) octaethylene glycol mono-n-dodecyl ether (C12E8, Wako)). The collected supernatant was loaded onto a HisTrap HP column (GE Healthcare), and the His-tagged F0F1 was eluted with buffer C (20 mm HEPES-KOH (pH 7.5), 500 mm NaCl, 5 mm MgCl2, 200 μm ADP, 500 mm imidazole, 20% glycerol (v/v), 1× protease inhibitor mixture, 5 mm PAB, 500 mm imidazole, 0.3% (w/v) C12E8, and 0.1% (w/v) E. coli total lipid) and further purified by size exclusion chromatography using a NAP-5 column (GE Healthcare) and buffer D (20 mm HEPES-NaOH (pH 7.5), 100 mm KCl, 2 mm MgCl2, 0.1× protease inhibitor mixture, 5 mm PAB, 0.3% (w/v) C12E8, and 0.1% (w/v) E. coli total lipid). The eluate was concentrated and further purified in a centrifugal filter (Amicon Ultra-4 100,000, Millipore). The protein concentration of the sample was determined by the BCA assay (Pierce) using bovine serum albumin (Sigma) as the standard.
ATPase Activity Measurements
The ATP hydrolysis activity of solubilized F0F1 was measured with an ATP regeneration system using a UV-visible spectrophotometer (VP-550, JASCO). 0.7 pmol of EF0F1 was suspended in 200 μl of buffer and incubated for a given time with various amounts of DCCD solubilized in ethanol at 25 °C. ATPase activity was started by introducing the F0F1 solution into buffer E (20 mm HEPES-NaOH (pH 7.5), 2 mm MgSO4, 100 mm KCl, 0.3% (w/v) C12E8, 0.1% (w/v) E. coli total lipid) containing 1.8 mm ATP, 2.5 mm phosphoenolpyruvate, 0.1 mg/ml pyruvate kinase, 0.1 mg/ml lactate dehydrogenase, and 0.2 mm NADH. The NADH absorbance at 340 nm was monitored for 900 s, and the ATP hydrolysis rate was calculated from the time course of the change in [NADH] using a molecular extinction coefficient value of 6,220 at 340 nm. After the ATP hydrolysis activity reached steady state, 0.2% (w/v) lauryldimethylamine oxide (LDAO) was added to confirm the reactivation of the activity. It has been reported that LDAO activates DCCD-inhibited F0F1 by inducing the uncoupling of F0 and F1 (32).
Rotation Assay
Coverslips coated with nickel-nitrilotriacetic acid were prepared as described previously (33). A flow cell of 10–30 μl in volume was made using two coverslips (bottom, 24 × 36 mm2; top, 24 × 24 mm2) separated by two spacers of 50-μm thickness. F0F1 was infused into the flow cell and incubated for 5 min. Unbound F0F1 was washed out with 100 μl of buffer E, and buffer E containing 1% BSA was infused into the flow cell to reduce nonspecific binding of the beads. A solution of streptavidin-coated magnetic beads of 200-nm diameter (Seradyn, Thermo Scientific) was infused into the flow cell. After a 10-min incubation, unbound beads were washed out by the infusion of 500 μl of buffer E containing 1.8 mm ATP. Rotation of the bead was observed under phase-contrast microscopy (IX70, Olympus) using a 60× objective lens. Images were captured with a charge-coupled device camera (FC300M; Takenaka) and recorded with a DV-CAM device (DSR-11; Sony) at 30 frames/s. Image analysis was performed using custom software (Digimo). Time-averaged rotation speed was calculated from over five consecutive revolutions. All experiments were carried out at 23 °C.
Mechanical Manipulation by Magnetic Tweezers
The experimental setup of the magnetic tweezers was previously described in detail by Hirono-Hara et al. (34). A schematic image of the magnetic tweezers is shown in Fig. 1A. The tweezers comprise four electromagnets, each constructed from a soft iron core (10 × 10 × 40 mm) and a copper wire with 100 turns around the core. Each pair of tweezers was electrically connected in series and separated by an interval of 15 mm. The two electromagnet pairs were crossed at each center space and positioned 10 mm above the microscope stage. The magnetic tweezers generated a magnetic field parallel to the stage, and the angle of the composite magnetic field was controlled by applying the sine components of the electric current to one pair (y axis) and the cosine components to the other (x axis). The field intensity was controlled by changing the current amplitude. The magnetic field was measured with a gauss meter (421 gaussmeter, Lake Shore Cryotronics, Westerville, OH) to confirm that the magnetic tweezers generated ∼200 G at the center of the focal plane with a precision of 4% for the intensity and 5° for the angle.
FIGURE 1.

Single-molecule observation of DCCD inhibition of F0F1. A, experimental system for the observation of the F0F1 rotation. After the immobilization of purified F0F1 on a nickel-nitrilotriacetic acid (Ni-NTA)-coated glass, magnetic beads were attached by fusing the biotin-binding domain to the β subunit of F0F1. The rotation of the bead was observed using optical microscopy. The rotor (red) rotates counterclockwise against the stator (blue). In stalling experiments, the rotational angle of the bead was controlled by magnetic tweezers. B, time courses of the revolutions of rotating molecules before and after adding 1 mm DCCD. After 5 min of recording the rotation, a buffer containing 1 mm DCCD was infused into the chamber. When the buffer change was finished (∼1 min), the recording was restarted. Of the eight rotating molecules, six terminated their rotations, and the remaining two (red lines) kept rotating. Of the former six molecules, four had already terminated their rotation before restarting the recording, and the other two stopped after restarting the video recording.
Manipulation of the magnetic tweezers was performed as follows. The tweezers were activated to trap the magnetic bead at the targeted angle. After a 3-s stall, the external magnetic field was turned off to observe whether the F0F1 rotated forward or backward. All procedures were controlled with custom-made software (Disimo). The image of the beads was recorded using the same method as that described under “Rotation Assay” above.
RESULTS
Rotation Behavior of F0F1 in the Presence of DCCD
To visualize the rotation of F0F1 using optical microscopy, a magnetic bead was attached to a biotin-binding domain introduced at the N terminus of the β subunit. Although the rotor complex of γϵc10 was kept immobilized, the stator α3β3δab2 complex rotated counterclockwise in the presence of ATP.
We selected smoothly rotating F0F1 molecules that had no obvious interference by steric interactions with the glass for the analysis. Typical time courses of the rotations are shown in Fig. 1B. Although F0F1 showed smooth consecutive rotation under saturating ATP (1.8 mm), they also showed short pauses during the rotation. The average duration time of these pauses was 4.9 s, which is consistent with a previous study (35) that showed EF0F1 transiently enters an inhibition state during the ATP-driven rotation, and the average duration time of this state is 2.57 ± 0.77 s. When 1 mm DCCD was introduced into the flow chamber, 27 of 29 F0F1 molecules eventually terminated their continuous rotations. F0F1 never resumed consecutive rotations within the observation time (more than 10 min). The other two molecules (Fig. 1B, red lines) showed rotations for more than 13 min after the DCCD injection. In contrast, when DCCD-free buffer was introduced, most molecules continued rotating (Fig. 2A, diamonds). Thus, we concluded that the terminations that appeared after the DCCD injection represent the DCCD inhibition state of F0F1. In the following analysis, we define the termination state as the DCCD inhibition state that appeared after DCCD injection and lasted more than 2 min.
FIGURE 2.
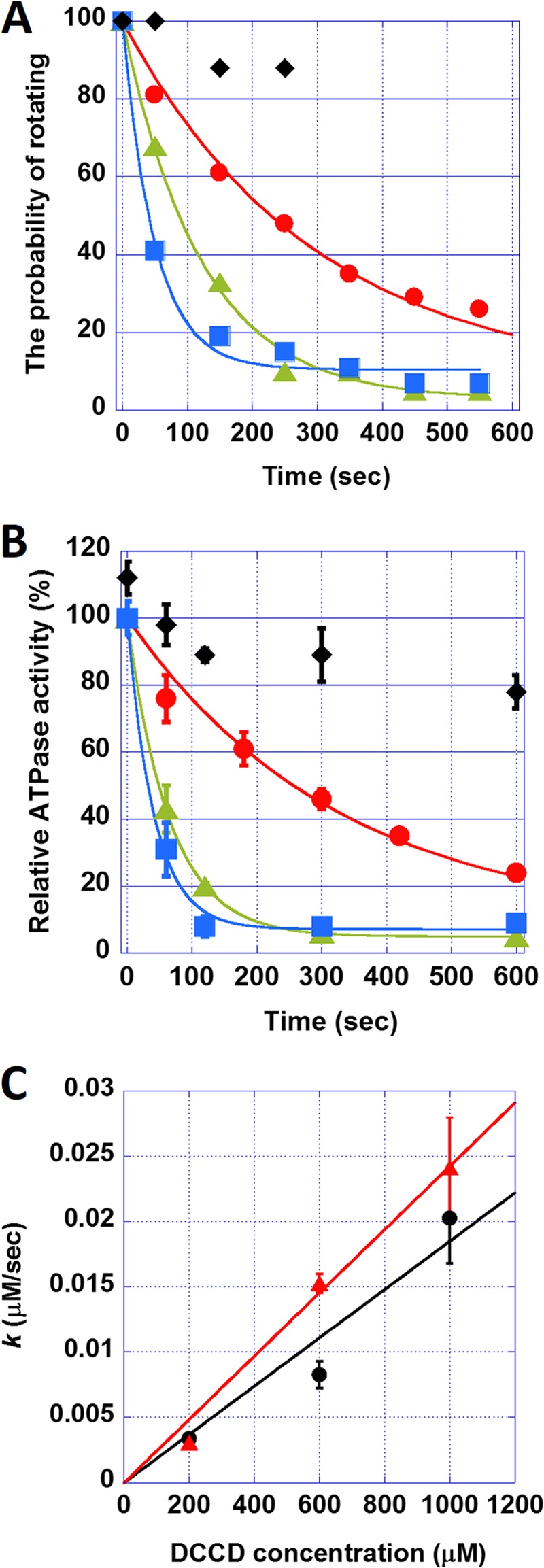
The kinetics of the DCCD inhibition of F0F1. A, the time course of the probability of rotating in the absence (♦) or presence of DCCD at 200 μm (●), 600 μm (▴), and 1 mm (■). Each plot was fitted with y = 100 − a)e−kt. B, the time course of bulk ATPase activities in the presence of DCCD at 200 μm (●), 600 μm (▴), and 1 mm (■). Diamonds (♦) represent ATPase activities in the presence of 0.2% (w/v) LDAO and 1 mm DCCD. Error bars represent the S.E. C, the relationship between the rate constant of DCCD inhibition k and DCCD concentration. The plot was fitted with y = m × x. The slope, m, was 1.9 × 10−5 and 2.4 × 10−5 s−1 according to the probability of rotating (black) and bulk ATPase activity (red) assays, respectively. Error bars represent the S.E.
Kinetic Analysis of DCCD Inhibition
To analyze the kinetics of the DCCD inhibition, we measured the rotating time, which was defined as the period from the DCCD injection until the time when the molecules lapsed into the DCCD-inhibited state, at various concentrations of DCCD (200 μm, 600 μm, and 1 mm). The time courses of the rotation probability were calculated from the rotating time (Fig. 2A, circles, triangles, and squares). The probability exponentially decayed to approach a constant value that corresponded to the DCCD-insensitive fraction. The time courses were fitted with a single exponential equation, y = 100 − a)e−kt (Fig. 2A, solid lines), where k and a represent the rate constant of the DCCD inhibition and the fraction of DCCD-insensitive molecules, respectively. We also conducted kinetic analysis of the DCCD inhibition on the ATPase activity in solution. ATPase activity decreased with the incubation time of DCCD, similar to the rotation assay results (Fig. 2B). It should be noted that it is reasonable to assume that preincubation of F0F1 with DCCD should show the same DCCD inhibition rate as real-time monitoring of DCCD inhibition because the concentration of the remaining DCCD in the ATPase assay mixture is low enough to prevent further F0F1 inhibition. In addition, we confirmed that the presence of ATP does not affect any DCCD inhibition rate of F0F1.
Fig. 2C shows k increased in proportion to the DCCD concentration, suggesting that DCCD inhibition is a first ordered reaction and that a single DCCD modification of the c-ring is sufficient to block the rotation. Additionally, the values of k are in good agreement with those values from the rotation assay. Finally, the rate constant of the DCCD inhibition standardized with the DCCD concentration was 2.4 × 10−5 and 1.8 × 10−5 m−1 s−1 according to the ATPase and rotation assays, respectively.
It is known that, along with the c subunit, DCCD can react with a highly conserved glutamate residue in the catalytic sites of F1 and inhibits the ATPase activity, although with a much lower reactivity (28, 36). To confirm that DCCD predominantly reacted with the c subunit in our experiments, we tested whether the bulk ATPase activities of F0F1 recovered from the DCCD inhibition by adding the detergent LDAO. LDAO is known to decouple the ATPase activity of F1 from the H+-conducting activity of F0 and thus restore the F0F1 ATPase activity in the presence of LDAO (32). As expected, when 0.2% (w/v) LDAO was added after a 10-min incubation of 1 mm DCCD, more than 80% of the ATPase activity was recovered (Fig. 2B, diamonds), demonstrating that DCCD predominantly reacted with F0. LDAO was not tested in the rotation assay, however, because it caused nonspecific attachment of the magnetic beads onto the glass coverslip, compromising the results.
Transient Activation from the DCCD Inhibition
We analyzed the rotary dynamics of the DCCD-inhibited state. To avoid additional DCCD binding to F0F1, DCCD in the flow chamber was washed out by injecting DCCD-free buffer after the DCCD-induced termination was observed.
Fig. 3A shows typical time courses of the revolutions of the molecules in the DCCD-inhibited state. Interestingly, some molecules showed transient rotations; that is, they rotated only once and abruptly stopped. In rare cases, inhibited molecules showed a maximum of two or three turns (the bottom time course in Fig. 3A). DCCD-inhibited F0F1 always stopped at the same position of the initial termination (Fig. 3A, green arrows), resulting in a single peak in the histogram of the angular positions (Fig. 3B). This result implies that the rotation of F0F1 was inhibited by a single DCCD modification. The angle distribution of the DCCD-inhibited state was fitted with a Gaussian function. The standard deviation of the angle distribution was relatively large (58.1°, Fig. 3B) when compared with that of the ATP-waiting state of F1 (37). Finally, the average frequency of the transient activation was determined as 0.018 s−1, which corresponds to an ATPase activity of 0.054 s−1, a value unlikely detectable in bulk ATPase measurements.
FIGURE 3.
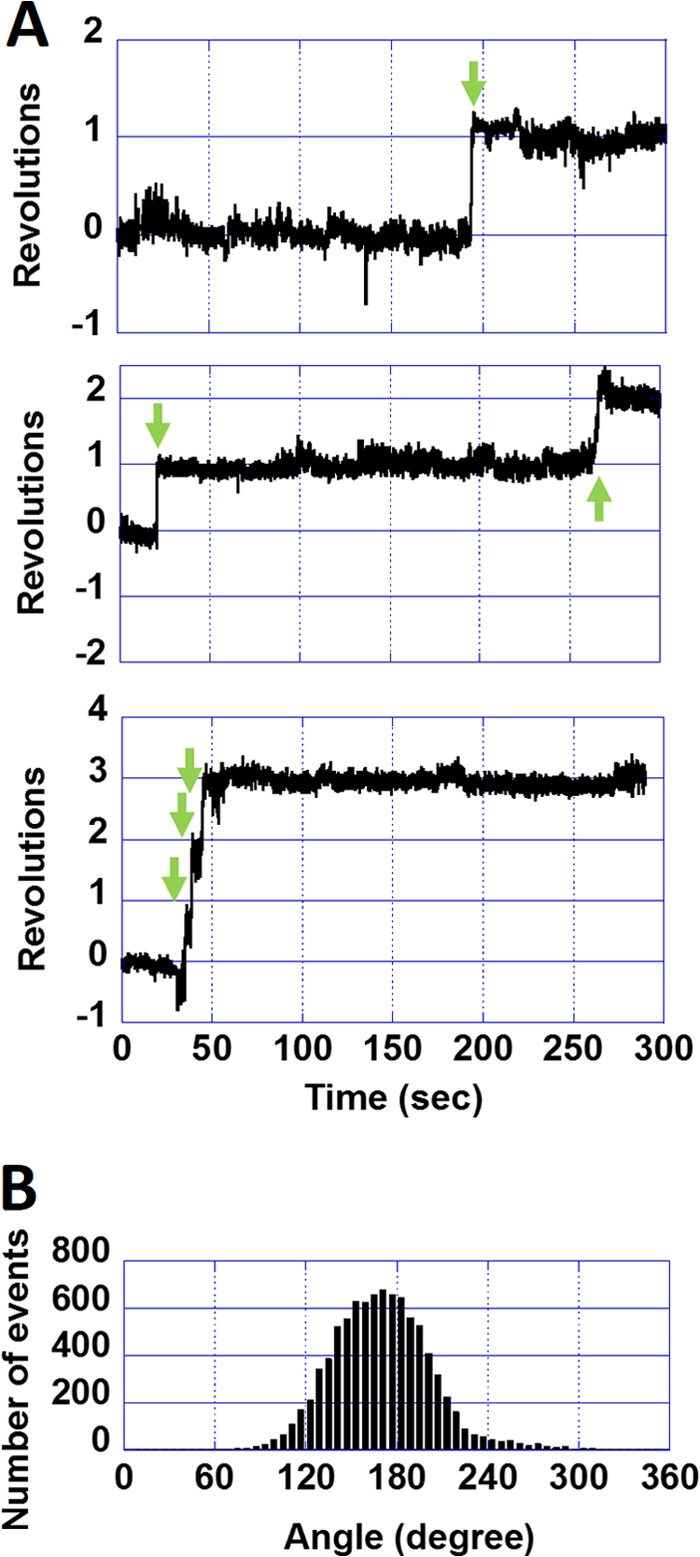
Positional shift during DCCD inhibition of wild-type F0F1. A, time courses of the revolutions of DCCD-inhibited wild-type F0F1. Green arrows indicate transient activations. B, histogram of angle rotations of DCCD-inhibited wild-type F0F1. When fitted with a Gaussian function, the standard deviation was 58.1°.
The Acceleration of Transient Activation by Mechanical Manipulation
We investigated whether the spontaneous transient activation could be accelerated by external force. After observing the DCCD-induced termination, the buffer was exchanged to the DCCD-free buffer. A magnetic field was applied to forcibly rotate and stall DCCD-inhibited F0F1 molecules at a target angle for 3 s. After these 3 s, the molecule was released from the tweezers, resulting in two behaviors (Fig. 4A). One behavior was deemed “activation,” in which F0F1 rotated forward one turn and terminated (blue arrow). The other was deemed “nonactivation,” in which F0F1 returned to the original termination angle (green arrow). In both cases, the angular position approximated the same angle as that before stalling. The inset in Fig. 4A shows histograms of the angle displacements for the two behaviors, with Gaussian distribution peaks at 3 ± 38 and 341 ± 57° for the activation and nonactivation states, respectively. It should be noted that F0F1 terminated at different positions from the original termination angle after being released from the magnetic tweezers when F0F1 was incubated with DCCD for a long time to allow multiple DCCD modifications of the c-ring (data not shown).
FIGURE 4.
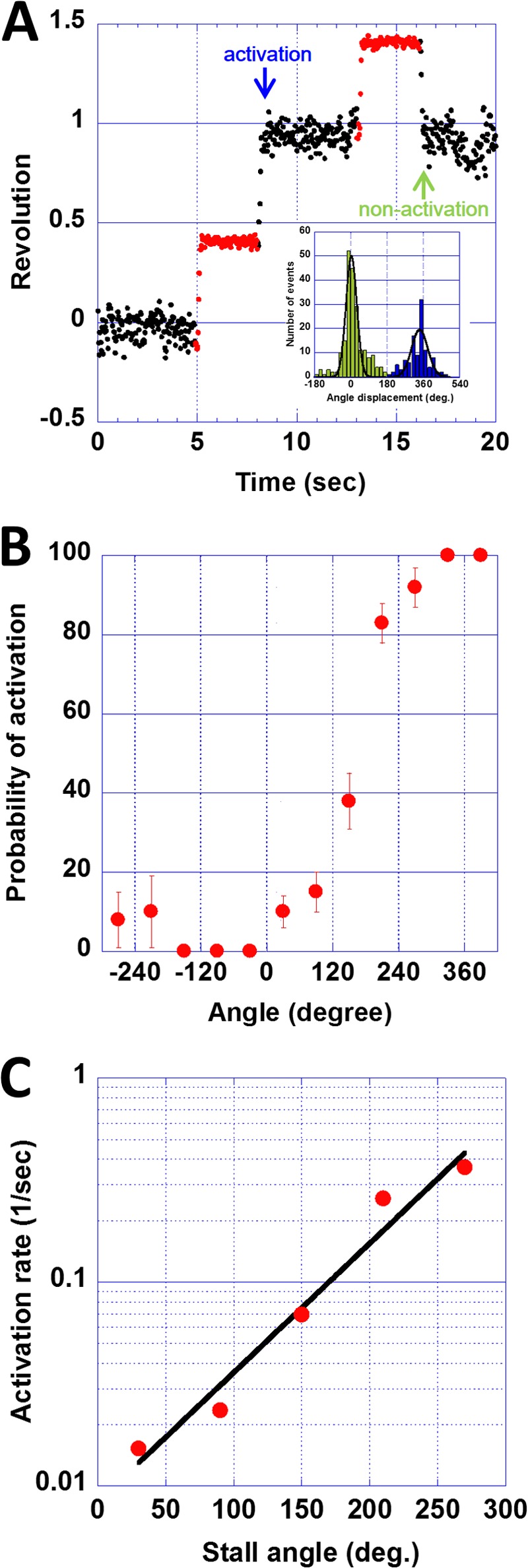
Mechanical activation of DCCD-inhibited wild-type F0F1. A, a typical time course of revolutions during mechanical activation. DCCD-inhibited F0F1 was forcibly rotated and stalled at a target angle with magnetic tweezers. After a 3-s window (red dots), the molecule was released from the magnetic tweezers. The released molecules showed two behaviors, activation and nonactivation. The inset shows a histogram of the angle displacements before and after stalling for activation (blue) and nonactivation (green) events. Gaussian peaks of these histograms were 341 ± 57 and 3 ± 38°, respectively. B, stall angle dependence on the probability of activation. Each column was obtained from 8–66 data sets. C, stall angle dependence on the activation rate, k, which was calculated using the equation p = 100(1 − e−kt) and the datasets at 30, 90, 150, 210, and 270° in B. The slope of the fitting line was −0.0146 kBT per 1°. deg., degrees.
The probability of mechanically induced transient activation is shown in Fig. 4B. The activation probability depended on the stall angle; stalling at over +0° significantly increased the activation probability, whereas stalling from 0° to −360° did not. The probability of activation reached 100% when the system stalled at over +300°. Based on these results, we estimated the activation rate constant assuming a simple first order reaction model.
The probability of activation, p, is given as a function of t as follows
where k is the rate constant of activation. In our experiments, the stall time was 3 s. k was determined from p values at 30, 90, 150, 210, and 270° stalls (Fig. 4C) and found to increase exponentially with the stall angle. Considering the Arrhenius equation (k = Ae (−ΔGa/kBT), where kB is the Boltzmann constant and T is the temperature, the increase in k means a decrease in the activation energy ΔGa. The slope of the fitting in Fig. 4C, which represents the activation energy change against the stall angle, was −0.0146 kBT per 1°. As described above, activation spontaneously occurred at a rate of 0.018 s−1 without any mechanical manipulation. This spontaneous activation rate corresponds to the activation rate at +44.1° in Fig. 4C, which suggests that spontaneous transient activation occurs when the α3β3δ-ab2 complex is thermally rotated in the forward direction. A similar role for thermal agitation in rate enhancement has been reported for the spontaneous activation of Mg-ADP inhibition and the ATP binding process of F1-ATPase (34, 38).
The Angular Position of the DCCD-inhibited State
In DCCD-inhibited molecules of the wild-type F0F1, the positional shifts of the angles were observed (Fig. 5A, red arrows). To determine the angular position of the DCCD-inhibited state in relation with the catalytic angle of F1, we conducted a rotation assay using the F1(βE181D) mutant in the presence of 200 μm DCCD. The corresponding mutant of F1 from thermophilic bacteria, TF1(βE190D), is known to have very low ATP hydrolysis activity (11). The catalytic dwell of these mutants is 100-fold longer than that of wild type, and hence the mutation causes clear stepwise rotations that pause at all catalytic angles, each 120° apart from the other (11). Fig. 5, B and C, show two typical time courses of the F1(βE181D) mutant before and after DCCD inhibition. The mutant showed a discrete 120° stepping rotation that paused at the catalytic angles and eventually lapsed into the DCCD-inhibited termination. Similar to the above experiments, after observing the termination, DCCD was washed out to avoid multiple DCCD modifications. During DCCD inhibition, the mutant showed the positional shifts in the termination angles as seen for the wild-type F0F1 (Fig. 5, A–C, red arrows). The positional shifts were, however, more obvious in the mutant, ranging from −94° to 139° with an average value of 58° (44 transitions for 11 molecules). The histograms of the angular distributions during the terminations showed multiple peaks (Fig. 5, B and C, bottom), with 2–4 peaks seen depending on the molecule.
FIGURE 5.

The rotational behaviors of the wild-type F0F1 and the mutant F0F1(βE181D) in the DCCD-inhibited state. A, three examples of the time courses of the rotational angles of the wild-type F0F1 in the DCCD-inhibited state. Red arrows indicate transitions. B and C, two examples of the time courses and corresponding histograms of the rotational angles of the βE181D mutant in the presence of 200 μm DCCD. Red arrows indicate the positional shift of the DCCD-inhibited state. Histograms of the angle rotations and the terminations are shown below the time courses. The histograms were fitted with a Gaussian function. D, a histogram of differences between catalytic angles and DCCD inhibition angles (20 peaks for 11 molecules). The angle difference between the two was calculated by taking the peak DCCD inhibition and peak catalytic angle to its left (red dotted lines in B).
Relative differences between the angle of DCCD-induced pause and the catalytic angle were determined for all molecules. When differences in the peak position between the catalytic angle histogram and DCCD inhibition angles histogram were calculated (Fig. 5B, red vertical dotted lines), no distinct peak was seen (Fig. 5D). Similarly, when the initial pause position was selected for this analysis, the histogram again had no obvious peak (data not shown). These results suggest that DCCD modifications are random among the 10 c subunits.
DISCUSSION
The DCCD-sensitive Rotation of F0F1
Several studies on the ATP-driven rotation of F0F1 have been reported since Sambongi et al. (39) first observed the rotation of purified E. coli F0F1 (8, 30, 40, 41). Although DCCD sensitivity has been the gold standard for ensuring that F0F1 molecules retain their competency when coupling ATP hydrolysis with H+ translocation, DCCD has not been used in the rotation assay for studying F0F1. In the early stage of the rotation assay, Triton X-100 has been often used to solubilize F0F1 (39). However, such a detergent risks impairing the DCCD sensitivity of F0F1, probably by loosening the subunit interaction, meaning that rotations observed in the early stage are not well coupled with the H+ translocation in F0 (36). Ueno et al. (30) attempted to solve this issue by using a lipid-like reagent, lysophosphatidylcholine, finding that the probability of rotating molecules was significantly decreased by DCCD-pretreatment prior to the rotation assay (41). However, DCCD inhibition during rotation observation remained elusive.
In the present study, we achieved more than 90% DCCD sensitivity of ATPase activity in solution by solubilizing F0F1 in buffer containing 0.1% (w/v) E. coli lipid total extract and 0.3% (w/v) of the detergent C12E8. In this buffer condition, most F0F1 molecules stopped the ATP-driven rotation after DCCD injection into the flow chamber. In addition, the time constant of the DCCD inhibition, as determined from the duration time of the rotation after DCCD injection, was consistent with that of the DCCD inhibition determined from ATP hydrolysis activity in solution. Thus, a lipid-like condition is critical for sustaining the integrity of the subunit interaction and the coupling nature of F0F1.
The Stoichiometry of DCCD for Inhibition
The rate of the DCCD inhibition was linearly proportional with the DCCD concentration, suggesting that the inhibition is a first order reaction and that a single DCCD modification is sufficient for the inhibition of the F0F1 coupling reaction, as reported previously (26, 27, 29). Interestingly, even after inhibition with DCCD, F0F1 showed transient activation by making at most three turns. The angular position after the transient activation was always the same as the initial termination position, although a slight positional shift during the termination was observed in some molecules. This result also suggests that a single DCCD modification is sufficient to block rotation.
No Preference of DCCD Incorporation
The angular position of the DCCD-induced terminations was analyzed in relation with the catalytic pause of F1 by using the mutant F0F1(βE181D). The distribution of the position of the DCCD-inhibited state showed no obvious peak between the catalytic angles of F1, indicating that DCCD randomly reacts with the c subunit regardless of the position of the c subunit relative to F1 and that there is obvious preference for the DCCD reactivity. This result is consistent with a crystal structure of the F1-c-ring complex (Protein Data Bank code 1QO1), where the c subunit showed identical conformations (20).
Transient Activation from DCCD Inhibition
We found that DCCD-inhibited F0F1 shows transient activation that can be largely enhanced when DCCD-inhibited molecules are forcibly rotated in the forward direction, but not in the backward direction. This asymmetric effect is explainable when considering a structural feature of F0. In the coupled rotation, H+ is translated between the a and c subunits of F0. Therefore, the c subunit involved in the H+ translocation should tightly interact with the a subunit, whereas other c subunits are exposed to the lipid bilayer. A crystal structure of a DCCD-modified c-ring from Spirulina platensis (Protein Data Bank codes 2XQU, 2XQS, and 2XQT) showed that covalently bound DCCD (N-acyl urea) protrudes outward from the H+-binding carboxylate (24). This observation implies that once the c-ring is modified by DCCD, its rotation is locked due to the steric hindrance of the protruding DCCD and a subunit (Fig. 6A). The transient activation then might be caused by a thermally induced looseness at the a-c interface. Forcible rotation in the forward direction enhances the looseness, whereas backward rotation causes F0 to be rotated until the DCCD clashes again with the a subunit.
FIGURE 6.
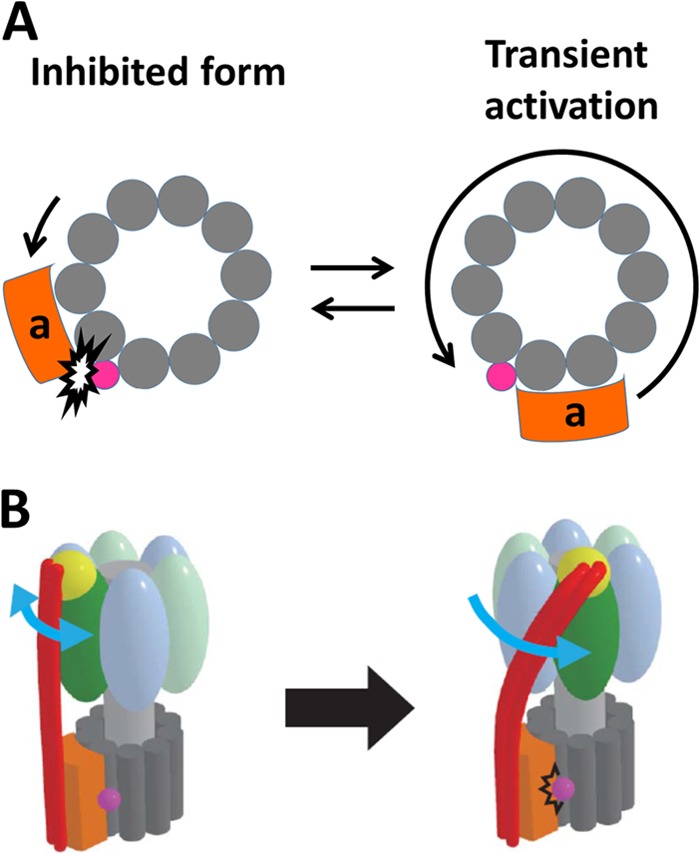
The models of DCCD inhibition of F0F1. A, the model of the transient activation of the DCCD-inhibited F0 when viewed from the F1 side. The rotation of the peripheral stalk of F0F1 is transiently activated and blocked again at the same angle after one rotation. B, the model of the DCCD-inhibited F0F1 showing the twisted peripheral stalk. The F1 part undergoes the ATP-driven rotation, whereas the F0 part is locked by DCCD, which provokes the twist of the peripheral stalk.
The Positional Shift of the DCCD-inhibited State
Surprisingly, we found that DCCD-inhibited F0F1 showed positional shifts in the termination angle (Fig. 5, A–C). Such positional shifts were not observed in other inhibitory states of F1 such as ADP inhibition and ϵ-subunit inhibition (34, 42). This difference is attributable to the inhibition target. The ADP and ϵ subunit inhibitions directly block the rotation of F1 by tightly binding to the catalytic site or extending the C-terminal helices of the ϵ subunit into the stator-rotor interface of F1 to cause steric hindrance (31, 43). On the other hand, in DCCD inhibition, DCCD blocks F0 rotation, whereas F1 is still catalytically active. This situation is similar to a stalling experiment using magnetic tweezers where actively rotating F1 molecules are stalled by an external torque when F1 reaches a dwelling pause while waiting for a particular catalytic reaction step such as ATP binding or ATP cleavage (38). The probability of the catalytic reaction step was measured as a function of the rotary angle of the γ subunit (38). It was revealed that over a wide range of stall angles, F1 can undergo both forward and reverse catalytic reactions. Forward reactions trigger torque generation to continue the rotation, whereas reverse reactions induce rotation back to the original dwelling position. Therefore, stalled F1 molecules show a positional shift in the termination angle that can be determined by a balance between the torque of F1 and an external torque generated by the magnetic tweezers.3
The positional shift of the DCCD-inhibited state can be explained as follows. The F0-locked state describes mechanical stalling for F1. Therefore, catalysis of F1 should be at equilibrium between the forward and reverse reactions, each accompanying the torque generation accordingly. Because F0 and F1 are connected via elastic stalk structures, the central rotor stalk and the peripheral stalk (3, 37, 44–47), F1 can change the angular position of the DCCD-induced termination; the position shifts forward when F1 conducts a forward reaction and shifts backward when the reaction is backward. In the schematic model shown in Fig. 6B, we attribute the elasticity to the peripheral stalk that is bent and stretched upon the torsional stress by F1 based on the studies of Cain and co-workers (48, 49) that showed that the peripheral stalk subunit, b, is exceptionally robust against the deletion or insertion mutation, suggesting structural flexibility of the peripheral stalk. It should be noted that Junge and co-workers (40) reported that the peripheral stalk is more rigid than the central rotor stalk against the torsional stress. Because the peripheral stalk bears mainly bending and stretching force in circumferential direction and thus, the torsional stress is relatively minor, we prefer the elastic peripheral stalk model. However, this issue remains to be tested experimentally.
The reason why F0F1(βE181D) showed obvious and large positional shifts is attributable to the slow kinetics and equilibrium of the catalysis. The catalytic reaction of the wild type is much faster (on a millisecond scale) than that of the mutant (∼200 ms) or the response time of the magnetic bead (∼100 ms). Therefore, it is highly likely that the relatively slow response of the magnetic tweezers obscured discrete positional shifts in wild-type F0F1.
Overall, the multiple positional shifts of the termination angles observed in DCCD-inhibited F0F1(βE181D) implies that several catalytic reactions and rotational steps are involved in this phenomenon and that F0F1 has very flexible components that can twist upon torque generation. For quantitative analysis of this phenomenon, F0F1 from thermophilic bacillus PS3 (TF0F1) would be more suitable because its step rotations and kinetics parameters have been well characterized.
Conclusions
We have demonstrated the DCCD inhibition of F0F1 using single-molecule techniques. The first order reaction kinetics of the inhibition and the essentially single position of the DCCD-induced termination indicate that a single modification of the c subunit in the c-ring is sufficient for blocking the rotation. We also found spontaneous transient activation upon DCCD inhibition that could be stimulated by forcibly rotating the complex to the forward direction. Finally, we showed a positional shift during DCCD inhibition, indicating a stochastic torque by F1 and large torsional flexibility in F0.
Acknowledgments
We thank all members of the Noji Laboratory for critical discussions and technical advice. We particularly thank R. Iino, R. Watanabe, and K. V. Tabata for valuable comments.
This work was supported in part by Grant-in-aid for Scientific Research 18074005 (to H. N.) and by a Special Education and Research Expenses grant (to H. N.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
M. Toei and H. Noji, unpublished data.
- DCCD
- N,N-dicyclohexylcarbodiimide
- C12E8
- octaethylene glycol monododecyl ether
- PAB
- 4-aminobenzamidine dihydrochloride
- EF0F1
- E. coli F0F1-ATPase
- LDAO
- lauryldimethylamine oxide.
REFERENCES
- 1. Boyer P. D. (1997) The ATP synthase–a splendid molecular machine. Annu. Rev. Biochem. 66, 717–749 [DOI] [PubMed] [Google Scholar]
- 2. Senior A. E., Nadanaciva S., Weber J. (2002) The molecular mechanism of ATP synthesis by F1F0-ATP synthase. Biochim. Biophys. Acta 1553, 188–211 [DOI] [PubMed] [Google Scholar]
- 3. Junge W., Sielaff H., Engelbrecht S. (2009) Torque generation and elastic power transmission in the rotary F0F1-ATPase. Nature 459, 364–370 [DOI] [PubMed] [Google Scholar]
- 4. Yoshida M., Muneyuki E., Hisabori T. (2001) ATP synthase–a marvellous rotary engine of the cell. Nat. Rev. Mol. Cell Biol. 2, 669–677 [DOI] [PubMed] [Google Scholar]
- 5. Okuno D., Iino R., Noji H. (2011) Rotation and structure of F0F1-ATP synthase. J. Biochem. 149, 655–664 [DOI] [PubMed] [Google Scholar]
- 6. Jiang W., Hermolin J., Fillingame R. H. (2001) The preferred stoichiometry of c subunits in the rotary motor sector of Escherichia coli ATP synthase is 10. Proc. Natl. Acad. Sci. U.S.A. 98, 4966–4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Düser M. G., Zarrabi N., Cipriano D. J., Ernst S., Glick G. D., Dunn S. D., Börsch M. (2009) 36 degrees step size of proton-driven c-ring rotation in F0F1-ATP synthase. EMBO J. 28, 2689–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ishmukhametov R., Hornung T., Spetzler D., Frasch W. D. (2010) Direct observation of stepped proteolipid ring rotation in E. coli F0F1-ATP synthase. EMBO J. 29, 3911–3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abrahams J. P., Leslie A. G., Lutter R., Walker J. E. (1994) Structure at 2.8 Ä resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628 [DOI] [PubMed] [Google Scholar]
- 10. Yasuda R., Noji H., Kinosita K., Jr., Yoshida M. (1998) F1-ATPase is a highly efficient molecular motor that rotates with discrete 120 degree steps. Cell 93, 1117–1124 [DOI] [PubMed] [Google Scholar]
- 11. Shimabukuro K., Yasuda R., Muneyuki E., Hara K. Y., Kinosita K., Jr., Yoshida M. (2003) Catalysis and rotation of F1 motor: cleavage of ATP at the catalytic site occurs in 1 ms before 40 degree substep rotation. Proc. Natl. Acad. Sci. U.S.A. 100, 14731–14736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adachi K., Oiwa K., Nishizaka T., Furuike S., Noji H., Itoh H., Yoshida M., Kinosita K., Jr. (2007) Coupling of rotation and catalysis in F1-ATPase revealed by single-molecule imaging and manipulation. Cell 130, 309–321 [DOI] [PubMed] [Google Scholar]
- 13. Watanabe R., Iino R., Noji H. (2010) Phosphate release in F1-ATPase catalytic cycle follows ADP release. Nat. Chem. Biol. 6, 814–820 [DOI] [PubMed] [Google Scholar]
- 14. Junge W., Lill H., Engelbrecht S. (1997) ATP synthase: an electrochemical transducer with rotatory mechanics. Trends Biochem. Sci. 22, 420–423 [DOI] [PubMed] [Google Scholar]
- 15. Angevine C. M., Fillingame R. H. (2003) Aqueous access channels in subunit a of rotary ATP synthase. J. Biol. Chem. 278, 6066–6074 [DOI] [PubMed] [Google Scholar]
- 16. Angevine C. M., Herold K. A., Fillingame R. H. (2003) Aqueous access pathways in subunit a of rotary ATP synthase extend to both sides of the membrane. Proc. Natl. Acad. Sci. U.S.A. 100, 13179–13183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Angevine C. M., Herold K. A., Vincent O. D., Fillingame R. H. (2007) Aqueous access pathways in ATP synthase subunit a: reactivity of cysteine substituted into transmembrane helices 1, 3, and 5. J. Biol. Chem. 282, 9001–9007 [DOI] [PubMed] [Google Scholar]
- 18. Watanabe R., Tabata K. V., Iino R., Ueno H., Iwamoto M., Oiki S., Noji H. (2013) Biased Brownian stepping rotation of F0F1-ATP synthase driven by proton motive force. Nat. Commun. 4, 1631. [DOI] [PubMed] [Google Scholar]
- 19. Girvin M. E., Fillingame R. H. (1994) Hairpin folding of subunit c of F1F0 ATP synthase: 1H distance measurements to nitroxide-derivatized aspartyl-61. Biochemistry 33, 665–674 [DOI] [PubMed] [Google Scholar]
- 20. Stock D., Leslie A. G., Walker J. E. (1999) Molecular architecture of the rotary motor in ATP synthase. Science 286, 1700–1705 [DOI] [PubMed] [Google Scholar]
- 21. Pogoryelov D., Yildiz O., Faraldo-Gómez J. D., Meier T. (2009) High-resolution structure of the rotor ring of a proton-dependent ATP synthase. Nat. Struct. Mol. Biol. 16, 1068–1073 [DOI] [PubMed] [Google Scholar]
- 22. Nakano T., Ikegami T., Suzuki T., Yoshida M., Akutsu H. (2006) A new solution structure of ATP synthase subunit c from thermophilic Bacillus PS3, suggesting a local conformational change for H+-translocation. J. Mol. Biol. 358, 132–144 [DOI] [PubMed] [Google Scholar]
- 23. Assadi-Porter F. M., Fillingame R. H. (1995) Proton-translocating carboxyl of subunit c of F1F0 H+-ATP synthase: the unique environment suggested by the pKa determined by 1H NMR. Biochemistry 34, 16186–16193 [DOI] [PubMed] [Google Scholar]
- 24. Pogoryelov D., Krah A., Langer J. D., Yildiz Ö., Faraldo-Gómez J. D., Meier T. (2010) Microscopic rotary mechanism of ion translocation in the F0 complex of ATP synthases. Nat. Chem. Biol. 6, 891–899 [DOI] [PubMed] [Google Scholar]
- 25. Sebald W., Machleidt W., Wachter E. (1980) N,N′-dicyclohexylcarbodiimide binds specifically to a single glutamyl residue of the proteolipid subunit of the mitochondrial adenosinetriphosphatases from Neurospora crassa and Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 77, 785–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fillingame R. H. (1975) Identification of the dicyclohexylcarbodiimide-reactive protein component of the adenosine 5′-triphosphate energy-transducing system of Escherichia coli. J. Bacteriol. 124, 870–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Negrin R. S., Foster D. L., Fillingame R. H. (1980) Energy-transducing H+-ATPase of Escherichia coli: reconstitution of proton translocation activity of the intrinsic membrane sector. J. Biol. Chem. 255, 5643–5648 [PubMed] [Google Scholar]
- 28. Yoshida M., Allison W. S. (1983) Modulation by ADP and Mg2+ of the inactivation of the F1-ATPase from the thermophilic bacterium, PS3, with dicyclohexylcarbodiimide. J. Biol. Chem. 258, 14407–14412 [PubMed] [Google Scholar]
- 29. Hermolin J., Fillingame R. H. (1989) H+-ATPase activity of Escherichia coli F1F0 is blocked after reaction of dicyclohexylcarbodiimide with a single proteolipid (subunit c) of the F0 complex. J. Biol. Chem. 264, 3896–3903 [PubMed] [Google Scholar]
- 30. Ueno H., Suzuki T., Kinosita K., Jr., Yoshida M. (2005) ATP-driven stepwise rotation of F0F1-ATP synthase. Proc. Natl. Acad. Sci. U.S.A. 102, 1333–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iino R., Hasegawa R., Tabata K. V., Noji H. (2009) Mechanism of inhibition by C-terminal α-helices of the epsilon subunit of Escherichia coli F0F1-ATP synthase. J. Biol. Chem. 284, 17457–17464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cook G. M., Keis S., Morgan H. W., von Ballmoos C., Matthey U., Kaim G., Dimroth P. (2003) Purification and biochemical characterization of the F1F0-ATP synthase from thermoalkaliphilic Bacillus sp. strain TA2.A1. J. Bacteriol. 185, 4442–4449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Itoh H., Takahashi A., Adachi K., Noji H., Yasuda R., Yoshida M., Kinosita K. (2004) Mechanically driven ATP synthesis by F1-ATPase. Nature 427, 465–468 [DOI] [PubMed] [Google Scholar]
- 34. Hirono-Hara Y., Ishizuka K., Kinosita K., Jr., Yoshida M., Noji H. (2005) Activation of pausing F1 motor by external force. Proc. Natl. Acad. Sci. U.S.A. 102, 4288–4293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sekiya M., Hosokawa H., Nakanishi-Matsui M., Al-Shawi M. K., Nakamoto R. K., Futai M. (2010) Single molecule behavior of inhibited and active states of Escherichia coli ATP synthase F1 rotation. J. Biol. Chem. 285, 42058–42067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gibbons C., Montgomery M. G., Leslie A. G., Walker J. E. (2000) The structure of the central stalk in bovine F1-ATPase at 2.4 Ä resolution. Nat. Struct. Biol. 7, 1055–1061 [DOI] [PubMed] [Google Scholar]
- 37. Okuno D., Iino R., Noji H. (2010) Stiffness of γ subunit of F1-ATPase. Eur. Biophys. J. 39, 1589–1596 [DOI] [PubMed] [Google Scholar]
- 38. Watanabe R., Okuno D., Sakakihara S., Shimabukuro K., Iino R., Yoshida M., Noji H. (2012) Mechanical modulation of catalytic power on F1-ATPase. Nat. Chem. Biol. 8, 86–92 [DOI] [PubMed] [Google Scholar]
- 39. Sambongi Y., Iko Y., Tanabe M., Omote H., Iwamoto-Kihara A., Ueda I., Yanagida T., Wada Y., Futai M. (1999) Mechanical rotation of the c subunit oligomer in ATP synthase (F0F1): direct observation. Science 286, 1722–1724 [DOI] [PubMed] [Google Scholar]
- 40. Sielaff H., Rennekamp H., Wächter A., Xie H., Hilbers F., Feldbauer K., Dunn S. D., Engelbrecht S., Junge W. (2008) Domain compliance and elastic power transmission in rotary F0F1-ATPase. Proc. Natl. Acad. Sci. U.S.A. 105, 17760–17765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tsunoda S. P., Aggeler R., Noji H., Kinosita K., Jr., Yoshida M., Capaldi R. A. (2000) Observations of rotation within the F0F1-ATP synthase: deciding between rotation of the F0c subunit ring and artifact. FEBS Lett. 470, 244–248 [DOI] [PubMed] [Google Scholar]
- 42. Konno H., Murakami-Fuse T., Fujii F., Koyama F., Ueoka-Nakanishi H., Pack C. G., Kinjo M., Hisabori T. (2006) The regulator of the F1 motor: inhibition of rotation of cyanobacterial F1-ATPase by the ϵ subunit. EMBO J. 25, 4596–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jault J. M., Matsui T., Jault F. M., Kaibara C., Muneyuki E., Yoshida M., Kagawa Y., Allison W. S. (1995) The α3β3 γ complex of the F1-ATPase from thermophilic Bacillus PS3 containing the α D261N substitution fails to dissociate inhibitory MgADP from a catalytic site when ATP binds to noncatalytic sites. Biochemistry 34, 16412–16418 [DOI] [PubMed] [Google Scholar]
- 44. Dickson V. K., Silvester J. A., Fearnley I. M., Leslie A. G., Walker J. E. (2006) On the structure of the stator of the mitochondrial ATP synthase. EMBO J. 25, 2911–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oster G., Wang H. (2000) Reverse engineering a protein: the mechanochemistry of ATP synthase. Biochim. Biophys. Acta 1458, 482–510 [DOI] [PubMed] [Google Scholar]
- 46. Stewart A. G., Lee L. K., Donohoe M., Chaston J. J., Stock D. (2012) The dynamic stator stalk of rotary ATPases. Nat. Commun. 3, 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cherepanov D. A., Mulkidjanian A. Y., Junge W. (1999) Transient accumulation of elastic energy in proton translocating ATP synthase. FEBS Lett. 449, 1–6 [DOI] [PubMed] [Google Scholar]
- 48. Sorgen P. L., Caviston T. L., Perry R. C., Cain B. D. (1998) Deletions in the second stalk of F1F0-ATP synthase in Escherichia coli. J. Biol. Chem. 273, 27873–27878 [DOI] [PubMed] [Google Scholar]
- 49. Sorgen P. L., Bubb M. R., Cain B. D. (1999) Lengthening the second stalk of F1F0 ATP synthase in Escherichia coli. J. Biol. Chem. 274, 36261–36266 [DOI] [PubMed] [Google Scholar]