

Key Points
Transfused murine RBCs expressing the KEL2 antigen induce polyclonal anti-KEL glycoprotein antibodies capable of fixing complement.
Complement plays a role in incompatible RBC clearance and modulation of the KEL2 antigen on transfused RBCs.
Abstract
Hemolytic transfusion reactions (HTRs) due to incompatible red blood cell (RBC) transfusions are a leading cause of transfusion associated death. Although many transfused incompatible RBCs are cleared, some remain in circulation despite the presence of RBC-specific antibodies, potentially due to “antigen modulation.” With a goal of better understanding incompatible RBC clearance, we generated a murine model with RBC-specific expression of a clinically significant human antigen (KEL2) known to be involved in antigen modulation as well as in HTRs. Wild-type (WT) recipients transfused with transgenic KEL2 RBCs generated anti-KEL glycoprotein alloantibodies, which fixed complement, led to intravascular hemolysis, and resulted in decreased levels of KEL2 antigen detectable on cells remaining in circulation. Antigen modulation did not appear to solely reflect removal of RBCs with higher antigen expression, because cells continued to display antigen modulation in the absence of significant clearance. Recipients genetically lacking complement exhibited lesser degrees of incompatible RBC clearance and antigen modulation in comparison with WT or FcγR knock-out (KO) animals, suggesting a role for complement in RBC clearance. In summary, this HTR model may serve as a platform to test strategies to downmodulate antigen and inhibit incompatible RBC clearance, thus potentially mitigating transfusion dangers.
Introduction
Complement is one of the major effector pathways by which antibodies destroy cellular targets to which they bind.1-4 C3 plays a pivotal role in this process both by serving as a direct opsonin after attaching to surfaces in the form of C3b and also by leading to downstream assembly of the membrane attack complex.5-7 In addition to fixation of C3, antibodies can opsonize cellular targets through ligation of Fcγ receptors (FcγRs).8,9 Although straightforward in concept, the destruction of targets by antigen-bound antibodies is a dynamic process, with multiple regulatory components.10,11 For example, self-tissues have several pathways that actively inhibit complement activation. Moreover, once deposited, C3b is broken down into iC3b, C3d, and C3dg.12 In this way, self-tissues have evolved methods to avoid destruction by binding of self-antibodies.
In addition to regulating the effector function of bound antibodies, the targets of antibody binding can undergo compensatory changes. Antigen modulation is a process by which target cells alter the antigens being recognized by the antibody in question. Antigen modulation occurs in multiple settings with different target tissues and antigens, including nicotinic cholinergic receptors in myasthenia gravis, desmogleins in pemphigus vulgaris, glycoproteins on platelets, and HLA on transplanted tissues.13 The phenomenology of antigen modulation in humans has been repeatedly described in the context of antibodies binding red blood cells (RBCs).14-20 This outcome has been termed depressed antigen, antigen suppression, weakened antigenicity, and antigen loss and has been observed for multiple blood group antigens, including Kell, RhD, RhC, Rhe, Jka, Jkb, Gerbich, LW, AnWJ, and Cromer. Among these, antigen modulation has been described most frequently for antigens in the Kell system. Nevertheless, despite the multiple settings in which antigen modulation occurs and its frequency in RBC biology, relatively little is known about its mechanistic underpinnings.
Although immune-mediated destruction of RBCs is often the result of auto- or alloantibody binding, it is not the inevitable outcome. In some cases, transfusion of a unit of RBCs against which a recipient has an alloantibody (ie, an “incompatible” unit) causes no clinical symptoms, with the transfused RBCs remaining in circulation and the hematocrit increasing appropriately.21,22 In other cases, all of the incompatible RBCs clear rapidly in a hemolytic transfusion reaction (HTR), potentially resulting in coagulopathy, renal failure, and death. In fact, HTRs are a leading cause of transfusion-associated death. The mechanism(s) by which anti-RBC antibodies have such different effects remain poorly understood; however, antigen modulation has been described in some RBCs that escape destruction from allo- or autoantibodies.
To investigate antigen modulation in a reductionist setting, we have recently described a transgenic mouse model with expression of the human Kell glycoprotein (KEL2) specifically on RBCs. The RBCs from KEL2 mice have a normal circulatory lifespan and are recognized by antibodies that bind to the main significant antigens in the Kell system, in particular KEL2, Kpb, and Jsb.23 We have recently reported that KEL2 RBCs transfused into wild-type (WT) recipients induce an anti-KEL glycoprotein response.24 Herein, we report that anti-KEL immunoglobulins induce antigen modulation on incompatible KEL2 RBCs, with C3 playing a role not only in this antigen modulation but also in cellular clearance. FcγRs likewise contribute to RBC clearance in this Kell model but are not required for antigen modulation. In aggregate, these studies provide the first report of complement-associated antigen modulation using an authentic human RBC antigen system, for which HTRs and antigen modulation have also been observed in humans.
Methods
Mice
C57BL/6 mice were purchased from the National Cancer Institute (Frederick, MD). KEL2 transgenic mice expressing the human KEL glycoprotein were generated by our laboratory. Donor KEL2 RBCs have approximately 2000 copies of the KEL2 antigen on their cell surface and have previously been published as “KEL2A.” 23 C3 knock-out (KO) and common γ chain (FcγR) KO (Fcer1g) mice were purchased from Jackson Laboratories (Bar Harbor, ME) and Taconic Farms (Hudson, NY), respectively. All mice used in these studies are on a C57BL/6 background, thus avoiding any potential major histocompatibility complex mismatch. All animals were housed in the Emory University Department of Animal Resources facilities, and all procedures and protocols were approved by the Emory University Institutional Care and Use Committee.
Antibodies and passive immunization
Antisera against the KEL glycoprotein was generated by transfusing KEL2 RBCs into recipients pretreated with an intraperitoneal injection of 100 μg poly (I:C) (Amersham/GE Healthcare, New York, NY) a total of 3 times, separated by 2 weeks. Pooled sera collected 2 weeks after the final transfusion was tested for KEL binding ability by flow crossmatch using KEL2 or control C57BL/6 RBCs as targets, using APC-conjugated goat anti-mouse IgG (BD Biosciences, San Jose, CA) or horseradish peroxidase–conjugated goat anti-mouse IgM or IgG subtypes (Bethyl Laboratories, Montgomery, TX), plus anti–horseradish peroxidase Cy5.5 (Jackson Immunoresearch Laboratories, West Grove, PA). Because the entire human KEL glycoprotein is present on donor RBCs but absent on recipient RBCs, the antibody response is “anti-KEL glycoprotein,” referred to herein as anti-KEL; however, this is not to be confused with antibodies specific for human polymorphisms (eg, anti-KEL1 or anti-KEL2). In passive immunization experiments, recipient mice were given 25 μL anti-KEL antiseruma intravenously 2 hours prior to transfusion.
Murine blood collection, fluorescent labeling, and transfusion
Donor KEL2 or WT C57BL/6 RBCs were collected into acid citrate dextrose and washed 3 times to remove residual citrate. Prior to transfusion, RBCs were labeled with chloromethylbenzamido 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (CM-DiI) or 3,3′-dihexadecyloxacarbocyanine perchlorate (DiO) according to the manufacturer’s instructions (Molecular Probes, Eugene, OR) and as previously described.25 After labeling, cells were washed at least 3 times to remove any unbound dye. Experimental and control RBCs were mixed at a 1:1 ratio, and recipient mice were transfused via lateral tail vein with 50-75 μL (equivalent of 1 human unit) of each type of blood. Survival of the transfused RBCs was determined by comparing the ratio of circulating KEL2 RBCs to control RBCs in recipients at select time points posttransfusion; in some experiments, posttransfusion blood smears were Wright-Giemsa stained and analyzed.
Cytokine and urine analysis
Serum was evaluated posttransfusion for interleukin-6, keratinocyte-derived chemokine, monocyte chemoattractant protein–1, and macrophage inflammatory protein–1β, using a Cytometric Bead Array Mouse Flex Kit (BD Biosciences); heme was measured by cyanomethemoglobin assay. Urine was evaluated posttransfusion, with absorbance measured at optical density (OD) 414 nm.
Flow cytometry
Transfused RBCs were analyzed for the presence of bound anti-KEL antibody by performing a direct antiglobulin test (DAT), using Alexafluor 488–conjugated goat anti-mouse IgG (Jackson Immunoresearch, West Grove, PA) or goat anti-mouse immunoglobulin conjugated to allophycocyanin (BD Biosciences PharMingen, La Jolla, CA). Antigen levels were determined by staining transfused RBCs with anti-KEL antisera, followed by the same secondary antibody. Transfused RBCs were analyzed for binding of all forms of C3 or active forms of C3 using biotinylated rat anti-mouse complement component C3 (clone RmC11H9) or mouse anti-human/mouse C3/C3b/iC3b (clone 10C7) (Cedarlane, ON, Canada), respectively, followed by Streptavidin conjugated to allophycocyanin secondary (BD Biosciences PharMingen). All antibodies were used at a 1:100 dilution, and samples were analyzed on a 4-color BD FACSCalibur.
Statistical analysis
All statistical analyses were performed using Graphpad Prism software (San Diego, CA). A Student t test was used to determine significant differences between 2 groups, and a 1-way analysis of variance with a Tukey post test was used to determine significance between three or more groups. Error bars represent 1 standard deviation, and significance was determined by a P value of ≤.05.
Results
Transfusion of KEL2 RBCs induces anti-KEL glycoprotein alloantibodies
To determine baseline immune responses to KEL2 RBCs, we transfused WT C57BL/6 (B6) mice with the volume-adjusted equivalent of 1 human unit of KEL2 RBCs. Immune responses were tracked serially posttransfusion by flow cytometry, using KEL2 or B6 RBCs as targets (Figure 1A). B6 recipients of KEL2 RBCs generated anti-KEL glycoprotein IgG, which was detectable in the serum approximately 5-7 days posttransfusion, and increased over time (Figure 1B). This was not a nonspecific antibody, because it bound to KEL2 but not B6 RBCs; furthermore, no anti-KEL was detected in syngeneic recipients following KEL2 RBC transfusion.
Figure 1.
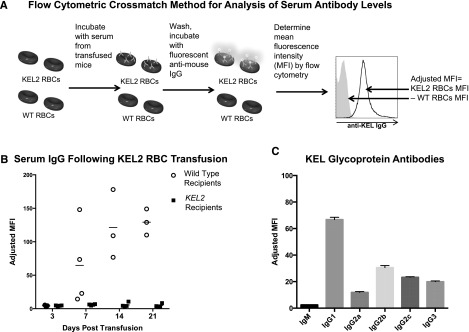
Anti-KEL glycoprotein antibodies are generated in response to transfused KEL2 RBCs. WT and KEL2 recipient mice were transfused with KEL2 RBCs, and anti-KEL glycoprotein levels were evaluated by flow cytometry posttransfusion. (A) Flow cytometric crossmatch method for analysis of serum antibody levels. MFI, mean fluorescence intensity. (B) Serum IgG levels were determined by flow cytometric crossmatch in WT (open circles) and KEL2 (solid squares) recipients at specific time points after transfusion. (C) Composition of antisera passively infused in subsequent experiments. These data are representative of 3 independent experiments, with 3-5 animals/group/experiment; error bars indicate standard deviation.
Although nearly 100% of transfused B6 mice generated anti-KEL glycoprotein antibodies following a single transfusion of KEL2 RBCs, the antibody levels varied between recipients. To study the mechanisms of RBC clearance in animals with uniform levels and composition of anti-KEL, we therefore switched to a passive immunization model. To generate antisera for this model, we transfused B6 mice 3 times with KEL2 RBCs, and antisera was pooled 2 weeks after the final transfusion. The IgM and IgG subtype composition of these antisera was determined by flow cytometric crossmatch (Figure 1C). These antisera were then injected IV into recipient mice to allow passive immunization with a defined titer and composition of anti-KEL.
Passive immunization of polyclonal anti-KEL results in incompatible RBC clearance
Clearance of the KEL2 RBCs in animals passively immunized with polyclonal anti-KEL was tracked by evaluating the ratio of DiI-positive KEL2 RBCs to DiO-positive B6 RBCs (Figure 2A shows gating strategy). By analyzing KEL2 RBC survival as a function of B6 RBC survival, we were able to control for factors such as injection volume, sera collection over time, and non-antigen-specific clearance due to normal cell turnover or damage during collection or labeling. A titration of anti-KEL showed essentially no clearance of KEL2 RBCs at very low doses, with maximal clearance noted using 25 µL of antisera (Figure 2B). No additional clearance was observed with higher amounts of antisera, and no KEL2 RBC clearance was observed in the saline control arms (Figure 2C). All subsequent studies were performed using a 25-μL dose of anti-KEL, with analyses limited to the first 24-48 hours posttransfusion to avoid the confounder of the recipients’ own anti-KEL responses.
Figure 2.
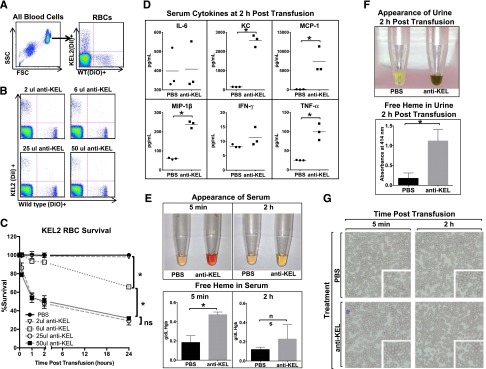
Anti-KEL antibodies induce clearance of KEL2 RBCs. Naïve WT recipients were passively immunized with phosphate-buffered saline (PBS) or titrated amounts of anti-KEL antisera and subsequently transfused with DiI-labeled KEL2 and DiO-labeled WT RBCs. (A) Gating strategy of DiI-positive KEL2 or DiO-positive B6 RBCs. (B-C) Clearance was determined by calculating the ratio of DiI-labeled KEL2 to DiO-labeled WT RBCs after passive antibody transfer. (D) Serum cytokines 2 hours posttransfusion. (E) Heme in serum and (F) heme in urine posttransfusion. (G) Wright-Giemsa–stained blood smears. These data are representative of 2 independent experiments, with 3-5 animals/group/experiment; error bars indicate standard deviation. *Statistical significance, P < .05.
Following incompatible KEL2 RBC transfusion, recipient serum was evaluated for cytokines, serum and urine were evaluated for free heme, and blood smears were generated. Elevations in keratinocyte-derived chemokine, monocyte chemoattractant protein –1, macrophage inflammatory protein –1β, and TNF-α were observed 2 hours after the transfusion of KEL2 RBCs in immunized but not nonimmunized recipients (Figure 2D). Additionally, free heme was noted in the serum visually as well as by cyanomethemoglobin assay (Figure 2E), and heme was noted in the urine visually as well as by OD414 absorbance (Figure 2F). Analysis of the Wright-Giemsa–stained blood smears under a brightfield microscope showed no obvious increase in spherocytes after incompatible transfusion (Figure 2G), though our detection abilities were limited given transfused blood volumes. Taken together, these data suggest that intravascular hemolysis is playing a significant role in the early clearance of incompatible KEL2 RBCs.
C3 binds to transfused KEL2 RBCs in immunized recipients and is rapidly inactivated
C3 deposition is a key component both in antibody-induced opsonization (through C3 receptors) and through generation of downstream complement components that lead to assembly of the membrane attack complex. To test the extent of C3 deposition in response to anti-KEL binding of KEL2 RBCs, we modified the DAT approach above by using anti-C3. Utilizing an antibody that recognizes all forms of C3, we observed substantial reactivity with anti-C3 on KEL2 but not B6 RBCs after an incompatible transfusion (Figure 3A; representative histograms are provided below graph). C3 deposition on KEL2 RBCs was detected as early as 10 minutes posttransfusion and then progressively diminished over time. This bound C3 was not an artifact of the passive immunization model, because C3 was also observed during primary active immunization (data not shown). To monitor C3b degradation, we performed DATs with an antibody that recognizes C3b and iC3b but not C3g or C3dg. C3/C3b/iC3b was detectable at early time points; however, unlike total C3 staining that was detected out to 24 hours posttransfusion, C3/C3b/iC3b staining became essentially undetectable by 2 hours (Figure 3B).
Figure 3.

C3 is rapidly inactivated after binding to transfused incompatible KEL RBCs. (A) Total C3 bound to circulating KEL2 RBCs in animals passively transferred with polyclonal anti-KEL (black bars, solid histograms) or saline (open bars, shaded histograms); representative plots are shown. (B) Active forms of C3 (C3/C3b/iC3b) bound to circulating KEL2 RBCs in animals passively transferred with polyclonal anti-KEL (black bars, solid histograms) or saline (open bars, shaded histograms). These data are representative of 3 independent experiments, with 3-5 animals/group/experiment. *P < .05.
Both C3 and FcγRs participate in clearance of KEL2 RBCs by anti-KEL
To assess the functional role that C3 plays in clearance of KEL2 RBCs by anti-KEL, we used recipients with a targeted deletion of the C3 gene (C3 KO). C3 KO recipients consistently had lower levels of KEL2 RBC clearance than did WT B6 mice (Figure 4A). However, clearance of RBCs was not eliminated in the C3 KO mice. To assess the role of FcγRs in KEL2 RBC clearance, we performed analogous studies in mice with a targeted deletion of the common γ chain for FcγRs (γ-chain KO mice). The γ-chain KO mice lack functional FcγRI, FcγRIII, and FcγRIV. Clearance of KEL2 RBCs by anti-KEL was also decreased in γ-chain KO mice in comparison with WT B6 (Figure 4A). Our interpretation of these data are that C3, FcγRI, FcγRIII, and FcγRIV are involved but not solely responsible for clearance of incompatible KEL2 RBCs.
Figure 4.

Clearance of transfused KEL2 RBCs occurs in part through FcγR-mediated or C3-mediated pathways. Naïve WT, C3KO, or FcγR KO recipients were passively immunized with PBS or 25 μL of anti-KEL antisera and subsequently transfused with DiI-labeled KEL2 and DiO-labeled WT RBCs. (A) Clearance curves of DiI-positive KEL2 RBCs remaining in circulation posttransfusion in WT or C3KO recipients passively transferred with polyclonal anti-KEL (dashed line) or saline (solid line). (B) Clearance curves of DiI-positive KEL2 RBCs remaining in circulation posttransfusion in WT or FcγR KO animals passively transferred with polyclonal anti-KEL (dashed line) or saline (solid line). These data are representative of 4 (panel A) or 5 (panel B) independent experiments, with 3-5 animals/group/experiment. *P < .05.
Anti-KEL induces antigen modulation of KEL2 in surviving RBCs by a mechanism involving C3
To test the extent to which IgG remained bound to KEL2 RBCs that survived incompatible transfusion, we performed DAT analysis using an anti-IgG reagent. Anti-IgG was readily detected after gating on KEL2 RBCs but not on B6 RBCs (Figure 5A shows representative histograms). This signal was specific for anti-KEL, because recipients transfused with saline instead of polyclonal anti-sera had essentially negative DATs, and B6 DiO–labeled RBCs also had essentially negative DATs. The intensity of anti-IgG staining progressively decreased over 24 hours in WT B6 mice, with a similar decline observed in γ-chain KO mice. In contrast, a lesser decrease in detectable IgG with slower kinetics was observed in C3 KO mice. (combined data in Figure 5B).
Figure 5.

Levels of detectable anti-KEL IgG bound to transfused KEL2 RBCs decreases in immunized WT and FcγR KO animals to 24 hours posttransfusion. (A) Detectable IgG bound to transfused KEL2 RBCs in WT, FcγR KO, or C3 KO animals passively transferred with polyclonal anti-KEL (solid line) or saline (shaded histogram); representative plots are shown; h, hour. (B) Compilation graphs of adjusted MFI of detectable IgG bound to KEL2 RBCs in WT, FcγR KO, or C3KO animals transferred with polyclonal anti-KEL, with background signal from animals transferred with saline subtracted out. These data are representative of 5 independent experiments, with 3-5 animals/group/experiment.
To test the hypothesis that antigen modulation may be induced by incompatible transfusion, we analyzed levels of KEL glycoprotein on circulating KEL2 RBCs by staining with the same polyclonal anti-KEL used for passive immunization. Similar to what was observed with respect to bound anti-KEL IgG in the DATs, the level of detectable KEL glycoprotein also decreased over time in both WT B6 as well as γ-chain KO recipients (Figure 6A shows representative histograms; combined data are shown in graph form in Figure 6B). In contrast to what was observed in WT or γ-chain KO mice animals, C3 KO recipients had fairly stable levels of KEL2 antigen detected out to 2 hours posttransfusion. However, a decrease in detectable KEL2 antigen was observed in C3 KO mice, albeit to a lesser degree and with slower kinetics than in WT animals. Taken together, these data suggest that antigen modulation occurs on KEL2 RBCs that survive incompatible transfusion and that C3 is likely involved in this process.
Figure 6.
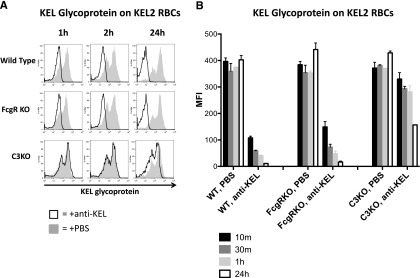
Amount of detectable KEL glycoprotein on RBCs transfused into immunized WT and FcγR KO animals decreases to 24 hours posttransfusion. (A) Detectable KEL glycoprotein on KEL2 RBCs transfused into in WT, FcγR KO, or C3 KO animals passively transferred with polyclonal anti-KEL (solid line) or saline (shaded histogram); representative plots are shown. (B) Compilation graphs of the MFI of detectable KEL glycoprotein on KEL2 RBCs transfused into WT, FcγR KO, or C3 KO animals, passively immunized with saline or polyclonal anti-KEL. These data are representative of 5 independent experiments, with 3-5 animals/group.
Selective destruction of RBCs with the highest level of KEL glycoprotein may occur at early but not later posttransfusion time points
A simple explanation for the observed changes in KEL2 antigen level following incompatible transfusion is that the cells with the highest levels of KEL2 antigen are the most susceptible to complement-mediated lysis. To test this hypothesis, we compared the percentage of incompatible KEL2 RBCs that cleared over discrete time intervals posttransfusion with the percentage of cells experiencing “antigen modulation” over the same period of time. In a representative experiment, 41.5% of KEL2 RBC transfused into a WT mouse in the presence of passively administered anti-KEL cleared in the initial 10 minutes after transfusion, 12.8% cleared between 10 minutes and 1 hour after transfusion, and 17% cleared between 1 and 24 hours after transfusion (Figure 7A). In contrast, there was a 54.8% change in KEL2 antigen detected at 10 minutes in comparison with pretransfusion, a 48.5% change in KEL2 antigen detected at 1 hour in comparison with 10 minutes, and a 65.4% change in KEL2 antigen detected at 24 hours in comparison with 1 hour posttransfusion (Figure 7B). Juxtaposition of graphs comparing clearance of circulating cells with a change in KEL2 antigen detectability (Figure 7C) suggests that although “antigen modulation” may simply reflect removal of the KEL2 RBCs with the highest level of expression, the percentage of cells undergoing clearance at the later time intervals is significantly outweighed by the percentage of cells experiencing antigen modulation, strongly suggesting that selective destruction of the highest-expressing KEL2 RBCs does not solely account for the antigen modulation observed at these later time points posttransfusion.
Figure 7.

Decrease in detectable KEL glycoprotein beyond 10 minutes cannot solely be attributed to clearance of KEL2 RBCs with the highest antigen level. (A) Following passive immunization and transfusion as previously described, we calculated clearance rates of DiI-labeled KEL2 RBCs between time points. (B) Changes in detectable KEL glycoprotein levels between time points was calculated by creating a gate that includes the cells from a particular time point and overlaying that gate onto the antigen level of the previous time point to determine the percentage change. (C) Representative bar graphs comparing the data from panels A and B. These data are representative of 5 independent experiments, with 3-5 animals/group/experiment. Error bars represent standard deviation. *Statistical significance, P < .01.
Discussion
These studies describe a murine model in which polyclonal anti-KEL immunoglobulins are generated through transfusion24 and are used to examine clearance following passive immunization. The RBC biology of incompatible transfusion in this model is multifaceted, with rapid intravascular clearance of a subset of incompatible RBCs, yet persistent circulation and apparent antigen modulation of other circulating transgenic RBCs. Similar findings have also been described in humans, with hemolysis of a subset of Kell RBCs yet antigen modulation of surviving Kell RBCs.14-20 Thus, the RBC clearance, RBC survival, and antigen modulation observed in our murine studies parallel what is known to occur in humans.
The current model is the first described in which C3 is involved not only in antibody-induced incompatible RBC clearance but also in apparent antigen modulation. C3 KO recipients not only have blunted RBC clearance rates but also have lesser degrees of antigen modulation than do WT recipients. It has been shown that C3 can be fixed after antibody binding to an extent that the bound C3 obfuscates binding of the detection immunoglobulin, and the most simplistic explanation of our findings is that C3 is merely masking the detectable antigen. However, a careful analysis of the kinetics seems to rule out this possibility. In particular, the C3-specific DAT detects peak C3 fixation by 10 minutes posttransfusion, yet antigen modulation is ongoing during a posttransfusion interval (2 hours to 24 hours) in which all detectable forms of C3 are decreasing. Thus, engagement of one of several distinct complement receptor pathways may be important in complement-mediated antigen modulation. Ongoing studies are exploring these possibilities.
It is theoretically possible that antigen modulation following incompatible transfusion simply reflects selective removal of antigen high cells within the transgenic antigen-positive population. Consistent with this, the percentage of cells undergoing clearance within the first 10 minutes posttransfusion parallel the percentage experiencing antigen modulation. However, close analysis of antigen modulation and clearance over subsequent time intervals demonstrates that a much higher percentage of cells experience antigen modulation than undergo clearance, strongly suggesting, at least for these later time intervals, that antigen modulation does not solely reflect preferential clearance of cells with higher levels of antigen. Furthermore, the percentage of cells experiencing antigen modulation is significantly attenuated in C3 KO recipients, despite clearance of over half the cells by 24 hours posttransfusion. In addition, antigen modulation in this incompatible RBC setting does not result in the presence of detectable spherocytes by blood smear or to obvious changes in RBC size by flow cytometry. Taken together, these data support a process of antigen modulation involving complement, and a process that is not solely an artifact of complement lysis-dependent selection of clearance of RBCs with the highest levels of KEL2 expression.
Analogous to the initiation of distinct effector pathways following antibody engagement, the results of the present study suggest that similarly unique pathways likewise evolved to regulate antibody–antigen interactions. For example, recent studies have demonstrated that antigen modulation also occurs after antibody engagement of membrane-bound hen egg lysozyme on transgenic RBCs following transfusion.25,26 However, in contrast to the present findings, antibody-induced modulation of membrane-bound hen egg lysozyme not only occurs in the absence of complement but also requires Fc γ receptors to be initiated. Although Fc receptors appear to play some role in antigen modulation following anti-KEL2 antibody engagement, C3 appears to play a primary role in antigen modulation in the present model, providing a unique, complement-dependent mechanism whereby cells experience significant alterations in antigen following antibody engagement. It remains possible that the primary antigen-modulation pathway engaged after antibody binding in various settings reflects the principle effector pathway used for clearance, because complement also appears to play a more significant role in anti-KEL2-mediated clearance. Regardless, these results suggest that different antibody effector mechanisms likely possess distinct regulatory pathways that may ultimately result in a similar outcome of antigen modulation following antibody engagement.
Although the vast majority of antibody contained in the polyclonal preparation used herein for passive transfer was IgG, a small amount of IgM was also present. Ongoing experiments are evaluating the ability of different class-switched monoclonal antibodies to bind to KEL2 RBCs, fix C3, and lead to clearance. It must also be stated that the polyclonal anti-KEL preparation used in these studies is against the entire KEL glycoprotein and not merely against the KEL1 or KEL2 epitopes, and it is known that antibody clustering impacts C3 binding efficiency. Thus, although this is a model of HTRs, the antigen/antibody characteristics and interactions are not exactly like those observed in the human setting involving anti-KEL1 or anti-KEL2 alloantibodies.21,27 Efforts are ongoing to generate transgenic founder animals with different levels of KEL expression, which will serve to further investigate the importance of antibody clustering in C3 binding and incompatible RBC clearance.
In addition to its destructive role, C3 has also been reported to be protective toward cells.28,29 The cells that remain in circulation beyond 10 minutes in our model have significant levels of bound “inactive” complement, staining positive for C3 but not the active C3, C3b, or iC3b. Thus one could consider a scenario in which active forms of complement play a destructive role with regard to incompatible KEL2 RBC clearance and yet inactive forms play a protective role. Ongoing studies are investigating whether complement binding or other methods of in vitro antigen modulation may allow for safer transfusion of RBCs in certain situations involving highly alloimmunized recipients.
Mechanistic analysis of clearance pathways in the current system results in a complex landscape with multiple participants. Deletion of the γ-chain (which results in lack of functional FcγRI, FcγRIII, or FcγRIV) or deletion of C3 decreases but does not eliminate clearance. This suggests that both FcγRs and C3 are involved in RBC clearance, yet other pathways potentially exist. Because the γ-chain KO mice have intact FcγRIIb signaling, it also remains possible that FcγRIIb is responsible for the remaining clearance in γ-chain KO mice; however, FcγRIIb has generally been observed to function as an inhibitory pathway and is not widely known to participate in active clearance.30,31 In addition, we have recently reported that some antibody-induced RBC clearance pathways function through mechanisms involving neither C3 nor Fcγ Rs.32 Thus, the residual clearance in this system may represent such alternative clearance biology.
The ability to prevent C3-mediated destruction of both allogeneic and self-RBCs may be clinically beneficial in certain situations. For example, at times it is impossible to locate compatible RBCs for transfusion in highly alloimmunized individuals, and the ability to “mask” incompatible antigens, either through inactive complement or other mechanisms, may allow for safer transfusion. The use of complement receptor inhibitors has in fact been proposed in such situations, though targeted therapy of the RBCs themselves may be an even more desirable strategy.33,34 Recently, the use of C1 esterase inhibitor concentrates has been described in a refractory case of C3-mediated autoimmune hemolytic anemia, with an increase in hemoglobin and a decrease in lactate dehydrogenase levels, suggesting a therapeutic benefit.35
In summary, our murine model with RBC-specific expression of a clinically significant human RBC antigen is the first to be described in which C3 plays a role in clearance as well as probable antigen modulation. This model begins to add to the understanding of antigen modulation, a phenomenon now reported multiple times in both humans and animals, in many different settings. In addition to providing evidence that C3 is likely playing a role in RBC antigen modulation, our model may also serve as a platform for the future development of strategies to minimize the dangers of RBC incompatibility through intentional antigen modulation or through alteration of clearance pathways involving C3.
Acknowledgments
The authors would like to thank Kate Henry, Ashley Bennett, Krystalyn Hudson, Linda Kapp, Justine Liepkalns, and Seema Patel for their contributions to discussions regarding this project.
The authors would also like to thank Immucor, whose funding was used in part to generate the KEL animals. This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute funding to J.E.H. (HL 092959, HL 115696) and to J.C.Z. (HL 086773).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: All authors designed the research and analyzed the data; K.R.G.-P., S.R.S., N.H.S., C.M.A., H.C.S., and J.E.H. conducted the research; and all authors contributed to the writing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: James C. Zimring, Puget Sound Blood Center Research Institute, 1551 Eastlake Ave East, Seattle, WA 98102; e-mail: jzimring@psbc.org.
References
- 1.Müller-Eberhard HJ. Complement. Annu Rev Biochem. 1969;38:389–414. doi: 10.1146/annurev.bi.38.070169.002133. [DOI] [PubMed] [Google Scholar]
- 2.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344(14):1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 3.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344(15):1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 4.Stowell SR, Winkler AM, Maier CL, et al. Initiation and regulation of complement during hemolytic transfusion reactions. Clin Dev Immunol. 2012;(2012):307093. [DOI] [PMC free article] [PubMed]
- 5.Mollison PL. The role of complement in antibody-mediated red-cell destruction. Br J Haematol. 1970;18(3):249–255. doi: 10.1111/j.1365-2141.1970.tb01440.x. [DOI] [PubMed] [Google Scholar]
- 6.Müller-Eberhard HJ. The membrane attack complex of complement. Annu Rev Immunol. 1986;4:503–528. doi: 10.1146/annurev.iy.04.040186.002443. [DOI] [PubMed] [Google Scholar]
- 7.Lambris JD. The multifunctional role of C3, the third component of complement. Immunol Today. 1988;9(12):387–393. doi: 10.1016/0167-5699(88)91240-6. [DOI] [PubMed] [Google Scholar]
- 8.Meryhew NL, Runquist OA. A kinetic analysis of immune-mediated clearance of erythrocytes. J Immunol. 1981;126(6):2443–2449. [PubMed] [Google Scholar]
- 9.Engelfriet CP. The immune destruction of red cells. Transfus Med. 1992;2(1):1–6. doi: 10.1111/j.1365-3148.1992.tb00128.x. [DOI] [PubMed] [Google Scholar]
- 10.Kim DD, Song WC. Membrane complement regulatory proteins. Clin Immunol. 2006;118(2-3):127–136. doi: 10.1016/j.clim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 11.Fujita T, Inoue T, Ogawa K, Iida K, Tamura N. The mechanism of action of decay-accelerating factor (DAF). DAF inhibits the assembly of C3 convertases by dissociating C2a and Bb. J Exp Med. 1987;166(5):1221–1228. doi: 10.1084/jem.166.5.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison RA, Lachmann PJ. The physiological breakdown of the third component of human complement. Mol Immunol. 1980;17(1):9–20. doi: 10.1016/0161-5890(80)90119-4. [DOI] [PubMed] [Google Scholar]
- 13.Zimring JC, Cadwell CM, Spitalnik SL. Antigen loss from antibody-coated red blood cells. Transfus Med Rev. 2009;23(3):189–204. doi: 10.1016/j.tmrv.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Bosco A, Xenocostas A, Kinney J, Cadwell CM, Zimring JC. An autoanti-Kp b immunoglobulin M that simulates antigen suppression. Transfusion. 2009;49(4):750–756. doi: 10.1111/j.1537-2995.2008.02045.x. [DOI] [PubMed] [Google Scholar]
- 15.Brendel WL, Issitt PD, Moore RE, et al. Temporary reduction of red cell Kell system antigen expression and transient production of anti-Kpb in a surgical patient. Biotest Bull. 1985;2:201–206. [Google Scholar]
- 16.Vengelen-Tyler V, Gonzalez B, Garratty G, et al. Acquired loss of red cell Kell antigens. Br J Haematol. 1987;65(2):231–234. doi: 10.1111/j.1365-2141.1987.tb02270.x. [DOI] [PubMed] [Google Scholar]
- 17.Williamson LM, Poole J, Redman C, et al. Transient loss of proteins carrying Kell and Lutheran red cell antigens during consecutive relapses of autoimmune thrombocytopenia. Br J Haematol. 1994;87(4):805–812. doi: 10.1111/j.1365-2141.1994.tb06741.x. [DOI] [PubMed] [Google Scholar]
- 18.van’t Veer MB, van Wieringen PM, van Leeuwen I, Overbeeke MA, von dem Borne AE, Engelfriet CP. A negative direct antiglobulin test with strong IgG red cell autoantibodies present in the serum of a patient with autoimmune haemolytic anaemia. Br J Haematol. 1981;49(3):383–386. doi: 10.1111/j.1365-2141.1981.tb07240.x. [DOI] [PubMed] [Google Scholar]
- 19.Marsh WL, Oyen R, Alicea E, Linter M, Horton S. Autoimmune hemolytic anemia and the Kell blood groups. Am J Hematol. 1979;7(2):155–162. doi: 10.1002/ajh.2830070208. [DOI] [PubMed] [Google Scholar]
- 20.Seyfried H, Górska B, Maj S, Sylwestrowicz T, Giles CM, Goldsmith KL. Apparent depression of antigens of the Kell blood group system associated with autoimmune acquired haemolytic anaemia. Vox Sang. 1972;23(6):528–536. doi: 10.1111/j.1423-0410.1972.tb03846.x. [DOI] [PubMed] [Google Scholar]
- 21.Reid ME, Lomas-Francis C. The Blood Group Antigen Facts Book. 2nd ed. Amsterdam, Netherlands: Elsevier Academic Press; 2004. [Google Scholar]
- 22.Janatpour KA, Kalmin ND, Jensen HM, Holland PV. Clinical outcomes of ABO-incompatible RBC transfusions. Am J Clin Pathol. 2008;129(2):276–281. doi: 10.1309/VXY1ULAFUY6E6JT3. [DOI] [PubMed] [Google Scholar]
- 23.Smith NH, Henry KL, Cadwell CM, et al. Generation of transgenic mice with antithetical KEL1 and KEL2 human blood group antigens on red blood cells. Transfusion. 2012;52(12):2620–2630. doi: 10.1111/j.1537-2995.2012.03641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stowell SR, Girard-Pierce KR, Smith NH, et al. Transfusion of murine red blood cells expressing the human KEL glycoprotein induces clinically significant alloantibodies [published online ahead of print April 29, 2013]. Transfusion. doi: 10.1111/trf.12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimring JC, Hair GA, Chadwick TE, et al. Nonhemolytic antibody-induced loss of erythrocyte surface antigen. Blood. 2005;106(3):1105–1112. doi: 10.1182/blood-2005-03-1040. [DOI] [PubMed] [Google Scholar]
- 26.Zimring JC, Cadwell CM, Chadwick TE, et al. Nonhemolytic antigen loss from red blood cells requires cooperative binding of multiple antibodies recognizing different epitopes. Blood. 2007;110(6):2201–2208. doi: 10.1182/blood-2007-04-083097. [DOI] [PubMed] [Google Scholar]
- 27.Westhoff CM, Reid ME. Review: the Kell, Duffy, and Kidd blood group systems. Immunohematol. 2004;20(1):37–49. [PubMed] [Google Scholar]
- 28.Moller G. Isoantibody-induced cellular resistance to immune haemolysis in vivo and in vitro. Nature. 1964;202:357–359. doi: 10.1038/202357a0. [DOI] [PubMed] [Google Scholar]
- 29.Logue GL, Rosse WF, Gockerman JP. Measurement of the third component of complement bound to red blood cells in patients with the cold agglutinin syndrome. J Clin Invest. 1973;52(2):493–501. doi: 10.1172/JCI107206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fong DC, Brauweiler A, Minskoff SA, et al. Mutational analysis reveals multiple distinct sites within Fc gamma receptor IIB that function in inhibitory signaling. J Immunol. 2000;165(8):4453–4462. doi: 10.4049/jimmunol.165.8.4453. [DOI] [PubMed] [Google Scholar]
- 31.Brauweiler AM, Cambier JC. Fc gamma RIIB activation leads to inhibition of signalling by independently ligated receptors. Biochem Soc Trans. 2003;31(Pt 1):281–285. doi: 10.1042/bst0310281. [DOI] [PubMed] [Google Scholar]
- 32.Liepkalns JS, Hod EA, Stowell SR, Cadwell CM, Spitalnik SL, Zimring JC. Biphasic clearance of incompatible red blood cells through a novel mechanism requiring neither complement nor Fcγ receptors in a murine model. Transfusion. 2012;52(12):2631–2645. doi: 10.1111/j.1537-2995.2012.03647.x. [DOI] [PubMed] [Google Scholar]
- 33.Yu J, Heck S, Debnath A, Yazdanbakhsh K. Identification of a complement receptor 1 peptide for inhibition of immune hemolysis. Biochem Biophys Res Commun. 2007;353(2):363–368. doi: 10.1016/j.bbrc.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yazdanbakhsh K. Review: complement receptor 1 therapeutics for prevention of immune hemolysis. Immunohematol. 2005;21(3):109–118. [PubMed] [Google Scholar]
- 35.Wouters D, Stephan F, Strengers P, et al. C1-esterase inhibitor concentrate rescues erythrocytes from complement-mediated destruction in autoimmune hemolytic anemia. Blood. 2013;121(7):1242–1244. doi: 10.1182/blood-2012-11-467209. [DOI] [PubMed] [Google Scholar]