

Abstract
Background
Cytokinins (CKs) have significant roles in various aspects of plant growth and development, and they are also involved in plant stress adaptations. The fine-tuning of the controlled CK levels in individual tissues, cells, and organelles is properly maintained by isopentenyl transferases (IPTs) and cytokinin oxidase/dehydrogenases (CKXs). Chinese cabbage is one of the most economically important vegetable crops worldwide. The whole genome sequencing of Brassica rapa enables us to perform the genome-wide identification and functional analysis of the IPT and CKX gene families.
Results
In this study, a total of 13 BrIPT genes and 12 BrCKX genes were identified. The gene structures, conserved domains and phylogenetic relationships were analyzed. The isoelectric point, subcellular localization and glycosylation sites of the proteins were predicted. Segmental duplicates were found in both BrIPT and BrCKX gene families. We also analyzed evolutionary patterns and divergence of the IPT and CKX genes in the Cruciferae family. The transcription levels of BrIPT and BrCKX genes were analyzed to obtain an initial picture of the functions of these genes. Abiotic stress elements related to adverse environmental stimuli were found in the promoter regions of BrIPT and BrCKX genes and they were confirmed to respond to drought and high salinity conditions. The effects of 6-BA and ABA on the expressions of BrIPT and BrCKX genes were also investigated.
Conclusions
The expansion of BrIPT and BrCKX genes after speciation from Arabidopsis thaliana is mainly attributed to segmental duplication events during the whole genome triplication (WGT) and substantial duplicated genes are lost during the long evolutionary history. Genes produced by segmental duplication events have changed their expression patterns or may adopted new functions and thus are obtained. BrIPT and BrCKX genes respond well to drought and high salinity stresses, and their transcripts are affected by exogenous hormones, such as 6-BA and ABA, suggesting their potential roles in abiotic stress conditions and regulatory mechanisms of plant hormone homeostasis. The appropriate modulation of endogenous CKs levels by IPT and CKX genes is a promising approach for developing economically important high-yielding and high-quality stress-tolerant crops in agriculture.
Background
Cytokinins (CKs) are a class of plant hormones that have significant roles in various aspects of plant growth and development, including apical dominance, shoot or root branching, leaf expansion, lateral bud growth, photosynthesis, seed germination, floral transition, and leaf senescence [1]. Moreover, these hormones participate in biomass distribution [2] and the response to environmental stimuli [3-5]. CKs signals are sensed and transduced by the two-component signal (TCS) system; the availability of CKs in suitable concentrations in the right place and at the right time is necessary for their interaction with a specific receptor. The fine-tuning of hormone levels in individual tissues, cells, and organelles must be properly maintained by the biosynthetic and metabolic enzymes [6]. CKs synthesis in plants is catalyzed by a family of isopentenyl transferases (IPTs) via the methylerythritol phosphate (MEP) and mevalonate (MVA) pathways [3,7,8]. Further studies reveal that ATP/ADP IPTs control the biosynthesis of isopentenyladenine (iP)- and trans-zeatin (tZ)-type CKs, whereas tRNA IPTs are responsible for the synthesis of cis-zeatin (cZ)-type CKs [3,7]. The irreversible degradation of CKs and their derivatives is catalyzed by CK oxidase/dehydrogenases (CKXs), which are encoded by a small gene family. The substrate specificities of IPTs have been proven to vary, depending on the origin and species [3]. Similarly, CKX enzymes have their own substrate specificity, with various spatial and temporal expression patterns [9].
The IPT and CKX gene families in various plants have been identified and cloned, including those in Arabidopsis thaliana[10,11], Oryza sativa[12,13], Zea mays[14,15], Glycine max[16], Solanum lycopersicum[17], Triticum aestivum[18], Hordeum vulgare[19], Sorghum bicolor[19], and Populus trichocarpa[20], among others. A putative ATP/GTP binding site (P-loop motif) exists in all of the deduced protein sequences of IPT, whereas CKX protein sequences have FAD- and CK-binding domains. The functions of IPT and CKX genes have likewise been extensively studied. The overexpression of AtIPT4 caused CK-independent shoot formation on calli [21]. The overexpression of ZmIPT2 in Arabidopsis produced transgenic plants with phenotypes indicative of CKs overproduction [14]. Transgenic tobacco with 35S-GmIPT1 similarly had the typical phenotypes of CKs overproduction [22]. The enhanced expression of an IPT gene designated as Sho showed CK-specific effects, including enhanced shooting and reduced apical dominance, as well as delayed senescence and flowering [23]. In Arabidopsis, the ckx3/ckx5 double mutant had higher CKs content, which caused the mutants to have more flower primordia, larger flowers, more siliques, and more seeds; the total seed yield of these mutants increased by 55%, as compared with the wild-type [24]. Ashikari et al. [25] conclusively showed that the reduced expression of Gn1a, a gene for OsCKX2, caused CKs accumulation in rice inflorescence meristems and increased the number of reproductive organs, thereby enhancing the grain yield by 21%. RNAi was more recently used in barley to silence HvCKX1, which increased both the grain number and grain weight [26]. The potential use of CK-related genes in seed size and seed mass determination will become an active field of study in the future. Meanwhile, increasing evidence suggests that CKs are involved in stress response [27-29]. Nishiyama et al. [30] proposed a model for the roles of bioactive CKs and antagonistic ABA under different stresses. All CK-deficient plants with reduced levels of different CKs exhibited strong stress-tolerant phenotypes; the majority of AtIPT and AtCKX genes were repressed by stress and ABA treatments. The IPT and CKX genes in maize and soybean similarly responded to drought and salt conditions [16,31]. Given that water deficit and high-salinity stresses have become limiting factors for crop and vegetable production and quality, these studies may provide new insights into breeding stress-resistant plants.
Brassica rapa is one of the most economically important vegetable crops worldwide. The whole genome sequencing of B. rapa (Chiifu-401-42), by The Brassica rapa Genome Sequencing Project Consortium (2011), enables us to undertake the genome-wide identification and functional analysis of the gene families related to the morphological diversity and agronomic traits of Brassica crops [32]. Furthermore, B. rapa serves as a crucial reference for understanding polyploidy-related crop genome evolution because of its agronomic importance and phylogenetic relationships [33].
Ando et al. [34] isolated cDNA fragments of five putative cytokinin synthase genes from B. rapa (BrIPT1, -3, -5, -7, and −8) and examined their expression levels using Northern blot analysis. O’Keefe et al. [35] identified BrIPT1, -3, and −5 as well as BrCKX1, -2, -3, and −5 in rapid-cycling Brassica; their expression levels during pod and seed development were examined. However, Brassicaceae genomes have undergone three rounds of whole genome duplication (WGD); these genomes are referred to as 1R, 2R, and 3R, which are equivalent to the γ, β, and α duplication events, and Brassica genomes have undergone another whole genome triplication (WGT) after speciation from Arabidopsis thaliana at approximately 17–20 MYA (million years ago) [36-38], leading to significantly increased duplicated gene numbers in B. rapa. A total of 41,174 protein-coding genes were identified in the B. rapa genome, which were roughly 1.5 times as many genes as those found in A. thaliana (27,411 genes in TAIR10) [32]. Accordingly, we hypothesized that the number of BrIPT and BrCKX genes would be greater than that of their Arabidopsis counterparts. The main objectives of our study were to identify all the IPT and CKX genes in B. rapa, to analyze the physiological and biochemical properties of their encoded proteins, to explore their gene expression patterns, to discover the mechanisms of their responses to abiotic stresses and exogenous plant hormones, and to identify several potential genes for breeding to increase plant production, quality, and stress-resistance. Furthermore, we intended to gain genome-level insights into the divergence, variation, and evolution of the BrIPT and BrCKX gene families.
Results
Identification and annotation of the BrIPT and BrCKX genes of B. rapa
The BrIPT and BrCKX genes were identified based on BLAST search results against the BRAD and NCBI databases. A total of 13 BrIPT genes and 12 BrCKX genes with confirmed conserved domains were obtained from the B. rapa genome (Tables 1, 2). The BrIPT and BrCKX genes were named according to their homologous genes in A. thaliana. Each AtIPT and AtCKX gene corresponded with approximately one to three BrIPT and BrCKX genes, except for AtIPT4 and AtIPT6.
Table 1.
IPTgene family in Brassica rapa , along with their molecular details and relevant genomic information
|
Gene namea |
Locusb |
Chrc |
Sub- genomed |
ORF lengthe(bp) |
Deduced polypeptidef |
TargetPg |
Glycosylation Sitesh |
||
|---|---|---|---|---|---|---|---|---|---|
| Length (aa) | MW (kDa) | PI | |||||||
|
BrIPT1-1 |
Bra004037 |
A07 |
MF2 |
1002 |
333 |
37.8 |
8.91 |
C/3 |
0 |
|
BrIPT1-2 |
Bra033933 |
A02 |
MF1 |
1095 |
364 |
41.6 |
8.64 |
C/1 |
0 |
|
BrIPT2 |
Bra034366 |
A04 |
MF1 |
1392 |
463 |
52.4 |
5.89 |
— |
1 |
|
BrIPT3-1 |
Bra040431 |
A04 |
MF1 |
1005 |
334 |
37.3 |
8.11 |
C/4 |
2 |
|
BrIPT3-2 |
Bra007728 |
A09 |
LF |
999 |
332 |
37.6 |
8.4 |
C/5 |
1 |
|
BrIPT5-1 |
Bra002204 |
A10 |
LF |
1005 |
334 |
37.9 |
6.35 |
— |
1 |
|
BrIPT5-2 |
Bra023701 |
A02 |
MF2 |
996 |
331 |
37.6 |
5.75 |
— |
1 |
|
BrIPT7-1 |
Bra014968 |
A07 |
LF |
999 |
332 |
37.1 |
8.68 |
C/5 |
1 |
|
BrIPT7-2 |
Bra028326 |
A01 |
MF1 |
906 |
301 |
33.6 |
8.71 |
C/3 |
1 |
|
BrIPT8-1 |
Bra037537 |
A01 |
MF1 |
975 |
324 |
36.9 |
8.5 |
C/2 |
0 |
|
BrIPT8-2 |
Bra001737 |
A03 |
MF2 |
984 |
327 |
37.0 |
9.26 |
C/4 |
1 |
|
BrIPT9-1 |
Bra006535 |
A03 |
MF1 |
1392 |
463 |
52.2 |
7.51 |
C/5 |
3 |
| BrIPT9-2 | Bra020081 | A02 | MF2 | 1395 | 464 | 52.2 | 7.96 | C/5 | 3 |
aGene Names given to BrIPTs in this work.
bLocus represented by the Brassica rapa genome database.
cChromosomal localization of the BrIPTs and BrCKXs, sca represents scaffold.
dThree sub-genomes of Brassica rapa genome.
eLength of open reading frame in base pairs.
fLength (number of amino acids), molecular weight (kDa), and isoelectric point (pI) of the deduced polypeptide.
gLocalization predicted with TargetP. M, mitochondria; C, chloroplast; S, secretory pathways; “—”, unknown location. (1 to 5), highest to lowest possibility.
hGlycosylation sites are predicted with NetNGly.
Table 2.
CKXgene family in Brassica rapa , along with their molecular details and relevant genomic information
|
Gene namea |
Locusb |
Chrc |
Sub- genomed |
ORF lengthe(bp) |
Deduced polypeptidef |
TargetPg |
Glycosylation Sitesh |
||
|---|---|---|---|---|---|---|---|---|---|
| Length (aa) | MW (kDa) | PI | |||||||
|
BrCKX1-1 |
Bra000229 |
A03 |
MF2 |
1323 |
440 |
49.3 |
8.71 |
— |
3 |
|
BrCKX1-2 |
Bra016928 |
A04 |
MF1 |
1149 |
382 |
42.8 |
7.25 |
S/5 |
4 |
|
BrCKX1-3 |
Bra004626 |
A05 |
LF |
4431 |
1476 |
162.7 |
8.47 |
M/1 |
12 |
|
BrCKX2-1 |
Bra036719 |
A09 |
MF2 |
1509 |
502 |
55.5 |
5.96 |
S/2 |
5 |
|
BrCKX2-2 |
Bra040677 |
sca |
LF |
1518 |
505 |
55.6 |
5.62 |
S/2 |
5 |
|
BrCKX3-1 |
Bra002777 |
A10 |
LF |
1101 |
366 |
50.0 |
5.63 |
C/5 |
3 |
|
BrCKX3-2 |
Bra035640 |
A02 |
MF1 |
1557 |
518 |
58.9 |
5.62 |
S/5 |
3 |
|
BrCKX4 |
Bra024135 |
A03 |
MF1 |
1575 |
524 |
58.0 |
5.71 |
S/1 |
5 |
|
BrCKX5 |
Bra015842 |
A07 |
LF |
1575 |
524 |
58.8 |
5.62 |
— |
2 |
|
BrCKX6 |
Bra007743 |
A09 |
LF |
1350 |
449 |
50.5 |
7.3 |
M/2 |
4 |
|
BrCKX7-1 |
Bra002371 |
A10 |
LF |
1578 |
525 |
57.9 |
4.92 |
— |
5 |
| BrCKX7-2 | Bra020157 | A02 | MF2 | 1554 | 517 | 57.6 | 5.2 | — | 3 |
aGene Names given to BrCKXs in this work.
bLocus represented by the Brassica rapa genome database.
cChromosomal localization of the BrIPTs and BrCKXs, sca represents scaffold.
dThree sub-genomes of Brassica rapa genome.
eLength of open reading frame in base pairs.
fLength (number of amino acids), molecular weight (kDa), and isoelectric point (pI) of the deduced polypeptide.
gLocalization predicted with TargetP. M, mitochondria; C, chloroplast; S, secretory pathways; “—”, unknown location. (1 to 5), highest to lowest possibility.
hGlycosylation sites are predicted with NetNGly.
The BrIPT genes were distributed on 7 out of 10 chromosomes (except for A05, A06, and A08), belonging to the three sub-genomes (LF, MF1, and MF2). The ORF lengths of the BrIPT genes ranged from 906 bp to 1395 bp, which encoded polypeptides of 301 aa to 464 aa with predicted molecular weights ranging from 33.6 kD to 52.4 kD. The theoretical pI ranged from 5.75 to 9.26. The BrIPT genes were all predicted to be localized in the chloroplasts. Similarly, the BrCKX genes were also distributed on 7 out of 10 chromosomes (except for A01, A06, and A08). The ORF lengths of the BrCKX genes were longer than those of the BrIPT genes; these ORFs ranged from 1101 bp to 4431 bp and encoded polypeptides of 366 aa to 1476 aa with predicted molecular weights ranging from 42.8 kD to 162.7 kD. The theoretical pI ranged from 4.92 to 8.71. The predicted subcellular localization of the BrCKX genes varied and included the chloroplast, mitochondria, and secretory pathways. Protein glycosylation contributes to regulation of enzymatic activity, translocation, and protein stability; thereby, adding an additional and important level of complexity to CKX regulation [39]. Similar to the ZmCKX genes, the BrCKX genes possessed 2 to 12 predicted glycosylation sites.
Gene structure and conserved domain analysis of BrIPT and BrCKX genes
To analyze the structural characteristics and conserved regions of the BrIPT and BrCKX gene families, their gene structures with exons and introns were mapped, their conserved regions and motifs were examined, and their putative protein sequences were aligned. Similar to the IPT gene families in other plants, BrIPT genes could be grouped into two types of IPT genes: plant adenylate IPT- or ADP/ATP-dependent genes (BrIPT1, -3, -5, -7 and −8) and plant tRNA IPT- or tRNA-dependent genes (BrIPT2 and BrIPT9). Plant tRNA IPT genes could be further divided into plant tRNA IPT genes of eukaryotic (BrIPT2) and prokaryotic (BrIPT9) origin. Seven plant adenylate BrIPT genes were notably composed of a single exon without introns and three plant adenylate BrIPT genes only had one intron. By contrast, the longer plant tRNA BrIPT genes had at least ten exons and nine introns, which was consistent with the more complicated tRNA IPT gene structures in other plants (Figure 1A). The multiple alignment of AtIPT protein sequences showed that AtIPT2 contained two inserted regions of approximately 20–80 amino acids in length; while the carboxyl-terminal region of the said protein sequences had an extra 40 amino acids. A similar structure was observed in BrIPT2. AtIPT9 and BrIPT9 had five shorter discontinuous inserted regions of approximately 8–14 amino acids instead of the two larger regions in AtIPT2 and BrIPT2. However, the carboxyl-terminal region with an extra 40 amino acids was conserved in both BrIPT2 and BrIPT9 (Additional file 1). Two types of IPT gene structures are closely related and had also unique conserved motifs. Seven conserved motifs were found for the BrIPT protein sequences (Figure 1A, Additional file 2). All BrIPT protein sequences contained one or two conserved regions that belonged to the P-loop NTPase superfamily (cl09099). Furthermore, three plant tRNA BrIPT protein sequences contained either PLNO2748 or PLNO2840 multi-domains, which were unique to tRNA IPTs. The multiple alignment of BrIPT protein sequences with AtIPT protein sequences likewise revealed two corresponding conserved regions, which were designated as Region a and Region b. Region a was found in all the BrIPT protein sequences; it was identified as a putative ATP/GTP-binding site (P-loop) motif, with a core sequence of TGxGKS (Motif 1). Region b was only present in three plant tRNA BrIPT protein sequences; this region was classified as a tRNA binding site. The two types of plant tRNA BrIPT proteins had different consensus sequences, which were denoted by a red box in Additional file 1.
Figure 1.

Gene structure and motif analysis of the BrIPT (A) and BrCKX (B) genes. Exons and introns were organized with green boxes and black lines, respectively. Motifs locations were shown with combined block diagrams.
The gene structures of the BrCKX genes were more diverse and contained several exons and introns (Figure 1B). A FAD-binding domain and a CK-binding domain were generally indispensable in a functional CKX gene; these domains belonged to the FAD-binding (cl14794) and CK-binding (cl07775) superfamilies, respectively. The unique BrCKX1-3 gene had a longer sequence length and more complex conserved domains (Figure 2). Based on the alignment and conserved domains analysis, the multi-domain BrCKX1-3 was composed of two regions, Region 1 and Region 2. Region 1 contained the typical conserved domains of the CKX gene family, namely, the FAD- and CK-binding domains. By contrast, Region 2 had the characteristics of a TPR protein and contained a TPR repeat and a Dnaj domain. However, the AtCKX1 (AT2G41510) and AtTPR15 (AT2G41520) genes in Arabidopsis are two completely different functional genes, even though these genes are located next to each other. Based on the gene structures analysis, as compared with that of A. thaliana, we proposed that BrCKX1-3 was formed by the fusion of the CKX and TPR genes. However, the actual mechanism of BrCKX1-3 formation is still unknown. Ten conserved motifs were found in the BrCKX genes (Figure 1B, Additional file 3). The sequence alignment showed that the FAD-binding domain was well conserved in the N-terminal halves of all known CKX sequences (Additional file 4). Moreover, the GHS motif (Motif 5) was perfectly conserved in this family protein sequences. We also identified several additional conserved motifs, including the VPHPWLNL motif (Motif 2) at position 450 and a PGQxIF signature (Motif 3) at the C-terminal end of the CKX protein sequences. These motifs appeared to be specific to CKX enzymes.
Figure 2.
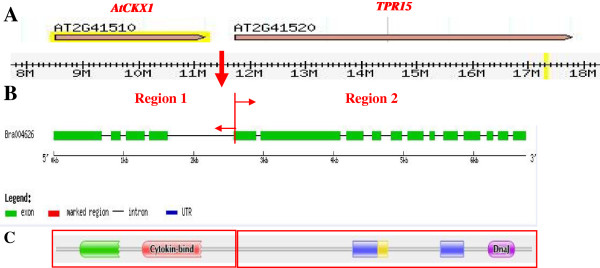
Gene structure and conserved domain analysis of BrCKX1-3. Physical positions of the AtCKX1 and AtTPR15 on the chromosome of A. thaliana (A). Gene structure of BrCKX1-3 (B). Conserved domains of BrCKX1-3 (C).
Chromosomal mapping and duplications of BrIPT and BrCKX genes
We mapped BrIPT and BrCKX genes on chromosomes (Figure 3A, 3B). BrCKX2-2 was not mapped due to its location on scaffold000232. Even genes with high similarity were not located on the same chromosome. Segmental and tandem duplications are important sources of gene duplication. Gu et al. [15] proposed that the CKX gene expansion occurred mainly through segmental duplications in maize, rice, and poplar. In this study, no tandem duplicated gene pairs were found. However, segmental duplicates existed in both BrIPT and BrCKX gene families (Additional file 5 and Additional file 6). Five pairs of segmental duplicates were found in 13 BrIPT genes: BrIPT1-1 (A07) and BrIPT1-2 (A02); BrIPT5-1 (A10) and BrIPT5-2 (A02); BrIPT7-1 (A07) and BrIPT7-2 (A01); BrIPT8-1 (A01) and BrIPT8-2 (A03); and BrIPT9-1 (A03) and BrIPT9-2 (A02). Three pairs of segmental duplicates were found in 12 BrCKX genes: BrCKX1-1 (A03), BrCKX1-2 (A04) and BrCKX1-3 (A04); BrCKX3-1 (A10) and BrCKX3-2 (A02); and BrCKX7-1 (A10) and BrCKX7-2 (A02). Each pair of segmental duplicates was distributed on different chromosomes. BrIPT3-1 and BrIPT3-2 were notably not considered as a pair of segmental duplicates, even though they shared high sequence similarity.
Figure 3.

Chromosomal mapping of BrIPT (A) and BrCKX (B) genes. The arrows next to gene names show the direction of transcription. BrCKX2-2 is not mapped for its location on scaffold000232. Segmental duplicates are connected by inter-chromosomal lines.
Phylogenetic relationships of the BrIPT and BrCKX gene families
To identify subgroups and reveal the evolutionary relationships of the two gene families, the phylogenetic analyses of the BrIPT and AtIPT genes, as well as that of the BrCKX and AtCKX genes, were performed with the putative protein sequences using MEGA (version 5.0) software. As shown in Figure 4A, the BrIPTs and AtIPTs were clustered into four subgroups: Group I (BrIPT1-1, -1-2, -8-1 and −8-2), Group II (BrIPT2), Group III (BrIPT3-1, -3-2, -5-1, -5-2, -7-1 and −7-2), and Group IV (BrIPT9-1 and BrIPT9-2). Group I and Group III included the majority of the BrIPT genes, which belonged to the plant adenylate IPT genes. The tRNA-dependent BrIPT genes and ADP/ATP-dependent BrIPT genes were highly diverse; these genes could be divided into two different subgroups. BrIPT2 in Group II, the AtIPT2 homolog, is a plant tRNA IPT gene of eukaryotic origin. BrIPT9-1, BrIPT9-2, and AtIPT9 all shared the properties of plant tRNA IPT genes with prokaryotic origins, and were classified into Group IV. Similarly, the BrCKX and AtCKX genes were also clustered into four subgroups: Group I (BrCKX1-1, -1-2, -1-3 and −6), Group II (BrCKX2-1, -2-2, -3-1, -3-2 and −4), Group III (BrCKX5), and Group IV (BrCKX7-1 and BrCKX7-2) (Figure 4B). Further phylogenetic reconstructions using IPT and CKX protein sequences from various species, including dicotyledons, monocotyledons, moss, algae, and bacteria, were performed to confirm the IPT and CKX genes subgroups, as well as to investigate the evolution of BrIPT and BrCKX genes. The corresponding plant IPT genes were divided into four groups: Groups I, II, III, and IV (Figure 5A). As shown in the figure, genes in dicotyledons and monocotyledons were randomly dispersed among the four groups. This phenomenon suggested that IPT genes originated from both dicotyledons and monocotyledons or were not lost after the divergence of the two plant groups. Furthermore, only the six IPT genes in the bryophyte P. patens (PpIPT) were found in Group IV, which also included the plant tRNA IPT genes of prokaryotic origin, implying that PpIPT genes possiblely originated from the tRNA IPT genes. Plant CKX genes were classified into five groups, with Group V as an additional group (Figure 5B). Group V contained only the six PpCKX genes.
Figure 4.

Phylogenetic relationships of BrIPTs and BrCKXs, together with their Arabidopsis conterparts, respectively. Trees were constructed using NJ method by the MEGA (version 5.0) program. Both BrIPTs(A) and BrCKXs(B) were divided into four groups indicated by differently colored dots.
Figure 5.

Phylogenetic relationships of IPT and CKX gene families from several species. Trees were constructed using NJ method by the MEGA (version 5.0) program. The phylogenetic tree of IPT gene family contained A. thaliana (9), A. lyrata (7), B. rapa (13), O. sativa (10), Z. mays (8), S. lycopersicum (6), P. trichocarpa (9), and P. patens (6), as well as NsIPT and RfIPT(A). The IPT genes were divided into four groups indicated by differently colored dots. BrIPT genes were highlighted by red rectangles. The phylogenetic tree of CKX gene family contained A. thaliana (7), A. lyrata (7), B. rapa (12), O. sativa (11), Z. mays (13), S. lycopersicum (6), P. trichocarpa (9), and P. patens (6), as well as NsCKX1 and RfCKX1(B). The CKX gene family was divided into five groups indicated by differently colored dots. BrCKX genes were highlighted by red rectangles.
Evolutionary patterns and divergence of IPT and CKX genes in the Cruciferae family
To elucidate the evolutionary patterns and divergence of IPT and CKX genes in Cruciferae family, the three sequenced cruciferous species (B. rapa, A. thaliana, and A. lyrata) were chosen to make a genome-level comparison. The genome size of the North American A. lyrata is 207 Mb, with eight chromosomes [40]. A. thaliana, with its 125 Mb genome and five chromosomes, has undergone a ~30% reduction in genome size [41] and nine to ten chromosomal rearrangements [42,43]. A. thaliana was estimated to have diverged from A. lyrata approximately 10 MYA [44-46]. B. rapa has approximately 529 Mb and ten chromosomes; this species is believed to have emerged from the Brassica progenitor approximately 8 MYA [33]. As shown in Figure 6A, 6B, the IPT and CKX genes were well dispersed on the chromosomes, except for the Sca1 chromosome in A. lyrata as well as A01, A06, and A08 in B. rapa. The corresponding relationships of the IPT and CKX genes on chromosomes were clearly evident: Sca2 corresponded to Chr1; Sca3 and Sca4 corresponded to Chr2; Sca3 and Sca5 corresponded to Chr3; Sca7 corresponded to Chr4; Sca6 and Sca8 corresponded to Chr5. This trend was consistent with a previous comparison of the A. lyrata and A. thaliana genomes [40]. Traces of chromosomal rearrangement and simplification in the A. thaliana genome were easily captured by the analysis. The corresponding relationships between B. rapa and A. thaliana were still regular, although relatively more complicated. Homologous genes were separately existed on Chr1 as paired with A02 and A07, Chr2 as paired with A03, A04, and A09, Chr3 as paired with A04, A07, and A09, Chr4 as paired with A03, and Chr5 as paired with A02, A03, and A10. This trend was consistent with the segmental collinearity of the B. rapa and A. thaliana genomes [47].
Figure 6.
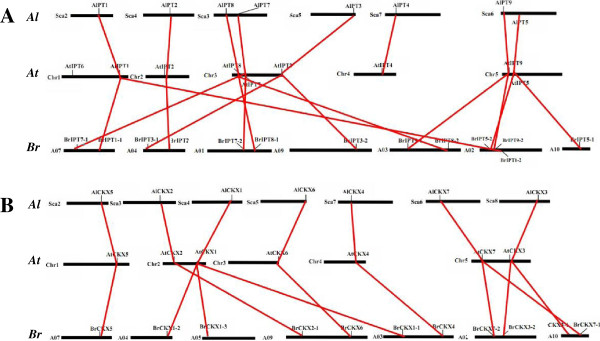
Relationship diagram of homologous IPT (A) and CKX (B) genes chromosomal distribution amongB. rapa (Br), A. thaliana (At), andA. lyrata(Al). Chromosomes are indicated by black lines with different lengths according to their own sizes. Homologous genes are linked by red lines.
To reveal the divergence of IPT and CKX genes in the Cruciferae family, the Ks and Ka modes for the paralogs and orthologs in the three genomes were compared (Additional file 7 and Additional file 8). Evidently, the Ks values of the paralogs in B. rapa were much smaller than the values of the orthologous genes between B. rapa, A. thaliana, and A. lyrata. The Ks values obtained from comparisons of the sets of putative orthologs were used to calculate the divergence time between B. rapa and A. thaliana. The Ks values ranged from 0.25 to 0.6, with a concentrated location between 0.4 and 0.5 (Figure 7). The calculation of divergence time was based on the neutral substitution rate of 1.5×10-8 substitutions per site per year for Chs[48]. Our results showed B. rapa diverged from A. thaliana at around 26–33 MYA, which coincided well with previous studies of speciation between the Brassica and Arabidopsis genomes [46,49-51].
Figure 7.
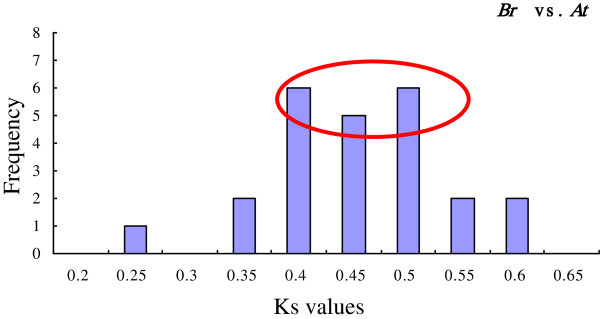
The Ks values distribution of IPT and CKX genes in the genomes of B. rapa and A. thaliana. The vertical axis indicates the frequency of paired sequences, whereas the horizontal axis denotes the Ks values with an interval of 0.05. The bars depict the positions of the modes of Ks distributions obtained from orthologous gene pairs. The peaks of the bars are marked with a red circle which represents the principal distribution extent of the Ks values.
Tissue or organ-specific expression of BrIPT and BrCKX genes
Tissue-specific and developmental stage-related expression data are useful in the identification of genes that are involved in defining the precise nature of individual tissues in a given developmental stage [16]. CKs have central regulatory functions during plant development and have essential functions during the adaptation to adverse environments. A clear expression profile of the genes of interest is an indispensable step to find and utilize agriculturally important genes. The homeostasis of the CK level in various tissues and organs is mainly regulated by the IPT and CKX enzymes. Thus, to obtain an initial picture of the functions of BrIPT and BrCKX genes during the vegetative and reproductive development, the transcript levels of these genes in the roots, stems, leaves, flowers, immature siliques, sepals, petals, stamens, pistils, little buds, medium-sized buds, and big buds were analyzed by qRT-PCR. As shown in Figure 8A, the tRNA IPT genes, BrIPT2, BrIPT9-1, and BrIPT9-2, were ubiquitously expressed, which was similar to the expression patterns of AtIPT2 and AtIPT9[7]. Other BrIPT genes demonstrated tissue or organ-specific expression levels. The expression of the BrIPT genes was similar to that of the AtIPT genes. BrIPT1-1 and BrIPT1-2 were highly expressed in the little and medium-sized buds, but reached very low levels in the big buds. BrIPT3-1 and BrIPT3-2 were highly expressed in the roots, and BrIPT3-2 was also expressed with very high levels in the leaves. BrIPT5-1 and BrIPT5-2 were mainly expressed in the roots. BrIPT7-1 and BrIPT7-2 were primarily transcribed in the stamens and roots. These segmental duplicated genes had uniform expression levels. BrIPT8-1 and BrIPT8-2 had unique expression patterns. BrIPT8-1 had the highest expression levels in siliques, whereas BrIPT8-2 was mainly expressed in stamens. The BrIPT genes were expressed abundantly in the reproductive organs. Maybe the roots alone could not supply sufficient CKs for the increasing demand in the reproductive organs. As can be seen from Figure 8B, the expression of BrCKX genes was more diverse, only BrCKX7-1 and BrCKX7-2 were uniformly expressed at high levels in sepals and petals. BrCKX1-1 was mainly expressed in the roots; BrCKX1-2 maintained a high level in the stamens, flowers, and petals. BrCKX1-3 seemed to be ubiquitously expressed, with relatively higher levels in the petals and pistils. BrCKX2-1 and BrCKX2-2 were mainly expressed in the reproductive organs, including the flowers, sepals, petals, and stamens. However, the expression of both genes was barely detected in vegetative organs. BrCKX3-1 was highly expressed in the petals, stamens, and flowers whereas BrCKX3-2 was mainly expressed in the floral buds. BrCKX4 and BrCKX5 had extremely high levels in the roots and stamens, respectively. BrCKX6 mostly transcribed in the roots, leaves, and sepals. The segmental duplicated pairs of BrIPT genes had similar expression profiles. However, this was the case in only one of the four segmental duplicated pairs of BrCKX genes. CKs could be rapidly and precisely transported from the synthetic positions to the functional parts via a multiple of putative purine permeases (PUPs) [52,53] and equilibrative nucleotide transporters (ENTs) [54,55] involved in CKs transport in plants. Thus, CKX enzymes are compulsory to be present in various organs to maintain the finely-tuned homeostasis of CKs levels. Although the segmental duplicated gene pairs originated from common ancestral genes after the WGT, they may have probably evolved into independent new genes with irreplaceable functions.
Figure 8.

Relative expression profiles of BrIPTs (A) and BrCKXs (B) in various tissues and organs. R, roots; S, floral stems; L, leaves; F, flowers; Si, immature siliques; Se, sepals; Pe, petals; St, stamens; Pi, pistils; LB, little buds; MB, medium-sized buds; and BB, big buds. Error bars represent ±SE of two biological replicates.
Stress-inducible cis-regulatory elements in the promoter regions of BrIPT and BrCKX genes
The cis-regulatory elements, which are located in the upstream regions of genes, act as the binding sites for TFs; thus, these sequences have essential roles in determining the tissue-specific or stress-responsive expression patterns of genes [16]. Increasing evidence has showed that genes responsive to multiple stimuli are closely correlated with cis-regulatory elements in their promoter regions [56,57]. To further understand transcriptional regulation and the potential functions of the BrIPT and BrCKX genes, putative promoter regions in the 2000 bp sequences upstream of the transcriptional start site were used to identify putative stress-responsive cis-regulatory elements. Several abiotic stress elements were found (Additional file 9 and Additional file 10). One salt-stress (S000453), one heat-stress (S00030), one cold-stress (S000407), one wound-stress (S000457), and three drought-stress (S000176, S000408, and S000415) cis-elements were common in the promoter regions of the BrIPT and BrCKX genes. Therefore, B. rapa may adapt to stressful environments by changing its CKs content. Particularly, BrIPT3-1, BrIPT3-2, and BrCKX3-2 had up to 32, 26, and 24 cold-stress elements (S000407), respectively. The number of drought-stress elements (S000415) in BrCKX5 reached 28. The other remaining genes likewise possessed abundant stress-inducible cis-regulatory elements. The tRNA IPT genes in Arabidopsis were notably unaffected by the mineral nutrients, auxin, and CK concentrations [7]. By contrast, three tRNA IPT genes in B. rapa, BrIPT2, BrIPT9-1, and BrIPT9-2 were also rich in the five types of cis-regulatory elements based on our results. Therefore, we hypothesized that the tRNA IPT genes might also have important functions in the adaptation to adverse environmental stimuli. Moreover, 0.4 and 1.2 abiotic stress-inducible cis-elements per promoter were found in the GmIPT and GmCKX genes, respectively [16]. Similarly, BrCKX genes had relatively more stress-responsive elements than BrIPT genes.
Expression profiles of BrIPT and BrCKX genes under drought and salt stress
Previous studies revealed that the CKs content was correlated with stress-resistance; changed transcripts of IPT and CKX genes were observed in plants when they were exposed to drought and high salinity conditions, including Arabidopsis[30], rice [10], maize, [31] and soybeans [16]. Theoretically, the BrIPT and BrCKX genes could also respond to various abiotic stresses because of the presence of stress-inducible cis-regulatory elements in their promoter regions. qRT-PCR was used to analyse the expression profiles of BrIPT and BrCKX genes under drought and salt stress conditions. The data were presented with clusters using fold-change values transformed to Log2 format. As shown in Figure 9, in drought conditions, the majority of the BrIPT and BrCKX genes were initially induced and upregulated before falling to basal levels during the time-course, despite the continuous application of the stressor. To be more specific, BrIPT7-1 and BrCKX3-2 were continuously induced and maintained at high levels in conditions of serious drought. BrIPT7-1 even showed remarkably more than a 40-fold increase in its expression levels. Three CKX genes, BrCKX1-1, BrCKX1-2, and BrCKX5 maintained their relatively high expressions throughout the experiment. Notably, in level III, genes such as BrIPT2, -3-1, -5-1, -9-1 and −9-2, as well as BrCKX4 and −7-2, eventually returned to the basal level, expressions of BrIPT8-2, BrCKX1-3 and BrCKX7-1 dropped below the basal level, transcripts of BrIPT3-1 and BrCKX2-2 were even hardly detectable. By contrast, salt treatment induced variable expression patterns. Still, the majority of BrIPT and BrCKX genes were primarily induced and upregulated. However, their transcripts fell with prolonged treatment time. The transcription of BrIPT9-2 seemed to be unaffected. Only BrIPT5-1 was continuously induced with a 7-fold increase in its expression levels, whereas BrCKX1-3 was suppressed and its expression levels dropped throughout the experiment. BrIPT7-1 and −9-1, as well as BrCKX4, -6 and −7-1, had relatively high levels after 16 h of salt treatment while several genes returned to the basal level, namely, BrCKX1-2, -3-2, -5, -6, and −7-2. Moreover, transcripts of BrIPT2, BrCKX1-1, BrCKX3-1, BrIPT3-1, BrIPT8-2, and BrCKX2-2 dropped below the basal level and were eventually undetectable at this time point. Except for a similar response to drought and salt treatments for both BrIPT and BrCKX genes, we noted that the transcripts of both BrIPT3-1 and BrCKX2-2 had almost disappeared with treatment time going on. Consistent with the results in maize [31], the tRNA IPT genes in Brassica responded to stressful environments as well.
Figure 9.

The heat map shows the real-time quantitative RT-PCR (qRT-PCR) analysis results of BrIPT and BrCKX genes under drought and salt treatments with two biological and three technical replicates. The expression levels of genes are presented using fold-change values transformed to Log2 format compared to control. The Log2 (fold-change values) and the color scale are shown at the bottom of heat map.
Effects of exogenous 6-BA and ABA on the expressions of BrIPT and BrCKX genes
Various phytohormones regulate the abiotic and biotic stress resistance. CKs and ABA are two important plant hormones involved in stress response. CKs and ABA have been postulated to exert antagonistic activities during the adaptation to environmental stress in plants [4,58,59]. To determine the effects of exogenous CKs and ABA on the expressions of BrIPT and BrCKX genes, we investigated the expression profiles of these genes using qRT-PCR analyses and data were also presented with clusters using fold-change values transformed to Log2 format. Viewing from Figure 10, five adenylate IPT genes in B. rapa were significantly repressed at 0.5 or 1 h, except for the unaffected BrIPT5-1. By contrast, the transcripts of BrIPT9-1 was relatively steady, but BrIPT2 and BrIPT9-2 were also moderately induced by 6-BA. We hypothesized that this phenomenon could be attributed to the dynamic balance of various CKs in B. rapa. 6-BA is an iP type CK, and exogenous 6-BA application repressed the transcriptions of adenylate IPT genes, whereas the tRNA IPT genes were upregulated by a feedback regulation. A remarkable increase was observed in the levels of BrCKX3-2 and BrCKX5 transcripts after 6-BA treatment. Nevertheless, the expression levels of BrCKX1-1, -1-2, -1-3, -2-2 and −4 were decreased by various degrees. For the ABA treatment, five adenylate IPT genes in B. rapa were significantly repressed at 0.5 or 1 h, except for BrIPT5-1. Nevertheless, the expression of BrIPT9-1 was relatively steady, whereas BrIPT2 and BrIPT9-2 was moderately induced by ABA, which was similar to the response patterns to 6-BA. The expressions of most of the BrCKX genes were down-regulated. However, the relative transcription levels of BrCKX3-1, BrCKX3-2, and BrCKX5 were upregulated at 0.5 or 1 h. The antagonisms between CKs and ABA in plants may account for this phenomenon, but this mechanism has yet to be explored.
Figure 10.

The heat map shows the real-time quantitative RT-PCR (qRT-PCR) analysis results of BrIPT and BrCKX genes with exogenous 6-BA and ABA treatments with two biological and three technical replicates. The expression levels of genes are presented using fold-change values transformed to Log2 format compared to control. The Log2 (fold-change values) and the color scale are shown at the bottom of heat map.
Discussion
The expansion and loss of BrIPT and BrCKX genes following hexaploidy formation by WGT
Brassicaceae genomes have undergone three rounds of whole genome duplication (WGD) and Brassica genomes have undergone another whole genome triplication (WGT) after speciation from Arabidopsis thaliana[36-38]. B. rapa is believed to have emerged from the Brassica progenitor approximately 8 MYA [33]. Considering WGT of the Brassica genomes, the IPT and CKX genes should have approximately three times as many members as that of Arabidopsis. However, we could only identify 13 IPT genes and 12 CKX genes in B. rapa, thereby suggesting a substantial loss of genes following hexaploidy formation by WGT. Gene loss was likewise observed for the CRF and PG gene families in B. rapa (unpublished data). Consistent with previous studies, the triplicated B. rapa genome contained only approximately twice the number of genes in Arabidopsis because of genome shrinkage [33]. Previous studies showed that AtIPT6 is non-functional in the Wassilewskija (Ws) ecotype because of a frame-shift mutation [60], but is a functional gene in the Columbia (Col) ecotype [7]. By contrast, AtIPT4 is a functional gene in all ecotypes [21]. Excluding a possible pseudogene, AtIPT6, the genomes of A. lyrata, and A. thaliana had equal numbers of IPT and CKX genes, thereby reflecting their relatively stable genome evolution because of a short separation time. However, both homologous genes of AtIPT4 and AtIPT6 in B. rapa were not found. Ando et al. [34] and O’Keefe et al. [35] identified several BrIPT and BrCKX genes, but still failed to obtain BrIPT4 and BrIPT6. Thus, the homologous genes of AtIPT4 and AtIPT6 were probably lost in B. rapa during the long evolutionary history. Gene loss was a more common phenomenon in distantly related species. Le et al. [16] reported that AtIPT1, AtIPT4, AtIPT6, AtIPT8, and GmIPT01 appeared to originate from a common ancestor. These genes expanded and were then retained in Arabidopsis but not in soybean after the speciation event. Genes with high homology to AtIPT1, AtIPT4, AtIPT6, and AtIPT8 were also not found in rice [12], maize [14], and tomato [17]. We did not find homologs for AtIPT4 and AtIPT6 in this study. Nevertheless, genes homologous with AtIPT1 and AtIPT8 were identified in B. rapa, namely, BrIPT1-1, BrIPT1-2, BrIPT8-1, and BrIPT8-2. These genes all belonged to Group I with close relationships. Another piece of supporting evidence was that another IPT gene was found in B. napus, BnIPT1, which was homologous to AtIPT1[35]. The evolutionary mechanism of IPT genes in various species is still waiting to be clarified.
The IPT and CKX genes in the bryophyte P. patens
The IPT and CKX genes have been discovered in the cyanobacteria Nostoc and the phytopathogen Rhodococcus fascians, respectively, with a large genetic distance from the other IPT and CKX genes. None of the IPT and CKX sequences have been found in green algae [15]. Schmuelling et al. [39] proposed that the presence of putative CKX sequences in cyanobacteria suggested that plant CKX genes might evolved from ancient chloroplast genes, which probably originated from the endosymbiosis of cyanobacteria. To date, the bryophyte P. patens is the earliest known terrestrial plant with IPT and CKX genes. Moreover, previous studies showed that the complete set of proteins in the CK signaling pathway first appeared in the said bryophyte [61]. Genes involved in the two-component system (TCS) of the P. patens were identified and characterized [62]. Thus, the CK metabolic pathway, and its signal transduction pathway imply that P. patens is the earliest species to have achieved a finely tuned control over CK homeostasis via biosynthesis that diverged to conquer land.
Evolutionary patterns of IPT and CKX genes in the Cruciferae family
Since the tetraploidization of the A. thaliana ancestor 30–35 MYA, a wave of chromosomal rearrangements has modified its genome architecture. The majority of rearrangements had occurred before the Arabidopsis–Brassica split at 20–24 MYA; thus, the segmental architecture of the A. thaliana genome was predominantly conserved in Brassica[49,51,63]. We made a genome-level comparison with the three sequenced cruciferous species (B. rapa, A. thaliana, and A. lyrata). By analyzing the corresponding relationships of the IPT and CKX genes on chromosomes, traces of chromosomal rearrangement were easily captured. Brassica genomes have undergone whole genome triplication (WGT) after speciation from Arabidopsis thaliana, leading to significantly increased duplicated genes. Duplicated genes occur as the tandem duplicated genes and segmental duplicated genes. For the IPT and CKX genes in B. rapa, no tandem duplicated genes were found, but five pairs of segmental duplicates were found among 13 BrIPT genes, and three pairs of segmental duplicates were found among 12 BrCKX genes, suggesting that the expansion of BrIPT and BrCKX genes after speciation from Arabidopsis thaliana is mainly attributed to segmental duplication events during the whole genome triplication (WGT). The Ka (non-synonymous substitution rates) and Ks (synonymous substitution rates) is a measure to explore the mechanism of gene divergence after duplication. Large-scale duplication events are defined as simultaneous duplications of genes. Assuming a molecular clock, the synonymous substitution rates (Ks) of these duplicates are expected to be similar over time. There are, however, substantial rate variations among genes [64]. We used the relative Ks measure as the proxy for time to evaluate the divergence time between B. rapa and Arabidopsis. The Ks values of the duplicated paralogs in B. rapa were notably much smaller than the values of the duplicated orthologous genes between B. rapa and Arabidopsis, further confirming that the duplicated IPT and CKX genes in B. rapa were produced during the whole genome triplication (WGT) after speciation from Arabidopsis.
Changed expression patterns of the duplicated BrIPT and BrCKX genes
Duplication occurs in an individual and may be retained or lost in the population, similar to a point mutation [65]. Unless the presence of an extra amount of gene product is advantageous, two genes with identical functions are unlikely to be stably maintained in the genome. Theoretical population genetics predicates that both duplicates can be stably maintained when they differ in some aspects of their functions [66]. Sub-functionalization and neo-functionalization are two evolutionary fates of duplicated genes. These may account for the varied expression patterns of paralogs. Our study showed BrIPT1-1 and 1–2, as well as BrCKX2-1, -2-2, -3-1, -3-2, -5, -7-1 and −7-2, were mainly expressed in the reproductive organs, whereas BrIPT3-1, -3-2, -5-1 and −5-2, as well as BrCKX4 and −6 had major transcripts in the vegetative organs. Even the segmental duplicated gene pairs had inconsistent expression profiles. For instance, BrCKX1-1 was mainly expressed in roots while BrCKX1-2 maintained at high levels in stamens, flowers and petals. Meanwhile, BrCKX1-3 was ubiquitously expressed, with relatively high levels in the petals and pistils. Likewise, a few duplicated gene pairs (GmIPT08 and GmIPT10; GmCKX05 and GmCKX06; GmCKX12 and GmCKX14) have undergone expression divergence in soybean [16]. The response mechanisms of the two gene families varied for the different types of stresses. In this study, the BrIPT5-1 was continuously induced by salt treatment, with 7-fold elevated expression levels. However, BrCKX1-3 was suppressed, with dropped expression levels. The expressions of the majority of the CKX family in Arabidopsis, namely, CKX2, CKX4, CKX5, and CKX7, decreased with salt treatment; whereas expressions of the remaining three genes (CKX1, CKX3 and CKX6) increased [30]. Meanwhile, increasing evidence indicated that the response to environmental stresses for plant IPT and CKX genes was tissue or organ-dependent. Stress induced ZmCKX1 expressions in leaves, but it had an opposite effect in roots [31]. The induction levels in the V6-stage leaves of soybean were correlated with the age of the trifoliate leaves; the older the leaf, the higher the induction. GmIPT09 and GmIPT13 were significantly induced by drought in the V6-stage leaves; the degree of induction of these genes was higher in the younger trifoliate leaves [16]. All these proofs strongly supported the hypothesis that daughter genes originated from a common parental gene might adopt part of the functions of their parental gene [67-69] or acquire new functions [70].
The possible roles of tRNA IPT genes in plants
Three tRNA IPT genes (BrIPT2, BrIPT9-1, and BrIPT9-2) were identified to have unique gene structures and expression patterns in B. rapa. AtIPT2, AtIPT9, ZmIPT1, ZmIPT10, OsIPT9, and OsIPT10 likewise belong to this group of tRNA IPT genes. The tRNA IPT genes are responsible for the synthesis of cZ-type CKs. Based on the data from various bioassays [71,72], the tZ was found to be a bioactive substance whereas cZ was reported to have a weak biological effect. Moreover, the 35S-linked AtIPT2 did not induce CK responses in calli when CKs were absent [21]. Despite the presumed inactivity of cZ as a free hormone, the presence of free cZ-type CKs in plant tissues has been repeatedly reported. Some plant species contain detectable levels of diverse cZ-type CKs, which are occasionally the predominant group of total CKs. Gajdosova et al. [9] hypothesized that the cZ-type CKs may function as delicate regulators of the CK responses in plants under growth-limiting conditions. Similar to AtIPT2 and AtIPT9, BrIPT2, BrIPT9-1, and BrIPT9-2, were ubiquitously expressed. The tRNA IPT genes in B. rapa notably responded to adverse environmental stresses, thereby indicating that the cZ biosynthesized by tRNA IPT genes may be also involved in stress resistance.
Conclusions
In summary, in this study we identified 13 IPT and 12 CKX genes in Chinese cabbage genome. The gene structures, conserved domains and phylogenetic relationships were analyzed. Duplications, evolutionary patterns and divergence of the IPT and CKX genes were investigated. We also characterized their expression patterns in various tissues and organs, their responses to abiotic stresses and exogenous phytohormones. To date, this is the first genome-wide study of the IPT and CKX gene families in B. rapa. Our results will help to enhance the understanding of the evolution of dynamic functions and fates for these BrIPT and BrCKX genes. Chinese cabbage is one of the most important vegetables and is widely cultivated. Unveiling the roles of BrIPT and BrCKX genes in plant growth, development and stress adaptation processes may help molecular breeders to develop economically important high-yielding and high-quality stress-tolerant crops in agriculture.
Methods
Identification of IPT and CKX gene families in B. rapa genome
The Arabidopsis IPT and CKX genes were used as seed sequences to search the Brassica Database (BRAD; version 1.1; http://brassicadb.org/brad/) and the NCBI database (http://www.ncbi.nlm.nih.gov). A total of 13 BrIPT and 12 BrCKX candidate genes were identified based on BLASTN and BLASTP searches with a cut-off expect value of 100. To ensure that no additional related genes were overlooked by the database search, the identified BrIPT and BrCKX genes were used as query sequences in additional multiple database searches to confirm the initial list of genes. Similarly, the IPT and CKX genes from Arabidopsis lyrata, Populus trichocarpa, and Physcomitrella patens were identified from the A. lyrata genome (version 1.0; http://genome.jgi-psf.org/Araly1/Araly1.info.html), the poplar genome assembly (version 1.1; http://genome.jgi-psf.org/Poptr1_1/Poptr1_1.home.html), and the P. patens genome assembly (version 1.1; http://genome.jgi-psf.org/Phypa1_1/Phypa1_1.info.html), respectively. The Pfam database (http://pfam.janelia.org/) [73] and the Conserved Domain Database (CDD) of the National Center for Biotechnology Information (NCBI) [74] were used to analyze all of the sequences that met the given requirements. Genes that did not contain the known conserved domains and motifs of the gene families were excluded from further analysis.
Multiple sequence alignment and phylogenetic analysis
The online MEME software (http://meme.sdsc.edu/meme/meme.html) was used to identify the BrIPT and BrCKX motifs, with an expected e-value of less than 2×10-30[75,76]. Multiple sequence alignments were conducted using ClustalX (version 1.81) [77]. Phylogenetic analysis was performed using the neighbor-joining (NJ) method of the MEGA (version 5.0) program [78]. The confidence limits of each branch in the phylogenetic tree were assessed by 1000 bootstrap replications and expressed as percentage values.
Physiological and biochemical property analysis of BrIPT and BrCKX proteins
The isoelectric point (pI) of the BrIPT and BrCKX proteins were predicted using Compute pI/Mwsoftware (http://www.expasy.ch/tools/pi_tool.html) [79]. Their subcellular localization was predicted by the TargetP software of the CBS database (http://www.cbs.dtu.dk/services/TargetP) [80]. The glycosylation sites were predicted using NetNGly (http://www.cbs.dtu.dk/services/NetNGlyc/) (unpublished database).
Cis-element analysis of the putative promoter regions of BrIPT and BrCKX genes
To identify cis-elements in the promoter regions of BrIPT and BrCKX genes, the 2000 bp sequences upstream of the transcriptional start site of each BrIPT and BrCKX gene were chosen. These sequences were used to query the PLACE database (http://www.dna.affrc.go.jp/PLACE/) [81], and the putative cis-regulatory elements of the promoter sequences were identified.
Chromosome mapping of the IPT and CKX genes in B. rapa, A. thaliana, and A. lyrata
Maps of the chromosomes were drawn using MS Office software, based on the location of each gene along the specific length of each chromosome. Tandem duplications were defined as genes located within 20 loci from each other [16]. Segmental duplications were identified by synteny analysis using an online tool of the Plant Genome Duplication Database (PGDD; http://chibba.agtec.uga.edu/duplication/) [82].
Evolutionary analysis of homologous genes between B. rapa and A. thaliana
Synteny analysis of the BrIPT and BrCKX genes were performed online using PGDD (http://chibba.agtec.uga.edu/duplication/). The occurrence of duplication events and homologous genes divergence, as well as the selective pressure on duplicated genes, were estimated by calculating synonymous (Ks) and non-synonymous substitutions (Ka) per site between the homologous genes using the LOCUS SEARCH utility of PGDD. The divergence time was calculated using the neutral substitution rate of 1.5×10-8 substitutions per site per year for the chalcone synthase gene (Chs) [48].
Plant growth and treatments
The plant materials (B. rapa ssp. pekinensis cv. Zhonghan No. 1) were grown at the experimental farm in Zhejiang University until flowering. Steps taken in growing the sampling plants were as follows: First, the seeds were sown on 10 Oct, 2012, and then were transplanted in the field outside after the seedlings with 5~6 pieces true leaves on 15 Nov, 2012. Subsequently, the seedlings were carried out with normal management for about 4 months. Finally, the plants produced flowers after bolting and materials were sampled on 10 Mar, 2013. The roots, floral stems, leaves, flowers, immature siliques, sepals, petals, stamens, pistils, little buds (<1.6 mm), medium-sized buds (1.6 mm to 2.8 mm), and big buds (>2.8 mm) were sampled to analyze the tissue- or organ-specific expression of the different genes. Total roots and main floral stems were chosen. For the leaves materials, young leaves at the second and third nodes from top of the main floral stems were chosen. For the flowers, new opened ones were selected. Immature siliques were defined as siliques about 3 days after pollination and fertilization. The diameters of the floral buds were measured using a Vernier caliper. All the materials were sampled from at least ten plants.
B. rapa ssp. pekinensis cv. Chiifu-401-42 plants were used in the different treatment conditions. All seedlings were grown under a 16 h/8 h light/dark photoperiod, at 25°C ± 1°C, for approximately 3 weeks. Only the second true leaves were sampled to minimize the differences between samples. The nutrient solution was supplemented with 200 mM NaCl for the salt treatments, and the leaves were collected at 0, 3, 8, and 16 h after stress induction. Water was withheld from three-week-old seedlings to initiate the drought treatment, and the leaves were subsequently classified into four levels, according to the degree of drought. Level 0 had normal leaves in well-watered seedlings. At level I (3 days after drought treatment), the leaves started to wither, whereas the leaves were severely withered at Level II (5 days after drought treatment). Level III (7 days after drought treatment) indicated that the whole seedlings had withered. The 3-week-old seedlings were sprayed with 100 μM 6-BA (6-Benzylaminopurine) for the cytokinin treatment and 100 μM ABA for the ABA treatment. The leaves were sampled at 0, 0.5, and 1 h after spraying, and the control was sprayed with double distilled water alone. All the materials sampled were immediately frozen in liquid nitrogen and stored in a refrigerator at −75°C.
RNA extraction and qRT-PCR analysis
The total RNA was extracted using the TRIZOL reagent (Invitrogen, Germany), according to the manufacturer’s instructions. The first cDNA strand was generated using the Takara Reverse Transcription System (Japan) by following the manufacturer’s protocol. A maximum of 1 ug RNA was used for each reverse-transcription reaction. gDNA eraser in the kits was used to eliminate DNA to prevent DNA contamination. qRT-PCR was performed using the primers listed in Additional file 11. The primers were designed using the Primer (version 5.0) software. The specificity of each primer to their target genes was checked using the BLASTN program of BRAD. cDNA templates were firstly homogenized with the EASY Solution in the kits. Up to 2 μL of diluted cDNA were subjected to each qRT-PCR reaction for a final volume of 15 μL, which contained 7.5 μL SYBR Green Master Mix Reagent (Takara, Japan) and 0.3 μL specific primers (3 pmol). The qRT-PCR reactions were performed using a StepOne real-time PCR machine (Bio-RAD, USA), programmed to heat for 30 s at 95°C, followed by 40 cycles of 5 s at 95°C and 45 s at 53–58°C, and at the end, 1 cycle of 1 min at 95°C, 30 s at 50°C and 30 s at 95°C. The specificity of the reactions was verified by melting curve analysis and products were further confirmed by agarose gel electrophoresis. Two biological replicates were performed with three technical replicates for each sample plus the negative control. RNA for each biological replicate pooled from a number of individuals per treatment/tissue. The BrActin1 gene was used as the reference gene. The comparative ΔΔCT method was used to calculate the relative expression levels of the different genes. The data of qRT-PCR were clustered using the average linkage method with Pearson correlation distance metric by Multiple Array Viewer [83].
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ZNL carried out the experiment, analyzed the data, and drafted the manuscript. YXL, MZ, and YPL performed the qRT-PCR analysis. LJK and MHZ performed the abiotic stress analysis and wrote the manuscript. GL, JSC, and XLY proposed and supervised the research. All authors read and approved the final manuscript.
Supplementary Material
Alignment of amino acid sequences of BrIPTs with AtIPTs. BrIPTs had two conserved regions, Region a and Region b. Region a, denoted by red boxes, contained a putative ATP/GTP-binding sites (P-loop) motif (prosite PS00017: consensus sequence TGxGKS), which were underlined with a black line. Region b was a putative tRNA binding site, denoted by red boxes.
Summary of conserved motifs of BrIPT protein sequences.
Alignment of amino acid sequences of BrCKXs with AtCKXs. The position of the motif for covalent binding of FAD cofactor (GHS, where H is the binding residue in all plant CKXs) was indicated. Amino acid residues predicted based on the structure of ZmCKX1 to bind the cytokinin substrate were marked with an asterisk. The cytokinin N9 hydrogen-bonding residue that affected the substrate specificity was marked with a red arrow.
Summary of conserved motifs of BrCKX protein sequences.
Synteny analysis of BrIPT genes in ±100 kb region with score greater than 1000. Synteny analysis revealed evidence of the segmental duplications among BrIPT genes. Synteny analysis of Bra036719 (BrIPT2-1) and Bra040677 (BrIPT2-2) were omitted owing to unknown scaffold location of Bra040677.
Synteny analysis of BrCKX genes in ±100 kb region with score greater than 1000. Synteny analysis revealed evidence of the segmental duplications among BrCKX genes.
Determination of Ka and Ks values of IPT genes. The Ka and Ks values of IPT genes were determined between duplicated genes in B. rapa and the homologous genes among the B. rapa, A. thaliana and A. lyrata. — means no duplicated genes were found.
Determination of Ka and Ks values of CKX genes. The Ka and Ks values of CKX genes were determined between duplicated genes in B. rapa and the homologous genes among the B. rapa, A. thaliana and A. lyrata. - means no duplicated genes were found.
Summary of the cis-elements found in the putative promoter regions of BrIPT genes. Cis-elements with larger numbers were marked red.
Summary of the cis-elements found in the putative promoter regions of BrCKX genes.Cis-elements with larger numbers were marked red.
Forward and Reverse Primers used in the qRT-PCR analysis.
Contributor Information
Zhenning Liu, Email: 11116061@zju.edu.cn.
Yanxia Lv, Email: 21116089@zju.edu.cn.
Mei Zhang, Email: 21116177@zju.edu.cn.
Yapei Liu, Email: 21016173@zju.edu.cn.
Lijun Kong, Email: 21216066@zju.edu.cn.
Minghua Zou, Email: 21216157@zju.edu.cn.
Gang Lu, Email: glu@zju.edu.cn.
Jiashu Cao, Email: jshcao@zju.edu.cn.
Xiaolin Yu, Email: xlyu@zju.edu.cn.
Acknowledgements
This research was partially supported by the Hi-Tech Research and Development Program of China (2012AA100104-4), the Breeding Project of the Sci-tech Foundation of Zhejiang Province (2012C12903), Zhejiang Provincial Natural Science Foundation of China (Y13C150001) and the Key Science & Technology Program of Zhejiang Province (2010C12004). The authors gratefully acknowledge Dr. Jian Wu for his helpful advice. We thank Dr. Youjian Yu, in particular, for the stimulating discussions and for critically reading the manuscript.
References
- Mok DWS, Mok MC. Cytokinin metabolism and action. Annu Rev Plant Physiol Plant Mol Biol. 2001;52:89–118. doi: 10.1146/annurev.arplant.52.1.89. [DOI] [PubMed] [Google Scholar]
- Takei K, Sakakibara H, Taniguchi M, Sugiyama T. Nitrogen-dependent accumulation of cytokinins in root and the translocation to leaf: Implication of cytokinin species that induces gene expression of maize response regulator. Plant and Cell Physiology. 2001;42:85–93. doi: 10.1093/pcp/pce009. [DOI] [PubMed] [Google Scholar]
- Sakakibara H. Cytokinins: Activity, biosynthesis, and translocation. Annual Review of Plant Biology. 2006;57:431–449. doi: 10.1146/annurev.arplant.57.032905.105231. [DOI] [PubMed] [Google Scholar]
- Werner T, Kollmer I, Bartrina I, Holst K, Schmulling T. New insights into the biology of cytokinin degradation. Plant Biology. 2006;8:371–381. doi: 10.1055/s-2006-923928. [DOI] [PubMed] [Google Scholar]
- Werner T, Schmulling T. Cytokinin action in plant development. Curr Opin Plant Biol. 2009;12:527–538. doi: 10.1016/j.pbi.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Frebort I, Kowalska M, Hluska T, Frebortova J, Galuszka P. Evolution of cytokinin biosynthesis and degradation. J Exp Bot. 2011;62:2431–2452. doi: 10.1093/jxb/err004. [DOI] [PubMed] [Google Scholar]
- Miyawaki K, Matsumoto-Kitano M, Kakimoto T. Expression of cytokinin biosynthetic isopentenyltransferase genes in Arabidopsis: tissue specificity and regulation by auxin, cytokinin, and nitrate. Plant Journal. 2004;37:128–138. doi: 10.1046/j.1365-313X.2003.01945.x. [DOI] [PubMed] [Google Scholar]
- Hwang I, Sakakibara H. Cytokinin biosynthesis and perception. Physiol Plant. 2006;126:528–538. doi: 10.1111/j.1399-3054.2006.00665.x. [DOI] [Google Scholar]
- Gajdosova S, Spichal L, Kaminek M, Hoyerova K, Novak O, Dobrev PI, Galuszka P, Klima P, Gaudinova A, Zizkova E. et al. Distribution, biological activities, metabolism, and the conceivable function of cis-zeatin-type cytokinins in plants. J Exp Bot. 2011;62:2827–2840. doi: 10.1093/jxb/erq457. [DOI] [PubMed] [Google Scholar]
- Takei K, Sakakibara H, Sugiyama T. Identification of genes encoding adenylate isopentenyltransferase, a cytokinin biosynthesis enzyme, in Arabidopsis thaliana. J Biol Chem. 2001;276:26405–26410. doi: 10.1074/jbc.M102130200. [DOI] [PubMed] [Google Scholar]
- Werner T, Motyka V, Laucou V, Smets R, Van Onckelen H, Schmulling T. Cytokinin-deficient transgenic Arabidopsis plants show multiple developmental alterations indicating opposite functions of cytokinins in the regulation of shoot and root meristem activity. Plant Cell. 2003;15:2532–2550. doi: 10.1105/tpc.014928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto T, Sakakibara H, Kojima M, Yamamoto Y, Nagasaki H, Inukai Y, Sato Y, Matsuoka M. Ectopic expression of KNOTTED1-like homeobox protein induces expression of cytokinin biosynthesis genes in rice. Plant Physiol. 2006;142:54–62. doi: 10.1104/pp.106.085811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YC, Weir NR, Hill K, Zhang WJ, Kim HJ, Shiu SH, Schaller GE, Kieber JJ. Characterization of genes involved in cytokinin signaling and metabolism from rice. Plant Physiol. 2012;158:1666–1684. doi: 10.1104/pp.111.192765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugiere N, Humbert S, Rizzo N, Bohn J, Habben JE. A member of the maize isopentenyl transferase gene family, Zea mays isopentenyl transferase 2 (ZmIPT2), encodes a cytokinin biosynthetic enzyme expressed during kernel development. Plant Mol Biol. 2008;67:215–229. doi: 10.1007/s11103-008-9312-x. [DOI] [PubMed] [Google Scholar]
- Gu RL, Fu JJ, Guo S, Duan FY, Wang ZK, Mi GH, Yuan LX. Comparative expression and phylogenetic analysis of maize Cytokinin Dehydrogenase/Oxidase (CKX) gene family. Journal of Plant Growth Regulation. 2010;29:428–440. doi: 10.1007/s00344-010-9155-y. [DOI] [Google Scholar]
- Le DT, Nishiyama R, Watanabe Y, Vankova R, Tanaka M, Seki M, Ham LH, Yamaguchi-Shinozaki K, Shinozaki K, Tran LSP. Identification and expression analysis of cytokinin metabolic genes in soybean under normal and drought conditions in relation to cytokinin levels. PLoS ONE. 2012;7:e42411. doi: 10.1371/journal.pone.0042411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo S, Kikuchi K, Fukuda M, Honda I, Imanishi S. Roles and regulation of cytokinins in tomato fruit development. J Exp Bot. 2012;63:5569–5579. doi: 10.1093/jxb/ers207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galuszka P, Frebortova J, Werner T, Yamada M, Strnad M, Schmulling T, Frebort I. Cytokinin oxidase/dehydrogenase genes in barley and wheat-cloning and heterologous expression. Eur J Biochem. 2004;271:3990–4002. doi: 10.1111/j.1432-1033.2004.04334.x. [DOI] [PubMed] [Google Scholar]
- Mameaux S, Cockram J, Thiel T, Steuernagel B, Stein N, Taudien S, Jack P, Werner P, Gray JC, Greenland AJ. et al. Molecular, phylogenetic and comparative genomic analysis of the cytokinin oxidase/dehydrogenase gene family in the Poaceae. Plant Biotechnol J. 2012;10:67–82. doi: 10.1111/j.1467-7652.2011.00645.x. [DOI] [PubMed] [Google Scholar]
- Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray) Science. 2006;313:1596–1604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- Kakimoto T. Identification of plant cytokinin biosynthetic enzymes as dimethylallyl diphosphate: ATP/ADP isopentenyltransferases. Plant and Cell Physiology. 2001;42:677–685. doi: 10.1093/pcp/pce112. [DOI] [PubMed] [Google Scholar]
- Ye CJ, Wu SW, Kong FN, Zhou CJ, Yang QK, Sun Y, Wang B. Identification and characterization of an isopentenyltransferase (IPT) gene in soybean (Glycine max L.) Plant Sci. 2006;170:542–550. doi: 10.1016/j.plantsci.2005.10.008. [DOI] [Google Scholar]
- Zubko E, Adams CJ, Machaekova I, Malbeck J, Scollan C, Meyer P. Activation tagging identifies a gene from Petunia hybrida responsible for the production of active cytokinins in plants. Plant Journal. 2002;29:797–808. doi: 10.1046/j.1365-313X.2002.01256.x. [DOI] [PubMed] [Google Scholar]
- Bartrina I, Otto E, Strnad M, Werner T, Schmulling T. Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana. Plant Cell. 2011;23:69–80. doi: 10.1105/tpc.110.079079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashikari M, Sakakibara H, Lin SY, Yamamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M. Cytokinin oxidase regulates rice grain production. Science. 2005;309:741–745. doi: 10.1126/science.1113373. [DOI] [PubMed] [Google Scholar]
- Zalewski W, Galuszka P, Gasparis S, Orczyk W, Nadolska-Orczyk A. Silencing of the HvCKX1 gene decreases the cytokinin oxidase/dehydrogenase level in barley and leads to higher plant productivity. J Exp Bot. 2010;61:1839–1851. doi: 10.1093/jxb/erq052. [DOI] [PubMed] [Google Scholar]
- Tran LSP, Urao T, Qin F, Maruyama K, Kakimoto T, Shinozaki K, Yamaguchi-Shinozaki K. Functional analysis of AHK1/ATHK1 and cytokinin receptor histidine kinases in response to abscisic acid, drought, and salt stress in Arabidopsis. Proc Natl Acad Sci U S A. 2007;104:20623–20628. doi: 10.1073/pnas.0706547105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havlova M, Dobrev PI, Motyka V, Storchova H, Libus J, Dobra J, Malbeck J, Gaudinova A, Vankova R. The role of cytokinins in responses to water deficit in tobacco plants over-expressing trans-zeatin O-glucosyltransferase gene under 35S or SAG12 promoters. Plant Cell and Environment. 2008;31:341–353. doi: 10.1111/j.1365-3040.2007.01766.x. [DOI] [PubMed] [Google Scholar]
- Argueso CT, Ferreira FJ, Kieber JJ. Environmental perception avenues: the interaction of cytokinin and environmental response pathways. Plant Cell and Environment. 2009;32:1147–1160. doi: 10.1111/j.1365-3040.2009.01940.x. [DOI] [PubMed] [Google Scholar]
- Nishiyama R, Watanabe Y, Fujita Y, Le DT, Kojima M, Werner T, Vankova R, Yamaguchi-Shinozaki K, Shinozaki K, Kakimoto T. et al. Analysis of cytokinin mutants and regulation of cytokinin metabolic genes reveals important regulatory roles of cytokinins in drought, salt and abscisic acid responses, and abscisic acid biosynthesis. Plant Cell. 2011;23:2169–2183. doi: 10.1105/tpc.111.087395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyroubalova S, Vaclavikova K, Tureckova V, Novak O, Smehilova M, Hluska T, Ohnoutkova L, Frebort I, Galuszka P. Characterization of new maize genes putatively involved in cytokinin metabolism and their expression during osmotic stress in relation to cytokinin levels. Plant Physiol. 2009;151:433–447. doi: 10.1104/pp.109.142489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun JH, Yu HJ, Shin JY, Oh M, Hwang HJ, Chung H. Auxin response factor gene family in Brassica rapa: genomic organization, divergence, expression, and evolution. Mol Genet Genomics. 2012;287:765–784. doi: 10.1007/s00438-012-0718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun JH, Kwon SJ, Yang TJ, Seol YJ, Jin M, Kim JA, Lim MH, Kim JS, Baek S, Choi BS. et al. Genome-wide comparative analysis of the Brassica rapa gene space reveals genome shrinkage and differential loss of duplicated genes after whole genome triplication. Genome Biol. 2009;10:R111. doi: 10.1186/gb-2009-10-10-r111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando S, Asano T, Tsushima S, Kamachi S, Hagio T, Tabei Y. Changes in gene expression of putative isopentenyltransferase during clubroot development in Chinese cabbage (Brassica rapa L.) Physiol Mol Plant Pathol. 2005;67:59–67. doi: 10.1016/j.pmpp.2005.09.005. [DOI] [Google Scholar]
- O'Keefe D, Song JC, Jameson PE. Isopentenyl transferase and Cytokinin Oxidase/Dehydrogenase gene family members are differentially expressed during pod and seed development in rapid-cycling Brassica. Journal of Plant Growth Regulation. 2011;30:92–99. doi: 10.1007/s00344-010-9171-y. [DOI] [Google Scholar]
- Blanc G, Wolfe KH. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 2004;16:1667–1678. doi: 10.1105/tpc.021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G, Hokamp K, Wolfe KH. A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res. 2003;13:137–144. doi: 10.1101/gr.751803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysak MA, Koch MA, Pecinka A, Schubert I. Chromosome triplication found across the tribe Brassiceae. Genome Res. 2005;15:516–525. doi: 10.1101/gr.3531105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmulling T, Werner T, Riefler M, Krupkova E, Manns IBY. Structure and function of cytokinin oxidase/dehydrogenase genes of maize, rice, Arabidopsis and other species. J Plant Res. 2003;116:241–252. doi: 10.1007/s10265-003-0096-4. [DOI] [PubMed] [Google Scholar]
- Hu TT, Pattyn P, Bakker EG, Cao J, Cheng JF, Clark RM, Fahlgren N, Fawcett JA, Grimwood J, Gundlach H. et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet. 2011;43(5):476–481. doi: 10.1038/ng.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JS, Pepper AE, Hall AE, Chen ZJ, Hodnett G, Drabek J, Lopez R, Price HJ. Evolution of genome size in Brassicaceae. Ann Bot. 2005;95:229–235. doi: 10.1093/aob/mci016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Kiefer M. Genome evolution among cruciferous plants: A lecture from the comparison of the genetic maps of three diploid species-Capsella rubella, Arabidopsis lyrata subsp Petraea, and A. thaliana. American Journal of Botany. 2005. pp. 761–767. [DOI] [PubMed]
- Yogeeswaran K, Frary A, York TL, Amenta A, Lesser AH, Nasrallah JB, Tanksley SD, Nasrallah ME. Comparative genome analyses of Arabidopsis spp.: Inferring chromosomal rearrangement events in the evolutionary history of A. thaliana. Genome Res. 2005;15:505–515. doi: 10.1101/gr.3436305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Lauga B, Charlesworth D. Rates and patterns of molecular evolution in inbred and outbred Arabidopsis. Mol Biol Evol. 2002;19:1407–1420. doi: 10.1093/oxfordjournals.molbev.a004204. [DOI] [PubMed] [Google Scholar]
- Ossowski S, Schneeberger K, Lucas-Lledo JI, Warthmann N, Clark RM, Shaw RG, Weigel D, Lynch M. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010;327:92–94. doi: 10.1126/science.1180677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilstein MA, Nagalingum NS, Clements MD, Manchester SR, Mathews S. Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2010;107:18724–18728. doi: 10.1073/pnas.0909766107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XW, Wang HZ, Wang J, Sun RF, Wu J, Liu SY, Bai YQ, Mun JH, Bancroft I, Cheng F. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat Genet. 2011;43:1035–U1157. doi: 10.1038/ng.919. [DOI] [PubMed] [Google Scholar]
- Koch MA, Haubold B, Mitchell-Olds T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae) Mol Biol Evol. 2000;17:1483–1498. doi: 10.1093/oxfordjournals.molbev.a026248. [DOI] [PubMed] [Google Scholar]
- Ziolkowski PA, Kaczmarek M, Babula D, Sadowski J. Genome evolution in Arabidopsis/Brassica: conservation and divergence of ancient rearranged segments and their breakpoints. Plant Journal. 2006;47:63–74. doi: 10.1111/j.1365-313X.2006.02762.x. [DOI] [PubMed] [Google Scholar]
- Yang YW, Lai KN, Tai PY, Li WH. Rates of nucleotide substitution in angiosperm mitochondrial DNA sequences and dates of divergence between Brassica and other angiosperm lineages. J Mol Evol. 1999;48:597–604. doi: 10.1007/PL00006502. [DOI] [PubMed] [Google Scholar]
- Town CD, Cheung F, Maiti R, Crabtree J, Haas BJ, Wortman JR, Hine EE, Althoff R, Arbogast TS, Tallon LJ. et al. Comparative genomics of Brassica oleracea and Arabidopsis thaliana reveal gene loss, fragmentation, and dispersal after polyploidy. Plant Cell. 2006;18:1348–1359. doi: 10.1105/tpc.106.041665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillissen B, Burkle L, Andre B, Kuhn C, Rentsch D, Brandl B, Frommer WB. A new family of high-affinity transporters for adenine, cytosine, and purine derivatives in Arabidopsis. Plant Cell. 2000;12:291–300. doi: 10.1105/tpc.12.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkle L, Cedzich A, Dopke C, Stransky H, Okumoto S, Gillissen B, Kuhn C, Frommer WB. Transport of cytokinins mediated by purine transporters of the PUP family expressed in phloem, hydathodes, and pollen of Arabidopsis. Plant Journal. 2003;34:13–26. doi: 10.1046/j.1365-313X.2003.01700.x. [DOI] [PubMed] [Google Scholar]
- Wormit A, Traub M, Florchinger M, Neuhaus HE, Mohlmann T. Characterization of three novel members of the Arabidopsis thaliana equilibrative nucleoside transporter (ENT) family. Biochem J. 2004;383:19–26. doi: 10.1042/BJ20040389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JP, Hirose N, Wang XC, Wen P, Xue L, Sakakibara H, Zuo JR. Arabidopsis SOI33/AtENT8 gene encodes a putative equilibrative nucleoside transporter that is involved in cytokinin transport in planta. J Integr Plant Biol. 2005;47:588–603. doi: 10.1111/j.1744-7909.2005.00104.x. [DOI] [Google Scholar]
- Walther D, Brunnemann R, Selbig J. The regulatory code for transcriptional response diversity and its relation to genome structural properties in A. thaliana. PLoS Genet. 2007;3:216–229. doi: 10.1371/journal.pgen.0030216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang YJ, You J, Xie KB, Xie WB, Xiong LZ. Systematic sequence analysis and identification of tissue-specific or stress-responsive genes of NAC transcription factor family in rice. Mol Genet Genomics. 2008;280:547–563. doi: 10.1007/s00438-008-0386-6. [DOI] [PubMed] [Google Scholar]
- Zdunek E, Lips SH. Transport and accumulation rates of abscisic acid and aldehyde oxidase activity in Pisum sativum L. in response to suboptimal growth conditions. Journal of Experimental Botany. 2001;52:1269–1276. doi: 10.1093/jexbot/52.359.1269. [DOI] [PubMed] [Google Scholar]
- Chow B, McCourt P. Hormone signalling from a developmental context. J Exp Bot. 2004;55:247–251. doi: 10.1093/jxb/erh032. [DOI] [PubMed] [Google Scholar]
- Miyawaki K, Tarkowski P, Matsumoto-Kitano M, Kato T, Sato S, Tarkowska D, Tabata S, Sandberg G, Kakimoto T. Roles of Arabidopsis ATP/ADP isopentenyltransferases and tRNA isopentenyltransferases in cytokinin biosynthesis. Proc Natl Acad Sci U S A. 2006;103:16598–16603. doi: 10.1073/pnas.0603522103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pils B, Heyl A. Unraveling the evolution of cytokinin Signaling. Plant Physiol. 2009;151:782–791. doi: 10.1104/pp.109.139188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida K, Yamashino T, Nakanishi H, Mizuno T. Classification of the genes involved in the two-component system of the moss Physcomitrella patens. Biosci Biotechnol Biochem. 2010;74:2542–2545. doi: 10.1271/bbb.100623. [DOI] [PubMed] [Google Scholar]
- Park JY, Koo DH, Hong CP, Lee SJ, Jeon JW, Lee SH, Yun PY, Park BS, Kim HR, Bang JW. et al. Physical mapping and microsynteny of Brassica rapa ssp pekinensis genome corresponding to a 222 kbp gene-rich region of Arabidopsis chromosome 4 and partially duplicated on chromosome 5. Mol Genet Genomics. 2005;274:579–588. doi: 10.1007/s00438-005-0041-4. [DOI] [PubMed] [Google Scholar]
- Shiu SH, Karlowski WM, Pan R, Tzeng YH, Mayer KF, Li WH. Comparative analysis of the receptor-like kinase family in Arabidopsis and rice. Plant Cell. 2004;16:1220–1234. doi: 10.1105/tpc.020834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JZ. Evolution by gene duplication: an update. Trends Ecol Evol. 2003;18:292–298. doi: 10.1016/S0169-5347(03)00033-8. [DOI] [Google Scholar]
- Nowak MA, Boerlijst MC, Cooke J, Smith JM. Evolution of genetic redundancy. Nature. 1997;388:167–171. doi: 10.1038/40618. [DOI] [PubMed] [Google Scholar]
- Jensen RA. Enzyme recruitment in evolution of new function. Annu Rev Microbiol. 1976;30:409–425. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- Orgel LE. Gene duplication an origin of proteins with novel functions. J Theor Biol. 1977;67:773–773. doi: 10.1016/0022-5193(77)90262-4. [DOI] [PubMed] [Google Scholar]
- Hughes AL. The evolution of functionally novel proteins after gene duplication. Proceedings of the Royal Society Biological Sciences. 1994;256:119–124. doi: 10.1098/rspb.1994.0058. [DOI] [PubMed] [Google Scholar]
- Zhang JZ, Rosenberg HF, Nei M. Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc Natl Acad Sci U S A. 1998;95:3708–3713. doi: 10.1073/pnas.95.7.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RY, Skoog F, Playtis AJ, Leonard NJ. Cytokinins: synthesis and biological activity of geometric and position isomers of zeatin. Plant Physiol. 1972;50:702–705. doi: 10.1104/pp.50.6.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminek M, Vanek T, Motyka V. Cytokinin activities of N-6-benzyladenosine derivatives hydroxylated on the side-chain phenyl ring. Journal of Plant Growth Regulation. 1987;6:113–120. [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J. et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Lu SN, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR. et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu ZX, Ma Q, Lin YX, Tang XL, Zhou YQ, Zhu SW, Fan J, Cheng BJ. Genome-wide identification, classification, and analysis of two-component signal system genes in maize. Genet Mol Res. 2011;10:3316–3330. doi: 10.4238/2011.December.8.3. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34:W369–W373. doi: 10.1093/nar/gkl198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method-A new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Nielsen H, Brunak S, Von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- Higo K, Ugawa Y, Iwamoto M, Korenaga T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999;27:297–300. doi: 10.1093/nar/27.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HB, Bowers JE, Wang XY, Ming R, Alam M, Paterson AH. Perspective - Synteny and collinearity in plant genomes. Science. 2008;320:486–488. doi: 10.1126/science.1153917. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Hagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li JW, Thiagarajan M, White JA, Quackenbush J. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Alignment of amino acid sequences of BrIPTs with AtIPTs. BrIPTs had two conserved regions, Region a and Region b. Region a, denoted by red boxes, contained a putative ATP/GTP-binding sites (P-loop) motif (prosite PS00017: consensus sequence TGxGKS), which were underlined with a black line. Region b was a putative tRNA binding site, denoted by red boxes.
Summary of conserved motifs of BrIPT protein sequences.
Alignment of amino acid sequences of BrCKXs with AtCKXs. The position of the motif for covalent binding of FAD cofactor (GHS, where H is the binding residue in all plant CKXs) was indicated. Amino acid residues predicted based on the structure of ZmCKX1 to bind the cytokinin substrate were marked with an asterisk. The cytokinin N9 hydrogen-bonding residue that affected the substrate specificity was marked with a red arrow.
Summary of conserved motifs of BrCKX protein sequences.
Synteny analysis of BrIPT genes in ±100 kb region with score greater than 1000. Synteny analysis revealed evidence of the segmental duplications among BrIPT genes. Synteny analysis of Bra036719 (BrIPT2-1) and Bra040677 (BrIPT2-2) were omitted owing to unknown scaffold location of Bra040677.
Synteny analysis of BrCKX genes in ±100 kb region with score greater than 1000. Synteny analysis revealed evidence of the segmental duplications among BrCKX genes.
Determination of Ka and Ks values of IPT genes. The Ka and Ks values of IPT genes were determined between duplicated genes in B. rapa and the homologous genes among the B. rapa, A. thaliana and A. lyrata. — means no duplicated genes were found.
Determination of Ka and Ks values of CKX genes. The Ka and Ks values of CKX genes were determined between duplicated genes in B. rapa and the homologous genes among the B. rapa, A. thaliana and A. lyrata. - means no duplicated genes were found.
Summary of the cis-elements found in the putative promoter regions of BrIPT genes. Cis-elements with larger numbers were marked red.
Summary of the cis-elements found in the putative promoter regions of BrCKX genes.Cis-elements with larger numbers were marked red.
Forward and Reverse Primers used in the qRT-PCR analysis.