

Abstract
Purpose
A previous report has indicated that over-expression of cofilin-1 (CFL-1), a member of the actin depolymerizing factor (ADF)/cofilin protein family, enhances cellular radiosensitivity. This study explores, the involvement of various DNA damage responses and repair systems in the enhanced cellular radiosensitivity as well as assessing the role of CFL-1 phosphorylation in radiosensitivity.
Materials and Methods
Human non-small lung cancer H1299 cells harboring a tet-on gene expression system were used to induce exogenous expression of wild-type CFL-1. Colony formation assays were used to determine cell survival after γ-ray exposure. DNA damage levels were determined by comet assay. DNA repair capacity was assessed by fluorescence-based DNA repair analysis and antibody detection of various repair proteins. The effects of CFL-1 phosphorylation on radiation responses were explored using two mutant CFL-1 proteins, S3D and S3A. Finally, endogenous CFL-1 phosphorylation levels were investigated using latrunculin A (LA), cytochalasin B (CB) and Y27632.
Results
When phosphorylatable CFL-1 was expressed, radiosensitivity was enhanced after exposure to γ-rays and this was accompanied by DNA damage. Phosphorylated histone H2AX (γ-H2AX) and p53-binding protein-1 (53BP1) foci, as well as Chk1/2 phosphorylation, were apparently suppressed, although ataxia telangiectasia mutated (ATM) kinase activation was apparently unaffected. In addition, two radiation induced double strand break (DSB) repair, systems, namely homologous recombination repair (HRR) and non-homologous end joining (NHEJ), were suppressed. Moreover, over-expression of CFL-1 S3D and CFL-1 S3A both enhanced radiosensitivity. However, enhanced radiosensitivity and reduced γ-H2AX expression were only detected in cells treated with LA which increased endogenous phospho-CFL-1, and not in cells treated with Y27632, which dephosphorylates CFL-1.
Conclusion
CFL-1 over-expression enhances radiosensitivity and this is associated with reduced DNA repair capacity. Although phosphorylated CFL-1 seems to be involved in radiosensitivity, further studies are required to address the importance of CFL-1 activity to the regulation of radiosensitivity.
Keywords: cofilin-1, radiosensitivity, DNA repair capacity, radiotherapy, γ-H2AX
Introduction
The intrinsic radiosensitivity of tumor cells is an important factor for determining radiotherapeutic efficacy. Intrinsic radiosensitivity is an addition to the conventional four Rs associated with radiotherapy and radiobiology, namely repair, repopulation, redistribution and reoxygenation (Steel et al. 1989). During tumor development, changes in the radiosensitivity of tumor cells may assist tumors in evading apoptosis, in avoiding cell cycle arrest and in activating oncogenes (Harrington et al. 2007). Enhancement of tumor radiosensitivity is believed to be important factor in improving the efficacy of current radiotherapeutic protocols.
The actin depolymerizing factor (ADF)/cofilin protein family consists of actin-binding proteins that are 15–19 kDa in size; they are important to actin dynamics in vitro and in vivo (Bamburg et al. 1999). Three isoforms of ADF/cofilin that are expressed in vertebrates have been identified, these are non-muscle cofilin (CFL-1), muscle cofilin (CFL-2) and ADF (Gillett et al. 1996). CFL-1 and ADF are primarily expressed in non-muscle mammalian cells, although CFL-1 predominates (Wang et al. 2007). The activity of CFL-1 is regulated by serine-3 phosphorylation via the Rho-associated protein kinase (ROCK)-LIM kinase (LIMK) signaling pathway, and protein phosphatase slingshot homolog 1 (SSH1) (Arber et al. 1998; Nishita et al. 2004). Phosphorylated CFL-1 is the inactive form of this protein and is unable to regulate actin dynamics either in vitro or in vivo. Exogenous over-expression of wild-type CFL-1 has been reported to alter cell shape and to enhance the radiosensitivity of human lung cancer cells (Lee et al. 2005). In contrast to these findings, a recent report has indicated that over-expression of CFL-1 and phosphoglycerate kinase 1 are both involved in the radioresistant phenotype of astrocytomas (Yan et al. 2012). Although the role of cofilin in cellular radiosensitivity remains unclear, the protein may influence ionizing radiation induced DNA unwinding via the regulation of actin architecture and cell shape (Olive & MacPhail 1992). However, whether the CFL-1 activity is essential for radiosensitivity remains to be addressed.
The capacity to carry out repair when DNA is damaged by ionizing radiation is a critical factor in determining cellular radiosensitivity. Ionizing radiation induces DNA double strand breaks (DSB), which is the most harmful type of DNA damage. These breaks are mainly repaired through homologous recombination repair (HRR) and nonhomologous end joining (NHEJ). Both mechanisms are initiated by the activation of ataxia telangiectasia mutated (ATM) kinase, which subsequently initiates a series of molecular events via regulation of the Mre11/Rad50/Nbs1 (MRN) protein complex (Czornak et al. 2008). This complex binds to and processes the broken DNA ends via inherent nuclease activity. Subsequently, during HRR, Rad51/Rad52 are recruited to DSB sites due to assistance from the BRCA1/2 tumor suppressor proteins. Alternatively, as part of the NHEJ machinery, the Ku70/Ku80/DNA protein kinase catalytic subunits (DNA-PKcs) from the protein complex are involved in the recognition of DSB sites. The Ku70/Ku80/DNA-PKc complex activates artemis endonuclease activity, which then further processes the DNA ends. The ends are then filled in by DNA polymerase family proteins and ligated together by the PNK/XRCC4/DNA ligase IV/XLF complex (Lieber 2010). Deregulation of Ku70, Ku80 and Rad51 has been reported to increase cellular radiosensitivity (Taki et al. 1996; Gu et al. 1997; Harima et al. 2003). In contrast, over-expression of Rad51 and Rad52 confers resistance to ionizing radiation (Park 1995; Vispe et al. 1998). The histone protein H2AX is a substrate of ATM kinase and is important for recruiting DNA repair molecules to damage sites after exposure to ionizing radiation (Paull et al. 2000; Keogh et al. 2006). Phosphorylated histone H2AXs (termed γ-H2AX) are promptly detected after ionizing radiation and are eventually down-regulated after the DNA damage sites have been repaired. Suppression of DNA repair capacity may lead to an enhancement of radiosensitivity.
In this study, we found that exogenous over-expression of CFL-1 enhanced cellular radiosensitivity. Phosphorylation of H2AX and related DNA repair machinery was inhibited by over-expressed phosphorylatable CFL-1. Moreover, expression of the phosphomimetic S3D form of CFL-1 or unphosphorylatable S3A form of CFL-1 both enhanced radiosensitivity. However, while treatment with an actin inhibitor was able to mediate the induction of endogenous phospho-CFL-1, causing enhanced radiosensitivity, when ROCK inhibitor mediated reduction of phospho-CFL-1 was carried out, this did not influence radiosensitivity. This leaves open the question as to whether the phosphocycle of CFL-1 is important to the regulation of radiosensitivity.
Materials and Methods
Cell lines
Human non-small lung cancer A549 cells, H1299 cells, and HCOXP (H1299 cells with stable cofilin over-expression) cells were cultured and maintained as reported previously (Lee et al. 2005). All of the cell lines were maintained at 37°C in a humidified incubator (5% CO2 and 95% air) and routinely subcultured every 2–3 days.
Reagents and antibodies
The reagents used in this study include latrunculin A (LA) (Calbiochem-Merck Millipore, Darmstadt, Germany), cytochalasin B (CB), doxycycline and Y27632 (Sigma-Aldrich Co. LLC, St. Louis, MO, USA). The antibodies used for Western blot analysis include the following: cofilin-1, profilin-1, actin, ARP2/3, vinculin, Wiskott-Aldrich Syndrome protein (WASP), RhoA, α-actinin, plastin, tropomyosin 2, formin-like protein 1 (FMNL1), p53 binding protein-1 (53BP1) and destrin (GeneTex, Inc., Irvine, CA, USA), p-cofilin, H2AX, Rad51, Rad52 and Ku70 (Santa Cruz Biotechnology, Inc., CA, USA), γ-H2AX (Sigma Aldrich Co. LLC, St. Louis, MO, USA), Ku-80 (Calbiochem-Merck Millipore, Darmstadt, Germany), Chk1, Chk2 and phospho-Chk1 (Ser 345), phospho-Chk2 (Tyr 68), and the Actin Reorganization Antibody Sampler Kit (Cell Signaling Technology, Inc., Danvers, MA, USA).
Western blot analysis
The cell lysates were collected, separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by Western blotting as described previously (Tsai et al. 2009)
Radiation exposure
Treatment with γ-rays was delivered by a cesium-137 source at a dose rate of 385 cGy/min (BIOBEAM 2000, MCP Medical International, Berlin, Germany).
Colony formation assay
The cells were trypsinized and resuspended in T25 flasks, which was followed by γ-ray exposure at room temperature at single doses of 2, 4, 6, and 8 Gy. After irradiation, the cells were seeded onto 6 cm dishes and incubated for 14 days without disturbance. The seeded cell number was increased as the radiation dosage increased. Formed colonies were visualized by staining with 0.02% crystal violet solution (w/v in 75% ethanol). The plating efficiency was determined as the ratio of the number of colonies divided by the number of cells seeded. The surviving fraction was determined from the ratio of plating efficiencies of irradiated cells compared to the unirradiated control. Each datum represents the mean of three independent experiments ± S.D.
Comet assay
Experimental evaluation of DNA damage and DNA repair capacity was conducted by comet assay as described previously (Lee et al. 2005). Briefly, cells were exposed to the indicated doses of ionizing radiation and then mixed with 10 μl Dulbecco’s Minimal Essential Medium (DMEM) (10,000 cells). The cell suspension was mixed with 75 μl of 0.75% low melting point agarose (LAMDA Biotech Inc., St. Louis, MO, USA) and loaded onto a non-frosted slide that had been pre-coated with 1% air-dried conventional agarose. The slides were incubated with lysis buffer (2.5 M NaCl; 100 mM Na2EDTA; 10 mM Tris-HCl, pH 10; and 1% Triton X-100) at 4°C for 1 hour and in an alkaline running buffer (300 mM NaOH and 1 mM Na2EDTA, pH > 13) for another hour. After electrophoresis, the slide was neutralized by placing in a neutralization buffer (0.4 mM Tris-HCl, pH 7.5), stained with ethidium bromide (20 μg/ml), and visualized using a fluorescent microscope (Leica DM IRB, Wetzlar, Germany). The tail moment of each single cell was determined as the percentage of DNA in the comet tail multiplied by the length of tail (that is [tail DNA/(head DNA + tail DNA)] × 100 × tail length). Between 50 and 200 cells were scored during each independent experiment.
Immunofluorescence staining
In brief, 3×103 cells were seeded on coverslips and incubated for 48 hours. The cells were then fixed with 4% paraformaldehyde in phosphate buffered saline (PBS) and stained with Alexa Fluor 488-conjugated phalloidin (1:200) for 30 minutes followed by 4′,6-diamidino-2-phenylindole (DAPI) for 10 minutes. The stained cells were examined using laser confocal microscopy (Olympus FV1000, Olympus Corporation, Japan). Foci of γ-H2AX were detected as described previously (Friesner et al. 2005).
Fluorescence-based analysis of HRR and NHEJ
Visualization of HRR and NHEJ was carried out using fluorescent reporter constructs as reported previously (Seluanov et al. 2010). In brief, the HRR and the NHEJ reporter cassettes each separately contain an engineered green fluorescent protein (GFP) sequence with an I-SceI restriction enzyme site; these were digested using the I-SceI endonuclease (New England Biolabs Inc., Ipswich, MA, USA). Linearized plasmid (10 μg) and linearized pDsRed2-N1 (0.2 μg, Clontech Laboratories, Inc., Mountain View, CA, USA) were co-transfected into HCOXP cells cultured in 24-well plates, and doxycycline was added 6 hours after transfection to induce CFL-1 expression or alternatively the cells were left untreated as the control. After 48 hours of incubation, the cells were exposed to 8Gy of γ-rays, and the fluorescent signals were visualized and captured using a fluorescent inverted microscope (CKX41 model, Olympus Inc., Center Valley, PA, USA).
Cell viability assay
Cell viability was measured by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide) assay. In brief, 4,000 cells were seeded per well in 96-well plates. MTT solution dissolved in the culture medium at a final concentration of 0.5 mmol/L was added to each well and incubated for 4 hours. Dimethyl sulfoxide was added to dissolve the MTT crystals and the optical density was measured at 570 nm using an enzyme-linked immunosorbent assay (ELISA) plate reader (Sunrise™; TECAN Group Ltd., Männedorf, Switzerland).
Statistical analysis
Independent experiments were conducted determine whether there were significant differences between the control and experimental groups. Significant differences were determined by the Student’s t-test (Prism 3.0, Graphpad software Inc., La Jolla, CA, USA); the results of the statistical analysis were considered significant when p<0.05.
Results
Over-expression of phosphorylatable CFL-1 enhances the radiosensitivity of human lung cancer cells
HCOXP cells are able to over-express exogenous wild-type CFL-1 when induced by doxycycline and become more sensitive to γ-rays (Lee et al. 2005). In this study, we further investigated serine-3 phosphorylation in this stable cell line after CFL-1 over-expression. Doxycycline induced CFL-1 and this induction resulted in a dose-dependent increase in phosphorylated CFL-1 (p-CFL) in the HCOXP cells (Figure 1A). The dose-dependent induction of CFL-1 was determined at different levels o of doxycycline, namely 0, 0.02, 0.04 and 0.1 μg/ml (Figure 1B). To investigate whether serine-3 phosphorylatable wild-type CFL-1 is associated with a change in radiosensitivity, HCOXP cells that had been treated with different concentrations of doxycycline were exposed to γ-rays were subjected to the colony formation assay. Compared to the parental H1299 cells, the CFL-1/pCFL over-expressing HCOXP cells exhibited a significant increase in sensitivity to γ-rays (Figure 1C). The enhancement in radiosensitivity of the cells expressing different levels of CFL-1 and p-CFL was then determined by calculating the survival fraction of the cells after exposure to γ-rays in a range 0 to 8 Gy. The derived survival curves demonstrated that HCOXP cells exposed to 0, 0.02, 0.04 and 0.1 μg/ml of doxycycline exhibited a dose-dependent increase in radiosensitivity (Figure 1D). The dose-modifying factor (DMF) of HCOXP cells expressing a 9-fold increase in CFL-1/p-CFL (induced by 0.1 μg/ml) compared to untreated cells were approximately 1.6 and 1.7 at survival fractions (SF) of 5% and 10%, respectively. The DMF was lower when cells were over-expressing lower levels of CFL-1; the results are summarized in Table 1. Therefore, based on the above finding, over-expression of phosphorylatable wild-type CFL-1 would seem to be able to alter cellular radiosensitivity.
Figure 1.

The effects of exogenous over-expression of phosphorylatable CFL-1 on cellular radiosensitivity. (A) Dose-dependent induction of phosphorylatable CFL-1 expression in HCOXP cells treated with doxycycline. The expression of serine-3 phosphorylated CFL-1 (p-CFL) and total CFL-1 were detected by Western blot analysis. (B) Densitometric measurement of doxycycline-induced CFL-1 expression. (C) The effects of over-expressed CFL-1 on cell viability after irradiation were compared by the colony formation assay. Dox: 0.1 μg/ml. (D) Survival curves of normal and concentration-dependent CFL-1 over-expressing cells were compared by calculating the survival fractions after irradiation. Each datum represented the mean of three independent experiments ± S.D.. The statistical analysis compared the survival fractions between untreated controls and cells treated with different concentrations of doxycycline. *, **, and *** represent p <0.05 for cells treated with 0.02, 0.04, and 0.1 μg/ml of doxycycline compared to untreated controls after irradiation.
Table 1.
The dose modifying factor (DMF) of cells with over-expressed CFL-1
| Induced CFL-1 level (Folds)a | Survival Fractions (SF) b
|
|
|---|---|---|
| 10 % | 5 % | |
| 3 | 1.26 0.077 | 1.23 0.064 |
| 6 | 1.46 0.047 | 1.40 0.058 |
| 9 | 1.72 0.082 | 1.62 0.03 |
Compared to normal status of HCOXP cells (without doxycycline treatment)
The radiation dose required for reaching preset SF before and after over- expression of CFL-1.
Radiation-induced DNA damage is enhanced and sustained in cells expressing phosphorylatable CFL-1
To investigate if the DNA damage level is influenced by the level of phosphorylated CFL-1 in cells, the comet assay was used to measure the tail moments of cells over-expressing phosphorylated CFL-1. It was found that over-expression of phosphorylatable CFL-1 in HCOXP cells did not induce DNA damage; nevertheless, the level of DNA damage in these cells was greatly increased compared with normal cells for up to 6 hours after irradiation (Figure 2A). DNA damage remained apparent in CFL-1 over-expressing cells at 24 hours after irradiation, but such damage was not apparent in normal cells at 24 hours after irradiation (Figure 2A). Additionally, the tail moment measurements were able to provided further evidence that the DNA damage found in CFL-1 over-expressing cells immediately after irradiation was significantly higher compared to normal cells immediately after irradiation and that this increase was sustained for up to 24 hours (Figure 2B). In contrast, the radiation-induced DNA damage found in normal cells was repaired within 6 hours. Therefore, it would seem that exogenous over-expression of phosphorylatable CFL-1 enhances radiation-induced DNA damage and delays DNA repair.
Figure 2.

The detection of γ-ray-induced DNA damage and repair responses in phosphorylatable CFL-1 over-expressing cells. (A) Comet assays were used to assess DNA damage levels in each individual cell with or without over-expressed CFL-1. The arrow heads indicate the comet tails that were detected by fluorescence microscopy. (B) The tail moments of the normal and CFL-1 over-expressing cells before and after irradiation were compared. Approximately 50 cells were measured for each experimental condition. Error bars indicate the standard deviation (S.D.) of the mean for 50 counted cells. *: p <0.05.
Exogenous over-expression of phosphorylatable CFL-1 suppresses ionizing radiation induced DNA damage responses
ATM kinase is important for sensing ionizing radiation-induced DSB and γ-H2AX formation (Burma et al. 2001). The appearance of nuclear γ-H2AX foci is an early response to DSB, and is important for Rad50, Rad51, and BRCA1 foci formation at the DSB sites for repair (Paull et al. 2000), Thus, we investigated whether exogenous over-expression of CFL-1 would influence ATM kinase activity and H2AX phosphorylation. The Western blot analysis demonstrated that the active form of ATM kinase (serine 1981 phosphorylated form) was not inhibited in normal and CFL-1 over-expressing cells after irradiation, but the level of γ-H2AX was greatly suppressed in irradiated CFL-1 over-expressing cells compared with normal cells (Figure 3A). ATM kinase mediated phosphorylation of the Chk1 and Chk2 kinases was also suppressed under the same conditions.
Figure 3.

The effect of over-expressed phosphorylatable CFL-1 on the formation of γ-H2AX after γ-ray exposure. (A) The effect of over-expressed phosphorylatable CFL-1 on ATM kinase activation, H2AX phosphorylation and Chk1/2 phosphorylation before and after γ-ray exposure were compared by Western blot analysis. (B) The visualization of the expression and distribution of phosphorylatable CFL-1 in HCOXP cells before and after doxycycline (0.1 μg/ml) treatment. DAPI was used for nuclear staining. (C) The detection of γ-H2AX foci formation in the nucleus under different conditions. (D) The quantification of cells expressing γ-H2AX foci.
Interestingly, confocal microscopy demonstrated that over-expressed CFL-1 and p-CFL could be detectable in nuclei of HCOXP cells (Figure 3B). Indirect immunofluorescence was subsequently used to examine the formation of γ-H2AX foci in the nuclei, and it was found that the formation of γ-H2AX foci after irradiation was significantly suppressed by over-expression of CFL-1 (Figure 3C). The formation of γ-H2AX foci under various conditions was quantified and is shown in Figure 3D. In a separate experiment, we also detected that the formation of Rad51 foci after exposure to ionizing radiation and found that this was also suppressed by over-expression of CFL-1 (Supplementary Figure 1). These observations suggested that the formation of γ-H2AX and any subsequent DNA damage responses are suppressed by over-expression of CFL-1.
CFL-1 over-expressing cells, when treated with ionizing radiation, show a reduction in DNA repair capacity
Ionizing radiation-induced DNA damage is known to initiate various DNA repair mechanisms, including HRR and NHEJ, which involve Rad51/Rad52 and Ku70/Ku80/DNA-PKc, respectively. Additionally, Mre11 is known to form a complex with Nbs1 and Rad50 in order to provide nuclease activity that allows DNA damage sites to be processed following irradiation (Buis et al. 2008). To determine whether over-expression of CFL-1 affect HRR and NHEJ, a GFP-based method was used to analyze DNA DSB repair (see materials and methods). The results showed that the HRR and NHEJ mediated repair of I-SceI endonuclease digested GFP expression cassettes was suppressed in HCOXP cells over-expressing CFL-1 (+dox) compared to untreated cells (-dox) after exposure to ionizing radiation (Figure 4A). The level of GFP expression was quantified and normalized against the co-transfected DsRed gene. The findings revealed that the repair capacity of both the HRR and NHEJ systems were reduced in irradiated CFL-1 over-expressing cells, although the suppression of HRR seemed be greater than the suppression of NHEJ (Figure 4B). The expression of the various DNA repair molecules involved in these repair systems were compared between normal and CFL-1 over-expressing cells collected at 3, 6, 12 and 24 hours after exposure to ionizing radiation. The results demonstrated that expression of Rad51, K70, and Ku80 were suppressed in CFL-1 over-expressing cells compared with normal cells after irradiation; interestingly, the reduction in Rad51 expression was greater than the suppression of Ku70/Ku80 expression (Figure 4C and 4D). In contrast, the expression levels of Rad52 and Mre11 did not show any significant change. Therefore, it would seem that the enhancement of radiosensitivity in cells expressing phosphorylatable CFL-1 seems to be associated with a reduction in DNA repair capacity.
Figure 4.

The effects of over-expressed phosphorylatable CFL-1 on HRR and NHEJ repair capacity after irradiation. (A) Fluorescence-based DNA repair analysis for HRR and NHEJ in HCOXP cells. Dox: 0.1 μg/ml for 48 hours; IR: 8 Gy. (B) Quantification of GFP ratio normalized against DsRed. (C) A time-course comparison of proteins involved in the HRR and NHEJ repair mechanisms with or without the over-expression of phosphorylatable CFL-1 followed by ionizing irradiation. (D) A densitometric analysis of the expression of DNA repair proteins in cells after γ-ray exposure. Each datum represented the mean of three independent experiments ± S.D.. *: p < 0.05 compared to cells without over-expressed CFL-1.
Over-expression of constitutively phosphorylated and unphosphorylated mutant CFL-1 proteins enhances radiosensitivity
To better understand whether the phosphorylated form of CFL-1 is important to the increase in cellular radiosensitivity, a constitutively phosphorylated mutant of CFL-1 (S3D-cofilin) and an unphosphorylated mutant of CFL-1 (S3A-cofilin) were used to investigate the cellular response to γ-rays. H1299 cells were independently transduced with S3D-cofilin and S3A-cofilin using a lentiviral infection system. Total cofilin expression was increased in the transduced cells. However, an increase in phosphorylated cofilin was only detected in cells transduced with the S3D cofilin mutant but not with the S3A cofilin mutant (Figure 5A). Subsequently, the transduced cells were exposed to different γ-ray dosages, and it was found that both the S3D mutant cofilin and the S3A mutant cofilin exhibited increased radiosensitivity (Figure 5B). These findings suggests that exogenous over-expression of both of these mutant forms of CFL-1 (constitutively phosphorylated and unphosphorylated) are able to influence cellular radiosensitivity.
Figure 5.
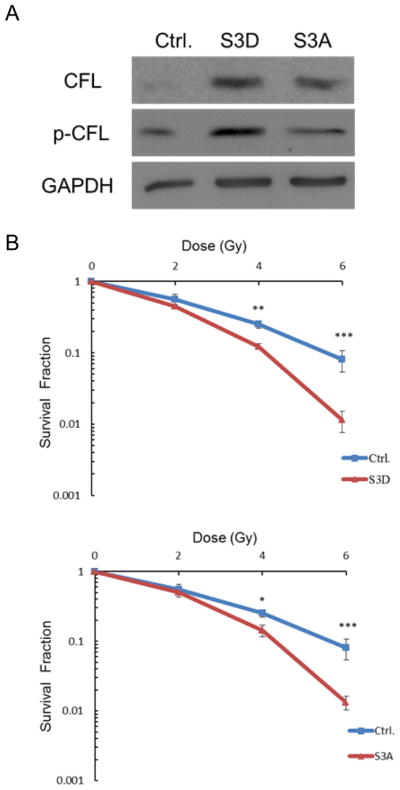
The effect of the constitutively phosphorylated mutant S3D CFL-1 and the unphosphorylated mutant S3A CFL-1 on cellular radiosensitivity. (A) Total and serine-3 phosphorylation levels of S3D and S3A CFL-1 in H1299 cells after transduction. (B) The survival curves of the vector, S3D and S3A transduced cells exposed to different γ-ray dosages. Each datum represented the mean of three independent experiments ± S.D. *: p < 0.05; **: p < 0.01; ***: p < 0.001.
1. Induction of cofilin-1 phosphorylation by treatment with the actin inhibitor LA but not by the actin inhibitor CB
CFL-1 activity is essential for actin organization. It has been found that an acute disruption of the actin cytoskeleton by an actin inhibitor leads to an increase in the phosphorylated form of cofilin in Drosophila S2R+ cells (Jovceva et al. 2007). In this study, we examined the effects of two different types of actin inhibitors, LA and CB, on cofilin-1 phosphorylation. The effect of latrunculin in terms of actin cytoskeleton disruption is at least 10-fold higher than that of cytochalasin (Spector et al. 1989). Both LA (1 μM) and CB (10 μM) were found to disrupt the actin architectures of H1299 cells compared to the situation in untreated control cells (Figure 6A). At the above concentrations, both actin inhibitors caused a similar reductions in cell viability (approximately 70% survival) (Figure 6B). A survey of actin-associated molecules, including total CFL-1, p-CFL, ADF, profilin, Rho A, vinculin, ARP2/3, vasodilator-stimulated phosphoprotein (VASP), p-VASP, α-actinin, plastin, ezrin/radixin/moesin (ERM), and p-ERM, demonstrated that p-CFL was the primary molecule significantly up-regulated by LA; but such up-regulation did not occur with CB (Figure 6C). Concentration-dependent treatment with the inhibitors confirmed that only LA and not CB was able to induce serine-3 phosphorylation of CFL-1 (Supplementary Figure 2). Subsequently, colony formation assays were performed and demonstrated that radiosensitivity was enhanced in the LA-treated cells but not in CB-treated cells (Figure 6D). The DMF of the LA-treated cells was 1.25 at 10% SF. Previously in this study we have shown that exogenous over-expression of phosphorylatable CFL-1 is able to repress γ-H2AX expression after irradiation (Figure 3A); a similar reduction in the level of γ-H2AX was found in LA-treated cells but not in CB-treated cells after irradiation (Figure 6E). The formation of γ-H2AX foci in the LA-treated cells was also reduced compared to untreated control cells after ionizing radiation (Supplementary Figure 3). However, whether phosphorylated cofilin-1 is essential for LA enhanced cellular radiosensitivity. requires further investigation.
Figure 6.

The association between phosphorylated CFL-1 and actin inhibitor-induced radiosensitivity. (A) The effects of LA and CB on the destabilization of actin cytoskeleton. (B) The MTT assay was used to determine the effect on cell viability of LA and CB. Each datum represented the mean of three independent experiments ± S.D. * and **: p <0.05. (C) A survey of the expression of actin-associated proteins in cells treated with LA or CB. *: >3-fold increase in expression compared with the untreated control cells by densitometry. (D) The effects of LA and CB on changes in radiosensitivity. Each datum represented the mean of three independent experiments ± S.D. *: p <0.05. (E) The expression of LA-treated or CB-treated γ-H2AX cells followed by irradiation. IR: 8 Gy of γ-rays. Time of drug treatment: 12 hours.
Radiosensitivity is unaffected by increased dephosphorylated of CFL-1 in cells
We next examined whether dephosphorylation of endogenous CFL-1 was able to influence radiosensitivity. To promote CFL-1 dephosphorylation, H1299 cells were treated with Y27632, a selective inhibitor for ROCK, which is important for CFL-1 phosphorylation. Y27632 was able to quickly dephosphorylate CFL-1 in only 5 to 10 minutes, and this dephosphorylation was sustained for up to 3 hours following treatment (Figures 7A and 7B). The total amount of CFL-1 present in the cells was not apparently affected by Y27632. Compared to the untreated controls, there was no significant change in radiosensitivity in the Y27632-treated cells (Figure 7C). These results suggest that an increase in the dephosphorylated form of CFL-1 but not in total CFL-1 does not seem to influence cellular radiosensitivity. However, additional investigations are needed to confirm this observation.
Figure 7.

The effects of a ROCK inhibitor on CFL-1 phosphorylation and cellular radiosensitivity. (A) Changes in CFL-1 and p-CFL levels in H1299 cells treated with Y27632 (10 μM) at different times. (B) The quantification of p-CFL levels normalized by GAPDH using densitometry. *: p<0.05 compared to the untreated control (ctrl). (C) The survival curves of Y27632-treated and untreated cells that had been irradiated. Each datum represented the mean of three independent experiments ± S.D.
Discussion
The significance of the effect of CFL-1 on the regulation of cellular radiosensitivity was further investigated in this study. The phosphocycle of CFL-1 is important for determining the functionality of CFL-1 in terms of actin dynamics. The current findings reveal that exogenous over-expression of phosphorylatable CFL-1 is able to enhance cellular radiosensitivity and suppress DNA repair capacity after irradiation by interfering with the formation of nuclear γ-H2AX foci, which subsequently affects the HRR and NHEJ pathways. These observations suggest that radiation-induced DSB are less frequently repaired in CFL-1 over-expressing cells compared to normal cells. Additionally, exogenous over-expression of both S3D mutant CFL-1 and S3A mutant CFL-1, which have opposite serine-3 phosphorylational phenotypes, also increases radiosensitivity, However, when endogenous phosphorylated CFL-1 was reduced by Y27632 treatment, this did not influence cellular radiosensitivity; while, in contrast, the actin inhibitor LA, which increased endogenous phosphorylated CFL-1, did enhance radiosensitivity in treated cells. Compared to the chemical treatments, exogenous over-expression of S3A mutant CFL-1 and S3D mutant CFL-1 not only increases the amount of CFL-1 in different phosphorylated states, but also increases the total amount of CFL-1 in cells. One possible explanation of this contradiction is that an increase in total CFL-1 may be sufficient to influence cellular radiosensitivity, whereas a change in the serine-3 phosphorylation state of CFL-1, but not total CFL-1, has a distinct and different influence on radiosensitivity. Additional studies are needed to investigate the role of CFL-1 phosphorylation on regulation of the cells responses to radiation response and their DNA repair capacity.
It has been reported previously that the level of phosphorylated CFl-1 is lower in tumorigenic cell lines compared with non-tumorigenic or resting peripheral T-lymphocytes (Nebl et al. 1996). Dephosphorylated CFL-1 is activated during the promotion of actin cytoskeletal turnover that occurs with the formation of membrane protrusions and invadopodia (Yamaguchi 2012). Both architectures are important during cancer invasion and metastasis. Wang et al. demonstrated that cofilin activity is directly related to the invasion, intravasation, and metastasis of mammary cancer cells (Wang et al. 2006). Therefore, it is plausable that dephosphorylated CFL-1 will be correlated with advanced malignancy, whereas an up-regulation of phospho-CFL-1 may inhibit tumor progression. Moreover, a recent study has reported that stable expression of the S3A mutant form of CFL-1 promotes local invasion and migration of astrocytoma in a xenograft tumor model (Nagai et al. 2011). In contrast, over-expression of LIMK2b, a p53-mediated molecule that increases the level of phospho-CFL-1, has been shown to suppress tumor cell growth (Hsu et al. 2010). In the present and earlier studies, we have demonstrated that CFL-1 expression has an influence radiosensitivity. Additionally, this study shows that the actin inhibitor LA increases phosphorylated CFL-1 and that this enhances radiosensitivity, while a second actin inhibitor CB does not exhibit a similar phenotype. However, an increase in the amount of dephosphorylated CFL-1 present in cells after treatment with Y27632 also did not alter radiosensitivity. Therefore, it appears that the phosphocycle of CFL-1 may not only be important to tumor progression but also, at least in part, to intrinsic radiosensitivity.
Although phosphorylated CFL-1 is regarded as biologically inactive during actin regulation, phosphorylated CFL-1 has been reported to bind and stimulate the activity of phospholipase D1 (PLD1) and affect this enzyme’s subsequent cellular functions (Han et al. 2007). PLD1 hydrolyzes phosphatidylcholine (PC), which is embedded in the cell membrane; this releases phosphatidic acid (PA), which is then converted into diacylglycerol (DAG), a secondary messenger that is required for intracellular signaling transduction. Radiation can promote PC hydrolysis via the stimulation of PLD1 activity, which suggests that PLD1 may mediate various pleiotropic effects of ionizing radiation (Avila et al. 1993). Moreover, oral administration of PC has been reported to confer protective effects in rats against radiation (Fittkau et al. 2001). Therefore, the enhancement of radiosensitivity by phosphorylated CFL-1 may be associated with the molecule’s potential interactions with PLD1. Whether the interactions between phosphorylated CFL-1 and PLD1 are essential for regulating intrinsic radiosensitivity is of interest and needs further investigation.
Whether actin inhibitors are able to influence radiosensitivity has not been widely reported. Disruption of the actin cytoskeleton by various actin inhibitors is largely dependent on their biochemical activity. In this study, we treated the cells with two commonly used actin inhibitors, namely LA and CB, which destabilize the actin cytoskeleton by sequestering monomeric actin and capping the barbed-ends of the actin filaments, respectively (Cooper 1987; Yarmola et al. 2000). Surprisingly, we found that phosphorylated CFL-1 was primarily up-regulated by LA but was unaffected by CB, but total CFL-1 was not affected by either agent. LA treated cells were found to exhibit enhanced radiosensitivity but this was not true for CB treated cells; these findings are interesting and there is a need for further investigations of the role of phospho-CFL-1 in the regulation of radiosensitivity.
Ionizing radiation-induced DSB are rapidly repaired by the HRR and NHEJ systems that are initiated by various ATM-related protein kinases, including ATM, ATR, and possibly DNA-dependent protein kinase catalytic subunit (DNA-PKcs). ATM kinase is known to phosphorylate a range of protein substrates, including H2AX, Chk1, Chk2, Rad17, NBS1, BRCA1, SMC1, 53BP1, and MDC1 after DSB are formed (Walworth & Bernards 1996; Lee & Paull 2007). Additionally, the formation of γ-H2AX is required for the retention of the foci formed by NBS1, 53BP1, MDC1 and BRCA1 at the DNA damage sites, which then allows subsequent repair procedures (Celeste et al. 2003). It has been reported that intermolecular autophosphorylation of ATM kinase at serine 1981 represents the initiation of ATM kinase activity after cells are exposed to ionizing radiation (Bakkenist & Kastan 2003). Our findings show that over-expressed CFL-1 does not reduce the level of ATM kinase phosphorylation after exposure to ionizing radiation, even though γ-H2AX formation and Chk1/2 phosphorylation are suppressed, Formation of nuclear 53BP1 foci was also found to decrease under these conditions (Supplementary Figure 4). This observation suggests that over-expressed CFL-1 does not suppress ATM kinase activity, but rather influences ATM kinase-mediated substrate phosphorylation during DNA repair. However, after exposure to ionizing radiation, H2AX is not only phosphorylated by ATM kinase but it is also phosphorylated by DNA-PKcs (Stiff et al. 2004). The involvement of DNA-PKcs in H2AX phosphorylation and a relationship with over-expressed CFL-1 cannot be excluded, and there is a need to further investigate whether CFL-1 is able to inhibit the activity of DNA-PKcs and affect the formation of γ-H2AX.
CFL-1 contains nuclear localization signals (NLS) and the protein is able to enter the nucleus in response to environmental stimuli (Yahara et al. 1996). Although the dephosphorylated form of cofilin is essential for nuclear entry under these conditions, Nagaoka et al. have proposed that exogenous phosphorylated cofilin (S3D) may be able to diffuse into the cell nucleus (Nagaoka et al. 1996). Consistent with their findings, we also found that over-expressed phosphorylatable CFL-1 in HCOXP cells accumulates in the nuclei. Although phosphorylatable CFL-1 may affect DNA repair capacity by suppressing γ-H2AX formation and thus affect the subsequent HRR and NHEJ repair pathways in our current results, it remains unclear whether unphosphorylated CFL-1 is able to exhibit a similar effect. In addition to exogenous over-expression of the non-phosphorylated mutant form of CFL-1 (S3A), knockdown of LIM kinase or over-expression of SSH1 may be able to reduce the phosphorylation level of CFL-1 and further investigations of the effects of such modifications on DNA repair after exposure to ionizing radiation are needed.
Conclusion
In summary, the current findings suggest that exogenous over-expression of phosphorylatable CFL-1 enhances cellular radiosensitivity. The DNA repair capacity after ionizing radiation treatment of these phosphorylatable CFL-1-expressing cells is significantly impaired. Specifically, the level of H2AX and Chk1/2 phosphorylation are reduced and the induction of the subsequent HRR and NHEJ DNA repair mechanisms are suppressed, although ATM kinase activity does not seem to be inhibited. Exogenous over-expression of the S3D mutant CFL-1 and the S3A mutant CFL-1 both enhance radiosensitivity. However, while the actin inhibitor LA is able to induce CFL-1 phosphorylation and enhance radiosensitivity, the actin inhibitor CB and the ROCK inhibitor Y27632 do not increase radiosensitivity in similar circumstances. It would be of interest to further investigate the involvement of the phosphocycle of CFL-1 in the regulation of cellular radiosensitivity and the induction of DNA repair machinery.
Supplementary Material
Acknowledgments
This study was supported by National Science Council of Taiwan, R.O.C. (NSC 99-2314-B-010-029-MY3 and 101-2623-E-010-002-NU), a grant from Taipei City Hospital (No.3), and a grant from the Ministry of Education, Aim for the Top University Plan, National Yang-Ming University.
Footnotes
Declaration of interest
The authors report no conflicts of interest.
References
- Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–9. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- Avila MA, Otero G, Cansado J, Dritschilo A, Velasco JA, Notario V. Activation of phospholipase D participates in signal transduction pathways responsive to gamma-radiation. Cancer Research. 1993;53:4474–6. [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Bamburg JR, McGough A, Ono S. Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends in Cell Biology. 1999;9:364–70. doi: 10.1016/s0962-8924(99)01619-0. [DOI] [PubMed] [Google Scholar]
- Buis J, Wu Y, Deng Y, Leddon J, Westfield G, Eckersdorff M, Sekiguchi JM, Chang S, Ferguson DO. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008;135:85–96. doi: 10.1016/j.cell.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. The Journal of Biological Chemistry. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nature Cell Biology. 2003;5:675–9. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- Cooper JA. Effects of cytochalasin and phalloidin on actin. The Journal of Cell Biology. 1987;105:1473–8. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czornak K, Chughtai S, Chrzanowska KH. Mystery of DNA repair: the role of the MRN complex and ATM kinase in DNA damage repair. Journal of Applied Genetics. 2008;49:383–96. doi: 10.1007/BF03195638. [DOI] [PubMed] [Google Scholar]
- Fittkau M, Gerlach R, Schmoll HJ. Protective effect of oral phosphatidylcholine on radiation-induced release of intestinal peptidases in rats. Journal of Cancer Research and Clinical Oncology. 2001;127:444–8. doi: 10.1007/s004320100244. [DOI] [PubMed] [Google Scholar]
- Friesner JD, Liu B, Culligan K, Britt AB. Ionizing radiation-dependent gamma-H2AX focus formation requires ataxia telangiectasia mutated and ataxia telangiectasia mutated and Rad3-related. Molecular Biology of the Cell. 2005;16:2566–76. doi: 10.1091/mbc.E04-10-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillett GT, Fox MF, Rowe PS, Casimir CM, Povey S. Mapping of human non-muscle type cofilin (CFL1) to chromosome 11q13 and muscle-type cofilin (CFL2) to chromosome 14. Annals of Human Genetics. 1996;60:201–11. doi: 10.1111/j.1469-1809.1996.tb00423.x. [DOI] [PubMed] [Google Scholar]
- Gu Y, Jin S, Gao Y, Weaver DT, Alt FW. Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8076–81. doi: 10.1073/pnas.94.15.8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Stope MB, de Jesus ML, Oude Weernink PA, Urban M, Wieland T, Rosskopf D, Mizuno K, Jakobs KH, Schmidt M. Direct stimulation of receptor-controlled phospholipase D1 by phospho-cofilin. The EMBO Journal. 2007;26:4189–202. doi: 10.1038/sj.emboj.7601852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harima Y, Sawada S, Miyazaki Y, Kin K, Ishihara H, Imamura M, Sougawa M, Shikata N, Ohnishi T. Expression of Ku80 in cervical cancer correlates with response to radiotherapy and survival. American Journal of Clinical Oncology. 2003;26:e80–5. doi: 10.1097/01.COC.0000077938.48974.59. [DOI] [PubMed] [Google Scholar]
- Harrington K, Jankowska P, Hingorani M. Molecular biology for the radiation oncologist: the 5Rs of radiobiology meet the hallmarks of cancer. Clinical Oncology : A Journal of the Royal College of Radiologists. 2007;19:561–71. doi: 10.1016/j.clon.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Hsu FF, Lin TY, Chen JY, Shieh SY. p53-Mediated transactivation of LIMK2b links actin dynamics to cell cycle checkpoint control. Oncogene. 2010;29:2864–76. doi: 10.1038/onc.2010.40. [DOI] [PubMed] [Google Scholar]
- Jovceva E, Larsen MR, Waterfield MD, Baum B, Timms JF. Dynamic cofilin phosphorylation in the control of lamellipodial actin homeostasis. Journal of Cell Science. 2007;120:1888–97. doi: 10.1242/jcs.004366. [DOI] [PubMed] [Google Scholar]
- Keogh MC, Kim JA, Downey M, Fillingham J, Chowdhury D, Harrison JC, Onishi M, Datta N, Galicia S, Emili A, et al. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature. 2006;439:497–501. doi: 10.1038/nature04384. [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 2007;26:7741–8. doi: 10.1038/sj.onc.1210872. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Sheu TJ, Keng PC. Enhancement of radiosensitivity in H1299 cancer cells by actin-associated protein cofilin. Biochemical and Biophysical Research Communications. 2005;335:286–91. doi: 10.1016/j.bbrc.2005.07.073. [DOI] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual Review of Biochemistry. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai S, Moreno O, Smith CA, Ivanchuk S, Romagnuolo R, Golbourn B, Weeks A, Seol HJ, Rutka JT. Role of the cofilin activity cycle in astrocytoma migration and invasion. Genes & Cancer. 2011;2:859–69. doi: 10.1177/1947601911431839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka R, Abe H, Obinata T. Site-directed mutagenesis of the phosphorylation site of cofilin: its role in cofilin-actin interaction and cytoplasmic localization. Cell Motility and the Cytoskeleton. 1996;35:200–9. doi: 10.1002/(SICI)1097-0169(1996)35:3<200::AID-CM3>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Nebl G, Meuer SC, Samstag Y. Dephosphorylation of serine 3 regulates nuclear translocation of cofilin. The Journal of Biological Chemistry. 1996;271:26276–80. doi: 10.1074/jbc.271.42.26276. [DOI] [PubMed] [Google Scholar]
- Nishita M, Wang Y, Tomizawa C, Suzuki A, Niwa R, Uemura T, Mizuno K. Phosphoinositide 3-kinase-mediated activation of cofilin phosphatase Slingshot and its role for insulin-induced membrane protrusion. The Journal of Biological Chemistry. 2004;279:7193–8. doi: 10.1074/jbc.M312591200. [DOI] [PubMed] [Google Scholar]
- Olive PL, MacPhail SH. Radiation-induced DNA unwinding is influenced by cell shape and trypsin. Radiation Research. 1992;130:241–8. [PubMed] [Google Scholar]
- Park MS. Expression of human RAD52 confers resistance to ionizing radiation in mammalian cells. The Journal of Biological Chemistry. 1995;270:15467–70. doi: 10.1074/jbc.270.26.15467. [DOI] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Current Biology. 2000;10:886–95. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- Seluanov A, Mao Z, Gorbunova V. Analysis of DNA double-strand break (DSB) repair in mammalian cells. Journal of Visualized Experiments : JoVE. 2010;43:2002. doi: 10.3791/2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector I, Shochet NR, Blasberger D, Kashman Y. Latrunculins--novel marine macrolides that disrupt microfilament organization and affect cell growth: I. Comparison with cytochalasin D. Cell Motility and the Cytoskeleton. 1989;13:127–44. doi: 10.1002/cm.970130302. [DOI] [PubMed] [Google Scholar]
- Steel GG, McMillan TJ, Peacock JH. The 5Rs of radiobiology. International Journal of Radiation Biology. 1989;56:1045–8. doi: 10.1080/09553008914552491. [DOI] [PubMed] [Google Scholar]
- Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Research. 2004;64:2390–6. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- Taki T, Ohnishi T, Yamamoto A, Hiraga S, Arita N, Izumoto S, Hayakawa T, Morita T. Antisense inhibition of the RAD51 enhances radiosensitivity. Biochemical and Biophysical Research Communications. 1996;223:434–8. doi: 10.1006/bbrc.1996.0911. [DOI] [PubMed] [Google Scholar]
- Tsai CH, Chiu SJ, Liu CC, Sheu TJ, Hsieh CH, Keng PC, Lee YJ. Regulated expression of cofilin and the consequent regulation of p27(kip1) are essential for G(1) phase progression. Cell Cycle. 2009;8:2365–74. doi: 10.4161/cc.8.15.9072. [DOI] [PubMed] [Google Scholar]
- Vispe S, Cazaux C, Lesca C, Defais M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Research. 1998;26:2859–64. doi: 10.1093/nar/26.12.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth NC, Bernards R. rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science. 1996;271:353–6. doi: 10.1126/science.271.5247.353. [DOI] [PubMed] [Google Scholar]
- Wang W, Eddy R, Condeelis J. The cofilin pathway in breast cancer invasion and metastasis. Nature Reviews. Cancer. 2007;7:429–40. doi: 10.1038/nrc2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Mouneimne G, Sidani M, Wyckoff J, Chen X, Makris A, Goswami S, Bresnick AR, Condeelis JS. The activity status of cofilin is directly related to invasion, intravasation, and metastasis of mammary tumors. The Journal of Cell Biology. 2006;173:395–404. doi: 10.1083/jcb.200510115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahara I, Aizawa H, Moriyama K, Iida K, Yonezawa N, Nishida E, Hatanaka H, Inagaki F. A role of cofilin/destrin in reorganization of actin cytoskeleton in response to stresses and cell stimuli. Cell Structure and Function. 1996;21:421–4. doi: 10.1247/csf.21.421. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H. [Molecular mechanisms of invadopodium formation by cancer cells]. Seikagaku. The Journal of Japanese Biochemical Society. 2012;84:35–8. [PubMed] [Google Scholar]
- Yan H, Yang K, Xiao H, Zou YJ, Zhang WB, Liu HY. Over-expression of cofilin-1 and phosphoglycerate kinase 1 in astrocytomas involved in pathogenesis of radioresistance. CNS Neuroscience & Therapeutics. 2012;18:729–36. doi: 10.1111/j.1755-5949.2012.00353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarmola EG, Somasundaram T, Boring TA, Spector I, Bubb MR. Actin-latrunculin A structure and function. Differential modulation of actin-binding protein function by latrunculin A. The Journal of Biological Chemistry. 2000;275:28120–7. doi: 10.1074/jbc.M004253200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.