

Abstract
Estrogen plays vital roles in human health and diseases. Estrogen mediates its actions almost entirely by binding to estrogen receptors (ER), alpha and beta which further function as transcription factors. Selective estrogen receptor modulators (SERMs) are synthetic molecules which bind to ER and can modulate its transcriptional capabilities in different ways in diverse estrogen target tissues. Tamoxifen, the prototypical SERM, is extensively used for targeted therapy of ER positive breast cancers and is also approved as the first chemo-preventive agent for lowering breast cancer incidence in high risk women. The therapeutic and preventive efficacy of tamoxifen was initially proven by series of experiments in the laboratory which laid the foundation of its clinical use. Unfortunately, use of tamoxifen is associated with de-novo and acquired resistance and some undesirable side effects. The molecular study of the resistance provides an opportunity to precisely understand the mechanism of SERM action which may further help in designing new and improved SERMs. Recent clinical studies reveal that another SERM, raloxifene, which is primarily used to treat post-menopausal osteoporosis, is as efficient as tamoxifen in preventing breast cancers with fewer side effects. Overall, these findings open a new horizon for SERMs as a class of drug which not only can be used for therapeutic and preventive purposes of breast cancers but also for various other diseases and disorders. Major efforts are therefore directed to make new SERMs with a better therapeutic profile and fewer side effects.
Keywords: breast cancer, osteoporosis, estrogen receptor, tamoxifen, raloxifene, SERMs, endocrine therapy, drug resistance
1. INTRODUCTION
Breast cancer incidences and death rates have dropped significantly during recent years, which is associated strongly with improvement in early detection methods and decrease of menopausal hormone replacement therapy (HRT) [1, 2]. HRT, in the form of estrogen alone or estrogen plus progesterone, had been widely used since the 1960s until recent years, to treat conditions associated with aging as well as unpleasant menopausal symptoms. HRT was also known to protect post-menopausal women from osteoporosis and also thought to protect women from heart disease and Alzheimer’s disease. However, the Women’s Health Initiative (WHI) study indicated that taking estrogen with or without progesterone for 5 or more years placed the women at higher risk of breast cancer, Alzheimer’s disease, heart disease, blood clot and stroke, although HRT is effective to reduce the risks of osteoporosis and colon cancer [3, 4]. The Million Women Study (MWS) conducted in the UK also showed that women taking HRT were more likely to develop breast cancer [5], endometrial cancer [6] and ovarian caner [7]. In the US, the use of estrogen-plus-progestin HRT has dropped almost 50% when the WHI announced their findings in 2002, and this was followed by a sharp 7% decrease of new breast cancers in 2003 [2]. Although the decrease of HRT uses is not the sole reason leading to less breast cancer incidences, much effort has been focused on finding more effective and safer compounds to replace HRT which not only relieve menopausal symptoms but also prevent and treat hormone-responsive cancers. One most promising approach is to use selective estrogen receptor modulators (SERMs).
SERMs are synthetic compounds that bind to estrogen receptors alpha and/or beta (ERα and/or ERβ) and exert estrogenic or antiestrogenic activities in a tissue/cell-specific manner. The first SERM that has been used successfully in the clinic to prevent and treat breast cancer is tamoxifen, a failed postcoital contraceptive that evolved into the “gold standard” for breast cancer treatment [8, 9]. Tamoxifen is estimated to have saved the lives of over 400,000 women with breast cancer [8]. The second generation SERM, raloxifene (formally called keoxifene), failed as a treatment for breast cancer but is effective against osteoporosis and prevents breast cancer at the same time. Raloxifene is as effective as tamoxifen to reduce invasive breast cancer risks without an increase in the risk of endometrial cancer observed with tamoxifen [10]. Indeed a recent study suggests that raloxifene might even be effective in preventing endometrial cancer [11]. These findings have acted as a catalyst for the search of new SERMs which are estrogen-like in bones and circulating lipids but antiestrogenic in women’s reproductive organs and therefore are anti-cancer agents. However, there are problems associated with the current SERMs such as drug resistance and side effects. For example, both tamoxifen and raloxifene increase both hot flashes and blood clots [12].
Besides SERMs, other endocrine therapies target the ER indirectly to prevent and treat breast cancer. Aromatase inhibitors (AIs) that block the synthesis of estrogen from androgen in peripheral tissues have been extensively studied and show efficacy equivalent or superior to tamoxifen to treat postmenopausal breast cancer [13]. Since the mechanism for AIs to treat ER-positive breast cancer is to deplete estrogen in postmenopausal patients, they do not increase risks of endometrium cancer or blood clot and may be a better choice for postmenopausal breast cancer patients than tamoxifen. However, AIs are not effective in premenopausal women with actively functioning ovaries because AIs do not inhibit ovarian estrogen production. In addition, AIs lack the estrogenic protective function for cardiovascular diseases or osteoporosis. As a result, the side effects of AIs are mostly consistent with estrogen deprivation. AIs are associated with a greater incidence of bone loss and musculoskeletal symptoms, and probably higher risk of cardiovascular disease suggested by adjuvant trials comparing AIs and tamoxifen [14]. However, AIs are associated with a lower incidence of gynecological symptoms, thromboembolic diseases and hot flashes compared to tamoxifen in adjuvant setting [14]. Another strategy is to use selective estrogen receptor down-regulators (SERDs), such as fulvestrant, that cause degradation of ERs. Fulvestrant has been approved to treat advanced breast cancer after tamoxifen failure, and a recent phase III trial indicated that fulvestrant and AI exemestane were equally effective with a similar safety profile [15].
Resistance is a common problem associated with endocrine therapy therefore alternative treatment strategies without cross-resistance are necessary. Compared to the pure anti-estrogenic actions like AIs or fulvestrant, an ideal SERM with beneficial estrogenic effects has great potential for breast cancer prevention and treatment, especially in postmenopausal women as they often suffer from unpleasant symptoms resulting from lower estrogen. A perfect SERM would reduce the risk of breast cancer, ovarian cancer and uterine cancer, as well as strengthen the bone, prevent coronary heart disease, strokes and Alzheimer’s disease, and relieve menopausal discomfort like hot flashes and vaginal atrophy [12].
The complicated outcome of SERMs action cannot just be explained by turning on or off the ERs and their downstream genes. Although much new knowledge is being developed, we are still evolving in our understanding of the detailed mechanism of SERMs and their interaction with the ERs. In the past decade, another group of protein factors, nuclear receptor coregulators, have been identified that are essential for modulating the functions of SERMs and ERs. In this article, we will review the evolving understanding of the molecular mechanisms of SERMs action in the context of other signal transduction pathways and nuclear receptor coregulators, as well as the problems associated with the application of SERMs as a treatment or preventative for breast cancer. Finally, the new SERMs with potential as new agents to treat or prevent breast cancer will be described.
2. MECHANISM OF ESTROGEN ACTION
2.1. Structure and Function of ER
The existence of estrogen binding protein was first predicted by Elwood Jensen and colleagues in early 1960’s [16]. The first ER cDNA, now known as ERα, was later cloned in the mid-1980’s [17, 18]. In 1996, an additional ER was cloned from rat prostate [19] and designated as ERβ. The action of estrogen in cells is therefore almost entirely mediated by these two related but distinct subtype of estrogen receptors, ERα and ERβ. Both receptors function as ligand activated transcription factors which can bind the cognate DNA sequences known as estrogen responsive elements (ERE), and activate transcription. The ER proteins can be structurally subdivided into six domains on the basis of the functions controlled by the region, as shown in Fig. (1). The A/B domain contains one of the two transcriptional activation functions (AFs), designated as AF1 which is involved in estrogen-independent activation of transcription. Another activation function domain, AF2, is located in the E domain which also harbors the ligand binding domain (LBD), and is involved in estrogen/ ligand dependent activation [20, 21]. The ERβ has 97% homology in the DBD and 61% homology in the LBD with ERα suggesting differential ligand binding capability of ERs [21].
Fig. (1).

Schematic comparison of human ER-α and ER-β structure. The structural domains are shown, and the percentage of amino acid identity shared by the two ERs is indicated for each domain. The horizontal bars highlight areas of different functions.
SERMs, the molecules which can bind to ERα and/or β and can either stimulate estrogen-like actions (agonist) or oppose estrogen actions (antagonist) in various estrogen target tissues and cells. This pharmacologic knowledge advanced studies to decipher the details of the molecular mechanism of estrogen action in different cell and tissue types.
The structural studies of a SERM complexed with the LBD of ERα and ERβ reveal that re-orientation of the AF2 helix (helix 12) after the binding of the SERM to the hydrophobic pocket of the LBD [22, 23]. The interaction of amino acid Asp351 of ERα with the alkylaminoethoxyphenyl side chain of tamoxifen or raloxifene is crucial to prevent the recruitment of coactivators to the SERM-receptor complex surface [22, 23]. Using different mutants of ERα for the amino acid Asp351, it was shown that shielding and neutralization of Asp351 by the side chain of raloxifene is critical in defining the antiestrogenicity of this SERM. Furthermore, it has been shown that changing the Asp351 from aspartate to glycine (D351G) abolishes the estrogen-agonist activity of the tamoxifen-ER complex, while retaining its antagonistic property. The AF2 region of the agonist-bound receptor is particularly important for the interactions of steroid receptor coactivators (SRCs 1–3) via the interacting amino acid motif LxxLL, known as nuclear receptor interacting domain (NRID). It is important to note that the affinity of ERs for these NRIDs of SRCs is highly dependent upon the ER subtype, α and β, and ligand bound to the ER [24–26]. Recruitment of these co-activator(s) is also responsible for facilitating the activation of estrogen responsive genes by modifying the chromatin structure and activating the transcriptional machinery. Additionally, SERMs may also show differential AF1 activity mediated by co-repressor binding. Using ERE-reporter constructs, it has been shown that the AF1 domain of ERα is actively involved in agonist-induced gene expression whereas the AF1 domain of ERβ is involved very weakly [27].
Estrogen can also modulate the expression of genes by another mechanism in which the receptor complex can interact with other transcription factors such as activating protein 1 (AP1) or stimulating protein 1 (Sp1) through a process known as a tethering mechanism. Intriguing differences are observed in the mechanism of action between ERα and ERβ through an AP1 site. In the presence of estrogen, ERα induces AP1 driven reporter activity but ERβ has no effect [28]. The raloxifene bound ERβ complex can induce transcriptional activity through the AP1 site but the activity through ERα bound to raloxifene is negligible.
ERs also act in a non-genomic manner initiated from the cell membrane. These actions are very fast (seconds to minutes) and occur without RNA or protein synthesis. They often mobilize second messenger molecules such as Ca2+ and cAMP, and are associated with protein kinase cascades such as PI3K/Akt and MAPK [29, 30]. Several explanations have been offered to explain these effects. There could be a subpopulation of nuclear ERs associate with the plasma membrane, either through posttranslational modification such as palmitoylation or mediated by scaffold proteins such as caveolin-1 and MNAR, since ERs do not have a transmembrane domain [29, 30]. Another membrane bound protein, G protein coupled receptor GPR30, was identified in recent years that mediates non-genomic actions of estrogen [31, 32]. The cellular localization of GPR30 is still controversial. Some evidence suggests it is at plasma membrane [33, 34] and other evidence suggests it is in the endoplasmic reticulum [32]. GPR30 binds to 17β-estradiol, tamoxifen and fulvestrant with high affinity [33] and is associated with breast cancer metastasis and transactivation of the epidermal growth factor receptor (EGFR) [35].
2.2. Co-Regulators
The co-regulators are protein molecules which can physically interact with the liganded or un-liganded ERs and modulate the transcription of the genes. The transcriptional activation or repression of the responsive genes is a combinatorial function of ligand-receptor interaction, recognition of cognate DNA sequence and recruitment of specific co-regulators onto the promoter of the gene. The assembly of the whole transcriptional complex is also dependent upon the affinity of the above mentioned individual components among themselves and their relative concentrations in the cell. Co-regulators play defining roles in the final tissue outcome in terms of transcriptional activation or repression mediated by estrogen or SERMs. The co-regulators can be broadly classified on the basis of their function, as co-activators which promote the activation of the transcriptional process, or co-repressors which are associated with repression of transcription of genes (Fig. (2)).
Fig. (2).

Schematic representation of different liganded-ER complexes interacting with co-regulators and consequent transcriptional activities. ERs that bind to estrogenic ligands interact with co-activators (CoA) and activate transcription. Anti-estrogen liganded-ER complexes interact with co-repressors (CoR) and inactivate transcription of responsive genes. Selective estrogen receptor modulators (SERMs) bind to ERs and interact with either co-activator or co-repressor complexes eliciting partial transcriptional activity depending upon the cellular context.
2.2.1 Co-Activators
Presently, around 200 co-activators are known, which are associated with 48 nuclear receptors [36]. The family of p160 proteins known as steroid receptor co-activators (SRCs) have been studied extensively. The relative abundance of SRC1 in uterine cells is responsible for the agonistic activity of tamoxifen, whereas in breast cancer cells, with low SRC1 levels, tamoxifen acts as an estrogen antagonist [37]. However, raloxifene, another related SERM, does not recruit SRC-1 even in the uterine cells [37], suggesting that the interaction with specific ligand which elicits a unique conformation of the receptor is critical for the interaction of co-regulators. These observations further provide an explanation for the earlier studies, where tamoxifen have been reported to induce growth of endometrial cancer cells but not of breast cancer cells in athymic mice [38] and also that estrogen agonistic properties of raloxifene is less in endometrial cancer cells [39]. These finding also translate very well to clinical experience [40]. In addition, the SERMs can enhance the stability of the co-activators (SRC1 and SRC3) and thereby influence the transcriptional capability of other nuclear receptors [41]. Post-translational modifications of the co-activators, including but not limited to phosphorylation, methylation, ubiquitylation, sumoylation and acetylation, can also regulate the gene activation by influencing the ability of the co-activators to interact with ER and other components of the transcriptional complex [34–36]. The understanding of structure-function relationship of ligands at the ER has formed the basis of designing effective new SERMs with fewer side effects.
2.2.2. Co-Repressors
Co-repressors are functional counterparts of co-activators, which are associated with transcriptionally inactive promoters and help repress the expression of genes [42]. Fewer co-repressors have been reported compared to the co-activators. In the case of ER, the co-repressors are known to interact with the un-liganded and/or antagonist bound receptor. The two most extensively studied co-repressors in connection with ER are Nuclear receptor corepressor (NCoR) and silencing mediator of retinoic acid and thyroid hormone receptor (SMRT). The ER bound to raloxifene or 4-hydroxytamoxifen (a potent antagonist metabolite of tamoxifen) is known to recruit NCoR and SMRT to the promoters of estrogen responsive genes and repress transcription [43–45]. It has been shown that inhibition of NCoR or SMRT with monoclonal antibodies can enhance the agonistic property of 4-hydroxytamoxifen [46]. Moreover, using fibroblasts from NCoR null mice, 4-hydroxytamoxifen was shown to be a relatively potent ERα agonist [47]. The critical role of NCoR and SMRT in 4-hydroxytamoxifen-induced arrest of cell proliferation of ERα positive breast cancer cells is confirmed because 4-hydroxytamoxifen-stimulated cell cycle progression now occurs in NCoR-and-SMRT-deficient breast cancer cells [48]. However not all estrogen responsive genes are activated by 4-hydroxytamoxifen in NCoR and SMRT deficient cells, clearly indicating that additional molecules are important in SERM-induced repression of estrogen responsive genes. Indeed, there are several other co-repressor proteins known for ER. Metastasis associated protein 1 (MTA1) is a corepressor found to mediate the ER transcriptional repression [49]. Another corepressor, known as repressor of estrogen action (REA) potentiates the inhibitory effects of anti-estrogens including 4-hydroxytamoxifen. Additionally, REA interacts with ER and competes with the co-activator SRC1 for binding to the estrogen bound ER [50, 51]. This again emphasizes the fact that the relative levels of co-regulators may be important in deciding the outcome of the SERM action. The proteasomal regulation of NCoR is another factor which may influence the SERM action. Degradation of NCoR occurs through the 26S proteasome, which is mediated by seven in absentia homologue 2 (Siah2) [52]. Interestingly, estrogen mediated upregulation of Siah2 in ER positive breast cancer cells has been implicated in the proteasomal degradation of NCoR, and subsequent de-repression of NCoR regulated genes [53].
In addition to acting as a “transcriptional adapter” between the receptors and the transcriptional machinery, the coregulator itself or its complex possess various enzymatic activities such as acetylation, phosphorylation, methylation or de-acetylation by which they are able to modify the local chromatin structure thereby making the local environment conducive for gene expression or repression. Intrinsic histone acetyl transferase activity was found to be associated with co-activator SRC1 which helps in the activation of transcriptional expression [54]. In contrast, the 4-hydroxytamoxifen bound ER complex which recruits the co-repressors NCoR and SMRT is associated with histone de-acetylases and other chromatin modifying enzymes [37, 55]. The deacetylase activity promotes transcriptional repression [37, 55]. Interestingly, another enzyme in the co-activator complex, CARM1 (coactivator associated arginine methyltransferase 1) has recently been implicated in modifying the coactivator itself and inducing the degradation of the complex [56]. This suggests the ability of the enzymes in the complex to modify other proteins in its own complex apart from a role in the modification of chromatin.
With this background of the molecular biology of SERM action, it is now appropriate to describe our evolving understanding about drug resistance. This is important not only because tumor drug resistance is the consequence of long term SERM administration, but also because new knowledge will aid patients with the development of novel treatment strategies for SERM-resistant breast cancer.
3. DRUG RESISTANCE TO SERMs
There are three types of resistance to SERMs based on the mechanism: metabolic resistance, intrinsic resistance and acquired resistance [57].
3.1. Metabolic Resistance
Metabolic resistance to tamoxifen is mostly related to CYP2D6, an enzyme product that metabolizes tamoxifen into its active forms 4-hydroxytamoxifen and endoxifen [58]. This has been extensively reviewed recently and will only be briefly mentioned here [13, 59]. CYP2D6 is genetically polymorphic and 5–8% of Caucasian subjects are CYP2D6 “poor metabolizers” thus are less likely to benefit from tamoxifen treatment, although it has been shown that these women tolerate tamoxifen better and tend to remain on the drug for longer [59]. The genotype of CYP2D6 has been shown in multiple clinical trials to be directly related to the outcome of tamoxifen use, however, the results are not always consistent. Eight studies indicated that CYP2D6 “poor metabolizer” genotypes have worse outcome of breast cancer patients who received tamoxifen but two studies contradicted this conclusion [60]. In addition to the genotype of CYP2D6, it is important to consider that other drugs may interact with the enzyme system and block the metabolic activation of tamoxifen. Unfortunately, selective serotonin reuptake inhibitors (SSRIs) that are used to relieve the menopausal side effects of tamoxifen are also metabolized by CYP2D6 and block the metabolic activation of tamoxifen. The proper choice of SSRI is therefore important so as not to impair tamoxifen metabolism. The SSRI of choice is venlafaxine that has only a low affinity for the CYP2D6 enzyme [61]. Although these emerging data about CYP2D6 genotypes and the drug interaction between tamoxifen and SSRIs are important, it is perhaps too early to use CYP2D6 status to routinely choose between tamoxifen and aromatase inhibitors to treat postmenopausal women with breast cancer. At present, an international consortium is evaluating the overall CYP2D6 status of completed clinical trials with tamoxifen to assemble a large scale retrospective analysis of the worth of genotyping. The aim is to answer the question of whether “poor metabolizers” should avoid tamoxifen use.
3.2. Intrinsic Resistance
Approximately 30% ER-positive breast cancer patients do not respond to tamoxifen [62]. This type of resistance is referred to as “de novo” resistance or intrinsic resistance. Clinical studies showed that only 40% patients with ER-positive, progesterone receptor (PR)-negative breast cancers are responsive to anti-estrogen treatment (tamoxifen or endocrine ablation) compared to 80% responsive rate in ER-and-PR-positive patients [58, 59]. Historically, the status of PR has been regarded as an indicator of a functional ER pathway, since expression of PR is regulated by estrogen. On the other hand, recent evidence suggested that the absence of PR is associated with excessive growth factor signaling such as overexpression of HER2 [63, 64], which has been known to impair estrogen induction of PR and reduce the effectiveness of tamoxifen treatment for breast cancer [65]. However, the negative association between PR and HER2 seems more evident in older women (> 45 yrs) [66] and it remains controversial that PR-status could be used for clinical decision on choosing between tamoxifen or AIs [67].
Growth factor signaling, especially through epidermal growth factor (EGF) pathway, has been studied extensively in the past two decades and linked to SERM resistance. This has been recently reviewed [68] and will only be briefly summarized here. EGF binds to ErbB family of cell surface receptors that include four closely related receptor tyrosine kinases: EGFR (ErbB-1), HER2/c-neu (ErbB-2), HER3 (ErbB-3) and HER4 (ErbB-4). Overexpression of HER2 has been clinically linked to less response to endocrine therapies and worse prognosis [69–71], so has the overexpression of EGFR [72]. Different ErbB family members can form heterodimers and activate multiple signalling pathways including PI3K/Akt and MAPK. The major molecular mechanisms leading to SERM resistance can be summarized as follows: 1. Activation of downstream kinase cascade results in the phosphorylation of ER at key residues (Ser106/107, 118, 167, 305 and Thr 311) which activates transcription in a ligand-independent manner. Phosphorylation may change the binding of ER with ligands, DNA and coregulators, which may ultimately alter the activity of SERMs [73]. For example, phosphorylation of ER at Ser167 by Akt and Ser118 by the MAPK pathway both cause ligand-independent activation [74–76]. A recent study showed that phosphorylation of ER at Ser305 altered the orientation between the C-terminus of ER and SRC-1 that led to the recruitment of ER transcription coactivators and RNA polymerase II even in the presence of tamoxifen [77]. 2. Phosphorylation of ER co-regulators is equally important as the phosphorylation of ER itself, since phosphorylated co-activators have increased activity in the presence of SERMs [78–80]. Phosphorylation of co-repressors such as SMRT is associated with the co-repressor’s nuclear export and impaired transcriptional suppressing function [81]. 3. Other than enhancing the transcriptional activity of the ER by phosphorylation, overexpression of EGFR or HER2 increases the non-genomic actions of ER, and SERMs may now act as estrogen agonists via the membrane effects of ER [82, 83]. In addition to the EGF signal pathway, the insulin-like growth factor (IGF) signal pathway is also involved in tamoxifen resistance [84]. It can activate PI3K/Akt pathway [71] and turn on genes that are otherwise activated by estrogen [85, 86].
Dysregulation of ER co-regulators is another major contributor to intrinsic SERM resistance. Overexpression of both AIB1 (SRC-3, ACTR, p/CIP, RAC3, TRAM-1) and HER2 have been shown to convert tamoxifen into an estrogen agonist in breast cancer cells [79]. Elevated AIB1 was found to associate with tamoxifen resistance, DNA-nondiploidy, high S-phase fraction and HER2 amplification in samples from clinical study [87]. Although a study indicated that high expression of AIB1 was not associated with relapse during tamoxifen treatment [88], AIB1 was shown to associate with tamoxifen resistance in breast cancers that overexpressed ErbB family proteins [88, 89]. AIB1 might be a predictor marker for tamoxifen ineffectiveness in ER-positive, HER2-positive and PR-negative breast cancer. On the other hand, low expression of ER co-repressor NcoR is associated with shorter relapse-free survival in breast cancer patients who only received tamoxifen after surgery [90]. Based on the emerging importance of co-regulators and tamoxifen resistance, one novel approach to overcome tamoxifen resistance is by the use of disulfide benzamide (DIBA) to disrupt the zinc finger in the ERα DNA binding domain. The approach facilitates ERα dissociation from coactivator AIB1 and concomitant association of corepressor NcoR without changing the phosphorylation of HER2, MAPK, Akt or AIB1 [91].
Another group of regulators associated with tamoxifen resistance are microRNAs (miRNA). These are naturally occurring single-stranded RNAs with the length of 21–23 nucleotides that do not code for proteins. They regulate gene expression mainly by inducing target mRNA degradation or inhibiting translation (protein synthesis). Dysregulation of miRNAs is associated with many cancers including breast cancer [92, 93]. Two recent studies show that miRNA-221/222 are upregulated in tamoxifen-resistant breast cancer cells and primary tumors, and they may contribute to tamoxifen resistance by down-regulating p27Kip1 or ERα [94, 95].
3.3. Acquired Resistance
Breast cancer patients who initially respond to tamoxifen later develop “acquired resistance” that is characterized by tamoxifen stimulated growth. This can be replicated in the laboratory with MCF-7 xenograft tumors implanted in ovariectomized athymic mice. Tamoxifen initially inhibits estrogen stimulated tumor growth but eventually some tumors start to grow during tamoxifen therapy [96]. These tumors now grow in response to either estrogen or tamoxifen and stop growing with no treatment or during treatment with fulvestrant [96]. The laboratory model is consistent with the clinic observation that aromatase inhibitor or fulvestrant are equally effective after the failure of tamoxifen treatment [97, 98]. It therefore appears that ER remains fully functional in the laboratory model of acquired tamoxifen resistance. In clinical studies, only 17–28% patients with acquired tamoxifen resistance have a loss of ER function [99, 100], and it is more likely that acquired resistance is associated with the stimulation of other growth/survival pathways [101]. For example, activated mammalian target of rapamycin (mTOR, downstream of PI3K/Akt and MAPK pathway) and c-Src (downstream of EGFR/HER2) were observed in breast cancer cells and mTOR and c-Src inhibitors can restore tamoxifen sensitivity in these cells, respectively [102, 103]. Several genes involved in cell proliferation and survival have altered expression level in breast cancer cells with acquired tamoxifen resistance. Examples of genes which down regulation is associated with acquired tamoxifen resistance include cyclin-dependent kinase inhibitors p21Cip [104] and p27Kip [105]. Examples of genes which upregulation is associated with tamoxifen acquired resistance include cyclin-dependent kinase 10 (CDK10) [106] and anti-apoptotic protein survivin [107].
Laboratory observation showed that acquired tamoxifen-resistant breast cancer cells/tumors respond differently to estrogen, and three phases of tamoxifen-resistance have been described, which seems to depend on the length of tamoxifen exposure [12]. Tumors with phase I resistance are stimulated by estrogen and tamoxifen but inhibited by AIs and fulvestrant; tumors with phase II resistance are stimulated by tamoxifen but are inhibited by estrogen due to apoptosis; tumors with phase III resistance (automatous growth) grow in a hormone-independent manner that is not responsive to either AIs or fulvestrant or SERMs, but estrogen still exerts apoptotic actions on those tumors [12]. The laboratory models suggest a new treatment strategy, in which limited duration, low-dose estrogen can be used to purge phase II- or phase III-resistant breast cancer cells so that the tumors will be responsive to antiestrogen therapy again. Phase II clinical study is ongoing to test this treatment plan [108].
Most studies on SERM-resistance are related to tamoxifen and little is known about raloxifene resistance. Based on a few studies on raloxifene resistance using cell culture and animal models, raloxifene-resistant tumors are likely to have similar properties as tamoxifen-resistant ones [109]. Raloxifene-resistant MCF7 cells generated by long-term exposure to raloxifene in vitro are also resistant to tamoxifen in vitro and in vivo. They exhibit phase II SERM-resistance as estradiol treatment causes tumor regression by inducing G2/M cell cycle arrest and apoptosis [110]. Another raloxifene-resistant breast tumor model generated by exposing MCF7 breast tumors to raloxifene in vivo exhibits phase I SERM-resistance whose growth is stimulated by tamoxifen, raloxifene and estrogen [109]. Interestingly, protein levels of EGFR and HER2 are also increased in this phase I raloxifene-resistant tumor model, which suggests raloxifene-resistant tumors share similar molecular mechanisms as tamoxifen-resistant ones [109].
Overall, the classifications of different forms of antihormonal drug resistance can be used as a basis to evaluate the pharmacology of new SERMs. The goal is to improve on tamoxifen, the pioneer that over the past 30 years found ubiquitous long term applications in the treatment and prevention of breast cancer.
4. NEW SERMs
The discovery of the first antibiotic penicillin initiated a search for further antibiotics to delay drug resistance and to target specific diseases. Similarly, the successful clinical application of tamoxifen in medicine has resulted in the investigation of numerous related molecules to develop the “ideal SERM”. However, it has been challenging to find a SERM that is superior to tamoxifen, which retains or extends its benefit to treat and prevent breast cancer but with fewer side effects. Tamoxifen maintains bone density in animals [111] and humans [112] so SERMs are being developed to treat osteoporosis, but the potential to prevent breast cancer and uterine cancer will also increase their clinical value and commercial success. The core structures of SERMs are diverse, including triphenylethylene, benzothiophene, chromene (benzopyran), naphthalene, indole and steroid, but each of the newer SERM is really a mimic of tamoxifen, raloxifene or estradiol. The development of dozens of SERMs have been discontinued due to ineffectiveness for human disease or severe side effects, but several new SERMs are under active investigations with great potential in breast cancer treatment and/or prevention, alone or in combination with other type of drugs. In addition, since the identification of ERβ in 1996 [19], ER subtype selective SERMs have been developed which could potentially be used as breast cancer preventives. Thus this area of medicinal chemistry remains an important topic of interest as new ER regulated targets emerge. We will review the current status of several agents that are either approved or in the process of drug development (summarized in Table).
Table.
Current status of new SERMs
| Drug Name | Category (Structure) | Effects | Preclinical Results | Clinical Status |
|---|---|---|---|---|
|
| ||||
| Toremifene | Tamoxifen-like | Breast cancer treatment | Fewer genotoxic effects than tamoxifen [113], bone effects similar to tamoxifen [119] | FDA approved for metastatic breast cancer |
| Heart protection | Phase II trial (65 women) better than tamoxifen regulating lipid metabolism [121, 122] | |||
| Mastalgia treatment | Phase II trials (62 and 195 women) effective [126, 127] | |||
| Prostate cancer prevention | Phase II trial (514 men) decreases prostate cancer incidence [128] | |||
| Relieve side effects of androgen deprivation therapy | Phase III trial (1,389 men) improves lipid profiles [130] | |||
| Phase III trial (1,392 men) increases bone mineral density [129] | ||||
|
| ||||
| Ospemifene | Tamoxifen-like | Vaginal atrophy treatment | Estrogenic effects on vaginal epithelium that is not observed with tamoxifen or raloxifene [134–136] | Phase III trial (826 women) relieves vaginal dryness |
| Osteoporosis treatment | Phase II trial (118 women): Comparable to or slightly better than raloxifene [139] | |||
| Phase III trial planned (detail not available) | ||||
| Breast cancer prevention | Inhibits tumor growth in animal models as effective as tamoxifen [140, 141] | Not available | ||
|
| ||||
| GW5638 (DPC974) & GW7604 | Tamoxifen-like | Breast cancer treatment (2nd line therapy) | Works as a SERM and as a SERD [148], effective in tamoxifen-resistant tumors [144, 145]; functions as an ER agonist in bone and cardiovascular system but an antagonist in breast and endometrium [142] | Phase I trial (9 patients who failed first-line hormone therapy) low toxicity [ASCO meeting 2002, abstract 452] |
|
| ||||
| Arzoxifene (LY353381) | Raloxifene-like | Breast cancer treatment | Antiestrogenic in breast and endometrium, estrogenic in bone and lipids [150] | Phase III trial (200 patients) inferior to tamoxifen [154] |
| Breast cancer prevention | Effective to prevent ER-positive and ER-negative mammary tumors especially in combination with LG100268 [140, 155] | Phase I trials (50 and 76 women) low toxicity and favorable biomarker profile [156] | ||
|
| ||||
| Lasofoxifene (CP-336156, Fablyn) | Raloxifene-like | Osteoporosis treatment and prevention | Higher potency than tamoxifen and raloxifene [158]; higher oral bioavailability than raloxifene [160] | Phase III trial (1,907 women) significantly increases bone mineral density compared to placebo, no endometrial effects, no association with thromboembolic disorder [159] |
| Phase III trial to compare with raloxifene (CORAL trial, details not available) | ||||
| Vaginal atrophy treatment | Phase III trail (445 patients) improves vaginal atrophy compared to placebo | |||
| Breast cancer treatment and prevention | Effects similar to tamoxifen to prevent and treat NMU-induced mammary tumor in rats [163] | Phase III trial (PEARL trial with 8,556 women), reduces ER-positive breast cancer incidence compared to placebo; slightly decreases major coronary disease risk; reduces vertebral and non-vertebral fractures; increases risks of venous thromboembolic events but not stroke; no endometrial effects [SABCS 2008, abstract 11] | ||
| Heart disease prevention | ||||
|
| ||||
| Pipendoxifene (ERA-923) | Raloxifene-like | Breast cancer treatment | Inhibits tamoxifen-sensitive and -resistant tumors in mice and rats no uterotrophic activities compared to raloxifene [167] | Phase II trial to treat tamoxifen-refractory breast cancer in postmenopausal women (details not available) |
|
| ||||
| Bazedoxifene (TSE-424 WAY-140424) | Raloxifene-like | Osteoporosis treatment and prevention | Increases bone density with little uterine or vasomotor effects | Phase III trial (7,492 women) reduces vertebral and non-vertebral fracture incidences, while raloxifene is not effective against non-vertebral fracture [171] |
| Phase III trial (497 women) reduces endometrial thickness, unique property among known SERMs [170] | ||||
| Breast cancer prevention | Inhibits estrogen-stimulated breast cancer cells growth [169] | Not available | ||
|
| ||||
| Acolbifene (EM-652, SCH57068) & EM-800 (SCH57050) | Raloxifene-like | Breast cancer treatment (2nd line therapy) | Highest affinity for ER, inhibit growth of multiple breast cancer cells in vitro and in vivo [180] | Phase III trial, less effective than anastrozole to treat tamoxifen-resistance breast cancer, study halted [182] |
| Breast cancer treatment (1st line therapy) | Phase III trial planned [182] | |||
| Breast cancer prevention | Phase II trial (started in February, 2009) for premenopausal women | |||
|
| ||||
| CHF4227 | Raloxifene-like | Breast cancer and osteoporosis prevention | Prevents DMBA-induced mammary tumors and preserves bone mass in rats [184, 185]; | Phase I trials (24 and 56 women) beneficial on bone markers and lipid metabolism; no effects on endometrium; not causing hot flashes |
|
| ||||
| SP500263 | Raloxifene-like | Breast cancer and osteoporosis treatment | Inhibits breast cancer cell growth in vitro and in vivo without stimulating uterine weight gain [188, 189], protects bone in vitro [190] | Not available |
|
| ||||
| HMR3339 | Steroidal | Osteoporosis and cardiovascular disease prevention | Better than raloxifene to protect cancellous bones [191] | Phase II trials (96 and 118 and 94 women) better than raloxifene at improving some beneficial cardiovascular markers [192–194] |
|
| ||||
| PSK3471 | Steroidal | Osteoporosis and breast cancer prevention and treatment | Prevents bone loss in vivo, inhibits growth of breast cancer cells in vivo [197] | Not available |
|
| ||||
| Trilostane (Modrenal) | ER subtype-selective | Breast cancer treatment | Increases estradiol binding to ERβ, increases ERβ expression, partially inhibits estrogen production [214, 215] | Approved in UK to treat advanced postmenopausal breast cancer after relapse to initial hormone therapy |
| Phase III trial (714 women with advanced breast cancer) effective for both ER-positive and ER-negative breast cancer, effective for endocrine therapy-resistant cancer | ||||
| Phase II trial for use in premenopausal breast cancer (details not available) | ||||
| Prostate cancer treatment | Phase II trial with hormone-refractory prostate cancer (details not available) | |||
|
| ||||
| TAS-108 (SR16234) | Steroidal, ER subtype-selective | Breast cancer treatment and prevention | Inhibits growth of tamoxifen- and AI-sensitive and resistant cancer cells in vitro and in vivo; inhibits DMBA-induced tumor growth in rats [216] | Phase I trials (16 and 15 women) effective and well tolerated [218, 219] |
| Phase II trails (145 and 97 postmenopausal women with advanced breast cancer) beneficial effects, well tolerated [SABCS, 2008, abstract 2131 and 2132] | ||||
4.1. Tamaoxifen-like SERMs
4.1.1. Toremifene (Fareston)
Toremifene (2) is a chlorinated tamoxifen analogue which has been approved in the US and several other countries for the treatment of metastatic breast cancer. Its structure is shown in Fig. (3). Toremifene is as effective as tamoxifen in the treatment of ER-positive breast cancer but with the potential of fewer genotoxic effects, since it does not produce DNA adducts in rat liver and human endometrium [113]. The mechanism for the reduced genotoxicity of toremifene can be explained as follows: Tamoxifen-DNA adducts are primarily formed via sulfonation of the α-hydroxylated tamoxifen metabolites, but the α-hydroxy metabolites of toremifene is poorly esterified or sulfonated, and even sulfonated α-hydroxy toremifene, α-sulfoxytoremifene, reacts poorly with DNA [114, 115]. However, there are some reports to show toremifene induces DNA damages and hepatocarcinogenesis in rats [116, 117].
Fig. (3).

SERMs with a structure mimicking tamoxifen containing a triphenylethylene core.
The effects of toremifene and tamoxifen on bones are similar [118], as are the endometrial effects. However, a recent safety evaluation demonstrates that secondary endometrial cancer incidence is lower with toremifene than with tamoxifen and is similar to that with raloxifene [119]. Nevertheless, toremifene stimulates the growth of human endometrial cancer implanted in athymic mice in the same way as tamoxifen [120]. The positive effects of toremifene on lipid profiles are superior to tamoxifen’s. Toremifene lowers the low-density lipoprotein (LDL) cholesterol to a level similar to that seen with tamoxifen, but unlike tamoxifen, toremifene slightly increases high-density lipoprotein (HDL) cholesterol and lowers triglycerides in the serum [121, 122].
Cross-resistance with tamoxifen is an important issue to consider when using toremifene for recurrent breast cancer because the majority of patients have received or failed adjuvant tamoxifen. Toremifene is completely cross-resistant with tamoxifen in human breast tumors implanted in athymic mice [123], as well as in breast cancer patients [124, 125]. Therefore, toremifene would not be effective as a second-line endocrine therapy after tamoxifen failure and may offer no therapeutic advantages over tamoxifen as an adjuvant therapy.
In recent years, toremifene has been developed to treat other estrogen-related diseases. Toremifene is effective to treat mastalgia in some small phase II trials [126, 127], and is also effective at decreasing prostate cancer incidences in a high-risk population [128]. In addition, a recent multicenter randomized phase III trial showed that toremifene increased bone density and improved lipid profile in men receiving androgen deprivation therapy for prostate cancer [129, 130].
4.1.2. Ospemifene (Deaminohydroxytoremifene, FC-1271a)
Ospemifene (3), or deaminohydroxytoremifene, is a metabolite of toremifene (Fig. (3)). Like toremifene, ospemifene is generally well tolerated and has a favorable safety profile. It does not induce DNA adducts in mice [131], rats [132] and monkey [133]. Ospemifene exerts a very weak estrogenic effect on endometrial histology, like raloxifene and decreases cholesterol [134]. However, unlike tamoxifen or raloxifene, ospemifene has significant estrogenic effects on vaginal epithelium [134–136] and is being developed for postmenopausal vaginal atrophy, a chronic condition experienced by about 40% postmenopausal women. Ospemifene is being evaluated in a phase III trial that has already recruited 826 women. Early results suggested that a 12-week course of ospemifene treatment significantly relieves symptoms of dryness in the vagina.
Ospemifene has showed promise in the prevention and treatment of osteoporosis. Cell culture studies indicated that ospemifene inhibits osteoclast formation and bone resorption and protects osteoblast-derived cells from apoptosis [137, 138]. In a recent phase II trial to compare effects of ospemifene and raloxifene on biochemical markers of bone turnover in postmenopausal women, ospemifene showed similar effects as raloxifene in regulating most of the bone markers examined, and at the 90-mg dose, ospemifene increased procollagen type I N propeptide (PINP) more than raloxifene [139]. Ospemifene is currently in phase III development for the treatment of postmenopausal osteoporosis.
Studies based on animal models suggest ospemifene might be effective in breast cancer prevention. Ospemifene prevented dimethylbenzanthracene (DMBA)-induced mammary tumors in female Sencar mice as effectively as tamoxifen, while raloxifene was not effective [140]. In a transplantable mouse model of ductal carcinoma in situ (DCIS), ospemifene had inhibitory effects equivalent to tamoxifen in terms of tumor growth and progression [141]. Nevertheless, the chemoprevention effects of ospemifene in breast cancer need to be further studied and substantiated by clinical trials.
4.1.3. GW5638 (DPC974) and GW7604
GW5638 (4) is a derivative of tamoxifen with an acrylate side chain in place of the dimethylaminoethoxy side chain in tamoxifen. GW7604 (5) is the 4-hydroxy version of GW5638, analogous to the major metabolite of tamoxifen, 4-hydroxytamoxifen. Their structures are shown in Fig. (3). GW5638 functions as a full ER agonist in bone and the cardiovascular system but as an antagonist in breast and endometrial system in rodent models [142].
Although the structures of tamoxifen and GW5638/GW7604 are similar, GW5638/GW7604 acts with a mechanism different from tamoxifen/4-hydroxy tamoxifen as suggested by the following evidence: 1. GW7604 acts as an antagonist in MDA-MB-231 cells transfected with wild-type ERα, but 4-hydroxytamoxifen acts as an agonist [143]; 2. Phage display experiments indicate that GW7604 bound ERα or ERβ is associated with different peptides from 4-hydroxytamoxifen, raloxifene or fulvestrant bound ERs [144]; 3. GW5638 inhibits the growth of tamoxifen-resistant breast tumor xenograft [144, 145]; 4. The crystal structure of the ERα LBD bound by GW5638 shows that GW5638 induces a distinct conformation of H12 in the ERα AF2 region, which increased exposure of hydrophobic residues and results in ERα destabilization in MCF7 cells [146].
GW5638 and GW7604 are also classified as selective estrogen receptor down-regulators (SERDs) because they induce ERα degradation, a property observed with the pure antiestrogen fulvestrant which was approved for the treatment of metastatic breast cancer [147]. However, a recent report [148] indicates that GW5638 induces ERα degradation through a different mechanism from fulvestrant and another SERD RU58,668, as the protein/protein interaction surface on ER required for fulvestrant-induced degradation is not necessary for GW5638-induced degradation. The fact that GW5638 has a unique mechanism to antagonize estrogen function and induces ER degradation in breast cancer cells makes it a possible second line therapy after tamoxifen failure and as an alternative to fulvestrant. Currently, GW5638 is under clinical development under the name DPC974 [148].
4.2. Raloxifene-like SERMs
4.2.1. Arzoxifene (LY353381)
Arzoxifene (7) is a derivative of raloxifene with the ketone group replaced by an ether group and the hydroxy group is replaced by a methoxy group (Fig. (4)). These modifications have improved the pharmokinetic properties [149]. Arzoxifene has antiestrogenic effects on breast and endometrium but pro-estrogenic effects on bone and lipids [150]. Arzoxifene is cross-resistant in some but not all tamoxifen-stimulated breast tumor xenografts [151]. Phase II clinical trials indicate that arzoxifene is effective to treat tamoxifen-sensitive or tamoxifen-refractory patients with advanced or metastatic breast cancer [152] and patients with recurrent or advanced endometrial cancer [153] with minimus toxicity. However, a phase III trial showed arzoxifene was inferior to tamoxifen to treat patients with locally advanced and metastatic breast cancer [154]. The main role of arzoxifene may reside in its chemoprevention potential since it is more potent than raloxifene in pre-clinical studies [149].
Fig. (4).

SERMs with a structure mimicking raloxifene.
The breast cancer chemoprevention property of arzoxifene has been studied with animal models and small short-term clinical trials. Arzoxifene effectively prevented nitrosomethylurea (NMU)-induced mammary tumor in rats [140] and induced apoptosis of breast cancer cells in rodent models especially when used in combination with rexinoid LG100268, a selective ligand for the retinoid X receptors (RXR) [155]. In two phase I clinical trails of women with newly diagnosed ductal carcinoma in situ or T1/T2 invasive cancer, arzoxifene did not demonstrate a significant reduction of tumor cell proliferation compared to placebo in 2–6 weeks treatment [156]. However, there were some favorable findings, such as a decrease of serum insulin like growth factor I (IGF-1) vs IGF binding protein 3 (IGFBP3) ratio and an increase of sex hormone binding globulin [156]. Another interesting aspect of the pharmacology of arzoxifene is that it might have chemopreventive properties for ER-negative breast cancer when used in combination with LG100268. A recent study showed that both SERMs arzoxifene and acolbifene alone prevent ER-negative mammary tumor in a mouse model and the effect is synergized with LG100268 [155]. Although the SERMs by themselves are not functional in the treatment of established tumors, together with LG100268 they inhibit proliferation and induce apoptosis in the ER-negative mammary tumors [155]. The mechanism how SERMs prevent tumorigenesis of ER-negative breast tissue is unknown, but the results suggest that arzoxifene has the potential for further clinical development as a chemoprevention drug of both ER-positive and negative breast cancer, especially in combination with rexinoids.
4.2.2 Lasofoxifene (CP-336156, Fablyn)
Lasofoxifene (8) has a naphthalene core structure, which is different from all the other SERMs discussed in this article (Fig. (4)). However, the crystal structure shows that lasofoxifene fits into the ERα LBD pocket in a similar manner as other ligands [157]. In addition, lasofoxifene-bound ERα LBD has similar conformational features as other SERM-bound ERα LBDs, such as tamoxifen or raloxifene, in which H12 in the “antagonist-bound” conformation and occludes the coactivator binding surface [157]. Lasofoxifene has a high affinity for ER with an IC50 of 1.5 nM, which is comparable to 17β-estradiol and higher than tamoxifen and raloxifene [158]. It preserves bone density and lowers serum cholesterol, and also has chemopreventive and chemotherapeutic effects in rat mammary tumor models without any uterine hypertrophic effects [159]. Lasofoxifene is currently undergoing an extensive clinical evaluation for the prevention and treatment of osteoporosis [159]. One advantage of lasofoxifene over raloxifene is its increased oral bioavailability due to the nonpolar naphthalene structure that makes it a poor substrate for intestinal wall glucuronidation [160]. In addition to its effects on bone, lasofoxifene significantly improves symptoms of vaginal atrophy [161] and a recently completed phase III trial indicated that lasofoxifene decreased vaginal pH and improved the vaginal-cell maturation index in osteoporotic postmenopausal women. These effects may be due to the increased vaginal ERβ and androgen receptor protein levels [162]. Lasofoxifene acts as a chemopreventive and treatment in the NMU-induced rat mammary tumor model. The results are similar to the comparator drug tamoxifen [163]. Phase III trials are currently ongoing to evaluate its ability to prevent breast cancer and cardiovascular diseases in postmenopausal women [164].
4.2.3. Pipendoxifene (ERA-923)
Pipendoxifene (9) has an indole core structure (Fig. (4)). It was designed by adding an alkylaminoethoxyphenyl side chain to zindoxifene (D-16726), a 2-phenylindol based SERM which failed as a treatment for breast cancer [165]. Pipendoxifene, also named ERA-923, mimics the structure of raloxifene and is devoid of uterotrophic activities in immature rats and ovariectomized mice compared to raloxifene [166]. It inhibits the growth of tamoxifen-sensitive and -resistant tumors in rats and mice [167] and is under phase II clinical development for the treatment of tamoxifen-resistant metastatic breast cancer. In a recent study, a combination of pipendoxifene and temsirolimus, which is a mammalian target of rapamycin (mTOR) inhibitor, synergistically inhibited growth of MCF-7 cells and xenograft models even at suboptimal doses, primarily by causing G1 cell cycle arrest [168]. This suggested that combination of a SERM and an mTOR inhibitor might be of clinic value as breast cancer treatments.
4.2.4. Bazedoxifene (TSE-424, WAY-140424)
Bazedoxifene (10) is another indole SERM, designed and synthesized at the same time as pipendoxifene with a slight structural difference, as shown in Fig. (4) [166]. This SERM is being actively developed to treat osteoporosis with the potential to prevent breast cancer. Bazodoxifene binds to ERα and ERβ with an affinity lower than raloxifene but is less selective for ERα [169]. It inhibits estrogen-mediated proliferation of breast cancer MCF7 cells and increases bone density with little uterine or vasomotor effects in rat models [169]. A Phase III trial with 497 healthy postmenopausal women showed that 6-month bazedoxifene treatment decreases endometrium thickness and uterine bleeding, suggesting antagonistic effects of bazedoxifene in endometrium [170]. Bazedoxifene is currently under review by the Food and Drug Administration (FDA) for the prevention and treatment of postmenopausal osteoporosis. The completed 3-year phase III trial which enrolled 7,492 postmenopausal women with moderate to severe osteoporosis showed bazedoxifene significantly reduced the incidences of vertebral and non-vertebral fracture compared to placebo, while raloxifene was not effective against non-vertebral fracture [171]. No safety concerns related to breast or endometrium were observed, however, a statistical insignificant increase of venous thromboembolic events was observed with groups treated with either bazedoxifene or raloxifene in the same study [172]. Based on studies using rodent models, combination of bazedoxifene and conjugated estrogens exerted positive vasomotor, lipid, and skeletal responses with minimal uterine stimulation [173]. This suggested that pairing SERMs and estrogen might be effective in the treatment of menopausal symptoms and prevention of osteoporosis. However, further studies are needed to examine the effectiveness of bazedoxifene in breast cancer prevention.
4.2.5. Acolbifene (EM-652, SCH57068) and EM-800 (SCH57050)
Acolbifene (EM-652) (11) and its orally active prodrug EM-800 (12) have a chromene core structure (Fig. (4)). They were initially misclassified as pure antiestrogens and their side chain was depicted by analogy with the pure antiestrogen fulvestrant [174]. However, the structure of acolbifene is actually similar to that of raloxifene, and unlike fulvestrant, the antiestrogenic side chain of acolbifene does not mask the mutant ER amino acid D351Y to produce an estrogenic action [175]. In addition, acolbifene and EM-800 act as antiestrogens in mammary and uterine tissues, but have estrogenic effects to prevent bone loss and have a favored function in the regulation of lipid metabolism by lowering plasma cholesterol and triglyceride in rodent models [176, 177]. Therefore, acolbifene and EM-800 should be classified as SERMs.
Acolbifene has the highest ER-binding affinity among all known compounds [178]. Preclinical studies indicated that acolbifene and EM-800 were more potent than tamoxifen, idoxifene, raloxifene, GW-5638, toremifene and droloxifene to inhibit the growth of breast cancer cell lines MCF-7, ZR-75-1, MCF-7 and T47D as well as ZR-75-1 xenograft in mice models [179, 180]. Interestingly, acolbifene caused disappearance of 65% ZR-75-1 xenograft in ovariectomized nude mice, while other SERMs tested (tamoxifen, toremifene, raloxifene, droloxifene, idoxifene and GW 5638) only decreased the tumor growth rate stimulated by estrone [180]. Acolbifene was evaluated as a second line therapy for tamoxifen-refractory breast cancers, since it was regarded as a pure anti-estrogen. In a small clinical trail involved 43 postmenopausal or ovariectomized women with breast cancer who had received tamoxifen for over a year but relapsed, the objective response to EM-800 was 12% with 1 complete response and 4 partial responses [181]. In a phase III trial to compare acolbifene with the aromatase inhibitor anastrozole in breast cancer patients who did not respond to tamoxifen, acolbifene did not show superior antitumor activity to anastrozole and the study was halted [182]. However, acolbifene and EM-800 may be more suitable as first line therapy and a phase III trial for untreated metastatic breast cancer patients is planned [182].
Recent studies indicate that acolbifene might be used in combination with other drugs. Acolbifene synergizes with rexinoid LG100268 in the prevention and treatment of mice with ER-negative mammary tumor [155]. It also synergizes with dehydroepiandrosterone (DHEA), which is a naturally produced prohormone for androgen and estrogen, in the prevention of dimethylbenzanthracene (DMBA)-induced mammary tumors in the rats [183]. A phase III trials of acolbifene plus DHEA for vaginal atrophy and uterine safety has been planned.
4.2.6. CHF4227
CHF4227 (13) is a SERM with a chromene (benzopyran) core structure, as shown in Fig. (4). Compared with raloxifene, CHF4227 binds to ERα and ERβ with higher affinity and inhibits the uterotropic action of 17alpha-ethynyl estradiol with more potency [184]. CHF4227 significantly prevents the development of DMBA-induced mammary tumors in rats [184]. It preserves bone mass without affecting uterine weight and decreases serum cholesterol and fat mass in ovariectomized rats [185]. A recent phase I study showed CHF4227 is well-tolerated, as 28 days of treatment has a positive effect on the serum lipid profile and bone markers without any negative effects on the endometrium or the fibrinolytic system. Additionally, CHF4227 does not cause vaginal bleeding or hot flashes [186]. These results suggest that CHF4227 is safe and worthy of further clinical development for osteoporosis and the chemoprevention of breast cancer.
4.2.7. SP500263
SP500263 (14) was discovered in a screen to identify ER agonist in bone cells [187]. It has a chromene core structure and binds to both ERα and ERβ with high affinity similar to raloxifene’s (Fig. (4)) [187]. SP500263 inhibits the growth of breast cancer MCF7 cells and xenografts in nude mice, and does not stimulate uterine weight gain in immature rats or ovariectomized adult rats [188, 189]. SP500263 also blocks osteoclastogenesis in human bone cell model [190]. These preclinical results suggest that SP500263 has potential for the treatment of both breast cancer and osteoporosis. However, clinical value of this drug has yet to be determined.
4.3. Steroidal SERMs
4.3.1. HMR3339
All of the SERMs described to this point are non-steroidal. Recently, steroidal SERMs have been described (Fig. (5)). In rats, HMP3339 (15) not only increases bone mineral density but also restores the mechanical strength at multiple sites even after ovariectomy, and it affects both cortical and cancellous bones, while raloxifene was effective only at cancellous sites [191]. HMR3339 has entered clinical investigation for the prevention of osteoporosis and cardiovascular diseases. In a series of small phase II trials with healthy postmenopausal women, HMR3339 was found to reduce total cholesterol, LDL cholesterol, C-reactive protein (CRP, a pro-inflammatory cytokine and a cardiovascular disease risk factor), asymmetric dimethylarginine (AMDA, a nitric oxide synthase inhibitor) and homocysteine [192–194]. Elevation of AMDA or homocysteine is linked to a high incidence of cardiovascular disease but raloxifene treatment does not reduce the level of either AMDA or homocysteine [192]. HMR3339 reduces concentrations of procarboxypeptidase U (pro-CpU, an inhibitor of fibrinolysis), antithrombin and fibrinogen to a degree similar to raloxifene and shows beneficial effects on some markers of fibrinolysis [195, 196]. Therefore, HMR3339 has potential to prevent cardiovascular diseases and possibly also osteoporosis. However, whether or not there is potential as a cancer preventive has not been determined.
Fig. (5).

Steroidal SERMs
4.3.2 PSK3471
PSK3471 (16) is a newly developed SERM with a structure similar to HMR3339 (Fig. (5)). It was reported to prevent gonadectomy-induced bone loss in male and female mice, and antagonize estradiol-stimulated MCF-7 cell proliferation [197].
4.4. ER Subtype Selective SERMs
ERα and ERβ have a different tissue distribution and have overlapping but distinct biological functions [198]. Unlike ERα, ERβ expression is not routinely examined in the clinic and its function in breast cancer remains unclear. ERβ expression is found in both normal and breast cancer specimens but does not correlate with ERα expression [199]. It seems that ERβ functions differently if it is expressed alone or co-expressed with ERα in breast cancers. In ERα-positive breast tumors, ERβ often antagonizes the pro-proliferation actions of ERα [200, 201] and its expression is associated with better response to endocrine therapy and a favorable clinical outcome in most cases [202]. Thus ERβ seems to function as a tumor suppressor. However in ERα-negative breast tumors, several studies indicated that the expression of ERβ correlates with proliferation markers such as Ki67 and cyclin A [202, 203], which suggested that ERβ might stimulate cancer growth. In the latter situation, ERβ could serve as an endocrine therapy target in those patients who would otherwise be regarded as ER-negative and have limited choice but chemotherapy. The presence of ERβ in ERα-negative breast cancers may partly explain why some “ER-negative” patients respond to SERMs. The reason that ERβ functions differently in the absence or presence of ERα might be due to the different activities between the ERα/β heterodimer and ERα or ERβ homodimers.
A new direction to consider is the estrogen related receptor (ERR) [204, 205]. There is emerging evidence that ERRα is critical for the growth of ER-negative breast-cancer MDA-MD-231 xenografts in mice [206], as ERRα appears to be involved in angiogenesis by inducing the expression of vascular endothelial growth factor (VEGF) [207, 208]. Novel therapeutic agents targeted to ERRα would be valuable to treat breast cancer.
Several ER-subtype selective SERMs have been reported, although it is difficult to design subtype selective ligands given the fact that only two amino acids are different in the ligand binding pocket between ERα and ERβ (despite that they have 61% amino acid identity in LBD). All the SERMs discussed previously were designed against ERα and have low subtype selection in terms of binding affinity. In contrast to the focus on ERα and breast cancer, most of ER subtype selective SERMs are developed for diseases other than breast cancer. In animal models, ERβ-selective agonists ERB041 and diaryl-propionitrile (DPN) have been shown to have anti-inflammatory properties and antidepressant-like effects, respectively [209, 210]. An ERβ agonist, 8-vinylestra-1,3,5 (10)-triene-3,17β-diol, stimulates ovarian follicular development in hypophysectomized rats and gonadotropin-releasing hormone a tagonist-treated mice [211], thus this drug could be used to enhance fertility [198]. A few ERβ agonists are being developed for clinical applications in Alzheimer’s disease and rheumatoid arthritis [212]. For breast cancer prevention and treatment, it is conceivable that ERβ agonist might have potential for ERα-and-β-positive tumors, especially in combination of an ERα selective antagonist, since the preclinical studies indicate a protective role of ERβ. However, this strategy poses a difficult pharmacologic issue of tissue pharmacodynamics. Nevertheless, a couple of ERβ modulators have been shown with positive effects to treat advanced postmenopausal breast cancer, which will be discussed below.
4.4.1. Trilostane (Modrenal)
Trilostane (17) (Fig. (6)) is an inhibitor of 3β-hydroxysteroid dehydrogenase, a critical enzyme in the conversion of DHEA to estradiol in breast tumors [213]. Trilostane increases the maximum binding of estradiol to ERβ but not ERα in MCF-7 breast cancer cells [214], and it increased the expression of ERβ in MCF-7 cells and rat uterine [215]. Trilostane is approved in UK to treat advanced postmenopausal breast cancer after relapse to initial hormone therapy and is currently under investigation to be used in prostate cancer and premenopausal breast cancer [213].
Fig. (6).
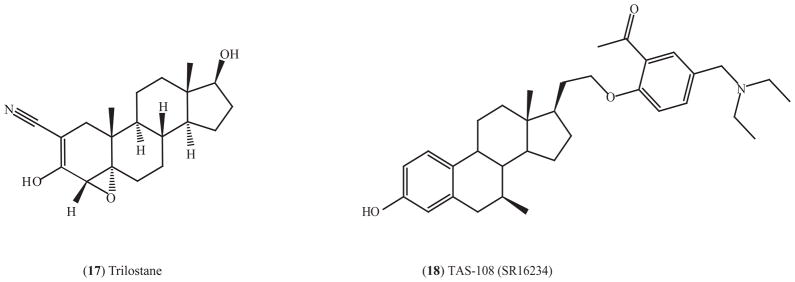
Structure of ERβ-selective agonists.
4.4.2. TAS-108 (SR16234)
Another type of subtype selective SERM that might be relevant to breast cancer is a combined ERα antagonist but ERβ agonist. TAS-108 (18) (Fig. (5)) is a steroidal antiestrogen for ERα and a partial agonist on ERβ [216]. TAS-108 has pure antiestrogenic effects for ERα in the presence or absence of estrogen but exhibits partial agonist activity on ERβ using in vitro reporter assay. TAS-108 inhibits the growth of tamoxifen-resistant breast cancer cells, DMBA-induced mammary tumor in rats and estrogen-stimulated growth of MCF7 xenografts with little uterotrophic effect [216, 217]. Phase I trial indicate that TAS-108 has anti-tumor activity, is well tolerated, and does not have effects on an endometrial thickness based on an evaluation using trans-vaginal ultrasound [218, 219]. Similar results were obtained in Phase II trials that recruited postmenopausal women with advanced breast cancer, according to presentations at San Antonio Breast Cancer Symposium (SABCS) in December, 2008. A phase III trial is planned [217]. TAS-108 did not increase bone loss like fulvestrant, which could be due to its agonistic property on ERβ. Another advantage over fulvestrant is that TAS-108 is orally administered [220]. TAS-108 is therefore a promising breast cancer drug, even for patients who have relapsed after tamoxifen.
5. CONCLUDING REMARKS
Endocrine therapy targeting to ERα has been very successful in the treatment and prevention of breast cancer [221, 222]. It is very effective and less toxic compared to combinational cytotoxic chemotherapy that was the only option 30 years ago. In the ensuing period, multiple strategies have been developed to antagonize estrogen action. Most experience has accumulated with the competitive inhibitor of estrogen action tamoxifen, but targeting aromatase to deplete estrogen with AIs in postmenopausal patients or to induce ER degradation with SERDs have been valuable innovations in therapies. The goal for treatment is to create a “no-estrogen environment”. However, SERMs that maintain the beneficial effects of estrogen but antagonize the harmful effects of estrogen have great potential in the prevention of multiple diseases in common. It is clear that many new SERMs are being developed that could provide better choices for patients in the future.
To overcome the unwanted side effects and problems with drug resistance, combination therapy might be another important direction in addition to the development of new SERMs. For example, combination of SERM acolbifene and DHEA could be protective against breast cancer and osteoporosis with beneficial effects to stimulate vaginal maturation and decrease skin dryness [182]. As traditional HRT is less acceptable to regulatory authorities because of the increased risk of breast cancer, a combination of HRT and a SERM may be a reasonable idea to relieve unpleasant menopausal effects while decrease breast cancer risks. With regards to avoiding drug resistance, combining a SERM and an inhibitor targeting significant survival signal transduction pathway is under active evaluation. By way of example, a combination of tamoxifen and inhibitors of the HER2 signal transduction pathway may prevent acquired tamoxifen resistance [223]. Similarly, SERM pipendoxifene and mTOR inhibitor temsirolimus synergistically inhibits the proliferation of MCF7 breast cancer cells and xenograft at suboptimal concentrations [168]. Additionally, combinations of a SERM (arzoxifene or acolbifene) and a rexinoid LG100268 are effective to prevent and treat ER-negative mammary tumors in animal models [155]. The potential combination seems endless but the marriage of molecular biology and medicine holds great promise for advances in targeted therapeutics based on the SERM model.
In summary, it is clear that the original idea of targeting specific hormone receptor with selective medicine has proven its worth by advancing medicine with the SERMs tamoxifen and raloxifene. Now there are a whole range of new SERMs poised for clinical applications. But this is not the end of the story. Novel selective modulators of all members of the nuclear receptor superfamily are under investigation addressing the treatment or prevention of diseases never before considered possible [57, 222].
Acknowledgments
Dr. Jordan is supported by the Department of Defense Breast Program under award number BC050277 Center of Excellence, FCCC Core Grant NIH P30 CA006927, the Genuardi’s Fund, the Weg Fund of Fox Chase Cancer Center and the Hollenbach Family Fund. The views and opinions of the author(s) do not reflect those of the US Army or the Department of Defense.
Footnotes
The authors have no potential conflict of interest.
References
- 1.Berry DA, Cronin KA, Plevritis SK, Fryback DG, Clarke L, Zelen M, Mandelblatt JS, Yakovlev AY, Habbema JD, Feuer EJ. Effect of screening and adjuvant therapy on mortality from breast cancer. N Engl J Med. 2005;353(17):1784–1792. doi: 10.1056/NEJMoa050518. [DOI] [PubMed] [Google Scholar]
- 2.Ravdin PM, Cronin KA, Howlader N, Berg CD, Chlebowski RT, Feuer EJ, Edwards BK, Berry DA. The Decrease in Breast-Cancer Incidence in 2003 in the United States. N Engl J Med. 2007;356(16):1670–1674. doi: 10.1056/NEJMsr070105. [DOI] [PubMed] [Google Scholar]
- 3.Writing Group for the Women’s Health Initiative I. Risks and Benefits of Estrogen Plus Progestin in Healthy Postmenopausal Women: Principal Results From the Women’s Health Initiative Randomized Controlled Trial. JAMA. 2002;288(3):321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 4.The Women’s Health Initiative Steering C. Effects of Conjugated Equine Estrogen in Postmenopausal Women With Hysterectomy: The Women’s Health Initiative Randomized Controlled Trial. JAMA. 2004;291(14):1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 5.Beral V. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362(9382):419–427. doi: 10.1016/s0140-6736(03)14065-2. [DOI] [PubMed] [Google Scholar]
- 6.Beral V, Bull D, Reeves G. Endometrial cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2005;365(9470):1543–1551. doi: 10.1016/S0140-6736(05)66455-0. [DOI] [PubMed] [Google Scholar]
- 7.Beral V, Bull D, Green J, Reeves G. Ovarian cancer and hormone replacement therapy in the Million Women Study. Lancet. 2007;369(9574):1703–1710. doi: 10.1016/S0140-6736(07)60534-0. [DOI] [PubMed] [Google Scholar]
- 8.Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2(3):205–213. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- 9.Veronesi U, Boyle P, Goldhirsch A, Orecchia R, Viale G. Breast cancer. Lancet. 2005;365(9472):1727–1741. doi: 10.1016/S0140-6736(05)66546-4. [DOI] [PubMed] [Google Scholar]
- 10.Cummings SR, Eckert S, Krueger KA, Grady D, Powles TJ, Cauley JA, Norton L, Nickelsen T, Bjarnason NH, Morrow M, Lippman ME, Black D, Glusman JE, Costa A, Jordan VC. The Effect of Raloxifene on Risk of Breast Cancer in Postmenopausal Women: Results From the MORE Randomized Trial. JAMA. 1999;281(23):2189–2197. doi: 10.1001/jama.281.23.2189. [DOI] [PubMed] [Google Scholar]
- 11.DeMichele A, Troxel AB, Berlin JA, Weber AL, Bunin GR, Turzo E, Schinnar R, Burgh D, Berlin M, Rubin SC, Rebbeck TR, Strom BL. Impact of Raloxifene or Tamoxifen Use on Endometrial Cancer Risk: A Population-Based Case-Control Study. J Clin Oncol. 2008;26(25):4151–4159. doi: 10.1200/JCO.2007.14.0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jordan VC. Selective estrogen receptor modulation: concept and consequences in cancer. Cancer Cell. 2004;5(3):207–213. doi: 10.1016/s1535-6108(04)00059-5. [DOI] [PubMed] [Google Scholar]
- 13.Jordan VC, Brodie AM. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids. 2007;72(1):7–25. doi: 10.1016/j.steroids.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perez EA. Safety profiles of tamoxifen and the aromatase inhibitors in adjuvant therapy of hormone-responsive early breast cancer. Ann Oncol. 2007;18(suppl_8):viii26–35. doi: 10.1093/annonc/mdm263. [DOI] [PubMed] [Google Scholar]
- 15.Chia S, Gradishar W, Mauriac L, Bines J, Amant F, Federico M, Fein L, Romieu G, Buzdar A, Robertson JF, Brufsky A, Possinger K, Rennie P, Sapunar F, Lowe E, Piccart M. Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: results from EFECT. J Clin Oncol. 2008;26(10):1664–1670. doi: 10.1200/JCO.2007.13.5822. [DOI] [PubMed] [Google Scholar]
- 16.Jensen EV, Jacobson HI. Basic guide to the mechanism of estrogen action. Recent Progress in Hormone Research. 1962;18:387–414. [Google Scholar]
- 17.Green S, Walter P, Kumar V, Krust A, Bornert JM, Argos P, Chambon P. Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature. 1986;320(6058):134–139. doi: 10.1038/320134a0. [DOI] [PubMed] [Google Scholar]
- 18.Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J. Sequence and expression of human estrogen receptor complementary DNA. Science. 1986;231(4742):1150–1154. doi: 10.1126/science.3753802. [DOI] [PubMed] [Google Scholar]
- 19.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A. 1996;93(12):5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai MJ, O’Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem. 1994;63:451–486. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- 21.MacGregor JI, Jordan VC. Basic guide to the mechanisms of antiestrogen action. Pharmacol Rev. 1998;50(2):151–196. [PubMed] [Google Scholar]
- 22.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389(6652):753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 23.Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson JA, Carlquist M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. Embo J. 1999;18(17):4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bramlett KS, Wu Y, Burris TP. Ligands specify coactivator nuclear receptor (NR) box affinity for estrogen receptor subtypes. Mol Endocrinol. 2001;15(6):909–922. doi: 10.1210/mend.15.6.0649. [DOI] [PubMed] [Google Scholar]
- 25.Wong CW, Komm B, Cheskis BJ. Structure-function evaluation of ER alpha and beta interplay with SRC family coactivators. ER selective ligands. Biochemistry. 2001;40(23):6756–6765. doi: 10.1021/bi010379h. [DOI] [PubMed] [Google Scholar]
- 26.Warnmark A, Almlof T, Leers J, Gustafsson JA, Treuter E. Differential recruitment of the mammalian mediator subunit TRAP220 by estrogen receptors ERalpha and ERbeta. J Biol Chem. 2001;276(26):23397–23404. doi: 10.1074/jbc.M011651200. [DOI] [PubMed] [Google Scholar]
- 27.Hall JM, McDonnell DP. The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology. 1999;140(12):5566–5578. doi: 10.1210/endo.140.12.7179. [DOI] [PubMed] [Google Scholar]
- 28.Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997;277(5331):1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 29.Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19(4):833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 30.Manavathi B, Kumar R. Steering estrogen signals from the plasma membrane to the nucleus: two sides of the coin. J Cell Physiol. 2006;207(3):594–604. doi: 10.1002/jcp.20551. [DOI] [PubMed] [Google Scholar]
- 31.Filardo EJ, Quinn JA, Frackelton AR, Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16(1):70–84. doi: 10.1210/mend.16.1.0758. [DOI] [PubMed] [Google Scholar]
- 32.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307(5715):1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 33.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146(2):624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 34.Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun. 2006;346(3):904–910. doi: 10.1016/j.bbrc.2006.05.191. [DOI] [PubMed] [Google Scholar]
- 35.Filardo EJ, Quinn JA, Sabo E. Association of the membrane estrogen receptor, GPR30, with breast tumor metastasis and transactivation of the epidermal growth factor receptor. Steroids. 2008;73(9–10):870–873. doi: 10.1016/j.steroids.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 36.Lonard DM, O’Malley BW. The expanding cosmos of nuclear receptor coactivators. Cell. 2006;125(3):411–414. doi: 10.1016/j.cell.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 37.Shang Y, Brown M. Molecular Determinants for the Tissue Specificity of SERMs. Science. 2002;295(5564):2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 38.Gottardis MM, Robinson SP, Satyaswaroop PG, Jordan VC. Contrasting actions of tamoxifen on endometrial and breast tumor growth in the athymic mouse. Cancer Res. 1988;48(4):812–815. [PubMed] [Google Scholar]
- 39.Gottardis MM, Ricchio ME, Satyaswaroop PG, Jordan VC. Effect of steroidal and nonsteroidal antiestrogens on the growth of a tamoxifen-stimulated human endometrial carcinoma (EnCa101) in athymic mice. Cancer Res. 1990;50(11):3189–3192. [PubMed] [Google Scholar]
- 40.Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, Bevers TB, Fehrenbacher L, Pajon ER, Jr, Wade JL, 3rd, Robidoux A, Margolese RG, James J, Lippman SM, Runowicz CD, Ganz PA, Reis SE, McCaskill-Stevens W, Ford LG, Jordan VC, Wolmark N. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. Jama. 2006;295(23):2727–2741. doi: 10.1001/jama.295.23.joc60074. [DOI] [PubMed] [Google Scholar]
- 41.Lonard DM, Tsai SY, O’Malley BW. Selective estrogen receptor modulators 4-hydroxytamoxifen and raloxifene impact the stability and function of SRC-1 and SRC-3 coactivator proteins. Mol Cell Biol. 2004;24(1):14–24. doi: 10.1128/MCB.24.1.14-24.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKenna NJ, Lanz RB, O’Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20(3):321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 43.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103(6):843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 44.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295(5564):2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 45.Smith CL, Nawaz Z, O’Malley BW. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol. 1997;11(6):657–666. doi: 10.1210/mend.11.6.0009. [DOI] [PubMed] [Google Scholar]
- 46.Lavinsky RM, Jepsen K, Heinzel T, Torchia J, Mullen TM, Schiff R, Del-Rio AL, Ricote M, Ngo S, Gemsch J, Hilsenbeck SG, Osborne CK, Glass CK, Rosenfeld MG, Rose DW. Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc Natl Acad Sci U S A. 1998;95(6):2920–2925. doi: 10.1073/pnas.95.6.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jepsen K, Hermanson O, Onami TM, Gleiberman AS, Lunyak V, McEvilly RJ, Kurokawa R, Kumar V, Liu F, Seto E, Hedrick SM, Mandel G, Glass CK, Rose DW, Rosenfeld MG. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell. 2000;102(6):753–763. doi: 10.1016/s0092-8674(00)00064-7. [DOI] [PubMed] [Google Scholar]
- 48.Keeton EK, Brown M. Cell cycle progression stimulated by tamoxifen-bound estrogen receptor-alpha and promoter-specific effects in breast cancer cells deficient in N-CoR and SMRT. Mol Endocrinol. 2005;19(6):1543–1554. doi: 10.1210/me.2004-0395. [DOI] [PubMed] [Google Scholar]
- 49.Mazumdar A, Wang RA, Mishra SK, Adam L, Bagheri-Yarmand R, Mandal M, Vadlamudi RK, Kumar R. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat Cell Biol. 2001;3(1):30–37. doi: 10.1038/35050532. [DOI] [PubMed] [Google Scholar]
- 50.Montano MM, Ekena K, Delage-Mourroux R, Chang W, Martini P, Katzenellenbogen BS. An estrogen receptor-selective coregulator that potentiates the effectiveness of antiestrogens and represses the activity of estrogens. Proc Natl Acad Sci U S A. 1999;96(12):6947–6952. doi: 10.1073/pnas.96.12.6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delage-Mourroux R, Martini PG, Choi I, Kraichely DM, Hoeksema J, Katzenellenbogen BS. Analysis of estrogen receptor interaction with a repressor of estrogen receptor activity (REA) and the regulation of estrogen receptor transcriptional activity by REA. J Biol Chem. 2000;275(46):35848–35856. doi: 10.1074/jbc.M001327200. [DOI] [PubMed] [Google Scholar]
- 52.Zhang J, Guenther MG, Carthew RW, Lazar MA. Proteasomal regulation of nuclear receptor corepressor-mediated repression. Genes Dev. 1998;12(12):1775–1780. doi: 10.1101/gad.12.12.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frasor J, Danes JM, Funk CC, Katzenellenbogen BS. Estrogen down-regulation of the corepressor N-CoR: mechanism and implications for estrogen derepression of N-CoR-regulated genes. Proc Natl Acad Sci U S A. 2005;102(37):13153–13157. doi: 10.1073/pnas.0502782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, McKenna NJ, Onate SA, Tsai SY, Tsai MJ, O’Malley BW. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. 1997;389(6647):194–198. doi: 10.1038/38304. [DOI] [PubMed] [Google Scholar]
- 55.Liu XF, Bagchi MK. Recruitment of distinct chromatin-modifying complexes by tamoxifen-complexed estrogen receptor at natural target gene promoters in vivo. J Biol Chem. 2004;279(15):15050–15058. doi: 10.1074/jbc.M311932200. [DOI] [PubMed] [Google Scholar]
- 56.Feng Q, Yi P, Wong J, O’Malley BW. Signaling within a coactivator complex: methylation of SRC-3/AIB1 is a molecular switch for complex disassembly. Mol Cell Biol. 2006;26(21):7846–7857. doi: 10.1128/MCB.00568-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jordan VC, O’Malley BW. Selective Estrogen-Receptor Modulators and Antihormonal Resistance in Breast Cancer. J Clin Oncol. 2007;25(36):5815–5824. doi: 10.1200/JCO.2007.11.3886. [DOI] [PubMed] [Google Scholar]
- 58.Jin Y, Desta Z, Stearns V, Ward B, Ho H, Lee KH, Skaar T, Storniolo AM, Li L, Araba A, Blanchard R, Nguyen A, Ullmer L, Hayden J, Lemler S, Weinshilboum RM, Rae JM, Hayes DF, Flockhart DA. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst. 2005;97(1):30–39. doi: 10.1093/jnci/dji005. [DOI] [PubMed] [Google Scholar]
- 59.Weinshilboum R. Pharmacogenomics of endocrine therapy in breast cancer. Adv Exp Med Biol. 2008;630:220–231. doi: 10.1007/978-0-387-78818-0_14. [DOI] [PubMed] [Google Scholar]
- 60.Flockhart D. CYP2D6 genotyping and the pharmacogenetics of tamoxifen. Clin Adv Hematol Oncol. 2008;6(7):493–494. [PubMed] [Google Scholar]
- 61.Borges S, Desta Z, Li L, Skaar TC, Ward BA, Nguyen A, Jin Y, Storniolo AM, Nikoloff DM, Wu L, Hillman G, Hayes DF, Stearns V, Flockhart DA. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: implication for optimization of breast cancer treatment. Clin Pharmacol Ther. 2006;80(1):61–74. doi: 10.1016/j.clpt.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 62.Harvell DM, Richer JK, Singh M, Spoelstra N, Finlayson C, Borges VF, Elias AD, Horwitz KB. Estrogen regulated gene expression in response to neoadjuvant endocrine therapy of breast cancers: tamoxifen agonist effects dominate in the presence of an aromatase inhibitor. Breast Cancer Res Treat. 2008 doi: 10.1007/s10549-008-9923-6. [DOI] [PubMed] [Google Scholar]
- 63.Kim H-J, Cui X, Hilsenbeck SG, Lee AV. Progesterone Receptor Loss Correlates with Human Epidermal Growth Factor Receptor 2 Overexpression in Estrogen Receptor-Positive Breast Cancer. Clin Cancer Res. 2006;12(3):1013s–1018. doi: 10.1158/1078-0432.CCR-05-2128. [DOI] [PubMed] [Google Scholar]
- 64.Arpino G, Weiss H, Lee AV, Schiff R, De Placido S, Osborne CK, Elledge RM. Estrogen receptor-positive, progesterone receptor-negative breast cancer: association with growth factor receptor expression and tamoxifen resistance. J Natl Cancer Inst. 2005;97(17):1254–1261. doi: 10.1093/jnci/dji249. [DOI] [PubMed] [Google Scholar]
- 65.Osborne CK, Schiff R, Arpino G, Lee AS, Hilsenbeck VG. Endocrine responsiveness: understanding how progesterone receptor can be used to select endocrine therapy. Breast. 2005;14(6):458–465. doi: 10.1016/j.breast.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 66.Huang HJ, Neven P, Drijkoningen M, Paridaens R, Wildiers H, Van Limbergen E, Berteloot P, Amant F, Christiaens MR, Vergote I. Association between HER-2/neu and the progesterone receptor in oestrogen-dependent breast cancer is age-related. Breast Cancer Res Treat. 2005;91(1):81–87. doi: 10.1007/s10549-004-8235-8. [DOI] [PubMed] [Google Scholar]
- 67.Jordan VC, Gelber RD. Problems With the Progesterone Receptor in Practice? J Clin Oncol. 2007;25(15):1957–1959. doi: 10.1200/JCO.2007.10.7342. [DOI] [PubMed] [Google Scholar]
- 68.Arpino G, Wiechmann L, Osborne CK, Schiff R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocr Rev. 2008;29(2):217–233. doi: 10.1210/er.2006-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 70.Konecny G, Pauletti G, Pegram M, Untch M, Dandekar S, Aguilar Z, Wilson C, Rong HM, Bauerfeind I, Felber M, Wang HJ, Beryt M, Seshadri R, Hepp H, Slamon DJ. Quantitative association between HER-2/neu and steroid hormone receptors in hormone receptor-positive primary breast cancer. J Natl Cancer Inst. 2003;95(2):142–153. doi: 10.1093/jnci/95.2.142. [DOI] [PubMed] [Google Scholar]
- 71.De Laurentiis M, Arpino G, Massarelli E, Ruggiero A, Carlomagno C, Ciardiello F, Tortora G, D’Agostino D, Caputo F, Cancello G, Montagna E, Malorni L, Zinno L, Lauria R, Bianco AR, De Placido S. A meta-analysis on the interaction between HER-2 expression and response to endocrine treatment in advanced breast cancer. Clin Cancer Res. 2005;11(13):4741–4748. doi: 10.1158/1078-0432.CCR-04-2569. [DOI] [PubMed] [Google Scholar]
- 72.Tsutsui S, Ohno S, Murakami S, Hachitanda Y, Oda S. Prognostic value of epidermal growth factor receptor (EGFR) and its relationship to the estrogen receptor status in 1029 patients with breast cancer. Breast Cancer Res Treat. 2002;71(1):67–75. doi: 10.1023/a:1013397232011. [DOI] [PubMed] [Google Scholar]
- 73.Likhite VS, Stossi F, Kim K, Katzenellenbogen BS, Katzenellenbogen JA. Kinase-Specific Phosphorylation of the Estrogen Receptor Changes Receptor Interactions with Ligand, Deoxyribonucleic Acid, and Coregulators Associated with Alterations in Estrogen and Tamoxifen Activity. Mol Endocrinol. 2006;20(12):3120–3132. doi: 10.1210/me.2006-0068. [DOI] [PubMed] [Google Scholar]
- 74.Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. 2001;276(13):9817–9824. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- 75.Bunone G, Briand PA, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. Embo J. 1996;15(9):2174–2183. [PMC free article] [PubMed] [Google Scholar]
- 76.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, et al. Activation of the estrogen receptor through phosphorylation by mitogen- activated protein kinase. Science. 1995;270(5241):1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 77.Zwart W, Griekspoor A, Berno V, Lakeman K, Jalink K, Mancini M, Neefjes J, Michalides R. PKA-induced resistance to tamoxifen is associated with an altered orientation of ERalpha towards co-activator SRC-1. Embo J. 2007;26(15):3534–3544. doi: 10.1038/sj.emboj.7601791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu RC, Qin J, Hashimoto Y, Wong J, Xu J, Tsai SY, Tsai MJ, O’Malley BW. Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) Coactivator activity by I kappa B kinase. Mol Cell Biol. 2002;22(10):3549–3561. doi: 10.1128/MCB.22.10.3549-3561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Font de Mora J, Brown M. AIB1 is a conduit for kinase-mediated growth factor signaling to the estrogen receptor. Mol Cell Biol. 2000;20(14):5041–5047. doi: 10.1128/mcb.20.14.5041-5047.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lopez GN, Turck CW, Schaufele F, Stallcup MR, Kushner PJ. Growth factors signal to steroid receptors through mitogen-activated protein kinase regulation of p160 coactivator activity. J Biol Chem. 2001;276(25):22177–22182. doi: 10.1074/jbc.M010718200. [DOI] [PubMed] [Google Scholar]
- 81.Hong SH, Privalsky ML. The SMRT corepressor is regulated by a MEK-1 kinase pathway: inhibition of corepressor function is associated with SMRT phosphorylation and nuclear export. Mol Cell Biol. 2000;20(17):6612–6625. doi: 10.1128/mcb.20.17.6612-6625.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96(12):926–935. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 83.Schiff R, Massarweh SA, Shou J, Bharwani L, Mohsin SK, Osborne CK. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin Cancer Res. 2004;10(1 Pt 2):331S–336S. doi: 10.1158/1078-0432.ccr-031212. [DOI] [PubMed] [Google Scholar]
- 84.Nicholson RI, Hutcheson IR, Knowlden JM, Jones HE, Harper ME, Jordan N, Hiscox SE, Barrow D, Gee JMW. Nonendocrine Pathways and Endocrine Resistance: Observations with Antiestrogens and Signal Transduction Inhibitors in Combination. Clin Cancer Res. 2004;10(1):346S–354. doi: 10.1158/1078-0432.ccr-031206. [DOI] [PubMed] [Google Scholar]
- 85.Lee AV, Weng CN, Jackson JG, Yee D. Activation of estrogen receptor-mediated gene transcription by IGF-I in human breast cancer cells. J Endocrinol. 1997;152(1):39–47. doi: 10.1677/joe.0.1520039. [DOI] [PubMed] [Google Scholar]
- 86.Creighton CJ, Casa A, Lazard Z, Huang S, Tsimelzon A, Hilsenbeck SG, Osborne CK, Lee AV. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol. 2008;26(25):4078–4085. doi: 10.1200/JCO.2007.13.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dihge L, Bendahl PO, Grabau D, Isola J, Lovgren K, Ryden L, Ferno M. Epidermal growth factor receptor (EGFR) and the estrogen receptor modulator amplified in breast cancer (AIB1) for predicting clinical outcome after adjuvant tamoxifen in breast cancer. Breast Cancer Res Treat. 2008;109(2):255–262. doi: 10.1007/s10549-007-9645-1. [DOI] [PubMed] [Google Scholar]
- 88.Kirkegaard T, McGlynn LM, Campbell FM, Muller S, Tovey SM, Dunne B, Nielsen KV, Cooke TG, Bartlett JM. Amplified in breast cancer 1 in human epidermal growth factor receptor - positive tumors of tamoxifen-treated breast cancer patients. Clin Cancer Res. 2007;13(5):1405–1411. doi: 10.1158/1078-0432.CCR-06-1933. [DOI] [PubMed] [Google Scholar]
- 89.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003;95(5):353–361. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 90.Girault I, Lerebours F, Amarir S, Tozlu S, Tubiana-Hulin M, Lidereau R, Bieche I. Expression Analysis of Estrogen Receptor {alpha} Coregulators in Breast Carcinoma: Evidence That NCOR1 Expression Is Predictive of the Response to Tamoxifen. Clin Cancer Res. 2003;9(4):1259–1266. [PubMed] [Google Scholar]
- 91.Wang LH, Yang XY, Zhang X, An P, Kim HJ, Huang J, Clarke R, Osborne CK, Inman JK, Appella E, Farrar WL. Disruption of estrogen receptor DNA-binding domain and related intramolecular communication restores tamoxifen sensitivity in resistant breast cancer. Cancer Cell. 2006;10(6):487–499. doi: 10.1016/j.ccr.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 92.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 93.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65(16):7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 94.Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL, Jacob S, Majumder S. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27(Kip1) J Biol Chem. 2008:M804612200. doi: 10.1074/jbc.M804612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhao J-J, Lin J, Yang H, Kong W, He L, Ma X, Coppola D, Cheng JQ. MicroRNA-221/222 negatively regulates ERalpha and associates with tamoxifen resistance in breast cancer. J Biol Chem. 2008:M806041200. doi: 10.1074/jbc.M806041200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 96.Gottardis MM, Jordan VC. Development of tamoxifen-stimulated growth of MCF-7 tumors in athymic mice after long-term antiestrogen administration. Cancer Res. 1988;48(18):5183–5187. [PubMed] [Google Scholar]
- 97.Osborne CK, Pippen J, Jones SE, Parker LM, Ellis M, Come S, Gertler SZ, May JT, Burton G, Dimery I, Webster A, Morris C, Elledge R, Buzdar A. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a North American trial. J Clin Oncol. 2002;20(16):3386–3395. doi: 10.1200/JCO.2002.10.058. [DOI] [PubMed] [Google Scholar]
- 98.Howell A, Robertson JF, Quaresma Albano J, Aschermannova A, Mauriac L, Kleeberg UR, Vergote I, Erikstein B, Webster A, Morris C. Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J Clin Oncol. 2002;20(16):3396–3403. doi: 10.1200/JCO.2002.10.057. [DOI] [PubMed] [Google Scholar]
- 99.Johnston SR, Saccani-Jotti G, Smith IE, Salter J, Newby J, Coppen M, Ebbs SR, Dowsett M. Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer. Cancer Res. 1995;55(15):3331–3338. [PubMed] [Google Scholar]
- 100.Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC, Schiff R, Osborne CK, Dowsett M. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol. 2005;23(11):2469–2476. doi: 10.1200/JCO.2005.01.172. [DOI] [PubMed] [Google Scholar]
- 101.Normanno N, Di Maio M, De Maio E, De Luca A, de Matteis A, Giordano A, Perrone F. Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr Relat Cancer. 2005;12(4):721–747. doi: 10.1677/erc.1.00857. [DOI] [PubMed] [Google Scholar]
- 102.de Graffenried LA, Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, Roth RA, Hidalgo M. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res. 2004;10(23):8059–8067. doi: 10.1158/1078-0432.CCR-04-0035. [DOI] [PubMed] [Google Scholar]
- 103.Chu I, Sun J, Arnaout A, Kahn H, Hanna W, Narod S, Sun P, Tan CK, Hengst L, Slingerland J. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell. 2007;128(2):281–294. doi: 10.1016/j.cell.2006.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Abukhdeir AM, Vitolo MI, Argani P, De Marzo AM, Karakas B, Konishi H, Gustin JP, Lauring J, Garay JP, Pendleton C, Konishi Y, Blair BG, Brenner K, Garrett-Mayer E, Carraway H, Bachman KE, Park BH. Tamoxifen-stimulated growth of breast cancer due to p21 loss. Proceedings of the National Academy of Sciences. 2008:0710887105. doi: 10.1073/pnas.0710887105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yuan Y, Qin L, Liu D, Wu R-C, Mussi P, Zhou S, Songyang Z, Xu J. Genetic Screening Reveals an Essential Role of p27kip1 in Restriction of Breast Cancer Progression. Cancer Res. 2007;67(17):8032–8042. doi: 10.1158/0008-5472.CAN-07-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Iorns E, Turner NC, Elliott R, Syed N, Garrone O, Gasco M, Tutt ANJ, Crook T, Lord CJ, Ashworth A. Identification of CDK10 as an Important Determinant of Resistance to Endocrine Therapy for Breast Cancer. Cancer Cell. 2008;13(2):91–104. doi: 10.1016/j.ccr.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 107.Moriai R, Tsuji N, Moriai M, Kobayashi D, Watanabe N. Survivin plays as a resistant factor against tamoxifen-induced apoptosis in human breast cancer cells. Breast Cancer Res Treat. 2008 doi: 10.1007/s10549-008-0164-5. [DOI] [PubMed] [Google Scholar]
- 108.Jordan VC, Lewis JS, Osipo C, Cheng D. The apoptotic action of estrogen following exhaustive antihormonal therapy: a new clinical treatment strategy. Breast. 2005;14(6):624–630. doi: 10.1016/j.breast.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 109.O’Regan RM, Osipo C, Ariazi E, Lee ES, Meeke K, Morris C, Bertucci A, Sarker MA, Grigg R, Jordan VC. Development and therapeutic options for the treatment of raloxifene-stimulated breast cancer in athymic mice. Clin Cancer Res. 2006;12(7 Pt 1):2255–2263. doi: 10.1158/1078-0432.CCR-05-2584. [DOI] [PubMed] [Google Scholar]
- 110.Liu H, Lee ES, Gajdos C, Pearce ST, Chen B, Osipo C, Loweth J, McKian K, De Los Reyes A, Wing L, Jordan VC. Apoptotic action of 17beta-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst. 2003;95(21):1586–1597. doi: 10.1093/jnci/djg080. [DOI] [PubMed] [Google Scholar]
- 111.Jordan VC, Phelps E, Lindgren JU. Effects of anti-estrogens on bone in castrated and intact female rats. Breast Cancer Res Treat. 1987;10(1):31–35. doi: 10.1007/BF01806132. [DOI] [PubMed] [Google Scholar]
- 112.Love RR, Mazess RB, Barden HS, Epstein S, Newcomb PA, Jordan VC, Carbone PP, DeMets DL. Effects of tamoxifen on bone mineral density in postmenopausal women with breast cancer. N Engl J Med. 1992;326(13):852–856. doi: 10.1056/NEJM199203263261302. [DOI] [PubMed] [Google Scholar]
- 113.Umemoto A, Lin CX, Ueyama Y, Komaki K, Santosh Laxmi YR, Shibutani S. Absence of DNA adduct in the leukocytes from breast cancer patients treated with toremifene. Chem Res Toxicol. 2006;19(3):421–425. doi: 10.1021/tx0503045. [DOI] [PubMed] [Google Scholar]
- 114.Shibutani S, Ravindernath A, Terashima I, Suzuki N, Laxmi YR, Kanno Y, Suzuki M, Apak TI, Sheng JJ, Duffel MW. Mechanism of lower genotoxicity of toremifene compared with tamoxifen. Cancer Res. 2001;61(10):3925–3931. [PubMed] [Google Scholar]
- 115.da Costa GG, Pereira PC, Churchwell MI, Beland FA, Marques MM. DNA adduct formation in the livers of female Sprague-Dawley rats treated with toremifene or alpha-hydroxytoremifene. Chem Res Toxicol. 2007;20(2):300–310. doi: 10.1021/tx600275d. [DOI] [PubMed] [Google Scholar]
- 116.Dragan VP, Vaughan J, Jordan VC, Pitot HC. Comparison of the effects of tamoxifen and toremifene on liver and kidney tumor promotion in female rats. Carcinogenesis. 1995;16(11):2733–2741. doi: 10.1093/carcin/16.11.2733. [DOI] [PubMed] [Google Scholar]
- 117.Sargent LM, Dragan YP, Sattler C, Bahnub N, Sattler G, Martin P, Cisneros A, Mann J, Thorgeirsson S, Jordan VC, Pitot HC. Induction of hepatic aneuploidy in vivo by tamoxifen, toremifene and idoxifene in female Sprague-Dawley rats. Carcinogenesis. 1996;17(5):1051–1056. doi: 10.1093/carcin/17.5.1051. [DOI] [PubMed] [Google Scholar]
- 118.Erkkola R, Mattila L, Powles T, Heikkinen J, Toivola B, Korhonen P, Mustonen M. Bone mineral density and lipid changes during 5 years of follow-up in a study of prevention of breast cancer with toremifene in healthy, high-risk pre- and post-menopausal women. Breast Cancer Res Treat. 2005;93(3):277–287. doi: 10.1007/s10549-005-5701-x. [DOI] [PubMed] [Google Scholar]
- 119.Harvey HA, Kimura M, Hajba A. Toremifene: an evaluation of its safety profile. Breast. 2006;15(2):142–157. doi: 10.1016/j.breast.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 120.O’Regan RM, Cisneros A, England GM, MacGregor JI, Muenzner HD, Assikis VJ, Bilimoria MM, Piette M, Dragan YP, Pitot HC, Chatterton R, Jordan VC. Effects of the antiestrogens tamoxifen, toremifene, and ICI 182,780 on endometrial cancer growth. J Natl Cancer Inst. 1998;90(20):1552–1558. doi: 10.1093/jnci/90.20.1552. [DOI] [PubMed] [Google Scholar]
- 121.Kusama M, Kaise H, Nakayama S, Ota D, Misaka T, Aoki T. Crossover trial for lipid abnormality in postmenopausal breast cancer patients during selective estrogen receptor modulators (SERMs) administrations. Breast Cancer Res Treat. 2004;88(1):9–16. doi: 10.1007/s10549-004-5449-8. [DOI] [PubMed] [Google Scholar]
- 122.Kusama M, Miyauchi K, Aoyama H, Sano M, Kimura M, Mitsuyama S, Komaki K, Doihara H. Effects of toremifene (TOR) and tamoxifen (TAM) on serum lipids in postmenopausal patients with breast cancer. Breast Cancer Res Treat. 2004;88(1):1–8. doi: 10.1007/s10549-004-4384-z. [DOI] [PubMed] [Google Scholar]
- 123.Lee ES, Schafer JM, Yao K, England G, O’Regan RM, De Los Reyes A, Jordan VC. Cross-resistance of triphenylethylene-type antiestrogens but not ICI 182,780 in tamoxifen-stimulated breast tumors grown in athymic mice. Clin Cancer Res. 2000;6(12):4893–4899. [PubMed] [Google Scholar]
- 124.Stenbygaard LE, Herrstedt J, Thomsen JF, Svendsen KR, Engelholm SA, Dombernowsky P. Toremifene and tamoxifen in advanced breast cancer--a double-blind cross-over trial. Breast Cancer Res Treat. 1993;25(1):57–63. doi: 10.1007/BF00662401. [DOI] [PubMed] [Google Scholar]
- 125.Vogel CL, Shemano I, Schoenfelder J, Gams RA, Green MR. Multicenter phase II efficacy trial of toremifene in tamoxifen-refractory patients with advanced breast cancer. J Clin Oncol. 1993;11(2):345–350. doi: 10.1200/JCO.1993.11.2.345. [DOI] [PubMed] [Google Scholar]
- 126.Gong C, Song E, Jia W, Qin L, Guo J, Jia H, Hu X, Su F. A double-blind randomized controlled trial of toremifen therapy for mastalgia. Arch Surg. 2006;141(1):43–47. doi: 10.1001/archsurg.141.1.43. [DOI] [PubMed] [Google Scholar]
- 127.Oksa S, Luukkaala T, Maenpaa J. Toremifene for premenstrual mastalgia: a randomised, placebo-controlled crossover study. Bjog. 2006;113(6):713–718. doi: 10.1111/j.1471-0528.2006.00943.x. [DOI] [PubMed] [Google Scholar]
- 128.Price D, Stein B, Sieber P, Tutrone R, Bailen J, Goluboff E, Burzon D, Bostwick D, Steiner M. Toremifene for the prevention of prostate cancer in men with high grade prostatic intraepithelial neoplasia: results of a double-blind, placebo controlled, phase IIB clinical trial. J Urol. 2006;176(3):965–970. doi: 10.1016/j.juro.2006.04.011. discussion 970–961. [DOI] [PubMed] [Google Scholar]
- 129.Smith MR, Malkowicz SB, Chu F, Forrest J, Price D, Sieber P, Barnette KG, Rodriguez D, Steiner MS. Toremifene increases bone mineral density in men receiving androgen deprivation therapy for prostate cancer: interim analysis of a multicenter phase 3 clinical study. J Urol. 2008;179(1):152–155. doi: 10.1016/j.juro.2007.08.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Smith MR, Malkowicz SB, Chu F, Forrest J, Sieber P, Barnette KG, Rodriquez D, Steiner MS. Toremifene improves lipid profiles in men receiving androgen-deprivation therapy for prostate cancer: interim analysis of a multicenter phase III study. J Clin Oncol. 2008;26(11):1824–1829. doi: 10.1200/JCO.2007.13.5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hellmann-Blumberg U, Taras TL, Wurz GT, DeGregorio MW. Genotoxic effects of the novel mixed antiestrogen FC-1271a in comparison to tamoxifen and toremifene. Breast Cancer Res Treat. 2000;60(1):63–70. doi: 10.1023/a:1006311214152. [DOI] [PubMed] [Google Scholar]
- 132.Qu Q, Zheng H, Dahllund J, Laine A, Cockcroft N, Peng Z, Koskinen M, Hemminki K, Kangas L, Vaananen K, Harkonen P. Selective estrogenic effects of a novel triphenylethylene compound, FC1271a, on bone, cholesterol level, and reproductive tissues in intact and ovariectomized rats. Endocrinology. 2000;141(2):809–820. doi: 10.1210/endo.141.2.7342. [DOI] [PubMed] [Google Scholar]
- 133.Wurz GT, Hellmann-Blumberg U, DeGregorio MW. Pharmacologic effects of ospemifene in rhesus macaques: a pilot study. Basic Clin Pharmacol Toxicol. 2008;102(6):552–558. doi: 10.1111/j.1742-7843.2008.00235.x. [DOI] [PubMed] [Google Scholar]
- 134.Komi J, Lankinen KS, Harkonen P, DeGregorio MW, Voipio S, Kivinen S, Tuimala R, Vihtamaki T, Vihko K, Ylikorkala O, Erkkola R. Effects of ospemifene and raloxifene on hormonal status, lipids, genital tract, and tolerability in postmenopausal women. Menopause. 2005;12(2):202–209. doi: 10.1097/00042192-200512020-00015. [DOI] [PubMed] [Google Scholar]
- 135.Voipio SK, Komi J, Kangas L, Halonen K, DeGregorio MW, Erkkola RU. Effects of ospemifene (FC-1271a) on uterine endometrium, vaginal maturation index, and hormonal status in healthy postmenopausal women. Maturitas. 2002;43(3):207–214. doi: 10.1016/s0378-5122(02)00206-2. [DOI] [PubMed] [Google Scholar]
- 136.Rutanen EM, Heikkinen J, Halonen K, Komi J, Lammintausta R, Ylikorkala O. Effects of ospemifene, a novel SERM, on hormones, genital tract, climacteric symptoms, and quality of life in postmenopausal women: a double-blind, randomized trial. Menopause. 2003;10(5):433–439. doi: 10.1097/01.GME.0000063609.62485.27. [DOI] [PubMed] [Google Scholar]
- 137.Kallio A, Guo T, Lamminen E, Seppanen J, Kangas L, Vaananen HK, Harkonen P. Estrogen and the selective estrogen receptor modulator (SERM) protection against cell death in estrogen receptor alpha and beta expressing U2OS cells. Mol Cell Endocrinol. 2008 doi: 10.1016/j.mce.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 138.Michael H, Harkonen PL, Kangas L, Vaananen HK, Hentunen TA. Differential effects of selective oestrogen receptor modulators (SERMs) tamoxifen, ospemifene and raloxifene on human osteoclasts in vitro. Br J Pharmacol. 2007;151(3):384–395. doi: 10.1038/sj.bjp.0707232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Komi J, Lankinen KS, DeGregorio M, Heikkinen J, Saarikoski S, Tuppurainen M, Halonen K, Lammintausta R, Vaananen K, Ylikorkala O, Erkkola R. Effects of ospemifene and raloxifene on biochemical markers of bone turnover in postmenopausal women. J Bone Miner Metab. 2006;24(4):314–318. doi: 10.1007/s00774-006-0689-9. [DOI] [PubMed] [Google Scholar]
- 140.Wurz GT, Read KC, Marchisano-Karpman C, Gregg JP, Beckett LA, Yu Q, Degregorio MW. Ospemifene inhibits the growth of dimethylbenzanthracene-induced mammary tumors in Sencar mice. J Steroid Biochem Mol Biol. 2005;97(3):230–240. doi: 10.1016/j.jsbmb.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 141.Namba R, Young LJ, Maglione JE, McGoldrick ET, Liu S, Wurz GT, DeGregorio MW, Borowsky AD, MacLeod CL, Cardiff RD, Gregg JP. Selective estrogen receptor modulators inhibit growth and progression of premalignant lesions in a mouse model of ductal carcinoma in situ. Breast Cancer Res. 2005;7(6):R881–889. doi: 10.1186/bcr1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Willson TM, Norris JD, Wagner BL, Asplin I, Baer P, Brown HR, Jones SA, Henke B, Sauls H, Wolfe S, Morris DC, McDonnell DP. Dissection of the molecular mechanism of action of GW5638; a novel estrogen receptor ligand, provides insights into the role of estrogen receptor in bone. Endocrinology. 1997;138(9):3901–3911. doi: 10.1210/endo.138.9.5358. [DOI] [PubMed] [Google Scholar]
- 143.Bentrem D, Dardes R, Liu H, MacGregor-Schafer J, Zapf J, Jordan V. Molecular mechanism of action at estrogen receptor alpha of a new clinically relevant antiestrogen (GW7604) related to tamoxifen. Endocrinology. 2001;142(2):838–846. doi: 10.1210/endo.142.2.7932. [DOI] [PubMed] [Google Scholar]
- 144.Connor CE, Norris JD, Broadwater G, Willson TM, Gottardis MM, Dewhirst MW, McDonnell DP. Circumventing tamoxifen resistance in breast cancers using antiestrogens that induce unique conformational changes in the estrogen receptor. Cancer Res. 2001;61(7):2917–2922. [PubMed] [Google Scholar]
- 145.Dardes RC, O’Regan RM, Gajdos C, Robinson SP, Bentrem D, De Los Reyes A, Jordan VC. Effects of a new clinically relevant antiestrogen (GW5638) related to tamoxifen on breast and endometrial cancer growth in vivo. Clin Cancer Res. 2002;8(6):1995–2001. [PubMed] [Google Scholar]
- 146.Wu YL, Yang X, Ren Z, McDonnell DP, Norris JD, Willson TM, Greene GL. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol Cell. 2005;18(4):413–424. doi: 10.1016/j.molcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 147.Fan M, Rickert EL, Chen L, Aftab SA, Nephew KP, Weatherman RV. Characterization of molecular and structural determinants of selective estrogen receptor downregulators. Breast Cancer Res Treat. 2007;103(1):37–44. doi: 10.1007/s10549-006-9353-2. [DOI] [PubMed] [Google Scholar]
- 148.Wittmann BM, Sherk A, McDonnell DP. Definition of functionally important mechanistic differences among selective estrogen receptor down-regulators. Cancer Res. 2007;67(19):9549–9560. doi: 10.1158/0008-5472.CAN-07-1590. [DOI] [PubMed] [Google Scholar]
- 149.Sporn MB. Arzoxifene: a promising new selective estrogen receptor modulator for clinical chemoprevention of breast cancer. Clin Cancer Res. 2004;10(16):5313–5315. doi: 10.1158/1078-0432.CCR-04-1377. [DOI] [PubMed] [Google Scholar]
- 150.Munster PN. Arzoxifene: the development and clinical outcome of an ideal SERM. Expert Opin Investig Drugs. 2006;15(3):317–326. doi: 10.1517/13543784.15.3.317. [DOI] [PubMed] [Google Scholar]
- 151.Schafer JM, Lee ES, Dardes RC, Bentrem D, O’Regan RM, De Los Reyes A, Jordan VC. Analysis of cross-resistance of the selective estrogen receptor modulators arzoxifene (LY353381) and LY117018 in tamoxifen-stimulated breast cancer xenografts. Clin Cancer Res. 2001;7(8):2505–2512. [PubMed] [Google Scholar]
- 152.Buzdar A, O’Shaughnessy JA, Booser DJ, Pippen JE, Jr, Jones SE, Munster PN, Peterson P, Melemed AS, Winer E, Hudis C. Phase II, randomized, double-blind study of two dose levels of arzoxifene in patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2003;21(6):1007–1014. doi: 10.1200/JCO.2003.06.108. [DOI] [PubMed] [Google Scholar]
- 153.McMeekin DS, Gordon A, Fowler J, Melemed A, Buller R, Burke T, Bloss J, Sabbatini P. A phase II trial of arzoxifene, a selective estrogen response modulator, in patients with recurrent or advanced endometrial cancer. Gynecol Oncol. 2003;90(1):64–69. doi: 10.1016/s0090-8258(03)00203-8. [DOI] [PubMed] [Google Scholar]
- 154.Deshmane V, Krishnamurthy S, Melemed AS, Peterson P, Buzdar AU. Phase III Double-Blind Trial of Arzoxifene Compared With Tamoxifen for Locally Advanced or Metastatic Breast Cancer. J Clin Oncol. 2007;25(31):4967–4973. doi: 10.1200/JCO.2006.09.5992. [DOI] [PubMed] [Google Scholar]
- 155.Liby K, Rendi M, Suh N, Royce DB, Risingsong R, Williams CR, Lamph W, Labrie F, Krajewski S, Xu X, Kim H, Brown P, Sporn MB. The combination of the rexinoid, LG100268; and a selective estrogen receptor modulator, either arzoxifene or acolbifene, synergizes in the prevention and treatment of mammary tumors in an estrogen receptor-negative model of breast cancer. Clin Cancer Res. 2006;12(19):5902–5909. doi: 10.1158/1078-0432.CCR-06-1119. [DOI] [PubMed] [Google Scholar]
- 156.Fabian CJ, Kimler BF, Anderson J, Tawfik OW, Mayo MS, Burak WE, Jr, O’Shaughnessy JA, Albain KS, Hyams DM, Budd GT, Ganz PA, Sauter ER, Beenken SW, Grizzle WE, Fruehauf JP, Arneson DW, Bacus JW, Lagios MD, Johnson KA, Browne D. Breast cancer chemoprevention phase I evaluation of biomarker modulation by arzoxifene, a third generation selective estrogen receptor modulator. Clin Cancer Res. 2004;10(16):5403–5417. doi: 10.1158/1078-0432.CCR-04-0171. [DOI] [PubMed] [Google Scholar]
- 157.Vajdos FF, Hoth LR, Geoghegan KF, Simons SP, LeMotte PK, Danley DE, Ammirati MJ, Pandit J. The 2.0 A crystal structure of the ERalpha ligand-binding domain complexed with lasofoxifene. Protein Sci. 2007;16(5):897–905. doi: 10.1110/ps.062729207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ke HZ, Paralkar VM, Grasser WA, Crawford DT, Qi H, Simmons HA, Pirie CM, Chidsey-Frink KL, Owen TA, Smock SL, Chen HK, Jee WS, Cameron KO, Rosati RL, Brown TA, Dasilva-Jardine P, Thompson DD. Effects of CP-336,156, a new, nonsteroidal estrogen agonist/antagonist, on bone, serum cholesterol, uterus and body composition in rat models. Endocrinology. 1998;139(4):2068–2076. doi: 10.1210/endo.139.4.5902. [DOI] [PubMed] [Google Scholar]
- 159.Gennari L. Lasofoxifene: a new type of selective estrogen receptor modulator for the treatment of osteoporosis. Drugs Today (Barc) 2006;42(6):355–367. doi: 10.1358/dot.2006.42.6.973583. [DOI] [PubMed] [Google Scholar]
- 160.Rosati RL, Da Silva Jardine P, Cameron KO, Thompson DD, Ke HZ, Toler SM, Brown TA, Pan LC, Ebbinghaus CF, Reinhold AR, Elliott NC, Newhouse BN, Tjoa CM, Sweetnam PM, Cole MJ, Arriola MW, Gauthier JW, Crawford DT, Nickerson DF, Pirie CM, Qi H, Simmons HA, Tkalcevic GT. Discovery and preclinical pharmacology of a novel, potent, nonsteroidal estrogen receptor agonist/antagonist, CP-336156, a diaryltetrahydronaphthalene. J Med Chem. 1998;41(16):2928–2931. doi: 10.1021/jm980048b. [DOI] [PubMed] [Google Scholar]
- 161.Gass M, Kagan R, Simon J, Spino C, Barcomb L. Objective Measures of Vaginal Atrophy Are Improved With Lasofoxifene, A Next-Generation Selective Estrogen Receptor Modulator. Obstetrics & Gynecology. 2005;105(4):71S. [Google Scholar]
- 162.Wang XN, Simmons HA, Salatto CT, Cosgrove PG, Thompson DD. Lasofoxifene enhances vaginal mucus formation without causing hypertrophy and increases estrogen receptor beta and androgen receptor in rats. Menopause. 2006;13(4):609–620. doi: 10.1097/01.gme.0000227337.73738.c9. [DOI] [PubMed] [Google Scholar]
- 163.Cohen LA, Pittman B, Wang CX, Aliaga C, Yu L, Moyer JD. LAS, a novel selective estrogen receptor modulator with chemopreventive and therapeutic activity in the N-nitroso-N-methylurea-induced rat mammary tumor model. Cancer Res. 2001;61(24):8683–8688. [PubMed] [Google Scholar]
- 164.Vogelvang TE, van der Mooren MJ, Mijatovic V, Kenemans P. Emerging selective estrogen receptor modulators: special focus on effects on coronary heart disease in postmenopausal women. Drugs. 2006;66(2):191–221. doi: 10.2165/00003495-200666020-00005. [DOI] [PubMed] [Google Scholar]
- 165.Stein RC, Dowsett M, Cunningham DC, Davenport J, Ford HT, Gazet JC, von Angerer E, Coombes RC. Phase I/II study of the anti-oestrogen zindoxifene (D16726) in the treatment of advanced breast cancer. A Cancer Research Campaign Phase I/II Clinical Trials Committee study. Br J Cancer. 1990;61(3):451–453. doi: 10.1038/bjc.1990.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Miller CP, Collini MD, Tran BD, Harris HA, Kharode YP, Marzolf JT, Moran RA, Henderson RA, Bender RH, Unwalla RJ, Greenberger LM, Yardley JP, Abou-Gharbia MA, Lyttle CR, Komm BS. Design, synthesis, and preclinical characterization of novel, highly selective indole estrogens. J Med Chem. 2001;44(11):1654–1657. doi: 10.1021/jm010086m. [DOI] [PubMed] [Google Scholar]
- 167.Greenberger LM, Annable T, Collins KI, Komm BS, Lyttle CR, Miller CP, Satyaswaroop PG, Zhang Y, Frost P. A new antiestrogen, 2-(4-hydroxy-phenyl)-3-methyl-1-[4-(2-piperidin-1-yl-ethoxy)-benzyl]-1H-in dol-5-ol hydrochloride (ERA-923), inhibits the growth of tamoxifen-sensitive and -resistant tumors and is devoid of uterotropic effects in mice and rats. Clin Cancer Res. 2001;7(10):3166–3177. [PubMed] [Google Scholar]
- 168.Sadler TM, Gavriil M, Annable T, Frost P, Greenberger LM, Zhang Y. Combination therapy for treating breast cancer using antiestrogen, ERA-923, and the mammalian target of rapamycin inhibitor, temsirolimus. Endocr Relat Cancer. 2006;13(3):863–873. doi: 10.1677/erc.1.01170. [DOI] [PubMed] [Google Scholar]
- 169.Komm BS, Kharode YP, Bodine PV, Harris HA, Miller CP, Lyttle CR. Bazedoxifene acetate: a selective estrogen receptor modulator with improved selectivity. Endocrinology. 2005;146(9):3999–4008. doi: 10.1210/en.2005-0030. [DOI] [PubMed] [Google Scholar]
- 170.Ronkin S, Northington R, Baracat E, Nunes MG, Archer DF, Constantine G, Pickar JH. Endometrial effects of bazedoxifene acetate, a novel selective estrogen receptor modulator, in postmenopausal women. Obstet Gynecol. 2005;105(6):1397–1404. doi: 10.1097/01.AOG.0000163253.27610.b9. [DOI] [PubMed] [Google Scholar]
- 171.Silverman SL, Christiansen C, Genant HK, Vukicevic S, Zanchetta JR, de Villiers TJ, Constantine GD, Chines AA. Efficacy of Bazedoxifene in Reducing New Vertebral Fracture Risk in Postmenopausal Women With Osteoporosis: Results From a 3-Year, Randomized, Placebo- and Active-Controlled Clinical Trial*. J Bone Miner Res. 2008 doi: 10.1359/jbmr.080710. [DOI] [PubMed] [Google Scholar]
- 172.Bazedoxifene: Bazedoxifene Acetate, TSE 424, TSE-424, WAY 140424. Drugs R D. 2008;9(3):191–196. doi: 10.2165/00126839-200809030-00007. No authors listed. [DOI] [PubMed] [Google Scholar]
- 173.Kharode Y, Bodine PVN, Miller CP, Lyttle CR, Komm BS. The Pairing of a Selective Estrogen Receptor Modulator, Bazedoxifene, With Conjugated Estrogens as a New Paradigm for the Treatment of Menopausal Symptoms and Osteoporosis Prevention. Endocrinology. 2008 doi: 10.1210/en.2008-0817. en.2008–0817. [DOI] [PubMed] [Google Scholar]
- 174.Tremblay A, Tremblay GB, Labrie C, Labrie F, Giguere V. EM-800, a novel antiestrogen, acts as a pure antagonist of the transcriptional functions of estrogen receptors alpha and beta. Endocrinology. 1998;139(1):111–118. doi: 10.1210/endo.139.1.5702. [DOI] [PubMed] [Google Scholar]
- 175.Schafer JI, Liu H, Tonetti DA, Jordan VC. The interaction of raloxifene and the active metabolite of the antiestrogen EM-800 (SC 5705) with the human estrogen receptor. Cancer Res. 1999;59(17):4308–4313. [PubMed] [Google Scholar]
- 176.Martel C, Picard S, Richard V, Belanger A, Labrie C, Labrie F. Prevention of bone loss by EM-800 and raloxifene in the ovariectomized rat. J Steroid Biochem Mol Biol. 2000;74(1–2):45–56. doi: 10.1016/s0960-0760(00)00087-x. [DOI] [PubMed] [Google Scholar]
- 177.Lemieux C, Gelinas Y, Lalonde J, Labrie F, Richard D, Deshaies Y. Hypocholesterolemic action of the selective estrogen receptor modulator acolbifene in intact and ovariectomized rats with diet-induced hypercholesterolemia. Metabolism. 2006;55(5):605–613. doi: 10.1016/j.metabol.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 178.Martel C, Provencher L, Li X, St Pierre A, Leblanc G, Gauthier S, Merand Y, Labrie F. Binding characteristics of novel nonsteroidal antiestrogens to the rat uterine estrogen receptors. J Steroid Biochem Mol Biol. 1998;64(3–4):199–205. doi: 10.1016/s0960-0760(97)00192-1. [DOI] [PubMed] [Google Scholar]
- 179.Simard J, Labrie C, Belanger A, Gauthier S, Singh SM, Merand Y, Labrie F. Characterization of the effects of the novel non-steroidal antiestrogen EM-800 on basal and estrogen-induced proliferation of T-47D, ZR-75–1 and MCF-7 human breast cancer cells in vitro. Int J Cancer. 1997;73(1):104–112. doi: 10.1002/(sici)1097-0215(19970926)73:1<104::aid-ijc16>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 180.Gutman M, Couillard S, Roy J, Labrie F, Candas B, Labrie C. Comparison of the effects of EM-652 (SCH57068), tamoxifen, toremifene, droloxifene, idoxifene, GW-5638 and raloxifene on the growth of human ZR-75–1 breast tumors in nude mice. Int J Cancer. 2002;99(2):273–278. doi: 10.1002/ijc.10302. [DOI] [PubMed] [Google Scholar]
- 181.Labrie F, Champagne P, Labrie C, Roy J, Laverdiere J, Provencher L, Potvin M, Drolet Y, Pollak M, Panasci L, L’Esperance B, Dufresne J, Latreille J, Robert J, Samson B, Jolivet J, Yelle L, Cusan L, Diamond P, Candas B. Activity and Safety of the Antiestrogen EM-800, the Orally Active Precursor of Acolbifene, in Tamoxifen-Resistant Breast Cancer. J Clin Oncol. 2004;22(5):864–871. doi: 10.1200/JCO.2004.05.122. [DOI] [PubMed] [Google Scholar]
- 182.Labrie F. Future perspectives of selective estrogen receptor modulators used alone and in combination with DHEA. Endocr Relat Cancer. 2006;13(2):335–355. doi: 10.1677/erc.1.00883. [DOI] [PubMed] [Google Scholar]
- 183.Luo S, Sourla A, Labrie C, Belanger A, Labrie F. Combined effects of dehydroepiandrosterone and EM-800 on bone mass, serum lipids, and the development of dimethylbenz(A)anthracene-induced mammary carcinoma in the rat. Endocrinology. 1997;138(10):4435–4444. doi: 10.1210/endo.138.10.5429. [DOI] [PubMed] [Google Scholar]
- 184.Galbiati E, Caruso PL, Amari G, Armani E, Ghirardi S, Delcanale M, Civelli M. Pharmacological actions of a novel, potent, tissue-selective benzopyran estrogen. J Pharmacol Exp Ther. 2002;303(1):196–203. doi: 10.1124/jpet.102.038034. [DOI] [PubMed] [Google Scholar]
- 185.Armamento-Villareal R, Sheikh S, Nawaz A, Napoli N, Mueller C, Halstead LR, Brodt MD, Silva MJ, Galbiati E, Caruso PL, Civelli M, Civitelli R. A new selective estrogen receptor modulator, CHF 4227.01, preserves bone mass and microarchitecture in ovariectomized rats. J Bone Miner Res. 2005;20(12):2178–2188. doi: 10.1359/JBMR.050801. [DOI] [PubMed] [Google Scholar]
- 186.Civelli M, Preti AP, Cenacchi V, Rondelli I, Guastalla D, Tarral A, Dostert P, Guillevic Y, Homery MC. Single and multiple ascending dose studies of a novel tissue-selective oestrogen receptor modulator, CHF 4227, in healthy postmenopausal women. Br J Clin Pharmacol. 2007;64(3):304–316. doi: 10.1111/j.1365-2125.2007.02870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Brady H, Doubleday M, Gayo-Fung LM, Hickman M, Khammungkhune S, Kois A, Lipps S, Pierce S, Richard N, Shevlin G, Sutherland MK, Anderson DW, Bhagwat SS, Stein B. Differential response of estrogen receptors alpha and beta to SP500263, a novel potent selective estrogen receptor modulator. Mol Pharmacol. 2002;61(3):562–568. doi: 10.1124/mol.61.3.562. [DOI] [PubMed] [Google Scholar]
- 188.Brady H, Desai S, Gayo-Fung LM, Khammungkhune S, McKie JA, O’Leary E, Pascasio L, Sutherland MK, Anderson DW, Bhagwat SS, Stein B. Effects of SP500263, a novel, potent antiestrogen, on breast cancer cells and in xenograft models. Cancer Res. 2002;62(5):1439–1442. [PubMed] [Google Scholar]
- 189.Sutherland MK, Brady H, Gayo-Fung LM, Leisten J, Lipps SG, McKie JA, O’Leary E, Patnaik N, Anderson DW, Bhagwat SS, Stein B. Effects of SP500263, a novel selective estrogen receptor modulator, on bone, uterus, and serum cholesterol in the ovariectomized rat. Calcif Tissue Int. 2003;72(6):710–716. doi: 10.1007/s00223-002-1029-2. [DOI] [PubMed] [Google Scholar]
- 190.Kung Sutherland MS, Lipps SG, Patnaik N, Gayo-Fung LM, Khammungkune S, Xie W, Brady HA, Barbosa MS, Anderson DW, Stein B. SP500263, a novel SERM, blocks osteoclastogenesis in a human bone cell model: role of IL-6 and GM-CSF. Cytokine. 2003;23(1–2):1–14. doi: 10.1016/s1043-4666(03)00179-0. [DOI] [PubMed] [Google Scholar]
- 191.Ammann P, Bourrin S, Brunner F, Meyer JM, Clement-Lacroix P, Baron R, Gaillard M, Rizzoli R. A new selective estrogen receptor modulator HMR-3339 fully corrects bone alterations induced by ovariectomy in adult rats. Bone. 2004;35(1):153–161. doi: 10.1016/j.bone.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 192.Vogelvang TE, Mijatovic V, Kenemans P, Teerlink T, van der Mooren MJ. HMR 3339, a novel selective estrogen receptor modulator, reduces total cholesterol, low-density lipoprotein cholesterol, and homocysteine in healthy postmenopausal women. Fertil Steril. 2004;82(6):1540–1549. doi: 10.1016/j.fertnstert.2004.05.093. [DOI] [PubMed] [Google Scholar]
- 193.Vogelvang TE, Mijatovic V, Kenemans P, Schalkwijk CG, van der Mooren MJ. Effect of HMR 3339, a novel selective estrogen receptor modulator, on C-reactive protein levels in healthy postmenopausal women. Am J Cardiol. 2004;94(9):1205–1208. doi: 10.1016/j.amjcard.2004.07.099. [DOI] [PubMed] [Google Scholar]
- 194.Verhoeven MO, Teerlink T, Kenemans P, Vogelvang TE, van der Mooren MJ. Effects on asymmetric dimethylarginine of HMR 3339, a novel selective estrogen receptor modulator: a 12-week, randomized, placebo-controlled, double-blind, dose-ranging study in healthy postmenopausal women. Menopause. 2007;14(2):235–242. doi: 10.1097/01.gme.0000235367.47350.2a. [DOI] [PubMed] [Google Scholar]
- 195.Vogelvang TE, Leurs JR, Mijatovic V, Willemse J, van der Mooren MJ. HMR 3339, a novel selective estrogen receptor modulator, reduces concentrations of procarboxypeptidase U, an inhibitor of fibrinolysis. A randomized, placebo-controlled study in postmenopausal women. J Thromb Haemost. 2005;3(5):1090–1092. doi: 10.1111/j.1538-7836.2005.01306.x. [DOI] [PubMed] [Google Scholar]
- 196.Vogelvang TE, Mijatovic V, Kenemans P, Emeis JJ, Heijst JA, van der Mooren MJ. The effects of 12 weeks of HMR 3339, a novel selective estrogen receptor modulator, on markers of coagulation and fibrinolysis: a randomized, placebo-controlled, double-blind, dose-ranging study in healthy postmenopausal women. Am J Obstet Gynecol. 2005;193(4):1384–1394. doi: 10.1016/j.ajog.2005.02.083. [DOI] [PubMed] [Google Scholar]
- 197.Windahl SH, Galien R, Chiusaroli R, Clement-Lacroix P, Morvan F, Lepescheux L, Nique F, Horne WC, Resche-Rigon M, Baron R. Bone protection by estrens occurs through non-tissue-selective activation of the androgen receptor. J Clin Invest. 2006;116(9):2500–2509. doi: 10.1172/JCI28809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhao C, Dahlman-Wright K, Gustafsson JA. Estrogen receptor beta: an overview and update. Nucl Recept Signal. 2008;6:e003. doi: 10.1621/nrs.06003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Skliris GP, Leygue E, Watson PH, Murphy LC. Estrogen receptor alpha negative breast cancer patients: estrogen receptor beta as a therapeutic target. J Steroid Biochem Mol Biol. 2008;109(1–2):1–10. doi: 10.1016/j.jsbmb.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 200.Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 2006;147(10):4831–4842. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- 201.Lin CY, Strom A, Li Kong S, Kietz S, Thomsen JS, Tee JB, Vega VB, Miller LD, Smeds J, Bergh J, Gustafsson JA, Liu ET. Inhibitory effects of estrogen receptor beta on specific hormone-responsive gene expression and association with disease outcome in primary breast cancer. Breast Cancer Res. 2007;9(2):R25. doi: 10.1186/bcr1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Murphy LC, Watson PH. Is oestrogen receptor-beta a predictor of endocrine therapy responsiveness in human breast cancer? Endocr Relat Cancer. 2006;13(2):327–334. doi: 10.1677/erc.1.01141. [DOI] [PubMed] [Google Scholar]
- 203.Rosa FE, Caldeira JR, Felipes J, Bertonha FB, Quevedo FC, Domingues MA, Moraes Neto FA, Rogatto SR. Evaluation of estrogen receptor alpha and beta and progesterone receptor expression and correlation with clinicopathologic factors and proliferative marker Ki-67 in breast cancers. Hum Pathol. 2008;39(5):720–730. doi: 10.1016/j.humpath.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 204.Ariazi EA, Jordan VC. Estrogen-related receptors as emerging targets in cancer and metabolic disorders. Curr Top Med Chem. 2006;6(3):203–215. doi: 10.2174/1568026610606030203. [DOI] [PubMed] [Google Scholar]
- 205.Stein RA, McDonnell DP. Estrogen-related receptor {alpha} as a therapeutic target in cancer. Endocr Relat Cancer. 2006;13(Supplement_1):S25–32. doi: 10.1677/erc.1.01292. [DOI] [PubMed] [Google Scholar]
- 206.Stein RA, Chang C-y, Kazmin DA, Way J, Schroeder T, Wergin M, Dewhirst MW, McDonnell DP. Estrogen-Related Receptor {alpha} Is Critical for the Growth of Estrogen Receptor-Negative Breast Cancer. Cancer Res. 2008;68(21):8805–8812. doi: 10.1158/0008-5472.CAN-08-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451(7181):1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 208.Ao A, Wang H, Kamarajugadda S, Lu J. Involvement of estrogen-related receptors in transcriptional response to hypoxia and growth of solid tumors. Proc Natl Acad Sci U S A. 2008;105(22):7821–7826. doi: 10.1073/pnas.0711677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Leventhal L, Brandt MR, Cummons TA, Piesla MJ, Rogers KE, Harris HA. An estrogen receptor-beta agonist is active in models of inflammatory and chemical-induced pain. Eur J Pharmacol. 2006;553(1–3):146–148. doi: 10.1016/j.ejphar.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 210.Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-beta potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem. 2001;44(24):4230–4251. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- 211.Hegele-Hartung C, Siebel P, Peters O, Kosemund D, Muller G, Hillisch A, Walter A, Kraetzschmar J, Fritzemeier KH. Impact of isotype-selective estrogen receptor agonists on ovarian function. Proc Natl Acad Sci U S A. 2004;101(14):5129–5134. doi: 10.1073/pnas.0306720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Barker S. Non-steroidal anti-estrogens in the treatment of breast cancer. Curr Opin Investig Drugs. 2006;7(12):1085–1091. [PubMed] [Google Scholar]
- 213.Puddefoot JR, Barker S, Vinson GP. Trilostane in advanced breast cancer. Expert Opin Pharmacother. 2006;7(17):2413–2419. doi: 10.1517/14656566.7.17.2413. [DOI] [PubMed] [Google Scholar]
- 214.Puddefoot JR, Barker S, Glover HR, Malouitre SD, Vinson GP. Non-competitive steroid inhibition of oestrogen receptor functions. Int J Cancer. 2002;101(1):17–22. doi: 10.1002/ijc.10547. [DOI] [PubMed] [Google Scholar]
- 215.Barker S, Malouitre SD, Glover HR, Puddefoot JR, Vinson GP. Comparison of effects of 4-hydroxy tamoxifen and trilostane on oestrogen-regulated gene expression in MCF-7 cells: up-regulation of oestrogen receptor beta. J Steroid Biochem Mol Biol. 2006;100(4–5):141–151. doi: 10.1016/j.jsbmb.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 216.Yamamoto Y, Shibata J, Yonekura K, Sato K, Hashimoto A, Aoyagi Y, Wierzba K, Yano S, Asao T, Buzdar AU, Terada T. TAS-108, a novel oral steroidal antiestrogenic agent, is a pure antagonist on estrogen receptor alpha and a partial agonist on estrogen receptor beta with low uterotrophic effect. Clin Cancer Res. 2005;11(1):315–322. [PubMed] [Google Scholar]
- 217.Buzdar AU. TAS-108: a novel steroidal antiestrogen. Clin Cancer Res. 2005;11(2 Pt 2):906s–908s. [PubMed] [Google Scholar]
- 218.Blakely LJ, Buzdar A, Chang HY, Frye D, Theriault R, Valero V, Rivera E, Booser D, Kuritani J, Tsuda M. A phase I and pharmacokinetic study of TAS-108 in postmenopausal female patients with locally advanced, locally recurrent inoperable, or progressive metastatic breast cancer. Clin Cancer Res. 2004;10(16):5425–5431. doi: 10.1158/1078-0432.CCR-04-0321. [DOI] [PubMed] [Google Scholar]
- 219.Saeki T, Noguchi S, Aogi K, Inaji H, Tabei T, Ikeda T. Evaluation of the safety and tolerability of oral TAS-108 in postmenopausal patients with metastatic breast cancer. Ann Oncol. 2009 doi: 10.1093/annonc/mdn714. [DOI] [PubMed] [Google Scholar]
- 220.Ariazi EA, Ariazi JL, Cordera F, Jordan VC. Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem. 2006;6(3):181–202. [PubMed] [Google Scholar]
- 221.Jensen EV, Jordan VC. The estrogen receptor: a model for molecular medicine. Clin Cancer Res. 2003;9(6):1980–1989. [PubMed] [Google Scholar]
- 222.Jordan VC. Tamoxifen: catalyst for the change to targeted therapy. Eur J Cancer. 2008;44(1):30–38. doi: 10.1016/j.ejca.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R. Tamoxifen Resistance in Breast Tumors Is Driven by Growth Factor Receptor Signaling with Repression of Classic Estrogen Receptor Genomic Function. Cancer Res. 2008;68(3):826–833. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]