

Abstract
Diabetic nephropathy (DN) is a leading cause of end stage renal disease. Diabetic vascular complications like DN can progress despite subsequent glycemic control, suggesting a metabolic memory of previous exposure to hyperglycemia. Diabetes profoundly impacts transcription programs in target cells through activation of multiple signaling pathways and key transcription factors leading to aberrant expression of pathological genes. Emerging evidence suggests that these factors associated with the pathophysiology of diabetic complications and metabolic memory might also be influenced by epigenetic mechanisms in chromatin such as DNA methylation, histone lysine acetylation and methylation. Key histone modifications and the related histone methyltransferases and acetyltransferases have been implicated in the regulation of inflammatory and pro-fibrotic genes in renal and vascular cells under diabetic conditions Advances in epigenome profiling approaches have provided novel insights into the chromatin states and functional outcomes in target cells affected by diabetes. Because epigenetic changes are potentially reversible, they can provide a window of opportunity for the development of much needed new therapies for DN in the future. In this review we discuss recent developments in the field of epigenetics and their relevance to diabetic vascular complications and DN pathogenesis.
Keywords: chromatin, epigenomics, diabetic nephropathy, histone modifications, metabolic memory
Introduction
A global surge in the incidence of diabetes and metabolic disorders has greatly increased the risk for multiple associated complications. Diabetes leads to severe complications in several major organs including eyes, heart, nerves and kidney that lead to reduced quality of life and increased mortality rates. More than 40% of patients with diabetes develop diabetic nephropathy (DN), a microvascular complication and chronic kidney disease that leads to progressive renal dysfunction and is a leading cause of end stage renal disease (ESRD). Furthermore, DN also increases the risk for cardiovascular diseases (CVD). Diabetes profoundly impacts transcription programs of cells in target tissues via the activation of multiple signaling pathways and key transcription factors (TFs) leading to aberrant expression of pro-inflammatory and pro-fibrotic genes involved in DN pathogenesis1–3. Emerging evidence suggests that these factors associated with the pathophysiology of diabetes and its complications might also be influenced by epigenetically regulated mechanisms in chromatin such as DNA methylation (DNAme), and histone lysine acetylation (HKac) and methylation (HKme)4, 5. Apart from genetic predisposition, it is now clear that additional factors, like changes in the environment and lifestyles that affect epigenetic states, can influence the etiology and progression of common human diseases like diabetes and DN. Thus, the study of epigenetic mechanisms can offer valuable and novel new insights into the pathophysiology of diabetes and susceptibility to the associated complications. Rapid developments in high throughput genomic approaches such as next generation DNA sequencing (NGS) and efforts of several world-wide consortia have yielded enormous and significant information about chromatin states and the epigenome under normal and disease states that also have the potential to reveal functions of genetic variants at various genome locations6, 7. These exciting advances in the field of epigenetics can be exploited to enhance our understanding of the molecular mechanisms involved in the DN pathogenesis. Furthermore, because, unlike genetic variants, epigenetic changes are potentially reversible, they could be used to not only identify novel biomarkers for early disease detection, but also for the development of new therapeutic targets for DN, a debilitating complication of diabetes. In this review we discuss the recent developments in the field of epigenetics and their relevance to the pathogenesis of DN.
Diabetic Nephropathy and Metabolic Memory
DN is clinically characterized by proteinuria, albuminuria, rising creatinine levels, and aberrant glomerular filtration rates. The key histological and pathological features of DN include renal glomerular hypertrophy, mesangial expansion and tubulointerstitial fibrosis due to accumulation of extracellular matrix (ECM) proteins such as collagens and fibronectin, thickening of the basement membrane, podocyte loss and foot process effacement. Endothelial dysfunction and inflammation due to macrophage infiltration also play important roles in DN pathogenesis1, 8.
Hyperglycemia as well as complex interactions between metabolic and hemodynamic factors are linked to the development of diabetic complications including DN1, 9. High glucose (HG) adversely impacts all cell types in the kidney including mesangial cells (MC), tubular cells, podocytes, endothelial cells (EC) and infiltrating monocytes/macrophages. It increases the formation of advanced glycation end products (AGEs) and levels of growth factors like transforming growth factor-beta1 (TGF-β1) and Angiotensin II (Ang II) in renal cells. TGF-β levels are increased in most renal cells in diabetes and is a major player in DN pathogenesis mainly due to its profibrotic actions1, 3. Cross-talk between these diabetogenic factors can amplify and perpetuate the expression of pathologic genes associated with the progression of DN (Fig. 1). Multiple signal transduction mechanisms and kinases including oxidant stress, protein kinase C, Akt kinase, receptor and non-receptor protein tyrosine kinases have been implicated in the activation of key effector TFs such as Smads and NF-κB downstream of HG and growth factors leading to increased production of pro-inflammatory cytokines, cell cycle genes, profibrotic and ECM genes involved in DN1, 3. Recently, microRNA (miRNA) mediated mechanisms have also been implicated10. Despite such advances in the understanding of the biochemical and molecular mechanisms leading to DN, currently available therapies are still not fully effective in preventing progression to ESRD, suggesting that additional mediators and mechanisms need to be explored.
Figure 1. Major pathways involved in the pathophysiology of diabetic nephropathy.

Complex interactions between metabolic and hemodynamic factors regulate the pathogenesis of diabetic nephropathy. Persistence of HG mediated damage including epigenetic modifications even after return to normoglycemia can lead to metabolic memory and increased risk for long term complications. TGF-β, transforming growth factor-β; AGEs, advanced glycation end product; RAAS, rennin angiotensin aldosterone system; PKC, protein kinase C; NF-κB, nuclear factor kappa-B; PTMs, posttranslational modifications; ECM, extracellular matrix.
One of the potential reasons for the long term progression of diabetic complications could be a ‘metabolic memory’ of prior exposure of target cells to HG leading to persistence of its deleterious effects long after glucose normalization11 (Fig. 1). This memory phenomenon has been identified in experimental models as well as clinical trials such as the Diabetes Control and Complications Trial (DCCT) and the follow-up observational Epidemiology of Diabetes Intervention and Complications (EDIC) study. The results of the DCCT demonstrated that patients with type 1 diabetes (T1D) placed on strict glycemic control with intensive insulin therapy had much lower incidence or severity of various complications, including nephropathy and neuropathy relative to those who were on standard/conventional therapy 12. Following the conclusion of the DCCT study, both conventional and intensive treatment DCCT groups were placed on intensive therapy and followed long-term in the EDIC study phase. Despite attainment of similar levels of HbA1c in both groups during EDIC, patients who were previously in the intensive therapy group during DCCT had significantly lower risks of developing microvascular complications (neuropathy and nephropathy) as well as macrovascular complications13, 14 relative to the original DCCT conventional treatment group, a phenomenon termed metabolic memory. Clinical trials of glycemic control in type 2 diabetes patients (UKPDS) have found a similar phenomenon referred to as a ‘legacy effect’15.
With respect to experimental models of metabolic memory, in early studies, Lorenzi and co-workers first reported a glycemic memory phenomenon in an animal and cell culture model 16. More recently, reports show that vascular smooth muscle cells (VSMC) from type 2 diabetic db/db mice exhibited enhanced pro-inflammatory responses relative to those from non-diabetic control db/+ mice even after culturing outside the animals for few weeks17. In cultured EC, HG induced changes in the expression of inflammatory and oxidant stress genes persisted even after returning to normal glucose18, 19. Furthermore, in experiments with type 1 diabetic dogs or rats, approaches to re-institute good glycemic control following several weeks of poor glycemic control did not prevent progression to retinopathy and nephropathy, demonstrating a memory of early hyperglycemia20–22. Since metabolic memory remains a major obstacle in the effective prevention and treatment of diabetic complications, there is much interest in determining the mechanisms underlying metabolic memory and in recent years, epigenetic factors have been implicated.
Epigenetics and the Epigenome: Why study this in Diabetic Nephropathy?
Epigenetics was originally defined by Waddington as ‘the causal interactions between genes and their products which bring the phenotype into being’ and mainly referred to changes during embryonic development23. A more commonly used definition of epigenetics is “the study of heritable changes in gene expression that occur without alterations in the underlying DNA sequences”. More recently this has been further refined as ‘the structural adaptation of chromosomal regions so as to register, signal, or perpetuate altered activity states’, to also account for alterations in the chromatin state and structure in response to various cues24, 25. Epigenetic mechanisms establish and maintain the chromatin structure to confer transcription memory important for the faithful transmission of gene expression patterns across multiple cell divisions even in the absence of signals that initiated them.. Epigenetic control of gene regulation plays an important role in embryogenesis, development, cell identity, stable inheritance of gene expression patterns in differentiated cells, genomic imprinting, X-chromosome inactivation, immune cell function, stem cell plasticity, differential disease susceptibility between monozygotic twins, and cellular responses to environmental signals26, 27.
In mammalian cells, chromosomal DNA is tightly packaged into ‘chromatin’, a higher order structure made up of arrays of subunits called nucleosomes. Each nucleosome consists of an octamer protein complex containing two copies each of core histone proteins H2A, H2B, H3 and H4, wrapped around by 147 bp chromosomal DNA. Posttranslational modifications (PTMs) of nucleosomal histones and DNA methylation (DNAme) represent epigenetic modifications (Fig. 2) 26, 27. These modifications, along with non-coding RNAs including short non-coding microRNAs (miRNAs) and long non coding RNAs (lncRNAs), regulate chromatin function and are collectively labeled as the ‘epigenome’, which stores the epigenetic information needed for the cell-type specific gene expression patterns. Recent advances in high throughput genomewide profiling and sequencing approaches have led to a broader understanding of various aspects of the epigenome and its correlations to phenotype6, 28. Alterations in epigenome states have a profound effect on gene regulation and biological outcomes and are implicated in the pathogenesis of various disorders including cancer29, 30. Furthermore, persistence of aberrant epigenetic marks, even after withdrawal of the original stimuli, can mediate disease progression and resistance to conventional therapies. Lifestyle choices like overeating can influence the epigenetic mechanisms leading to aberrant expression of genes involved in metabolic disorders and cardiovascular diseases4, 11. Notably, intrauterine environment and maternal nutrition can promote epigenetic changes to control the onset of metabolic abnormalities in adult life31. The heritable nature of epigenetic marks could also predispose future generations for metabolic abnormalities and reduced lifespan32. It is worth examining whether they also affect the development of complications like DN later in life. The importance of epigenetic mechanisms in diabetes and its complications has only recently been appreciated Understanding how diet, physical activity and environmental factors influence the epigenome could provide novel new insights into the pathogenesis of diabetes complications like DN and metabolic memory, and hence to newer therapeutic modalities and diagnostic biomarkers for early intervention. Since genome wide association studies (GWAS) have uncovered only limited candidate susceptible genes for DN, evaluation of epigenotypes by epigenome wide association studies (EWAS) may provide critical new information.
Figure 2. Schematic diagram showing enrichment of histone modifications at various regulatory elements in chromatin.
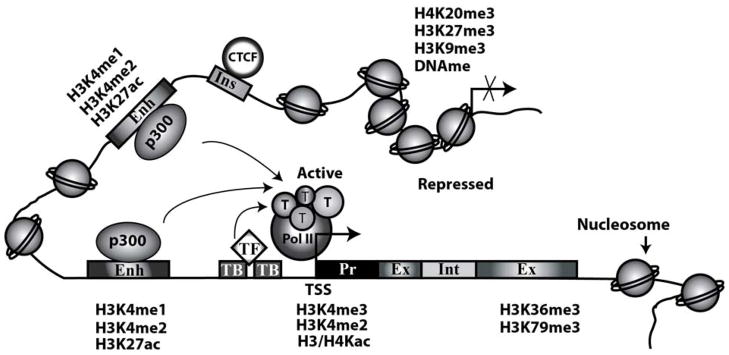
Transcriptionally active chromatin is characterized by open chromatin states with nucleosome depleted regions providing increased access to transcription factors (TF), RNA polymerase II (Pol II) and other components of the transcription machinery (T). Whereas repressed chromatin has a compact structure with higher density of nucleosomes and restricted accessibility. In general, active gene promoters are enriched with H3K4me3, H3K4me2 and H3/H4Kac, transcribed Exons (Ex) and Introns (Int) are enriched with H3K36me3 and H3K79me3. Enhancers (Enh) are enriched with H3K4me1 and the histone acetyl transferase p300 and active enhancers are marked by H3K4me2 and H3K27ac. Repressed promoters are enriched with H3K9me2/3, H3K27me3, H4K20me3 and DNA methylation (DNAme). Insulators (Ins) are enriched with the CCCTC-binding factor (CTCF) and demarcate active and inactive chromatin regions. TB-TF binding sites; Kme-lysine methylation; Kac-lysine acetylation.
DNA methylation and its role in diabetes and DN
DNAme is the most well established epigenetic mark. It occurs at 5′-Cytosines of ‘CpG’ dinucleotides, which tend to form clusters known as CpG islands at key genome regions. Both DNA methyl transferase 3A (DNMT3A) and DNMT3B mediate denovo DNAme, whereas DNMT1 acts as a maintenance methyltransferase using S-Adenosyl-methionine as a co-factor 27, 33. Emerging evidence shows the presence of active DNA demethylation suggesting DNAme is more dynamic than thought earlier. While key oxidases have been implicated, the mechanisms mediating DNA de-methylation in mammalian systems are not yet fully understood27, 33. Generally DNAme at promoter regions leads to gene repression, whereas at gene bodies it might regulate transcription elongation and alternative splicing. Studies also implicate DNAme in the activity of other transcription regulatory elements such as enhancers and insulators27. DNAme is recognized by methyl binding proteins including methyl-CpG binding protein 2 (MeCP2), which can recruit transcriptional co-repressors via protein-protein interactions to inhibit gene expression. Furthermore, DNAme itself can inhibit binding of TFs at promoters to block gene expression27.
In the context of diabetes, the role of DNAme has been studied in the transgerational inheritance of metabolic diseases which have led to the hypothesis that environment and diet may influence epigenetic modifications at imprinted genes, transposable elements and repeat elements in genomic DNA to regulate disease pathogenesis31. A role for DNAme has also been suggested in Intrauterine growth restriction (IUGR) induced effects on the fetal epigenome that can reprogram the timing and intensity of gene expression leading to metabolic abnormalities later in adult life34. Aberrant DNAme has been reported in the reduced expression of genes involved in insulin expression, signaling and energy metabolism in islets and skeletal muscle in diabetes and the metabolic syndrome35–39. HG and free fatty acids could regulate DNAme suggesting nutrient excess can lead to dysregulation of DNAme and alter expression of genes linked with susceptibility to obesity and diabetes36, 38. Variations in DNAme have also been recently implicated in the expression of diabetes susceptibility genes40, 41. Such studies may help confer functionality to disease associated single nucleotide polymorphisms42.
A few studies have investigated a role for DNAme in the pathogenesis of DN and ESRD5, 43, 44. Assessment of DNAme in whole blood genomic DNA obtained from diabetic patients with DN relative to those without DN revealed differential methylation in a number of genes including UNC13B which has been previously linked with DN43. Another study identified a number of regions with differential DNAme in the saliva samples of patients with ESRD relative to those with CKD alone44. On the other hand some studies in renal cell culture models under diabetic conditions or DN animal models did not show significant changes in DNAme5. Another interesting report showed that DNAme can play a role in a model of fibrosis and in TGF-β1 actions. The authors found that hypermethylation of RASAL1 can increase Ras activation in fibroblasts, leading to proliferation and fibrosis45. Sequencing approaches in EC showed that HG could induce alterations in DNAme at key genes involved in endothelial cell dysfunction46. Because fibrotic gene expression and EC dysfunction are involved in DN pathogenesis, abnormalities in DNAme in EC and other renal cells in vivo might contribute to changes in the expression of genes associated with DN. Additional studies are needed with key renal cells including podocytes, mesangial and tubular cells, as well as with renal tissues and biopsies obtained from animal models of DN and humans at various stages of DN, to identify abnormalities in DNAme patterns that may be specifically linked with DN.
Histone Modifications
Covalent posttranslational modifications (PTMs) of nucleosomal histone proteins are now known to play important roles in gene regulation26. Several histone PTMs have been identified including lysine (K) acetylation (Kac), methylation (Kme) and ubiquitination, Ser/Thr phosphorylation and Arginine methylation. Histone PTMs occur mostly in the exposed amino-terminal tails with some exceptions. The involvement of histone Kac and Kme in gene transcription has been extensively studied and will be discussed here26, 28. In general, histone Kac (such as H3K9ac, H3K14ac, H4K5ac) at gene promoters correlates with transcriptional activation whereas its removal is associated with gene repression. Histone Kme can be associated with either gene activation or repression depending on the amino acid residue modified and the extent of methylation i. e., mono (Kme), di (Kme2) or tri (Kme3) methylation. H3K4me1/2/3 and H3K36me2/3 are generally associated with transcriptionally active genome regions, whereas H3K9me3, H3K27me3 and H4K20me3 with repressed regions26. Genomewide mapping has led to the discoveries that distinct patterns of specific histone modifications can distinguish key regulatory regions of the genome including promoters, enhancers, exons, introns, intergenic regions and repetitive elements6, 28 (Fig. 2). Transcriptionally active gene promoters are enriched with H3K9Ac, H3K4me2 and H3K4me3 while gene bodies and transcribed regions are enriched with H3K36me3 and H3K79me3. On the other hand, inactive or silent gene promoters are enriched with repressive marks H3K9me3 and H3K27me36, 28. In general, enhancers are difficult to identify, but this has changed with the discoveries that they are enriched with H3K4me1 and also H3K27ac47 (Fig. 2). Bivalent promoters in developmental genes are enriched with both active (H3K4me3) and repressive (H3K27me3) marks and are therefore in a poised state6, 28. Furthermore, enrichment of H3K27me3 at the beginning of gene bodies is associated with inhibition of transcription elongation48. Histone H3K4me3 and H3K36me3 chromatin marks are recognized to be associated with active transcription. Hence, mapping of their enrichment profiles genome-wide is now a well established method to identify known and novel transcripts including non coding RNAs (ncRNAs) that do not have a coding potential but can modulate the epigenetic machinery 49.
Histone Kac is mediated by histone acetyl transferases (HAT) such as p300, CBP and TIP60 which can also act as transcription co-activators. Conversely, histone deacetylases (HDAC) including HDAC1-11 and Sirtuins remove acetylation marks and in general act as co-repressors with some exceptions. Sirtuins have generated lot of interest since they are regulated by energy metabolism50. Kme is mediated by histone lysine methyl transferases (HMTs) and erased by histone lysine demethylases (HDMs). HMTs and HDMs are highly specific to the lysine residue being modified and the extent of methylation (Kme1/2/3). SUV39H1, the first HMT to be identified, mediates H3K9me3 (a repressive mark), whereas Lysine specific demethylase 1 (LSD1), the first demethylase identified, removes H3K4me1/me2. H3K27me3 is mediated by Ezh2 and the Polycomb Repressive Complex (PRC), while H3K4me is mediated by SET7 (H3K4me1) and members of the MLL family (H3K4me1/2/3). SET2 mediates H3K36me3 and DOT1a regulates H3K79me3. Among other demethylases, JMJD1A demethylates H3K9me2, but H3K9me3 is removed by JMJD2A. JMJD3 removes H3K27me3, while JARID1A removes H3K4me3. The SET domain present in most HMTs catalyzes Kme using S-adenosyl methionine (SAM) as a co-factor, whereas HDMs demethylate via an amino oxidase domain (in LSD1) or JmjC domain (JMJD2A) and use FAD and α-ketoglutarate as co-factors respectively51.
Histone modifying enzymes can be recruited to promoters by binding to specific DNA sequences in the promoters such as PRC response elements, or by binding to pre-existing modifications or via interaction with RNA Polymerase II and TFs52. Recent studies demonstrated that ncRNAs can also bind to HMTs and HDMs and target them to specific genome locations53. Histone modification enzymes can also modify lysine residues on non-histone proteins including TFs, adding another level of complexity54. Therefore, a new nomenclature has been proposed for HMTs and HDMs based on their enzymatic activity and the order of discovery, e.g., SUV39H1 and MLL have been renamed as KMT1A and KMT2A respectively54. The specificity of HMTs and HDMs can be modified by altering key amino acids in their catalytic domains suggesting that such mutations in humans can potentially be involved in disease pathogenesis. Furthermore, because histone modifying enzymes use metabolites such as acetyl-CoA (HATs), SAM (HMTs) and α-ketoglutarate (HDMs) as cofactors, they might act as metabolic sensors. As such, mis-regulation of their function by factors such as diet and environment can lead to metabolic abnormalities55. The role of DNAme in epigenetic transmission is more widely studied than that of histone PTMs, although recent studies have suggested that the histone modifiers like PRC proteins and histone recycling can be involved56, 57. Overall, the cross-talk between histone PTMs, DNAme and ncRNAs provides another layer of epigenetic regulation and their impact on transcription processes is being increasingly appreciated53, 58. The continuous interplay between these epigenetic modifications and the environment regulates multitudes of responses from a single eukaryotic genome in diverse cell types, which if dysregulated, can result in diabetes and its cardiovascular and renal complications.
Histone PTMs and Diabetes
Histone PTMs regulate chromatin structure and gene expression by recruiting chromatin remodeling proteins, transcription co-activators, and co-repressors26. Emerging evidence shows the involvement of key histone PTMs in the regulation of genes associated with the pathogenesis of diabetes. Regulation of insulin gene expression as well as its secretion from islets in response to changing glucose levels is a key process in glucose homeostasis, one that is dysregulated in diabetes. Studies show that the islet specific TF Pdx-1 can modulate this process of insulin regulation through epigenetic mechanisms59. In response to elevated glucose conditions, Pdx1 recruits co-activator HATs p300 and CBP and a HMT SET7/9 (SET7), which increases activation marks H3/H4Kac and H3K4me2 respectively at the insulin promoter to promote open chromatin formation accessible to transcription machinery and enhance insulin transcription59, 60. In contrast, under low glucose conditions, Pdx1 recruits co-repressors HDAC1 and HDAC2, promoting chromatin compaction and inhibition of insulin expression59. Interestingly, Pdx-1 also controls the islet specific expression of SET7 by direct interaction with its promoter60. Genomewide mapping of HK4me1, H3K4me3, H3K79me2 in islets revealed several islet specific promoters and enhancers. Furthermore, several regulatory elements located near diabetes susceptible loci exhibited allele specific differences in their activity61. Another study also mapped open chromatin regions in islets and identified association of allele specific differences in enhancer activity with genetic variations near diabetes susceptible loci62 further highlighting how genetic variations in non-coding regions might affect chromatin structure in diabetes. Histone PTMs along with DNAme were also found to play an important role in epigenetic regulation of Pdx1 and insulin expression in islets of diabetic offspring from IUGR rats, suggesting that histone PTMs can be affected by maternal malnutrition34.
Adipogenesis plays an important role in the pathogenesis of metabolic abnormalities and is tightly controlled by the transcription factors C/EBPβ and PPARγ. Dynamic changes in histone PTMs and recruitment of the corresponding modifiers can regulate C/EBPβ and PPARγ induced gene expression involved in adipocyte differentiation63, 64. Interestingly, epigenetic inactivation of PPARγ has been demonstrated in adipocytes from type 2 diabetes (T2D) animals65 further supporting a role for epigenetic processes in adipocyte dysfunction and T2D. Another study reported increased predisposition to obesity and metabolic syndrome in mice deficient in Jhdm2a, a H3K9me2 demethylase, demonstrating that deficiency in key histone modifying enzymes might contribute to metabolic abnormalities66. Overall, these studies highlight how alterations in chromatin structure can contribute to diabetes development. This is clearly a research area likely to show increased activity in the upcoming years. It is possible that epigenetic changes that contribute to the pathology of diabetes can also directly or indirectly affect target organs leading to complications.
Histone PTMs and gene regulation in Diabetic Nephropathy
Accumulating evidence shows that epigenetic regulation of gene expression plays important roles in kidney development and other renal disorders such as acute kidney injury 5, 67 that will not be discussed here. Because very few genetic loci have been found to be associated with CKD and DN, it is increasingly felt that epigenetics could also be involved. Reports from studies in cell culture and animal models show that epigenetic histone PTMs are involved in the expression of key genes associated with DN pathogenesis. TGF-β signaling plays an important role in the expression of fibrotic and ECM genes such as Collagen 1alpha 2 (Col1a2), plasminogen activator inhibitor 1 (PAI-1) and cell cycle inhibitor p21 in renal cells, which contribute to DN pathogenesis1. TGF-β regulates gene expression mostly through activation of the TFs Smad 2, Smad3 and Smad4, which can collaborate with HATs and chromatin remodeling factors1, 68. TGF-β can also mediate the effects of HG1, 3. Recent studies examined these mechanisms as well as histone Kac and Kme in rat MC (RMC) treated with TGF-β and HG. TGF-β increased H3K9/14ac near Smad and SP1 binding sites by recruiting the HATs p300 and CBP to the PAI-1 and p21 promoters. Co-transfection experiments showed that CBP and p300, but not p/CAF, increased transcriptional activity of PAI-1 and p21 promoters and enhanced TGF-β induced gene expression. In contrast, inhibition of these HATs by overexpressing dominant negative mutants lacking HAT activity blocked TGF-β induced gene expression. Furthermore, over expression of HDAC1 and HDADC5 inhibited TGF-β induced gene expression, whereas, inhibition of HDACs increased H3K9/14ac and gene expression further supporting a key role for histone Kac and HATs in TGF-β induced gene expression69. TGF-β also increased association of p300 with Smads2/3 and SP1, and increased acetylation of Smads in RMC69. In a similar MC culture model, TGF-β induced Col1a1, CTGF and PAI-1 was associated with increased levels of activation Kme marks (H3K4me1, H3K4me2 and H3K4me3) and reduced levels of repressive marks (H3K9me2 and H3K9me3) at their promoters70. TGF-β also induced the expression of SET7 HMT and enhanced its recruitment to ECM gene promoters. SET7 gene silencing abolished TGF-β induced gene expression further confirming a key role of SET7 in fibrotic gene expression in MC. HG treatment of RMC also led to similar changes in histone PTMs at fibrotic and cell cycle gene (p21) promoters69, 70. Interestingly, the epigenetic effects of HG, including similar changes in promoter H3Kac and H3Kme as well as SET7 recruitment, were significantly blocked by a TGF-β antibody, demonstrating a key mediatory role of TGF-β in HG induced epigenetic histone modifications69, 70. Together these results from in vitro studies support a critical role for epigenetic mechanisms in TGF-β and HG induced pathological gene expression relevant to DN (Fig. 3).
Figure 3. Epigenetic mechanisms involved in Diabetic Nephropathy.

High glucose (HG), and TGF-β-transforming growth factor-beta1 (TGF-β) regulate Extracellular matrix (ECM) genes and cell cycle genes by increasing active modifications H3K9/14ac and H3K4me1/2/3, and inhibiting repressive marks H3K9me2/3 at these gene promoters. TGF-β promotes the recruitment of CBP/p300 which increases H3K9/14ac and chromatin access to Smads and SP1 transcription factors. CBP/p300 also regulates Smad activity by direct acetylation. TGF-β mediated inhibition of HDAC1 and HDAC5 may also play a role in increased H3K9/14ac. TGF-β induces SET7 expression and promotes its recruitment to gene promoters, which increases H3K4me1/2. Similar epigenetic mechanisms are induced by HG and they are blocked by TGF-β antibodies (TGF-β Ab) implicating TGF-β as a major mediator of epigenetic events in Diabetic Nephropathy. R-Corepressors. HMT-histone methyl transferases; HDAC-histone deacetylases; SP1B-SP1 binding sites; SBE-Smad binding elements; Pr-promoter; Pol II-RNA Polymerase II; T- components of transcription machinery.
Evidence for epigenetic changes in DN also comes from studies in animal models. One report showed changes in global histone modifications are associated with the expression of fibrotic and cell cycle genes involved in DN pathogenesis5. HDAC inhibitors showed renoprotective effects, but it was not clear if these were due to inhibition of epigenetic or non-epigenetic effects mediated by HDACs71. A recent study used ChIP assays for a more focused assessment of genes associated with DN. Glomeruli from streptozotocin injected type 1 diabetic (T1D) mice and T2D db/db mice exhibited increased expression of PAI-1 and p21 genes and this was associated with elevated levels of H3K9/14ac near Smad and SP1 binding sites at their promoters compared to the respective non-diabetic controls. These results demonstrate the in vivo relevance of HG and TGF-β induced H3K9/14ac in cultured MC69. However, it clear that a single histone PTM does not fully represent the chromatin status at these genes, but more likely a histone code of multiple PTMs. With this in mind, another recent study profiled several histone PTMs in glomeruli from diabetic db/db mice and control db/+ mice72. Results showed that relative to db/+ mice, glomeruli from db/db mice exhibited increased RNA Polymerase II (Pol II) recruitment, enhanced levels of several activation marks, and decreased levels of key repressive marks at the promoters of PAI-1 and RAGE (receptor for advanced glycation end products). These results suggest that epigenetic mechanisms regulated by diabetes in vivo can co-operate to promote open chromatin formation around the PAI-1 and RAGE promoters resulting in enhanced access to transcription machinery and transcription of these genes in db/db mice. Furthermore, the expression of several HATs, HDACs and HMTs were also increased, the most notable being KAT5 (TIP60), HDAC7, HDAC9, SET2, SET4 and SET7. In this study, db/db mice were also treated with or without Losartan, an Ang II type 1 receptor blocker (ARB) to test whether the renoprotective effects of ARBs are associated with the inhibition of epigenetic mechanisms. Losartan treatment for 10 weeks ameliorated key indices of DN, but did not reverse all the epigenetic changes observed in the db/db mice72. Incomplete inhibition of epigenetic changes associated with DN might be one explanation for the relative inefficiency of ARBs to prevent progression to ESRD in many patients73. This study also highlights the complexity of performing epigenome profiling with renal tissues from animal models of a chronic progressive disease like DN and the need to evaluate various time periods during disease progression. Similar studies with other treatment modalities, TGF-β antibodies and RAGE inhibitors might provide novel information about the epigenetic mechanisms in DN that may or may not be inhibited by these interventions.
Histone PTMs in vascular cells and monocytes under diabetic conditions. Potential connections between epigenetics and metabolic memory
Chronic inflammation is a hallmark of diabetes complications, including DN where macrophage infiltration and increased inflammatory gene expression have been observed in the kidney. HG increased activation of NF-κB is a major mechanism of inflammatory gene expression in vascular cells and monocytes relevant to diabetes complications and recently several studies have demonstrated the involvement of epigenetic modifications in these events74, 75. HG induced NF-κB mediated inflammatory gene expression in THP-1 monocytes and promoted NF-κB binding in EC, key events in vascular diseases76, 77. In monocytes, HG increased the recruitment of co-activator HATs CBP/p300, and augmented the levels of active marks H3Kac and H4Kac to promote open chromatin formation at inflammatory gene promoters78. HG actions on NF-κB activation were enhanced by HDAC inhibitors and blocked by overexpression of HDACs, further supporting the role of histone Kac78. Interestingly, profiling approaches with chromatin immunoprecipitation linked to microarrays (ChIP-on-chip) revealed differential regulation of H3K4me2 and H3K9me2 marks at gene bodies of several genes in HG treated THP-1 monocytes79. Similar changes in H3Kac, H3K4me2 and H3K9me2 at key genes were also found in blood monocytes obtained from diabetic patients thereby demonstrating disease relevance78, 79. Furthermore, ChIP-on-chip epigenome profiling of blood lymphocytes from T1D patients versus healthy controls demonstrated significant variations in the repressive H3K9me2 mark at a subset of genes associated with T1D, inflammation and autoimmunity 80. Recently, profiling of several histone PTMs at chromosome 6 regions revealed key variations in H3K9ac at the enhancer regions of two HLA genes whose single nucleotide polymorphisms are closely linked to T1D81. These data support the involvement of epigenetic modifications in the etiology of diabetes and potential cross talk between genetic and epigenetic variations that however need further testing in bigger cohorts. In another in vitro study with monocytes, the H3K4me transferase SET7, which regulates insulin gene expression in islets, was also shown to be required for the maximal activation of a subset of NF-κB inducible inflammatory genes in monocytes suggesting that SET7 may act as a co-activator of NF-κB82. In vivo, enhanced inflammatory gene expression in macrophages from type 1 diabetic mice was associated with increased SET7 recruitment and H3K4me2 at these gene promoters82. Interestingly, HG induced expression of key inflammatory genes and increase in histone Kac at their promoters in THP-1 monocytes were attenuated by the anti-inflammatory agent Curcumin via inhibiting p300 and increasing HDAC2 levels83, suggesting that certain natural products like curcumin can exert protective effects in diabetic complications by attenuating epigenetic modifications. Further studies are needed to determine how these data can be used for the development of epigenetic therapies for DN and other diabetic complications.
HG also regulates epigenetic mechanisms in vascular cells including those related to DN like ECs. Short term exposure of EC to HG increased the expression of p65, the active subunit of NF-κB, and inflammatory genes via increased recruitment of SET7 and H3K4me1 at these gene promoters 18. Notably, HG treatment triggered nuclear localization and activity of SET7 in EC84, further demonstrating that HG can directly affect epigenetic modulators. Diabetic retinopathy is associated with repression of the key anti oxidant gene mitochondrial superoxide dismutase (SOD) in retinal EC. HG was shown to inhibit the SOD gene by increasing promoter levels of the repressive mark H4K20me3 through upregulation and increased recruitment of the corresponding methyl transferase SUV420H2 to the SOD gene promoter19. Furthermore, HG also up-regulated inflammatory genes via inhibition of the repressive mark H3K9me3 at their promoters in vascular smooth muscle cells, which can play a key role in atherosclerosis and hypertension85. Overall, these studies clearly demonstrate the epigenetic regulation of gene expression by HG in cells relevant to DN and related vascular complications (Fig. 4).
Figure 4. Histone Modifications and gene regulation in vascular complications.

Diabetes enhances expression of NF-κB induced inflammatory genes (TNF-α, IL-6 and MCP-1) and inhibits antioxidant stress genes superoxide dismutase (SOD) in monocytes and vascular cells via multiple epigenetic mechanisms: A. in vascular smooth muscle cells, diabetes increases miR-125b, which blocks SUV39H1 expression, leading to inhibition of H3K9me3 (repressive mark) and recruitment of co-repressor HP1 at inflammatory gene promoters resulting in increased inflammatory gene expression; B. In retinal endothelial cells, expression of anti-oxidant genes such as SOD is repressed by HG induced SUV420H2, which mediates a repressive modification H4K20me3. 3. In monocytes and endothelial cells, diabetes conditions promote recruitment of co-activator Histone acetyl transferase (p300) and H3K4methyl transferase SET7, which increase activation marks H3/H4Kac and H3K4me respectively leading to enhanced NF-κB mediated inflammatory gene expression. Persistence of these epigenetic modifications even after removal of the diabetic stimuli might be an underlying mechanism involved in metabolic memory of diabetic complications. Inf genes-inflammatory genes; SOD-Super oxide dismutase; Pr-promoter; TSS-transcription start site; Pol II-RNA Polymerase II; T- components of transcription machinery.
As discussed earlier, prior exposure to hyperglycemia has been related to a metabolic memory of long term sustained complications like despite subsequent glycemic control. Since Histone Kme is a relatively stable epigenetic mark, a number of studies have examined the hypothesis that persistently altered histone Kme at key pathological genes might lead to their sustained up-regulation and metabolic memory. In support of this, it was demonstrated that enhanced inflammatory gene expression and migration in VSMC obtained from T2D db/db mice relative to control db/+ mice even after being cultured for several passages in vitro was associated with a persistent loss of the repressive mark H3K9me3 at these gene promoters. Furthermore, in parallel there was a down-regulation of the corresponding H3K9me3 methyl transferase SUV39H1 which was attributed at least in part to increased levels of miR-125b in db/db mice, revealing a novel cross-talk between miRNA actions and epigenetic components in diabetes85, 86. Similarly, sustained changes in H3K4me1 and SET7 were implicated in the prolonged upregulation of p65 in EC that were previously exposed to HG for short time periods, and these changes were mediated by oxidant stress dependent mechanisms18. Furthermore, in a rat model of diabetic retinopathy and metabolic memory, sustained downregulation of the SOD gene was attributed to persistent increases in promoter levels of the repressive mark H4K20me3 and increased expression of its methyl transferase SUV420H219. Together, these studies strongly suggest that epigenetic mechanisms related to alterations in histone PTMs may be involved in metabolic memory of DN and other complications (Fig 4). Further studies are needed to understand how HG and diabetes drive these epigenetic events in vitro and in vivo, over short and long time periods, and how they can be reversed to prevent the progression of complications even after glycemic control. Importantly, additional studies with human diabetic subjects experiencing metabolic memory are needed to extrapolate these observations to clinical metabolic memory and glycemic variations.
High throughput Genomic and Epigenomic Approaches
Several techniques have been developed to analyze gene expression patterns and epigenetic modifications in mammalian cells using microarrays and massively parallel next generation DNA sequencing (NGS) platforms87–89. DNAme is analyzed using bisulfite conversion of genomic DNA, digestion with methyl sensitive enzymes, immunoprecipitation of methylated DNA, and affinity capture of methylated cytosines in DNA. DNA extracted from these methods is hybridized to microarrays or sequenced using NGS platforms to obtain genome-wide distribution of DNAme (methylome)89. Histone modifications are analyzed using Chromatin immunoprecipitation (ChIP) assays in which DNA and proteins are cross linked by fixing the tissue samples or treated cells with formaldehyde. DNA in the cross-linked chromatin is sheared by sonication to smaller DNA fragments and immunoprecipitated with antibodies against specific histone modifications. Immunoprecipitated DNA is reverse cross-linked and then analyzed using PCR primers specific to promoters or other genomic regions of interest to determine the genome locations enriched with specific histone PTMs. ChIP can also be used to analyze enrichment of TFs, histone modification enzymes and other epigenetic factors that interact with chromatin. This has been extended to genomewide analysis by hybridization of ChIP-enriched DNA to microarrays (ChIP-on-chip)90 or by NGS (sequencing) methods (ChIP-Seq)87. Advances in NGS technologies have also led to the development of other novel techniques including FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements)-Seq which allows detection of accessible chromatin and regulatory elements based on differential cross-linking efficiency of nucleosome enriched and depleted regions91. FAIRE-Seq and DNAse-Seq have extended the capabilities of the widely used DNAse hypersensitivity assays to determine regulatory or open chromatin regions genomewide92. Furthermore, RNA-Seq has revolutionized transcriptome analysis in diverse cell types and disease conditions and led to the detection of novel transcripts88. The major advantage of NGS is that, unlike microarrays, no prior information about genomic DNA sequence is needed and the data obtained can be used to quantitatively estimate the changes in gene expression and epigenetic modifications genomewide. Integration of the transcriptome, DNA-methylome and ChIP-seq data can yield extensive and comprehensive information about the epigenome state in diverse pathophysiological conditions93, 94. Recently, the Encyclopedia of DNA Elements (ENCODE) project completed high-quality whole-genome functional annotations of the human and mouse genomes and deposited the data in publicly available databases6, 95, 96. This information along with resources from NIH Roadmap Epigenomics consortium (http://www.roadmapepigenomics.org) are valuable reference tools to accelerate and catalyze new research in epigenomics of human disease.
High through put approaches using microarray and NGS platforms have been successfully used to profile epigenetic modifications or open chromatin (accessible) regions in monocytes, lymphocytes79–81, pancreatic islets61, 62, 97, 98, or EC46 under diabetic conditions. Similar studies in renal tissues from mouse models of DN and from diabetic subjects are being performed in several laboratories and the results, when available, are likely to yield novel insights into epigenome variations linked with DN pathogenesis.
Summary and Perspectives
The pathogenesis of DN involves complex interactions between metabolic and hemodynamic factors with major roles being played by HG, TGF-β, AGEs and Ang II. Signal transduction pathways and TFs regulated by these factors that lead to pathologic gene expression have been extensively studied. Emerging evidence shows that epigenetic mechanisms in chromatin including histone PTMs, DNAme and miRNAs might also play key role in the etiology of diabetes and DN. Persistence of epigenetic modifications triggered by diabetic stimuli could be one of the key mechanisms underlying metabolic memory. A role for several HMTs and the corresponding histone PTMs has been demonstrated in the expression of fibrotic and inflammatory genes associated with DN. But the involvement of many others and mechanisms of their regulation by upstream signal transduction pathways are still unknown. However, this is a rapidly expanding and dynamic field and it is likely that, aided by the recent advances made by the Human Epigenome project and ENCODE 6, 95, 96, other chromatin factors and epigenetic mechanisms related to diabetes and DN will be revealed in the upcoming years. Epigenomics or EWAS may also enable us to determine the functional significance of genetic variants in certain non-coding and coding regions as well as cross talk between the genetic and epigenetic machinery. Because epigenetic changes are potentially reversible in nature, there is an opportunity to develop combination therapies with epigenetic drugs30 and antagomirs (miRNA inhibitors)99 to complement the current treatments for DN. However, there are also challenges to overcome. Since epigenetic patterns are cell specific, data from EWAS using heterogeneous kidney tissues and biopsies could be difficult to interpret. Furthermore, obtaining glomerular and tubular biopsies from affected individuals and matched controls poses another major obstacle. Because inflammation is closely associated with most diabetic complications including DN, another approach is to examine inflammatory cells like blood monocytes that are obtained easily and non-invasively. Overall, it is anticipated that further research in the field of epigenetics may lead to the identification of much needed new biomarkers and drug targets for early detection and treatment of DN.
Acknowledgments
Financial support: National Institutes of Health, R01 DK081705, R01 DK058191 and R01 DK065073 and Juvenile Diabetes Research Foundation (to RN)
Footnotes
Conflict of interest statement: None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kanwar YS, Sun L, Xie P, et al. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. 2011;6:395–423. doi: 10.1146/annurev.pathol.4.110807.092150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woroniecka KI, Park AS, Mohtat D, et al. Transcriptome analysis of human diabetic kidney disease. Diabetes. 2011;60:2354–2369. doi: 10.2337/db10-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanchez AP, Sharma K. Transcription factors in the pathogenesis of diabetic nephropathy. Expert Rev Mol Med. 2009;11:e13. doi: 10.1017/S1462399409001057. [DOI] [PubMed] [Google Scholar]
- 4.Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes. 2009;58:2718–2725. doi: 10.2337/db09-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy MA, Natarajan R. Epigenetics in diabetic kidney disease. J Am Soc Nephrol. 2011;22:2182–2185. doi: 10.1681/ASN.2011060629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunham I, Kundaje A, Aldred SF, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Human Epigenome Task Force. Moving AHEAD with an international human epigenome project. Nature. 2008;454:711–715. doi: 10.1038/454711a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Harris DC. Macrophages in renal disease. J Am Soc Nephrol. 2011;22:21–27. doi: 10.1681/ASN.2010030269. [DOI] [PubMed] [Google Scholar]
- 9.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 10.Kato M, Natarajan R. MicroRNA circuits in transforming growth factor-beta actions and diabetic nephropathy. Semin Nephrol. 2012;32:253–260. doi: 10.1016/j.semnephrol.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villeneuve LM, Natarajan R. The role of epigenetics in the pathology of diabetic complications. Am J Physiol Renal Physiol. 2010;299:F14–25. doi: 10.1152/ajprenal.00200.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA. 2002;287:2563–2569. doi: 10.1001/jama.287.19.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. JAMA. 2003;290:2159–2167. doi: 10.1001/jama.290.16.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nathan DM, Cleary PA, Backlund JY, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colagiuri S, Cull CA, Holman RR. Are lower fasting plasma glucose levels at diagnosis of type 2 diabetes associated with improved outcomes?: U.K. prospective diabetes study 61. Diabetes Care. 2002;25:1410–1417. doi: 10.2337/diacare.25.8.1410. [DOI] [PubMed] [Google Scholar]
- 16.Roy S, Sala R, Cagliero E, et al. Overexpression of fibronectin induced by diabetes or high glucose: phenomenon with a memory. Proc Natl Acad Sci U S A. 1990;87:404–408. doi: 10.1073/pnas.87.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li SL, Reddy MA, Cai Q, et al. Enhanced proatherogenic responses in macrophages and vascular smooth muscle cells derived from diabetic db/db mice. Diabetes. 2006;55:2611–2619. doi: 10.2337/db06-0164. [DOI] [PubMed] [Google Scholar]
- 18.El-Osta A, Brasacchio D, Yao D, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205:2409–2417. doi: 10.1084/jem.20081188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong Q, Kowluru RA. Epigenetic changes in mitochondrial superoxide dismutase in the retina and the development of diabetic retinopathy. Diabetes. 2011;60:1304–1313. doi: 10.2337/db10-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engerman RL, Kern TS. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes. 1987;36:808–812. doi: 10.2337/diab.36.7.808. [DOI] [PubMed] [Google Scholar]
- 21.Kowluru RA, Abbas SN, Odenbach S. Reversal of hyperglycemia and diabetic nephropathy: effect of reinstitution of good metabolic control on oxidative stress in the kidney of diabetic rats. J Diabetes Complications. 2004;18:282–288. doi: 10.1016/j.jdiacomp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 22.Kowluru RA. Effect of reinstitution of good glycemic control on retinal oxidative stress and nitrative stress in diabetic rats. Diabetes. 2003;52:818–823. doi: 10.2337/diabetes.52.3.818. [DOI] [PubMed] [Google Scholar]
- 23.Waddington CH. The epigenotype. 1942. Int J Epidemiol. 2012;41:10–13. doi: 10.1093/ije/dyr184. [DOI] [PubMed] [Google Scholar]
- 24.Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–616. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 28.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 29.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 30.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Wu Z, Li D, et al. Nutrition, Epigenetics, and Metabolic Syndrome. Antioxid Redox Signal. 2012 doi: 10.1089/ars.2011.4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen ZX, Riggs AD. DNA methylation and demethylation in mammals. J Biol Chem. 2011;286:18347–18353. doi: 10.1074/jbc.R110.205286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simmons R. Epigenetics and maternal nutrition: nature v. nurture. Proc Nutr Soc. 2011;70:73–81. doi: 10.1017/S0029665110003988. [DOI] [PubMed] [Google Scholar]
- 35.Ling C, Del Guerra S, Lupi R, et al. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia. 2008;51:615–622. doi: 10.1007/s00125-007-0916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barres R, Osler ME, Yan J, et al. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab. 2009;10:189–198. doi: 10.1016/j.cmet.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 37.Nikoshkov A, Sunkari V, Savu O, et al. Epigenetic DNA methylation in the promoters of the Igf1 receptor and insulin receptor genes in db/db mice. Epigenetics. 2011;6:405–409. doi: 10.4161/epi.6.4.14791. [DOI] [PubMed] [Google Scholar]
- 38.Yang BT, Dayeh TA, Kirkpatrick CL, et al. Insulin promoter DNA methylation correlates negatively with insulin gene expression and positively with HbA(1c) levels in human pancreatic islets. Diabetologia. 2011;54:360–367. doi: 10.1007/s00125-010-1967-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang BT, Dayeh TA, Volkov PA, et al. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol Endocrinol. 2012;26:1203–1212. doi: 10.1210/me.2012-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toperoff G, Aran D, Kark JD, et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum Mol Genet. 2012;21:371–383. doi: 10.1093/hmg/ddr472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bell CG, Finer S, Lindgren CM, et al. Integrated genetic and epigenetic analysis identifies haplotype-specific methylation in the FTO type 2 diabetes and obesity susceptibility locus. PLoS One. 2010;5:e14040. doi: 10.1371/journal.pone.0014040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leung A, Schones DE, Natarajan R. Using epigenetic mechanisms to understand the impact of common disease causing alleles. Curr Opin Immunol. 2012;24:558–563. doi: 10.1016/j.coi.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bell CG, Teschendorff AE, Rakyan VK, et al. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genomics. 2010;3:33–42. doi: 10.1186/1755-8794-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sapienza C, Lee J, Powell J, et al. DNA methylation profiling identifies epigenetic differences between diabetes patients with ESRD and diabetes patients without nephropathy. Epigenetics. 2011;6:20–28. doi: 10.4161/epi.6.1.13362. [DOI] [PubMed] [Google Scholar]
- 45.Bechtel W, McGoohan S, Zeisberg EM, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16:544–550. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pirola L, Balcerczyk A, Tothill RW, et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res. 2011;21:1601–1615. doi: 10.1101/gr.116095.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin F, Li Y, Ren B, et al. Enhancers: multi-dimensional signal integrators. Transcription. 2011;2:226–230. doi: 10.4161/trns.2.5.17712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen S, Ma J, Wu F, et al. The histone H3 Lys 27 demethylase JMJD3 regulates gene expression by impacting transcriptional elongation. Genes Dev. 2012;26:1364–1375. doi: 10.1101/gad.186056.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guttman M, Amit I, Garber M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baur JA, Ungvari Z, Minor RK, et al. Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov. 2012;11:443–461. doi: 10.1038/nrd3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8:307–318. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- 52.Smith E, Shilatifard A. The chromatin signaling pathway: diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol Cell. 2010;40:689–701. doi: 10.1016/j.molcel.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–346. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Allis CD, Berger SL, Cote J, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 55.Sassone-Corsi P. Physiology. When metabolism and epigenetics converge. Science. 2013;339:148–150. doi: 10.1126/science.1233423. [DOI] [PubMed] [Google Scholar]
- 56.Groth A, Corpet A, Cook AJ, et al. Regulation of replication fork progression through histone supply and demand. Science. 2007;318:1928–1931. doi: 10.1126/science.1148992. [DOI] [PubMed] [Google Scholar]
- 57.Abmayr SM, Workman JL. Holding on through DNA replication: histone modification or modifier? Cell. 2012;150:875–877. doi: 10.1016/j.cell.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 58.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 59.Babu DA, Deering TG, Mirmira RG. A feat of metabolic proportions: Pdx1 orchestrates islet development and function in the maintenance of glucose homeostasis. Mol Genet Metab. 2007;92:43–55. doi: 10.1016/j.ymgme.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deering TG, Ogihara T, Trace AP, et al. Methyltransferase Set7/9 maintains transcription and euchromatin structure at islet-enriched genes. Diabetes. 2009;58:185–193. doi: 10.2337/db08-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stitzel ML, Sethupathy P, Pearson DS, et al. Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab. 2010;12:443–455. doi: 10.1016/j.cmet.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gaulton KJ, Nammo T, Pasquali L, et al. A map of open chromatin in human pancreatic islets. Nat Genet. 2010;42:255–259. doi: 10.1038/ng.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Musri MM, Gomis R, Parrizas M. A chromatin perspective of adipogenesis. Organogenesis. 2010;6:15–23. doi: 10.4161/org.6.1.10226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lefterova MI, Steger DJ, Zhuo D, et al. Cell-specific determinants of peroxisome proliferator-activated receptor gamma function in adipocytes and macrophages. Mol Cell Biol. 2010;30:2078–2089. doi: 10.1128/MCB.01651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fujiki K, Kano F, Shiota K, et al. Expression of the peroxisome proliferator activated receptor gamma gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol. 2009;7:38. doi: 10.1186/1741-7007-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tateishi K, Okada Y, Kallin EM, et al. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature. 2009;458:757–761. doi: 10.1038/nature07777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dressler GR. Epigenetics, development, and the kidney. J Am Soc Nephrol. 2008;19:2060–2067. doi: 10.1681/ASN.2008010119. [DOI] [PubMed] [Google Scholar]
- 68.Ross S, Cheung E, Petrakis TG, et al. Smads orchestrate specific histone modifications and chromatin remodeling to activate transcription. EMBO J. 2006;25:4490–4502. doi: 10.1038/sj.emboj.7601332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yuan H, Reddy MA, Sun G, et al. Involvement of p300/CBP and Epigenetic Histone Acetylation in TGF-beta1 Mediated Gene Transcription in Mesangial Cells. Am J Physiol Renal Physiol. 2012 doi: 10.1152/ajprenal.00523.2012. Epub Dec 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun G, Reddy MA, Yuan H, et al. Epigenetic histone methylation modulates fibrotic gene expression. J Am Soc Nephrol. 2010;21:2069–2080. doi: 10.1681/ASN.2010060633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee HB, Noh H, Seo JY, et al. Histone deacetylase inhibitors: a novel class of therapeutic agents in diabetic nephropathy. Kidney Int Suppl. 2007:S61–66. doi: 10.1038/sj.ki.5002388. [DOI] [PubMed] [Google Scholar]
- 72.Reddy MA, Putta S, Lanting LL, Yuan H, Wang M, Alpers CE, Bomsztyk K, Natarajan R. Effect of Losartan on Epigenetic Changes in the Renal Glomeruli of Diabetic db/db Mice. [abstract] J Am Soc Nephrol. 2012;23:TH-PO467. [Google Scholar]
- 73.Ruggenenti P, Cravedi P, Remuzzi G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat Rev Nephrol. 2010;6:319–330. doi: 10.1038/nrneph.2010.58. [DOI] [PubMed] [Google Scholar]
- 74.Reddy MA, Natarajan R. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc Res. 2011;90:421–429. doi: 10.1093/cvr/cvr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guha M, Bai W, Nadler JL, et al. Molecular mechanisms of tumor necrosis factor alpha gene expression in monocytic cells via hyperglycemia-induced oxidant stress-dependent and -independent pathways. J Biol Chem. 2000;275:17728–17739. doi: 10.1074/jbc.275.23.17728. [DOI] [PubMed] [Google Scholar]
- 77.Shanmugam N, Reddy MA, Guha M, et al. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes. 2003;52:1256–1264. doi: 10.2337/diabetes.52.5.1256. [DOI] [PubMed] [Google Scholar]
- 78.Miao F, Gonzalo IG, Lanting L, et al. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J Biol Chem. 2004;279:18091–18097. doi: 10.1074/jbc.M311786200. [DOI] [PubMed] [Google Scholar]
- 79.Miao F, Wu X, Zhang L, et al. Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J Biol Chem. 2007;282:13854–13863. doi: 10.1074/jbc.M609446200. [DOI] [PubMed] [Google Scholar]
- 80.Miao F, Smith DD, Zhang L, et al. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes. 2008;57:3189–3198. doi: 10.2337/db08-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miao F, Chen Z, Zhang L, et al. Profiles of epigenetic histone post-translational modifications at type 1 diabetes susceptible genes. J Biol Chem. 2012;287:16335–16345. doi: 10.1074/jbc.M111.330373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li Y, Reddy MA, Miao F, et al. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J Biol Chem. 2008;283:26771–26781. doi: 10.1074/jbc.M802800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yun JM, Jialal I, Devaraj S. Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin. J Nutr Biochem. 2011;22:450–458. doi: 10.1016/j.jnutbio.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Okabe J, Orlowski C, Balcerczyk A, et al. Distinguishing hyperglycemic changes by set7 in vascular endothelial cells. Circ Res. 2012;110:1067–1076. doi: 10.1161/CIRCRESAHA.112.266171. [DOI] [PubMed] [Google Scholar]
- 85.Villeneuve LM, Reddy MA, Lanting LL, et al. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc Natl Acad Sci U S A. 2008;105:9047–9052. doi: 10.1073/pnas.0803623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Villeneuve LM, Kato M, Reddy MA, et al. Enhanced levels of microRNA-125b in vascular smooth muscle cells of diabetic db/db mice lead to increased inflammatory gene expression by targeting the histone methyltransferase Suv39h1. Diabetes. 2010;59:2904–2915. doi: 10.2337/db10-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 89.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 90.Huebert DJ, Kamal M, O’Donovan A, et al. Genome-wide analysis of histone modifications by ChIP-on-chip. Methods. 2006;40:365–369. doi: 10.1016/j.ymeth.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 91.Simon JM, Giresi PG, Davis IJ, et al. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat Protoc. 2012;7:256–267. doi: 10.1038/nprot.2011.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hesselberth JR, Chen X, Zhang Z, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat Methods. 2009;6:S22–32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet. 2010;11:476–486. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stamatoyannopoulos JA, Snyder M, Hardison R, et al. An encyclopedia of mouse DNA elements (Mouse ENCODE) Genome Biol. 2012;13:418. doi: 10.1186/gb-2012-13-8-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rosenbloom KR, Dreszer TR, Long JC, et al. ENCODE whole-genome data in the UCSC Genome Browser: update 2012. Nucleic Acids Res. 2012;40:D912–917. doi: 10.1093/nar/gkr1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mutskov V, Felsenfeld G. The human insulin gene is part of a large open chromatin domain specific for human islets. Proc Natl Acad Sci U S A. 2009;106:17419–17424. doi: 10.1073/pnas.0909288106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bhandare R, Schug J, Le Lay J, et al. Genome-wide analysis of histone modifications in human pancreatic islets. Genome Res. 2010;20:428–433. doi: 10.1101/gr.102038.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Putta S, Lanting L, Sun G, et al. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol. 2012;23:458–469. doi: 10.1681/ASN.2011050485. [DOI] [PMC free article] [PubMed] [Google Scholar]