

Abstract
Macrophages play an important role in innate and adaptive immunity as professional phagocytes capable of internalizing and degrading pathogens to derive antigens for presentation to T cells. They also produce pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α) that mediate local and systemic responses and direct the development of adaptive immunity. The present work describes the use of label-free quantitative proteomics to profile the dynamic changes of proteins from resting and TNF-α-activated mouse macrophages. These analyses revealed that TNF-α activation of macrophages led to the down-regulation of mitochondrial proteins and the differential regulation of several proteins involved in vesicle trafficking and immune response. Importantly, we found that the down-regulation of mitochondria proteins occurred through mitophagy and was specific to TNF-α, as other cytokines such as IL-1β and IFN-γ had no effect on mitochondria degradation. Furthermore, using a novel antigen presentation system, we observed that the induction of mitophagy by TNF-α enabled the processing and presentation of mitochondrial antigens at the cell surface by MHC class I molecules. These findings highlight an unsuspected role of TNF-α in mitophagy and expanded our understanding of the mechanisms responsible for MHC presentation of self-antigens.
Macrophages are professional phagocytes that internalize large particles such as dead cells or microorganisms and play important roles in immunity, inflammation, and tissue repair (1). In mammals, the internalization of microorganisms at sites of infection by macrophages proceeds via a sequential chain of events that leads to the sequestration of pathogens in phagosomes, where they are killed and degraded by hydrolytic enzymes. The functional properties of phagosomes appeared relatively recently in the evolution of multicellular organisms through the acquisition of molecular machineries that transformed phagosomes from lytic vacuoles into organelles fully competent for antigen presentation (2). Indeed, the processing of proteins from internalized microorganisms to derive antigens for presentation at the cell surface on major histocompatibility complex (MHC)1 class I and class II molecules is a key mechanism of adaptive immunity (3).
Macrophages are immune effector cells that mediate defense of the host against a variety of bacteria, viruses, and other microorganisms. Classical activation of macrophages involves Toll-like receptor ligands (e.g. lipopolysaccharides) and pro-inflammatory cytokines such as interferon-γ (IFN-γ) produced by natural killer cells or activated T-helper 1 lymphocytes (4, 5). IFN-γ activation results in the transcriptional regulation of hundreds of genes including nitric oxide synthase-2 and phagocyte oxidase that are associated with the production of reactive oxygen species (ROS) and provide enhanced killing abilities to macrophages (6). This cytokine also mediates phagosome maturation and antigen loading on MHC class I and class II molecules (7–11). Alternate activation of macrophages by interleukin 4 (IL-4) and IL-13 cytokines produced by T-helper 2 cells has also been proposed to account for allergic, cellular, and humoral responses to parasitic and extracellular pathogens (12). These cytokines can promote the development of wound-healing macrophages, though this activation results in poor antigen-presenting cells that are less efficient at producing ROS or at killing intracellular pathogens than classically activated macrophages (13).
Classically activated macrophages can also secrete pro-inflammatory cytokines such as IL-1, IL-6, and IL-23 that can lead to the expansion of T-helper 17 cells associated with autoimmune responses (14). Interestingly, macrophages activated in a MyD88-dependent manner through Toll-like receptor ligand stimulation produce tumor necrosis factor alpha (TNF-α), another important cytokine that synergizes with IFN-γ to enhance macrophage activation. Exogenous stimulation of macrophages by TNF-α can also arise from the secretion of this cytokine by antigen presenting cells. The significance of TNF-α in mounting an appropriate immune response is of particular importance in Leishmania infections, as macrophages stimulated with IFN-γ alone are less efficient at clearing this parasite because of a lack of Toll-like receptor ligand expression. TNF-α plays an important role in inflammatory cell activation and recruitment and is associated with the development of many chronic inflammatory diseases such as rheumatoid arthritis (15) and Crohn's disease (16).
Relatively few studies have investigated the molecular mechanisms and signaling associated with the activation of macrophages by TNF-α. Previous studies using tandem affinity purification and mass spectrometry have provided a physical and functional map of the human TNF-α pathway (17). Stable isotope labeling by amino acids in cell culture has been used to identify changes in the phosphoproteome of HeLa cells in response to TNF-α (18) and to determine the dynamic profiles of TNF-α-induced nuclei-associated proteins in HEK293 cells (19). More recently, label-free quantitative proteomics was used to identify secreted proteins from human-adipose-tissue-derived mesenchymal stem cells during inflammation (20), and Choi et al. utilized bio-orthogonal non-canonical amino acid tagging in combination with proteomics and isobaric tags to identify newly synthesized proteins induced by TNF-α and IL-1β in human monocytic THP-1 cells (21).
To investigate the molecular mechanisms of TNF-α, we profiled the changes in protein abundance in resting and activated RAW264.7 mouse macrophages using label-free quantitative proteomics. We evaluated three independent separation protocols enabling the fractionation of intact proteins (GELFREE and macroporous reverse-phase chromatography) and peptides (strong cation exchange chromatography (SCX)) prior to the analysis of the corresponding tryptic digests via LC-MS/MS on an LTQ-Orbitrap mass spectrometer to obtain a comprehensive view of the macrophage proteome. Importantly, quantitative proteomics analyses of TNF-α-activated macrophages highlighted the down-regulation of several mitochondria proteins. This observation was also correlated by flow cytometry, biochemical assays, and immunofluorescence microscopy experiments, and indicated that TNF-α impaired mitochondrial functions in activated macrophages. The Atg5-dependent degradation of mitochondria and the presentation of mitochondrial antigens by MHC class I molecules suggest that TNF-α macrophage stimulation led to mitophagy and contributed to the modification of the MHC class I peptide repertoire.
EXPERIMENTAL PROCEDURES
Cell Lines
Raw264.7 macrophage cell lines and RAW-Kb-Mito gB30–694 cell lines were grown in DMEM containing 10% FBS supplemented with glutamine and penicillin/streptomycin. Mouse embryonic fibroblast (MEF) wild-type and Atg5−/− cells were grown in DMEM containing 10% FBS supplemented with glutamine and penicillin/streptomycin. The lacZ-inducible gB HSV-specific CD8+ T cell hybridoma HSV-2.3.2E2 was maintained in RPMI 1640 medium supplemented with 5% fetal bovine serum, 2 mm glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin.
Raw-Kb Construction
cDNA was prepared from purified mRNA extracted (NucleoSpin RNA II, Macherey-Nagel Bethlehem, PA) from the BMA3.1A7 cell line (haplotype H-2Kb). cDNA of H-2Kb was amplified via PCR using the primers GTGAATTCGCCACCATGGTACCGTG and GATCTCGAGTCACGCTAGAGAATG, and the 1125-bp PCR product was cloned into the pUB6/V5-His A vector (Invitrogen) using the EcoRI and XhoI restriction sites. This plasmid was then transfected into RAW264.7 cells, and stably transfected cells were selected using blasticidin 3 μg/ml added 24 h after transfection for a period of 1 week. Resistant cells were scraped and surface labeled with anti-H-2Kb mouse antibody PE (BD Biosciences) using conditions that preserved cell integrity. Cells exhibiting the highest levels of fluorescence were sorted into 96-well plates (one cell per well) (BD FACs Vantage cell sorter) and amplified in culture for 2 weeks. Cell surface expression levels of H-2Kb in each clone were then tested by means of surface labeling (see above) followed by flow cytometry analysis, and the clone showing the highest fluorescence levels was selected, amplified, and used in the subsequent experiments (supplemental Fig. S1).
pIRES-gB30–694-Mito Vector
The sequence coding for amino acids 30 to 694 of HSV-1 gB (gB30–694) was cloned from purified HSV-1 DNA (strain F) kindly provided by Johanne Duron (Université de Montréal) using the primers GTAACTAGTGCTCCGACTTCCCCCG and GTAGATATCCTTGATCTCGTGGCGGGTGTA containing, respectively, the restriction sites SpeI and EcoRV. gB30–694 lacked both the signal peptide and the transmembrane domain of the viral gB, but it included the sequence gB498–505 coding for the H-2Kb restricted SSIEFARL peptide.
gB30–694 was cloned in pIRES2-EGFP-Mito kindly provided by Claude Perreault (Université de Montréal). The resulting vector displayed the backbone of pIRES2-EGFP including, in the multiple cloning site, a cassette containing the mitochondrial matrix-targeting sequence (from human cytochrome c oxidase, MSVLTPLLLRGLTGSARRLPVPRAKIHSL) followed by gB30–694.
RAW-Kb-Mito gB30–694 Cell Line
The pIRES-gB30–694-Mito vector was transfected in RAW-Kb cells. Stably transfected cells were selected via the addition of G418 at 0.5 mg/ml in the culture medium 24 h after transfection. After 8 days, cells displaying high GFP levels were sorted in 96-well plates (one cell per well; BD FACs Vantage cell sorter). After 2 weeks of culture, expression and proper processing of the endogenous fusion protein in the different clones were tested in presentation assays. Each clone was co-cultured with gB HSV-specific CD8+ T cell hybridoma HSV-2.3.2E2 overnight, and the level of activation of the gB-specific hybridoma was tested as described below. The clone displaying the highest presentation levels of the gB SSIEFARL epitope was amplified and used in subsequent experiments.
Crude Membrane Preparation
Control cells or cells stimulated with TNF-α (10 ng/ml) for 24 h were lysed in HB buffer (8.5% sucrose, 3 mm imidazole, protease and phosphatase inhibitors, pH 7.4). The cell homogenate was centrifuged at 3000 rpm (4 °C) for 5 min to remove the nuclei. The crude membrane extract was isolated via ultracentrifugation at 50,000 × g at 4 °C for 30 min, and the pellet was resuspended in 8 m urea, 10 mm ammonium bicarbonate, 1 mm tris(2-carboxyethyl)phosphine.
Protein and Peptide Fractionation
Three different methods were used to separate proteins or peptides prior to LC-MS/MS analyses. First, membrane proteins (200 μg/replicate; 6 m urea, 1% acetic acid) were separated via macroporous reversed-phase C18-chromatography (mRP-C18) using an Agilent 1200 LC system (Agilent Technologies, Santa Clara, CA). The LC column was maintained at 80 °C, and chromatographic separations were achieved using a multi-segment elution gradient with eluents A (0.1% trifluoroacetic acid in water, v/v) and B (0.1% trifluoroacetic acid in acetonitrile v/v). The gradient conditions consisted of two steps, with increasing concentrations of eluent B (3%–80% B over 6–49 min; 80%–100% B over 49–59 min) followed by a re-equilibration at 3% B for 15.0 min. The flow rate was maintained at 0.75 ml/min, and a total of 12 fractions were collected.
The second method used a gel-eluted liquid fraction entrapment electrophoresis (GELFREE) 8100 system (Expedeon, San Diego, CA) that enabled the separation of membrane proteins (200 μg/replicate in Laemmli buffer) into 12 fractions across the mass range of 30–150 kDa. SDS was removed from fractions via buffer exchange to 8 m urea using Ultracel-10 multiscreen filterplates (Millipore, Billerica, MA) before proteolytic digestion.
In the third method, membrane protein extracts (200 μg/replicate) were digested with trypsin (sequencing grade; Promega, Madison, WI), and the resulting peptides were separated on an SCX column (Polysulfoethyl A, PolyLC Inc.) Columbia, MD. Chromatographic separations were achieved on an Agilent 1200 LC system using a multi-segment elution gradient with 10 mm ammonium formate, 25% acetonitrile, pH 3.1 (buffer A) and 500 mm ammonium formate, 25% acetonitrile, pH 6.7 (buffer B). The gradient conditions consisted of two steps with increasing concentrations of eluent B (over 10–65 min; 65%–100% B over 65–71 min) followed by a re-equilibration at 0% B for 15.0 min. The flow rate was maintained at 0.3 ml/min, and a total of 12 fractions were collected.
Proteolytic Digestion
Proteins fractionated via mRP or GELFREE were digested by Lys-C and trypsin as follows. Lyophilized protein samples (10 to 20 μg) were resolubillized in 40 μl of 8 m urea, 50 mm ammonium bicarbonate and reduced by means of incubation with 3 mm tris(2-carboxyethyl)phosphine (Thermo Scientific) for 20 min at room temperature. Reduced cysteines were alkylated with 10 mm chloroacetamide (Sigma) and incubated for 15 min at room temperature in the dark. Samples were diluted to a final concentration of 2 m urea with 50 mm ammonium bicarbonate, 1 mm CaCl2 and digested with 0.5 μg of trypsin overnight at 37 °C. The digestion was subsequently quenched by the addition of formic acid to a final concentration of 5%.
Mass Spectrometry
Peptides were separated on a 150-μm inner diameter, 10 cm reversed-phase nano-LC column (Jupiter C18, 3 μm, 300 Å, Phenomenex, Torrance, CA) with a loading buffer of 0.2% formic acid. Peptide elution was achieved using a linear gradient of 5%–40% acetonitrile over 90 min on an Eksigent 2D-nanoLC (Dublin, CA) with a flow rate of 600 nl/min. The nano-LC was coupled to an LTQ-Orbitrap XL mass spectrometer (Thermo-Electron, Bremen, Germany), and samples were injected in an interleaved manner. The mass spectrometer was operated in data-dependent acquisition mode with a 1-s full range mass scan at 60,000 resolution, followed by five product ion scans (MS/MS) of the most abundant precursors above a threshold of 10,000 counts. Collision-induced dissociation was performed in the LTQ at 35% collision energy and an Activation Q of 0.25.
Protein Identification and Quantitative Analysis
The centroided MS/MS data were merged into single peak-list files for each of the three fractionation platforms used (Distiller, V2.4.2.0) and searched with the Mascot search engine, v2.3.01 (Matrix Science, Boston, MA), against a concatenated forward and reversed mouse International Protein Index database (IPI mouse rel. 3.54) containing 55,987 forward protein sequences. Mascot was searched with a parent ion tolerance of 10 ppm and a fragment ion mass tolerance of 0.5 Da. Carbamidomethylation of cysteine; oxidation of methionione; deamidation; phosphorylation of serine, threonine, and tyrosine residues; and ubiquitination of lysine residues (GlyGly) were specified as variable modifications. The confidence in ubiquitination site assignments was adapted from a probability score function based on the method of Olsen et al. (22) and integrated in the ProteoConnections platform (23). The false discovery rate was calculated as the percentage of positive hits in the decoy database versus the target database, and a false discovery rate of 2% was considered for both proteins and peptides. Protein identifications are reported only for those assigned with a minimum of two peptides per protein.
Label-free quantitative proteomics was used to profile protein abundance across sample sets, as reported previously (8, 24). Briefly, Mascot peptide identifications were matched to ion intensity (MS peak intensity, minimum threshold: 8000 counts) extracted from the aligned MS raw data files (tolerances set to m/z = 15 ppm and reaction time = 1 min). For each LC-MS run, we normalized peptide ratios so that the median of their logarithms was 0, to account for unequal protein amounts across conditions and replicates. Intensities were summed across fractions for peptides of identical sequences, and protein ratios were calculated as the median of all peptide ratios, while minimizing the effect of outliers. Only proteins defined by two or more peptide quantification events were considered. The relative standard deviation was below 58% for 95% of the detected proteins. Proteins with a 2-fold variation and a p value below 0.1 were considered differentially regulated.
Bioinformatics Analysis
Transmembrane proteins were predicted using TMHMM 2.0 (25). Gene Ontology annotations for cellular component, biological process, and molecular function were obtained from the Gene Ontology (GO) project using DAVID Bioinformatics resources (26, 27). To identify cellular components or biological processes that were statistically overrepresented in our protein list, we used the binomial statistics tool to compare classifications of multiple clusters of lists to a reference list (Mus musculus total proteome). Only terms that were significantly enriched/depleted with a p value of less than 0.05 were used for the analysis. A global protein–protein interaction network was generated using Cytoscape Version 2.8.0 (28) by submitting all MS-identified proteins to the Cytoscape plugin Bisogenet Version 1.41 (29). Using the gene names of MS-identified proteins, this plugin allowed us to query simultaneously six human protein–protein interaction databases—Biogrid, Intact, Mint, Dip, Bind, and HPRD—in order to generate a global interactome that contained neighbors of MS-identified proteins up to a distance of 1. Subnetworks were created by manually annotating the MS-identified proteins with the UniProt database (30) and by functionally clustering the proteins of the global protein–protein interaction network.
Western Blot
Protein samples were separated by means of 4%–12% SDS-PAGE (Invitrogen) and transferred onto nitrocellulose membrane (Pall Corporation, Port Washington, NY). Proteins of interest were detected using a rabbit polyclonal anti-cytosolic phospholipase A2 (cPLA2) antibody (Cell Signaling, Danvers, MA) or a mouse monoclonal anti-glyceraldehyde 3-phosphate dehydrogenase antibody (Millipore), followed by a secondary antibody coupled to horseradish peroxidase (Millipore) for ECL detection (GE Healthcare).
Flow Cytometry Analysis
A JC-1 Mitochondrial Membrane Potential Assay Kit (Cayman Chemical Company, Ann Arbor, MI) was used to quantify the amount of mitochondria in control and TNF-α-stimulated cells (24 h) via flow cytometry on a FACSCalibur flow cytometer (BD Biosciences). RAW 267.4 macrophages or MEFs were incubated with the JC-1 dye for 30 min. Cells were harvested and analyzed immediately after incubation. Healthy, functional mitochondria with a high potential contained red JC-1 J-aggregates that were quantified in the FL2 channel (excitation: 520–570 nm; emission: 570–610 nm). Apoptotic, unhealthy mitochondria with low potential contained mainly green JC-1 monomers detectable in the FL1 channel (excitation: 485 nm; emission: 535 nm). Mitochondria were quantified using relative fluorescence intensities on a gated population of a uniform cell population. Autophagy was blocked using 3-methyl adenine (3-MA) (10 mm; Sigma-Aldrich) for the last 3 h of the TNF-α stimulation. LysoSensor (Invitrogen) was used to quantify the acidity of lysosomes via flow cytometry on a FACSCalibur flow cytometer (BD Biosciences). RAW 267.4 macrophages were incubated with LysoSensor. Cells were harvested and analyzed immediately after incubation. The acidification of lysosomes was quantified using relative fluorescence intensities on a gated population of a uniform cell population.
Immunofluorescence
Cells were fixed and permeabilized according to the manufacturer's indications (Cytofix/Cytoperm Kit, BD Biosciences). Mitochondria were labeled using Tim23 or Tom20 antibodies (BD Biosciences), and we used antibodies against light chain 3 (LC3) (Thermo Scientific Life Science, Pittsburg, MA) and gB (Santa Cruz Biotechnology, Santa Cruz, CA) to label autophagosomes and gB glycoprotein, respectively. Antibodies were revealed using IgG Alexa-488- and 568-coupled secondary antibodies (Invitrogen). Samples were analyzed using a confocal laser scanning microscope (Zeiss LSM510) with a 63× objective.
Electron Microscopy
For morphological analyses, cells were fixed overnight at 4 °C in 2.5% glutaraldehyde (Canemco) in 0.1 M sodium cacodylate (Canemco Canton de Gore, QC, Canada) buffer, followed by a post-fixation in 1% osmium tetroxyde (Canemco) in 0.1 M sodium cacodylate buffer for 1 h at 4 °C. Contrast of cell membranes was enhanced by uranyl acetate (Mecalab Montreal, QC, Canada) treatment. Cells were dehydrated in ethanol followed by a 1:1 mixture of ethanol/Epon and then embedded in Epon and polymerized at 60 °C for 3 days. Sections were examined on a Phillips CM 100 transmission electron microscope.
Antigen Presentation Assay
RAW-Kb-Mito gB30–694 cells (75 × 104 cells) treated with TNF-α (10 ng/ml) for 24 h and 3-MA (10 mm; Sigma Aldrich), rapamycin (10 μg/ml; Calbiochem), or carbonyl cyanide m-chlorophenyl hydrazone (20 μm; Sigma Aldrich) for 3 h were fixed for 10 min at 23 °C with 1% (w/vol) paraformadehyde. Fixation was followed by three washes in complete DMEM. Antigen presentation assays were performed as described elsewhere (31). Briefly, antigen-presenting cells were cultured for 12 h at 37 °C together with 4 × 105 HSV-2.3.2E2 cells (β-galactosidase-inducible, gB-specific CD8+ T cell hybridoma) for analysis of the activation of T cells. Cells were then lysed (0.125 m Tris base, 0.01 m cyclohexane diaminotetraacetic acid, 50% glycerol (v:v), 0.025% (v:v) Triton X-100, and 0.003 m dithiothreitol, pH 7.8). A β-galactosidase substrate buffer (0.001 m MgSO4 × 7 H2O, 0.01 m KCl, 0.39 m NaH2PO4 × H2O, 0.6 m Na2HPO4 × 7 H2O, 100 mm 2-mercaptoethanol, and 0.15 mm chlorophenol red β-d-galactopyranoside, pH 7.8) was added for 2 to 4 h at 37 °C. Cleavage of the chromogenic substrate chlorophenol red-β-d-galactopyranoside was quantified in a Gemini plate reader (Molecular Devices) at 595 nm. In figures, the data and error bars are shown as the means of three replicate experiments with their respective relative standard deviation.
RESULTS
To obtain a full repertoire of proteins associated with TNF-α-activated macrophages, we isolated crude membrane extracts from the post-nuclear supernatant of mouse macrophage RAW264.7 cells using ultracentrifugation and monitored the fractionation efficiency using immunoblots for several cellular markers (supplemental Fig. S2). This cell fractionation afforded protein extracts of reduced sample complexity while simultaneously enriching for potentially interesting organellar and vesicular proteins. Preliminary proteomics analyses of crude membrane extracts (200-μg aliquots each) were performed using three independent separation platforms, namely, mRP-C18, GELFREE, and SCX fractionation, to obtain a comprehensive identification of the corresponding samples. Whereas the first two platforms separated intact proteins, SCX enabled efficient separation of peptides following tryptic digestion of the crude membrane extract. In each case, three biological replicates were separated into 12 fractions. Fractionated proteins (mRP-C18, GELFREE) were digested by trypsin, and fractions from all three separation platforms were analyzed on an LTQ-Orbitrap mass spectrometer. We compared the distribution of quantifiable peptides detected in one, two, or three replicates using label-free quantitative proteomics (supplemental Fig. S3) and observed that mRP-C18 provided the highest number of reproducibly detected peptides in all replicates (91%) relative to SCX (88%) and GELFREE (47%). The reproducibility of protein fractionation across biological replicates and the enhanced sequence coverage observed for mRP-C18 relative to the other two platforms provided significant advantages for quantitative proteomics. Supplemental Fig. S4 shows the overlap in protein identification and sequence coverage obtained for each platform, and lists of protein and peptide identifications are provided as supplemental Tables S1 and S2, respectively. Phosphopeptide identifications obtained from these analyses are provided as supplemental Fig. S5. These experiments indicated that mRP-C18 enabled more accurate measurements of peptide abundance than the other two platforms examined, and it was selected in subsequent quantitative proteomics experiments. The experimental workflow used in the present study is summarized in Fig. 1.
Fig. 1.

Workflow for large-scale quantitative proteomics analyses of RAW264.7 macrophages. Crude membrane extracts were obtained from resting or TNF-α-stimulated (24 h) macrophages via ultracentrifugation. Protein extracts (n = 3) were fractionated by means of macroporous reversed-phase chromatography followed by tryptic digestion. Peptides were analyzed via LC-MS/MS on an LTQ-Orbitrap XL mass spectrometer. Label-free quantitative proteomics was used to correlate changes in protein abundances across control and TNF-α-activated macrophage extracts. GO term enrichment and protein networks were obtained from identified proteins. The proteomics data set was validated functionally by several biochemical assays.
Quantitative Proteomics of TNF-α-activated Macrophages
Label-free quantitative proteomics experiments were performed on mRP-C18 fractions to identify differentially regulated proteins upon TNF-α stimulation of macrophages. The database search enabled the identification of 13,808 peptides and 1516 proteins with a 2% false discovery rate (see “Experimental Procedures”). We identified 1373 proteins with at least two or more unique peptides with quantifiable abundance measurements (supplemental Table S3). Scatter plots of abundance measurements for peptide ions identified in either control or TNF-α-stimulated extracts indicated that 95% of all ions had relative standard deviation values less than 58% across all three biological replicates, attesting to the reproducibility of the method (supplemental Fig. S6). The consistency of fold-change measurements is also shown in supplemental Fig. S7 for citrate synthase and ATP synthase subunit b, each identified with nine peptides. The distribution of fold-change versus p values is represented in the volcano plot of Fig. 2. Based on the reproducibility of abundance measurements, we used a fold-change of ≥2 and p values of ≤0.1 to define differential regulation across biological replicates. Peptides assigned to the same protein were regrouped together to determine the overall fold-change of abundance upon TNF-α stimulation. Out of 1373 proteins, we identified 174 and 15 proteins that were down- and up-regulated upon TNF-α stimulation, respectively. Several proteins previously reported to be modulated by TNF-α included TNF receptor-associated protein 1 (TRAP1) and cPLA2 (Table I). The overexpression of cPLA2 upon TNF-α stimulation was validated by immunoblots (Fig. 3A) and confirmed the abundance measurement obtained via mass spectrometry (Figs. 3B and 3C).
Fig. 2.

Large-scale membrane proteome analysis of resting and TNF-α-activated macrophages. Volcano plot representation of protein abundance changes upon TNF-α activation. A total of 189 differentially regulated proteins with fold-changes ≥ 2 and p values < 0.1 were identified upon TNF-α stimulation.
Table I. Changes in abundance of selected macrophage membrane proteins upon TNF-α stimulation.
| Expression log2 (TNF-α/control) | Sequence coverage (%) | |
|---|---|---|
| Known TNF-α-modulated proteins | ||
| Cytosolic phospholipase A2 | 1.2 | 9 |
| Keratin 17 | 1.4 | 11 |
| TNFR-associated protein 1 (HSP75) | −1.3 | 43 |
| Mitochondrial proteins | ||
| Enoyl-CoA hydratase | −1.9 | 31 |
| Mitochondrial 2-oxoglutarate/malate carrier | −2.1 | 40 |
| Trifunctional enzyme subunit alpha | −1.1 | 37 |
| Mitochondrial import receptor subunit TOM22 homolog | −1.3 | 47 |
| Cytoskeleton proteins | ||
| Actin cytoplasmic 1 | −1.1 | 75 |
| Tubulin alpha-4A chain | −1.3 | 52 |
| Vesicle trafficking and lysosome | ||
| Cathepsin Z | 1.2 | 9 |
| Vesicle trafficking protein SEC22b | −1.6 | 32 |
| Translocon-associated protein subunit beta (Ssr2) | 1.1 | 5 |
| Sorting nexin-1 | −1.1 | 18 |
| Lysosome-associated membrane glycoprotein 1 (LAMP1) | −1.3 | 13 |
| Immune response | ||
| Osteopontin | 1.1 | 15 |
| H-2 class I histocompatibility antigen K-D alpha chain | −1.1 | 29 |
| Protein degradation | ||
| Proteasome activator complex subunit 1 | −1.0 | 26 |
| Ubiquitin carrier protein | −1.2 | 26 |
| Autophagy-related proteins | ||
| HSP90 α | −1.4 | 49 |
| HSP90 β | −1.3 | 53 |
| 14–3-3 protein epsilon | −1.7 | 60 |
Fig. 3.
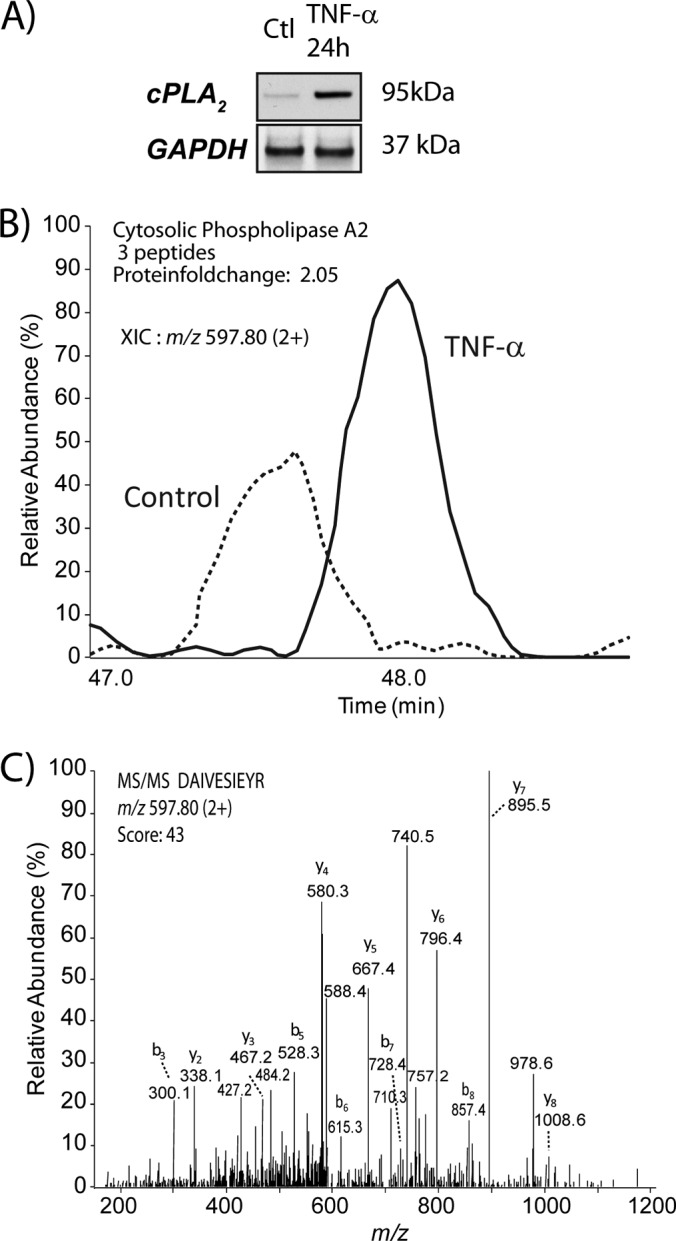
Quantitative proteomics analysis of membrane proteins in TNF-α-stimulated macrophages identified the overexpression of cPLA2. A, immunoblot showing the increased abundance of cPLA2 upon TNF-α stimulation. B, extracted ion chromatogram of m/z 597.82+ corresponding to the peptide DAIVESIEYR from cPLA2 in control (black) and TNF-α-activated macrophage extracts (red). C, MS/MS spectrum of m/z 597.82+ confirming the peptide identification.
Our quantitative proteomics analyses also revealed the differential regulation of several proteins involved in lysosomal degradation (e.g. cathepsin Z) and vesicle trafficking (e.g. Sec22b, sorting nexin1). Previous studies have indicated a role for TNF-α in stimulating autophagy in different cell types, although the exact mechanism is presently unknown (32–34). Interestingly, we observed that 37 mitochondrial proteins, accounting for 21% of all down-regulated proteins, were modulated by TNF-α. An example is shown in supplemental Fig. S7 for citrate synthase, an acetyl-CoA-dependent mitochondrial enzyme involved in the conversion of oxaloacetate into citrate. We obtained a sequence coverage of 33%, and all corresponding peptides showed a consistent decrease in abundance following the activation of macrophages with TNF-α. Interestingly, we also noted that a number of mitochondrial proteins, such as DNA topoisomerase 1 mitochondrial and ATP-dependent Clp protease ATP-binding subunit clpX-like mitochondrial, were ubiquitinated in response to TNF-α (supplemental Table S4, supplemental Fig. S8). This result is consistent with a recent report indicating the activation of the ubiquitin-proteasome system in mitophagy (35).
Protein Interaction Network Analysis of TNF-α-activated Macrophages Uncovers the Regulation of Mitochondrial Proteins
To obtain additional functional insights into proteins and pathways that are differentially regulated in macrophages, we conducted bioinformatics analyses of our proteomics data sets. First, we grouped differentially abundant proteins according to their GO terms compared with those of the mouse reference proteome. GO terms were considered significant when they had p values < 0.05 in a Fisher exact test, resulting in 168 significant terms. Protein groups were then sorted according to “Cellular Component,” “Biological Processes,” and “Molecular Function” GO categories (Fig. 4A).
Fig. 4.

Bioinformatics analyses of the membrane proteome from TNF-α-activated macrophages reveals the down-regulation of mitochondria proteins. A, Gene Ontology enrichment analysis of up-regulated proteins shows a strong enrichment for terms related to structural molecule and hydrolase activities whereas terms associated with vesicle trafficking and mitochondrial functions are down-regulated (numbers represent distinct proteins). B, global protein–protein interaction network comprising 7439 nodes and 80,449 edges. Subnetworks were created by manually annotating the MS-identified proteins with the UniProt database and functionally clustering the proteins from the global protein–protein interaction network. Subnetworks affected by TNF-α activation, such as immune response, ROS and mitochondria, and vesicular trafficking, are displayed.
Up-regulated proteins were associated with GO terms related to cytoskeleton and structural molecule activity. Interestingly, we also observed enrichment for hydrolase activity and cellular nitrogen metabolic processes. Proteins that did not show any significant change in abundance upon TNF-α activation were enriched in mostly basal processes and non-redundant pathways (data not shown). Down-regulated proteins were mostly associated to vesicles and mitochondrial functions. Interestingly, proteins located at the mitochondrial inner membrane showed a more pronounced enrichment than those located at the mitochondrial outer membrane, suggesting that specific subsets of proteins are down-regulated upon TNF-α stimulation. More specifically, we noted that proteins implicated in energy generation or cellular respiration were represented in the subset of down-regulated proteins. Taken together, these analyses suggest that TNF-α activation resulted in extensive cytoskeleton remodeling and vesicular trafficking and contributed to the down-regulation of mitochondrial proteins involved in metabolism and in energy generation.
Next, we used the protein expression data to develop a protein interaction network using Cytoscape. The combination of interactions found in mouse and their human orthologs resulted in a network of 7439 proteins (nodes) and 80,449 connections (edges). From this complex network we extracted sub-networks using UniProt annotations. We identified several groups of membrane proteins modulated by TNF-α, including subnetworks comprising proteins involved in TNF-α signaling, vesicular trafficking, immune response, ROS and mitochondria, and mitophagy and autophagy (Fig. 4B). This network highlights the perturbation of different mitochondrial functions in response to TNF-α activation of macrophages. However, not all mitochondrial proteins were affected equally by TNF-α. Several mitochondrial proteins involved in ROS were down-regulated upon TNF-α activation, whereas mitochondrial ribosomes were unaffected. This interaction network further underscores the modulation of different Rab and effector proteins involved in the control of membrane trafficking, such as Rab5, Rab10, and Rab11, together with the down-regulation of several proteins participating in SNARE fusion, including AnxA7, Vti1b, Vapb, and Vat1. Importantly, we also identified subnetworks related to immune response (e.g. proteasome, antigen processing) and autophagy that were regulated by TNF-α, suggesting a potential association between autophagy and antigen presentation and processing.
TNF-α Induces Specific Autophagic Elimination of Mitochondria in Macrophages
Our quantitative proteomics analyses indicated that mitochondria proteins were down-regulated in TNF-α-activated macrophages. This cytokine is known to induce several types of cell signaling events, including apoptosis, activation of NF-kB, and activation of different kinases (e.g. p38, c-Jun, and the extracellular signal regulated kinase ERK) (36). Mitochondria are key components in a pathway to programmed cell death through the release of death-promoting factors (e.g. cytochrome c, Smac, and endonuclease G) from the intermembrane space (37). To determine whether reduced mitochondrial proteins arose from pre-apoptotic signal, we monitored the abundance of fluorescently labeled annexin A5 at the plasma membrane using flow cytometry (supplemental Fig. S9). Annexin A5 is used as a probe to detect cells expressing phosphatidylserine on the cell surface, an event associated with apoptosis and other forms of cell death (38). Flow cytometry analyses revealed that apoptotic and dead cells represented ∼15% of the cell population in both control and TNF-α-activated macrophages. Similar results were also obtained when cells were stained with 7-amino actinomycin D, a compound that intercalates with double-stranded DNA and penetrates cell membranes of necrotic or dead cells but is excluded from viable cells (supplemental Fig. S9). Furthermore, no significant change in cell count was observed between control and activated macrophages, confirming that TNF-α does not impair cell viability (data not shown).
To determine the extent of changes in mitochondrial functions associated with TNF-α, we examined the effect of this cytokine on the opening of the mitochondrial permeability transition pore, a property that reflects the integrity of the mitochondrial membrane and its transmembrane potential, ΔΨmt. We used JC-1 (5,5′,6,6′-tetrachloro1,1′,3,3′-tetramethyl benzimidazolyl carbocyanine iodide), a fluorescent dye that is taken up by cells and specifically accumulates inside mitochondria. The fluorescence of JC-1 shifts from 527 nm to 590 nm upon aggregation, and the ratio of the green/red fluorescence is used to probe changes in ΔΨmt and provide a direct measurement of the population of healthy (high-potential) versus impaired (low-potential) mitochondria. We first compared the flow cytometry profile of control macrophages stained with JC-1 to the profiles of cells stimulated for 24 h with different cytokines. We observed that macrophages activated by IL-4, Il-13, IL-10, and IL-1β had ratios of healthy to impaired mitochondria comparable to those of control cells (Fig. 5A). However, macrophages stimulated with TNF-α and IFN-γ showed a 30% decrease and 25% increase in the ratio of healthy to impaired mitochondria, respectively (Fig. 5A). The increase in mitochondrial activity upon IFN-γ stimulation is consistent with previous reports indicating that activated macrophages utilize significant amounts of glycolytically generated ATP to maintain high ΔΨmt and prevent apoptosis (39). In contrast, the decrease in mitochondrial potential in non-apoptotic macrophages suggests that TNF-α induced selective degradation of mitochondria, in agreement with our quantitative proteomics experiments. Importantly, these results also indicated that TNF-α is the only major immune-response-modulating cytokine leading to a decrease in healthy mitochondria in activated macrophages.
Fig. 5.
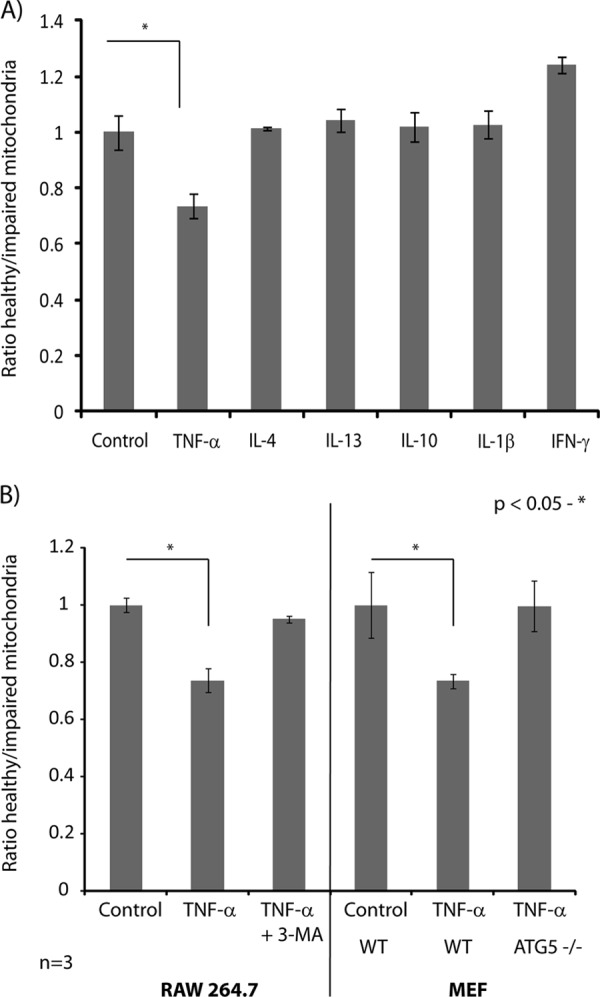
Changes in mitochondrial functions associated with TNF-α-activated macrophages. A, flow cytometry analysis of changes in mitochondrial membrane potential using JC-1 in RAW264.7 macrophages following stimulation with TNF-α, IL-4, IL-13, IL-10, IL-1β, and IFN-γ. Macrophages were stimulated with cytokines for 24 h, and the proportion of healthy versus functionally impaired mitochondria in macrophages was determined using mean fluorescence values of monomeric JC-1 or J-aggregates. TNF-α strongly reduced the ratio of healthy to functionally impaired macrophages, whereas the other cytokines had no significant effect on the mitochondrial membrane potential. B, flow cytometry analysis of changes in mitochondrial membrane potential using JC-1 in RAW264.7 macrophages and MEFs stimulated with TNF-α for 24 h. The ratio of healthy versus functionally impaired mitochondria in macrophages was determined using mean fluorescence values of monomeric JC-1 or J-aggregates. Treatment with the autophagy inhibitor 3-methyl adenine for 3 h abolished the effect of TNF-α on mitochondrial potential. Knock-down of Atg5 restored the original ratio of healthy mitochondria in TNF-α-activated MEF cells.
The selective degradation of mitochondria proteins upon TNF-α activation prompted us to examine the mechanism by which this takes place and the effect of this cytokine on other cell types. We used MEF cells that were morphologically and functionally similar to mouse macrophages. MEF cells stimulated with TNF-α led to a decrease in the ratios of healthy to impaired mitochondria comparable to those observed for RAW 264.7 macrophages (Fig. 5B). Also, TNF-α activation of MEF cells did not lead to decreased cell counts or increased apoptosis (data not shown). We surmised that the impaired mitochondrial activities observed in both TNF-α-activated macrophages and MEF cells might arise from an autophagy-mediated degradation of mitochondria, or mitophagy (40). To verify this proposal, we compared the change in mitochondrial potential of TNF-α-activated macrophages with and without 3-MA, a compound that prevents autophagy by blocking the formation of autophagosomes via the inhibition of phosphatidylinositol 3-kinases (PI-3Ks) (41). These experiments indicated that 3-MA restored normal mitochondrial activities in TNF-α-activated macrophages (Fig. 5B). Furthermore, the effects of TNF-α on mitochondrial functions were also abrogated in MEF cells isolated from Atg5 −/− mice (Fig. 5B). The autophagy-related protein Atg5 is required in the formation of the autophagosome, a vacuole in which intracellular components such as mitochondria are sequestered before their degradation in the lysosome (42). Consistent with this observation, we also noted that macrophages stimulated with TNF-α displayed increased lysosomal degradation activities when stained with LysoSensor, a pH-sensitive fluorescent probe that accumulates in acidic organelles (supplemental Fig. S10).
We used immunofluorescence microscopy to confirm that mitochondria were degraded in autophagosomes upon TNF-α activation. Macrophage cells were stained for the mitochondrion inner membrane protein Tim23 and the autophagic marker LC-3 II to monitor their respective subcellular distributions. In unstimulated cells, Tim23 was evenly distributed across cells, whereas LC3 II was barely detectable (Fig 6A). A significant increase in LC3 II abundance was observed following 24 h of stimulation with TNF-α, confirming the autophagic activation and the formation of characteristic cellular autophagosome punctae containing LC3 II (Fig. 6A). Interestingly, we noted the co-localization of mitochondria and autophagosomes and the striking disappearance of mitochondria in regions where autophagosomes were more densely distributed, consistent with increased mitophagy activities. A salient feature of autophagy is the sequestration of cytosolic organelles and protein aggregates in double membrane-bound compartments to be transported to and degraded in the lysosomal vacuoles (43). Morphological analyses performed using electron microscopy revealed the encapsulation of mitochondria by a double membrane in TNF-α-activated macrophages (Fig. 6B). Under control conditions, mitochondria appeared intact, whereas TNF-α activation of macrophages led to impaired mitochondrial structure. In addition, we observed that mitochondria fused with lysosomes upon TNF-α stimulation. Collectively, these results confirmed that TNF-α selectively led to the degradation of mitochondria via the induction of autophagy.
Fig. 6.
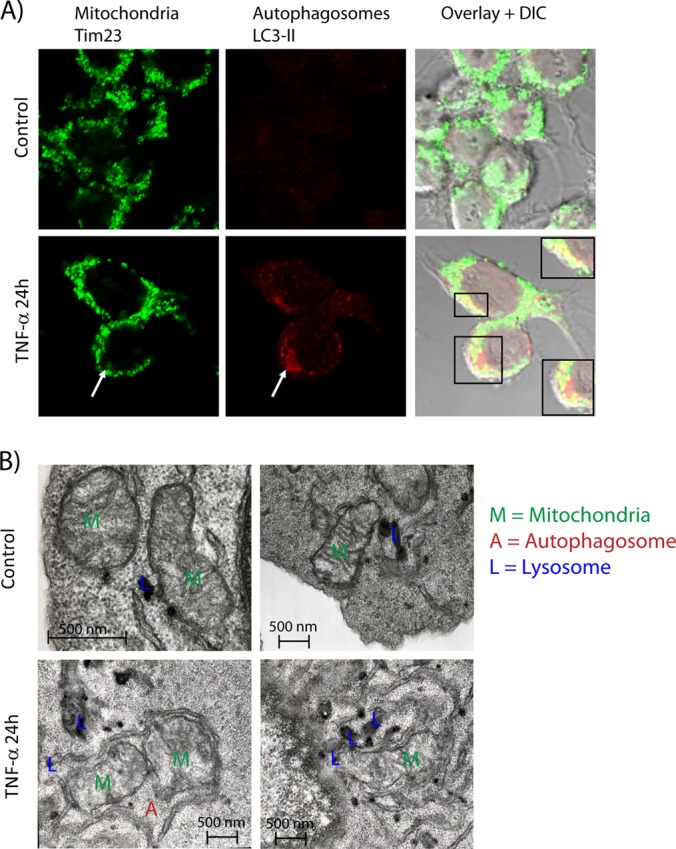
TNF-α induces mitophagy. A, TNF-α stimulation for 24 h induced LC3-II punctae (red) and confirmed the co-localization of autophagosomes and mitochondria. In control cells, no LC3-II punctae were detected, and mitochondria were distributed throughout the cell. B, morphological analyses using electron microscopy of control and TNF-α-stimulated macrophages. Electron micrograph images showed the presence of functionally impaired mitochondria in TNF-α-stimulated RAW264.7 macrophages. Mitochondria from TNF-α-activated macrophages were enclosed by autophagosomal-like double-membrane vesicles, whereas healthy and functional mitochondria were observed in control cells.
Macrophage Activation by TNF-α Increases MHC Class I Presentation of Mitochondrial Antigens
Autophagy is not limited to the clearance of intracellular components; it also plays important roles in both innate and adaptive immunity (44, 45). Previous reports highlighted the significance of autophagy in promoting the presentation of endogenous antigens on MHC class II molecules, thus leading to the activation of CD4+ T cells (46, 47). Interestingly, recent studies also proposed the regulation of macroautophagy and MHC class II expression by TNF-α (34) and a potential interplay between the vacuolar and MHC class I presentation pathways in IL-1β-activated autophagy (31). Results from this proteomics study suggest a strong modulation of proteins involved in MHC class I antigen presentation in TNF-α-activated macrophages. In particular, in TNF-α-regulated proteins we observed several proteins involved in vesicular trafficking (e.g. sorting nexin-1 and -2, vesicle-trafficking protein SEC22b) and degradation (e.g. LAMP1 and beta-hexosaminidase α and β subunits), as well as proteins directly implicated in antigen presentation (e.g. H2 class I histocompatibility antigen K-D, d-D and l-D alpha chain, protein transport protein SEC61β) and proteasomal degradation (e.g. proteasome activator complex subunit I, 26S proteasome non-ATPAse regulatory subunit 14, proteasome subunit beta type-3). To our knowledge, the role of the MHC class I presentation machinery in displaying intracellular antigens following the activation of autophagy by TNF-α has not been described thus far. To further investigate the influence of TNF-α on the presentation of mitochondrial antigens via the MHC class I pathway, we developed a system allowing us to study and compare the molecular mechanisms involved in the processing and presentation of endogenous antigens by macrophages. We produced a RAWKb macrophage cell line that stably expressed a truncated form of glycoprotein B (gB) from HSV-1 and targeted its localization to mitochondria using a mitochondrial matrix targeting sequence from cytochrome c oxidase. The vacuolar response initiated by autophagy increases the processing and presentation of gB antigens on MHC class I molecules that can be monitored via the activation of CD8+ T cells.
The subcellular distribution of HSV-1 gB was determined via immunofluorescence microscopy, and we confirmed its co-localization with the mitochondrial marker Tom20 (Fig. 7A). Next, we cultured these cells for 24 h with TNF-α and/or specific pharmacological inhibitors to evaluate their functional effects on antigen presentation. Following stimulation, cells were fixed with paraformaldehyde and then co-cultured with lacZ-inducible gB-specific CD8+ T cell hybridoma to measure T cell activation against the HSV-1 gB antigen 12 h later. We observed a 30% increase in MHC class I presentation of gB antigens for macrophages stimulated by TNF-α (Fig. 7B). Stimulation of the CD8+ T cell hybridoma was decreased after treatment of activated macrophages with PI-3K inhibitor 3-MA, further supporting the proposal that autophagy contributes to the vacuolar processing and presentation of gB antigen on MHC class I molecules. The effect of mitophagy on antigen presentation was confirmed separately using carbonyl cyanide m-chlorophenyl hydrazone, a protonophore that leads to rapid membrane depolarization and degradation of mitochondria (Fig. 7B). Interestingly, macrophages treated with rapamycin, an inhibitor of the kinase mTOR that normally induces autophagy, had no effect on CD8+ T cell stimulation. Together, these results indicate that TNF-α promoted the selective vacuolar processing of mitochondria proteins and improved the ability of activated macrophages to cross-present mitochondrial antigens to CD8+ T cells.
Fig. 7.

Influence of TNF-α on antigen presentation. A, gB antigen peptide (green) expressed in RAW264.7 cells is targeted to mitochondria and co-localized with Tom20 (red). B, TNF-α enhanced MHC class I cross-presentation of gB antigen. RAW macrophages (control or 24 h following TNF-α activation) were incubated with different pharmacological inhibitors. Macrophages were fixed and co-cultured with lacZ-inducible gB HSV-specific CD8+ T cell hybridoma for 12 h, and cell activation was measured using a UV-visible spectrometer following the hydrolysis of β-Gal.
DISCUSSION
In this report, we describe a comprehensive proteomics study aimed at characterizing the proteome of resting and TNF-α-activated mouse macrophages. Label-free quantitative proteomics analysis of crude membrane extracts from TNF-α-stimulated macrophages revealed the differential regulation of several proteins involved in vesicular trafficking, protein degradation, and immune response (Table I). These analyses also confirmed the identification of several proteins known to be modulated by TNF-α, such as cPLA2 and TRAP1. Previous reports have described the activation of cPLA2 upon TNF-α activation of macrophages (48, 49). This protein is activated by calcium and comprises both lysophospholipase and transacylase activities. cPLA2 selectively hydrolyzes arachidonyl phospholipids in the sn-2 position, releasing arachidonic acid, and this enzyme plays a major role in the initiation of the inflammatory response. In comparison, TRAP1, also referred to as heat shock protein 75, is a member of the HSP90 family (50) that interacts with the intracellular domain of type I TNF receptor. TRAP1 was found to localize to mitochondria and exhibited ATPase activities, but it does not form stable complexes with classic HSP90 co-chaperones (51).
It is noteworthy that the activation of cPLA2 increases the intracellular levels of arachidonic acid and mediates the production of ROS. Although low levels of ROS act as signaling molecules, their prolonged production can impair mitochondrial functions through oxidative damage (52) and depolarization of the transmembrane potential, ΔΨmt (Fig. 5). Interestingly, we observed a consistent down-regulation of mitochondrial proteins in response to TNF-α activation (Fig. 4 and supplemental Fig. S4). We also noted that mitochondrial proteins were not all affected in a similar manner upon TNF-α activation, and down-regulated proteins represented mostly enzymes (65%) and transporters (30%), whereas proteins associated with mitochondrial ribosomes remained largely unaffected.
Importantly, we observed that TNF-α was the only molecule among all cytokines examined that led to the selective degradation of mitochondria in lytic vesicles through autophagy. Several studies have suggested a role for TNF-α in stimulating autophagy in human and murine macrophages (32–34, 53), but the exact mechanism by which TNF-α stimulates autophagy is not fully understood, and it might differ depending on the cell type. This process is initiated by damaged mitochondria, which, upon the loss of ΔΨmt, stabilize the voltage-sensitive kinase Pink1 on the outer mitochondrial membrane, leading to the recruitment of the E3 ubiquitin ligase Parkin and the ubiquitination of mitochondrial proteins (54, 55). It is noteworthy that the regulation of Pink, Parkin, or Nix upon TNF-α stimulation was not determined in the present study, and that macroautophagy might be involved in the elimination of mitochondria. Interestingly, we noted that a number of mitochondrial proteins were ubiquitinated, including DNA topoisomerase 1 mitochondrial. The accumulation of ubiquitylated mitochondrial proteins is thought to facilitate the recruitment of the ubiquitin-binding protein p62 (sequestosome-1), an adaptor protein that binds to Lys-63 polyubiquitin chains of ubiquitylated substrates and mediates the interaction with LC3 to facilitate the autophagosomal degradation of the damaged mitochondria (56). A schematic representation of the autophagic pathways is presented in supplemental Fig. S11.
Impaired mitochondria are first encapsulated in a characteristic double-membrane structure known as the autophagosome prior to fusing with lysosome, at which point their cargo is degraded (Fig. 6B). This double membrane can arise from an extension of endoplasmic reticulum known as the omegasome that contributes important components to the formation of autophagosomes (57). These vesicles are coated with the autophagosome marker microtubule associated protein 1 LC3, a ubiquitin-like protein that is covalently attached to phosphatidylethanolamine during the vesicles' biogenesis. Consistent with this notion, immunofluorescence microscopy experiments indicated that LC3-II punctae were present in TNF-α-stimulated macrophages but not in resting macrophages (Fig. 6A). We also confirmed that the selective degradation of mitochondria was dependent on both PI-3K and Atg5 (Fig. 5B). During the induction of autophagy, Atg5 conjugates to the ubiquitin-like protein Atg12 and interacts with Atg16 to form an oligomeric complex that localizes to nascent autophagosomes (58). The formation of the Atg12–Atg5–Atg16 complex is a prerequisite for the lipidation of LC3 and its targeting to autophagosomes prior to their fusion with either late endosomes (amphisomes) or lysosomes (autolysosomes) (59, 60).
In addition to the elimination of dysfunctional mitochondria, autophagy also plays important roles in innate and adaptive immunity via the processing and presentation of endogenously expressed antigens by MHC class I and class II molecules (61–63). The notion that the degradation of intracellular antigens after autophagy is used by the mammalian immune system to display intracellular antigens on MHC class II for CD4+ T cell stimulation was recently expanded to include antigen processing via MHC class I presentation (31). Indeed, previous results from our group indicated that in addition to the classical MHC class I pathway, viral proteins can be engulfed during HSV-1 infection by autophagosomes formed from the membrane of the outer nuclear envelope. The inhibition of autophagy was confirmed via siRNA silencing of the Atg5 gene and abrogated HSV-1 specific CD8+ T cell stimulation (31). In the present study, we further expanded the role of autophagy in the processing of intracellular antigens and their presentation by MHC class I molecules. Using a RAWKb macrophage cell line that stably expressed a truncated form of gB from HSV-1 targeted to mitochondria, we showed that autophagy increased the processing and presentation of mitochondrial viral antigens on MHC class I molecules following TNF-α activation (Fig. 7). CD8+ T cell activation was modulated by PI-3K inhibition and a loss of ΔΨmt, consistent with the notion that autophagy contributes to the processing and presentation of mitochondria-specific gB antigen on MHC class I molecules. This pathway is independent of autophagy induced by mTORC1 inhibition, as macrophages treated with rapamycin showed no increase in CD8+ T cell stimulation. Taken together, these findings highlight a novel role for TNF-α in mitophagy and in the processing and presentation of mitochondrial antigens by MHC class I molecules. It is interesting to note that increased ROS production and oxidative stress induced by the accumulation of damaged mitochondria can lead to a host danger signal initiated by NLRP3 inflammasomes often associated with many chronic inflammatory diseases (64). The NLRP3 inflammasome is negatively regulated by autophagy, and the targeted removal of dysfunctional mitochondria following TNF-α prevents progressive cell damage and inflammation.
Supplementary Material
Acknowledgments
We thank Christiane Rondeau and Eric Bonneil for assistance with electron microscopy and mass spectrometry analyses and G. Arthur (University of Manitoba) for the wild-type and Atg5−/− mouse embryonic fibroblasts produced by N. Mizushima (Medical and Dental University, Tokyo). M.D. and P.T. hold Canada Research Chairs in Cellular Microbiology and Proteomics and Bioanalytical Spectrometry, respectively. C.B. holds a Vanier Canada Graduate Scholarship from the Natural Science and Engineering Research Council (NSERC). IRIC is supported in part by the Canadian Center of Excellence in Commercialization and Research, the Canada Foundation for Innovation (CFI), and the Fonds de Recherche du Québec en Santé (FRQS).
Footnotes
* This work was supported by a Canadian Institutes of Health Research grant to M.D. and P.T.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are::
- 3-MA
- 3-methyl adenine
- cPLA2
- cytosolic phospholipase A2
- gB
- glycoprotein B
- GELFREE
- gel-eluted liquid fraction entrapment electrophoresis
- GO
- Gene Ontology
- HSV-1
- herpes simplex virus 1
- IFN-γ
- interferon-γ
- JC-1
- 5,5′,6,6′-tetrachloro1,1′,3,3′-tetramethyl benzimidazoyl carbocyanine iodide
- LC3
- light chain 3
- LTQ
- linear trap quadrupole
- MEF
- mouse embryonic fibroblast
- MHC
- major histocompatibility complex
- mRP-C18
- macroporous reversed-phase C18
- MS/MS
- tandem mass spectrometry
- PI-3K
- phosphatidylinositol-3 kinase
- ROS
- reactive oxygen species
- SCX
- strong cation exchange
- TNF-α
- tumor necrosis factor-α
- TRAP1
- TNF receptor-associated protein 1.
REFERENCES
- 1. Mosser D. M., Edwards J. P. (2008) Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boulais J., Trost M., Landry C. R., Dieckmann R., Levy E. D., Soldati T., Michnick S. W., Thibault P., Desjardins M. (2010) Molecular characterization of the evolution of phagosomes. Mol. Syst. Biol. 6, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jutras I., Desjardins M. (2005) Phagocytosis: at the crossroads of innate and adaptive immunity. Annu. Rev. Cell Dev. Biol. 21, 511–527 [DOI] [PubMed] [Google Scholar]
- 4. Platanias L. C. (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386 [DOI] [PubMed] [Google Scholar]
- 5. Murray P. J., Wynn T. A. (2011) Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ehrt S., Schnappinger D., Bekiranov S., Drenkow J., Shi S., Gingeras T. R., Gaasterland T., Schoolnik G., Nathan C. (2001) Reprogramming of the macrophage transcriptome in response to interferon-gamma and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J. Exp. Med. 194, 1123–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Watts C., Amigorena S. (2001) Phagocytosis and antigen presentation. Semin. Immunol. 13, 373–379 [DOI] [PubMed] [Google Scholar]
- 8. Trost M., English L., Lemieux S., Courcelles M., Desjardins M., Thibault P. (2009) The phagosomal proteome in interferon-γ-activated macrophages. Immunity 30, 143–154 [DOI] [PubMed] [Google Scholar]
- 9. Jutras I., Houde M., Currier N., Boulais J., Duclos S., LaBoissière S., Bonneil E., Kearney P., Thibault P., Paramithiotis E., Hugo P., Desjardins M. (2008) Modulation of the phagosome proteome by interferon-γ. Mol. Cell. Proteomics 7, 697–715 [DOI] [PubMed] [Google Scholar]
- 10. Houde M., Bertholet S., Gagnon E., Brunet S., Goyette G., Laplante A., Princiotta M. F., Thibault P., Sacks D., Desjardins M. (2003) Phagosomes are competent organelles for antigen cross-presentation. Nature 425, 402–406 [DOI] [PubMed] [Google Scholar]
- 11. Guermonprez P., Saveanu L., Kleijmeer M., Davoust J., Van Endert P., Amigorena S. (2003) ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 425, 397–402 [DOI] [PubMed] [Google Scholar]
- 12. Gordon S. (2003) Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35 [DOI] [PubMed] [Google Scholar]
- 13. Edwards J. P., Zhang X., Frauwirth K. A., Mosser D. M. (2006) Biochemical and functional characterization of three activated macrophage populations. J. Leukoc. Biol. 80, 1298–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kolls J. K., Linden A. (2004) Interleukin-17 family members and inflammation. Immunity 21, 467–476 [DOI] [PubMed] [Google Scholar]
- 15. Maini R., Elliott M., Brennan F., Feldmann M. (1995) Beneficial effects of tumour necrosis factor-alpha (TNF-alpha) blockade in rheumatoid arthritis (RA). Clin. Exp. Immunol. 101, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Dullemen H. M., van Deventer S. J. H., Hommes D. W., Bijl H. A., Jansen J., Tytgat G. N. J., Woody J. (1995) Treatment of Crohn's disease with anti-tumor necrosis factor chimeric monoclonal antibody (cA2). Gastroenterology 109, 129–135 [DOI] [PubMed] [Google Scholar]
- 17. Bouwmeester T., Bauch A., Ruffner H., Angrand P.-O., Bergamini G., Croughton K., Cruciat C., Eberhard D., Gagneur J., Ghidelli S., Hopf C., Huhse B., Mangano R., Michon A.-M., Schirle M., Schlegl J., Schwab M., Stein M. A., Bauer A., Casari G., Drewes G., Gavin A.-C., Jackson D. B., Joberty G., Neubauer G., Rick J., Kuster B., Superti-Furga G. (2004) A physical and functional map of the human TNF-[alpha]/NF-[kappa]B signal transduction pathway. Nat. Cell Biol. 6, 97–105 [DOI] [PubMed] [Google Scholar]
- 18. Cantin G. T., Venable J. D., Cociorva D., Yates J. R. (2005) Quantitative phosphoproteomic analysis of the tumor necrosis factor pathway. J. Proteome Res. 5, 127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ma D.-j., Li S.-J., Wang L.-S., Dai J., Zhao S.-l., Zeng R. (2009) Temporal and spatial profiling of nuclei-associated proteins upon TNF-[alpha]/NF-[kappa]B signaling. Cell Res. 19, 651–664 [DOI] [PubMed] [Google Scholar]
- 20. Lee M. J., Kim J., Kim M. Y., Bae Y. S., Ryu S. H., Lee T. G., Kim J. H. (2010) Proteomic analysis of tumor necrosis factor-alpha-induced secretome of human adipose tissue-derived mesenchymal stem cells. J. Proteome Res. 9, 1754–1762 [DOI] [PubMed] [Google Scholar]
- 21. Choi K. Y., Lippert D. N., Ezzatti P., Mookherjee N. (2012) Defining TNF-alpha and IL-1beta induced nascent proteins: combining bio-orthogonal non-canonical amino acid tagging and proteomics. J. Immunol. Methods [DOI] [PubMed] [Google Scholar]
- 22. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 23. Courcelles M., Lemieux S., Voisin L., Meloche S., Thibault P. (2011) ProteoConnections: a bioinformatics platform to facilitate proteome and phosphoproteome analyses. Proteomics 11, 2654–2671 [DOI] [PubMed] [Google Scholar]
- 24. Trost M., Sauvageau M., Herault O., Deleris P., Pomies C., Chagraoui J., Mayotte N., Meloche S., Sauvageau G., Thibault P. (2012) Posttranslational regulation of self-renewal capacity: insights from proteome and phosphoproteome analyses of stem cell leukemia. Blood 120, e17-e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sonnhammer E., Von Heijne G., Krogh A. (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. 175–182 [PubMed] [Google Scholar]
- 26. Sherman B. T., Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Da Wei Huang B. T. S., Lempicki R. A. (2008) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 28. Cline M. S., Smoot M., Cerami E., Kuchinsky A., Landys N., Workman C., Christmas R., Avila-Campilo I., Creech M., Gross B. (2007) Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2, 2366–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Martin A., Ochagavia M. E., Rabasa L. C., Miranda J., Fernandez-de-Cossio J., Bringas R. (2010) BisoGenet: a new tool for gene network building, visualization and analysis. BMC Bioinformatics 11, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. UniProt Consortium (2012) Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 40, D71-D75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. English L., Chemali M., Duron J., Rondeau C., Laplante A., Gingras D., Alexander D., Leib D., Norbury C., Lippe R., Desjardins M. (2009) Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 10, 480–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baregamian N. (2009) Tumor necrosis factor-α and apoptosis signal-regulating kinase 1 control reactive oxygen species release, mitochondrial autophagy and c-Jun N-terminal kinase/p38 phosphorylation during necrotizing enterocolitis. Oxid. Med. Cell. Longev. 2, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jia G., Cheng G., Gangahar D. M., Agrawal D. K. (2006) Insulin-like growth factor-1 and TNF-α regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol. Cell Biol. 84, 448–454 [DOI] [PubMed] [Google Scholar]
- 34. Keller C. W., Fokken C., Turville S. G., Lünemann A., Schmidt J., Münz C., Lünemann J. D. (2011) TNF-α induces macroautophagy and regulates MHC class II expression in human skeletal muscle cells. J. Biol. Chem. 286, 3970–3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chan N. C., Salazar A. M., Pham A. H., Sweredoski M. J., Kolawa N. J., Graham R. L. J., Hess S., Chan D. C. (2011) Broad activation of the ubiquitin–proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aggarwal B. B., Gupta S. C., Kim J. H. (2012) Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 119, 651–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saelens X., Festjens N., Vande Walle L., van Gurp M., van Loo G., Vandenabeele P. (2004) Toxic proteins released from mitochondria in cell death. Oncogene 23, 2861–2874 [DOI] [PubMed] [Google Scholar]
- 38. Vermes I., Haanen C., Steffens-Nakken H., Reutelingsperger C. (1995) A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 184, 39–51 [DOI] [PubMed] [Google Scholar]
- 39. Garedew A., Henderson S. O., Moncada S. (2010) Activated macrophages utilize glycolytic ATP to maintain mitochondrial membrane potential and prevent apoptotic cell death. Cell Death Differ. 17, 1540–1550 [DOI] [PubMed] [Google Scholar]
- 40. Kim I., Rodriguez-Enriquez S., Lemasters J. J. (2007) Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 462, 245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Petiot A., Ogier-Denis E., Blommaart E. F., Meijer A. J., Codogno P. (2000) Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 275, 992–998 [DOI] [PubMed] [Google Scholar]
- 42. Mizushima N., Sugita H., Yoshimori T., Ohsumi Y. (1998) A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 273, 33889–33892 [DOI] [PubMed] [Google Scholar]
- 43. Nakatogawa H., Suzuki K., Kamada Y., Ohsumi Y. (2009) Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol. 10, 458–467 [DOI] [PubMed] [Google Scholar]
- 44. Levine B., Deretic V. (2007) Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 7, 767–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schmid D., Munz C. (2007) Innate and adaptive immunity through autophagy. Immunity 27, 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dengjel J., Schoor O., Fischer R., Reich M., Kraus M., Muller M., Kreymborg K., Altenberend F., Brandenburg J., Kalbacher H., Brock R., Driessen C., Rammensee H. G., Stevanovic S. (2005) Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 7922–7927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Paludan C., Schmid D., Landthaler M., Vockerodt M., Kube D., Tuschl T., Munz C. (2005) Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307, 593–596 [DOI] [PubMed] [Google Scholar]
- 48. Lee C.-W., Lin C.-C., Lee I. T., Lee H.-C., Yang C.-M. (2011) Activation and induction of cytosolic phospholipase A2 by TNF-α mediated through Nox2, MAPKs, NF-κB, and p300 in human tracheal smooth muscle cells. J. Cell. Physiol. 226, 2103–2114 [DOI] [PubMed] [Google Scholar]
- 49. McPhillips K., Janssen W. J., Ghosh M., Byrne A., Gardai S., Remigio L., Bratton D. L., Kang J. L., Henson P. (2007) TNF-alpha inhibits macrophage clearance of apoptotic cells via cytosolic phospholipase A2 and oxidant-dependent mechanisms. J. Immunol. 178, 8117–8126 [DOI] [PubMed] [Google Scholar]
- 50. Song H. Y., Dunbar J. D., Zhang Y. X., Guo D., Donner D. B. (1995) Identification of a protein with homology to hsp90 that binds the type 1 tumor necrosis factor receptor. J. Biol. Chem. 270, 3574. [PubMed] [Google Scholar]
- 51. Felts S. J., Owen B. A. L., Nguyen P. M., Trepel J., Donner D. B., Toft D. O. (2000) The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J. Biol. Chem. 275, 3305–3312 [DOI] [PubMed] [Google Scholar]
- 52. Lee J., Giordano S., Zhang J. (2012) Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem. J. 441, 523–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ling Y. M., Shaw M. H., Ayala C., Coppens I., Taylor G. A., Ferguson D. J. P., Yap G. S. (2006) Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J. Exp. Med. 203, 2063–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chan N. C., Salazar A. M., Pham A. H., Sweredoski M. J., Kolawa N. J., Graham R. L., Hess S., Chan D. C. (2011) Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Geisler S., Holmstrom K. M., Skujat D., Fiesel F. C., Rothfuss O. C., Kahle P. J., Springer W. (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131 [DOI] [PubMed] [Google Scholar]
- 56. Okatsu K., Saisho K., Shimanuki M., Nakada K., Shitara H., Sou Y., Kimura M., Sato S., Hattori N., Komatsu M., Tanaka K., Matsuda N. (2010) p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15, 887–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Walker S., Chandra P., Manifava M., Axe E., Ktistakis N. T. (2008) Making autophagosomes. Autophagy 4, 1093–1096 [DOI] [PubMed] [Google Scholar]
- 58. Mizushima N., Kuma A., Kobayashi Y., Yamamoto A., Matsubae M., Takao T., Natsume T., Ohsumi Y., Yoshimori T. (2003) Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 116, 1679–1688 [DOI] [PubMed] [Google Scholar]
- 59. Fujita N., Itoh T., Omori H., Fukuda M., Noda T., Yoshimori T. (2008) The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 19, 2092–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hanada T., Noda N. N., Satomi Y., Ichimura Y., Fujioka Y., Takao T., Inagaki F., Ohsumi Y. (2007) The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 282, 37298–37302 [DOI] [PubMed] [Google Scholar]
- 61. Levine B., Mizushima N., Virgin H. W. (2011) Autophagy in immunity and inflammation. Nature 469, 323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Deretic V. (2011) Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol. Rev. 240, 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Münz C. (2010) Antigen processing via autophagy—not only for MHC class II presentation anymore? Curr. Opin. Immunol. 22, 89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhou R., Yazdi A. S., Menu P., Tschopp J. (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.