

Abstract
Soluble factors that promote survival and differentiation of glia and neurons during development are likely to play key roles in neurodegeneration and demyelinating diseases such as multiple sclerosis (MS) and have the potential to be important therapeutic targets. We examined the effect of trkB signaling and the expression patterns of neurotrophic and gliotrophic factors in the mouse brain in MOG-induced experimental allergic encephalomyelitis (EAE). With induction of mild disease, trkB heterozygous mice were more severely affected compared to their wild type littermates. However, with more potent disease induction, trkB heterozygotes fared similar to their wild type littermates, suggesting complex modulatory roles for trkB signaling. One possible explanation for this difference is that the expression patterns of neurotrophic factors correlate with disease severity in individual mice with mild disease, but not in more severe disease. With the less potent induction in C57BL/6 mice, we found that BDNF was consistently increased at EAE onset, while the soluble gliotrophic factor neuregulin (NRG1) was increased only in the chronic phase of the disease. Treatment of these animals with glatiramer acetate (GA) to decrease disease severity resulted in lower levels of both BDNF and NRG1 expression in some mice at 35 days after immunization compared to those in untreated EAE mice, but had no direct effect on these factors in the absence of EAE. Our results suggest a complex interplay between neurotrophic and gliotrophic factors in EAE that is dependent on disease stage and severity. While signaling by BDNF through trkB is protective in mild disease, this effect was not seen in more severe disease. The late induction of NRG1 in the chronic stage of disease could also worsen disease severity through its known ability to activate microglial, inflammatory pathways. While complex, these studies begin to define underlying axoglial trophic activities that are likely involved in both disease pathogenesis and repair.
Introduction
Based on the efficacy of immunomodulatory therapies in patients with MS, much of the research and thinking about pathogenesis and treatment has traditionally focused on suppressing or modulating the immune system. One of the most successful therapeutic strategies in combating this disorder is a mixture of basic peptides, randomly synthesized at specific molar ratios, called glatiramer acetate (GA) (Kieseier and Stuve, 2011; Lalive et al., 2011; Racke and Lovett-Racke, 2011; Ragheb et al., 2001). Recent reports suggest that in addition to its immunoregulatory anti-inflammatory effects, GA may also have neuroprotective effects and that this protection may be conferred through the local expression of neurotrophic factors such as BDNF in the brain (Aharoni et al., 2005). BDNF is involved in neuronal survival, differentiation, and function, as well as glial development in the central nervous system (CNS) (Chao, 2003). BDNF and its receptor truncated trkB tyrosine kinase receptor (gp145trkB) were found in lesions of MS patients (Stadelmann et al., 2002) and also in EAE (Aharoni et al., 2005; Colombo et al., 2012; De Santi et al., 2009). The therapeutic effect of GA was reported previously to have neuroprotective effects with the potential to modulate BDNF expression in the diseased brain (Aharoni et al., 2005). The most recent report from Lee et al (Lee et al., 2012) indicates that the CNS damage is increased in the early stages of the disease when BDNF is lacking, but there was no significant effect in later stages of the disease.
Recent studies have rekindled the notion that neurological dysfunction in patients with MS relates best to the degree of neuronal (axonal) and glial damage (Amor et al., 2010; Derfuss et al., 2010; Ilieva et al., 2009; Loeb, 2007; Siffrin et al., 2010). Therefore, to understand the pathogenesis of MS and to develop biologically targeted therapeutics, a greater understanding of axoglial biology is needed in the context of inflammatory modulation. In addition to neurotrophic factors, such as BDNF, central to axoglial biology are protein regulatory factors that signal in the opposite direction, from neurons to glia, and are referred to as “gliotrophic” factors. One of these is called neuregulin derived from the neuregulin1 gene (NRG1) that is important for peripheral and central nervous system development (De Santi et al., 2009; De Santi et al., 2011; Hohlfeld, 2008; Linker et al., 2009; Loeb, 2007; Nagahara and Tuszynski, 2011). Despite their importance in development, little is known of the interrelated roles of neurotrophic and gliotrophic factors in neurodegeneration and demyelinating diseases, such as MS (Loeb and Fischbach, 1997; Loeb, 2007).
Our recent work suggests that NRG1 is tightly regulated by neurotrophic factors, including BDNF and GDNF, both at the transcriptional and post-translational levels (Esper and Loeb, 2004; Loeb and Fischbach, 1997; Loeb et al., 2002; Loeb, 2007; Ma et al., 2011). NRG1 has been shown to be critical for peripheral nerve development, myelination, and more recently, spinal cord microglial activation in a model of chronic pain (Calvo et al., 2010; Esper et al., 2006; Falls, 2003). The NRG1 gene has also been implicated as a susceptibility factor in families with schizophrenia (Buonanno, 2010; Mei and Xiong, 2008). The NRG1 gene carries out these important activities through a large repertoire of both secreted (type I) and membrane-bound (type III) forms that are highly expressed in brain and spinal cord neurons (Loeb et al., 1999; Meyer et al., 1997). Evidence from a number of studies suggests a positive feedback loop between neuronal NRG1 and glial- and muscle-derived, neurotrophic factors (Esper and Loeb, 2004; Esper et al., 2006; Falls, 2003; Loeb and Fischbach, 1997; Loeb et al., 2002; Ma et al., 2011). To date, this important reciprocal signaling pathway has not been explored in MS or the EAE mouse model where marked inflammation, demyelination and glial activation are observed.
EAE is an inflammatory autoimmune disease of the CNS that serves as a useful animal model for testing treatment strategies for MS (Lisak and Behan, 1975; Steinman and Zamvil, 2006). The disease is inducible in the susceptible mouse strain C57BL/6 by immunization with myelin oligodendrocyte glycoprotein (MOG) peptide 35-55 (O’Neill et al., 2006; Papenfuss et al., 2004). Here, we examined the expression patterns of neurotrophic and gliotrophic factors in the mouse brain in MOG-induced EAE in C57BL/6 mice as well as the effects of reduced BDNF signaling in mice heterozygous for the BDNF receptor, trkB. We also examined the effect of GA treatment. We found a complex pattern of gliotrophic and neurotrophic gene expression that varied with disease severity and could be suppressed by GA treatment. Understanding the changes of gliotrophic- and neurotrophic- factors as a function of disease severity will be important to optimize neuroprotective treatment strategies in MS.
Materials and Methods
Mice
Adult female C57BL/6 mice (JAX stock 000664), trkB heterozygous (trkB+/−, 5 generations of backcrossing into C57BL/6 background; JAX stock 002544) (Klein et al., 1993) and age-matched female wild type littermates were purchased from Jackson Laboratories (Bar Harbor, ME). A second series of experiments utilized trkB+/− mice with 7 generations of backcrossing into C57BL/6 background (JAX stock 003098), also from Jackson Laboratories. Wild type littermates for 5 and 7 generations of trkB heterozygous (JAX stock 002544 and 003098) were also from Jackson Laboratories.
Induction and evaluation of EAE
C57BL/6, trkB+/− and trkB+/+ (wt littermates) mice (5 generations of backcrossing, JAX stock 002544) were immunized subcutaneously on day zero in four sites over the back (left and right shoulder, left and right flank) with 200 μg of MOG35–55 (MEVGWYRSPFSRVVHLYRNGK) (Princeton Biomolecules, Langhorne, PA) emulsified in a volume of 100 μl of complete Freund’s adjuvant (CFA) containing 1 mg/ml of M. tuberculosis (M. Tbc) (CFA, Difco Laboratories, Detroit, MI, USA) (O’Neill et al., 2006; Papenfuss et al., 2004). On days 0 and 2 post-immunization, 200 ng of pertussis toxin (PT) (List Biological, Campbell, CA) was administered intraperitoneally (i.p.) in 0.2 ml of PBS (Sigma, St. Louis, Minnesota). Mice were observed daily for clinical signs and scored as follows: limp tail or waddling gait with decreased tail tonicity +1, waddling gait +2, partial hind limb paralysis +3, full hind limb paralysis +4, death +5.
In some of experiments (Figure 1C and 1D), to induce more severe disease, trkB+/− and trkB+/+ (wt littermates) mice (7 generations of backcrossing, JAX stock 003098) were injected as above except that M. Tbc was increased to 5 mg/ml in CFA and PT was increased to 300 ng on days 0 and 2 post-immunization.
Figure 1. Decreased TrkB expression increases disease severity in less potent immunization conditions.

Female trkB+/− and age- and gender-matched female wild type littermates (trkB+/+) were immunized with MOG35-55(200μg)/CFA containing M. Tbc (1mg/ml or 5mg/ml)/PT(200X2 ng or 300X2 ng) using two immunization protocols resulting in mild (A) or severe (C) disease induction in trkB+/+ mice. Mice were scored daily for 48 days. Each line represents disease course for an individual mouse. There is increased disease incidence and disease severity in trkB+/− mice compared to trkB+/+ mice with the less potent immunization conditions (Figures 1A-1B). However, there is no difference in disease severity between the trkB+/− and trkB+/+ mice with the more potent immunization conditions (Figures 1C-1D).
GA treatment
GA (Copaxone, Copolymer 1) from batch 242900110 with an average Mr of 7 kDa, was obtained from Teva Pharmaceutical Industries. GA treatment was administered by consecutive daily i.p. injections (1.5 mg per mouse per day) at different stages of disease: 1) at EAE induction (day 0), 8 injections daily (suppression); or 2) after appearance of first clinical sign, 8 injections daily (treatment) (Aharoni et al., 2008).
RNA isolation and real-time quantitative RT-PCR (qPCR)
Mice designated for qPCR were anesthetized (ketamine 80 mg/kg, i.p. and xylazine 40 mg/kg, i.p.) and then had intracardiac perfusion with PBS (0.1 m, pH 7.4). Perfused mouse brains were dissected and rapidly frozen and stored at the minus 80°C. RNA was extracted using the Qiagen RNeasy Lipid Tissue Mini Kit (Qiagen, Valencia, CA, USA) (Rakhade et al., 2005). Quantification of RNA was carried out using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). The quality of RNA was determined on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) using an RNA 6000 Nano chip kit, RNA ladder and Agilent analysis software (Agilent Technologies). All samples had RIN (RNA integrity number) values above 8.0 and 260/280 ratios near 2.0.
The expression of mouse type I and III NRG1, BDNF and GDNF were each measured relative to GAPDH using Taqman Assays-On-Demand primers (Applied Biosystems, Foster City, CA, U.S.A.). 1.5 μg of total RNA was used in a 20 μl reverse transcription synthesis reaction primed with oligo-dT primers (Superscript First Strand Synthesis System, Invitrogen, Carlsbad, CA, U.S.A.). PCR was performed in triplicate using 1X Taqman Universal PCR master mix (Applied Biosystems) with the DNA Engine Opticon Continuous Fluorescence Detection System (MJ Research, Waltham, MA, U.S.A.) utilizing the following primers and Taqman probes: type I NRG1: Mm00626552_m1; type III NRG1: Mm01212129_m1; BDNF: Mm01334047_m1; GDNF: Mm00599849_m1 and GAPDH: Mm99999915_g1)(Song et al., 2012). Cycle threshold (Ct) values were calculated using Opticon monitor software, with the threshold set at 40 standard deviations above background. The relative expression was calculated by normalizing the expression of individual genes to GAPDH and using the 2-ΔΔCt method (Livak and Schmittgen, 2001).
Statistical analysis
A nonparametric ANOVA with Tukey’s post hoc test was performed for disease severity: 1) between groups of wt (trkB+/+) and trkB+/− mice (Table 1), and, 2) groups of control and GA-suppression as well as GA-treatment mice (Table 2).
Table 1.
Disease severity and incidence were significantly increased in trkB +/− mice compared to their wt littermates (trkB+/+) with less potent disease induction, however, trkB +/− mice fared similar to wt littermates with more potent disease inductiona
| Immunization conditions |
Mice | Incidence of EAE (%) |
Mean Day of Onsetb |
Mean cumulative Clinical Scorec |
Mean Maximum Severityd |
Mortality |
|---|---|---|---|---|---|---|
| Less potent | trkB +/+ | 2/5 (40%) | 11.0 ± 0.0 | 32.8 ± 6.7 | 1.0 ± 1.4 | 0/5 (0%) |
| trkB +/− | 5/5 (100%)* | 13.2 ± 2.7 | 56.6 ± 36.6 | 3.6 ± 1.5* | 1/5 (20%) | |
| More potent | trkB +/+ | 5/5 (100%) | 14.4 ± 0.5 | 79.1 ± 29.5 | 3.1 ± 1.3 | 0/5 (0%) |
| trkB +/− | 4/4 (100%) | 14.8 ± 2.5 | 54.5 ± 12.3 | 2.5 ± 0.0 | 0/4 (0%) |
trkB+/− and age- and gender-matched female wide type littermates (trkB+/+) (n=4-5 each group) were immunized with MOG35-55(200μg)/CFA containing M. Tbc (1mg/ml ‘less potent’ or 5mg/ml ‘more potent’)/PT(400ng ‘less potent’ or 600ng ‘more potent’) using two immunization protocols resulting in mild or severe disease induction in wild type littermates trkB+/+ mice. Mice (n=4-5 each group) were scored daily, and the mean score of each group ± SD is shown.
Mean of the first sign of clinical scores for each animal.
Mean of the sum of scores for each animal over the entire observation period (48 days).
Mean of the highest clinical score exhibited by individual animals within a group during the entire course of EAE.
p< 0.05 by t test between trkB+/− and +/+ (wt) under the same immunization conditions.
Table 2.
GA treatment reduces disease severity in EAEa
| Treatment | Incidence of EAE (%) |
Mean Day of Onsetb |
Mean cumulative Clinical Scorec |
Mean Maximum Severityd |
Mortality |
|---|---|---|---|---|---|
| EAE | 4/4 (100%) | 14.25 ± 1.26 | 37.63 ± 17.89 | 1.88 ± 0.75 | 0/4 (0%) |
|
EAE + GA
suppression |
1/4 (25%)* | 17.00 | 7.00 ± 14.00* | 2.00 | 0/4 (0%) |
|
EAE + GA
treatment |
4/4 (100%) | 14.25 ± 0.82 | 13.50 ± 13.80* | 1.75 ± 0.96 | 0/4 (0%) |
C57BL/6 mice were immunized with MOG35-55(200 μg)/CFA containing M. Tb (1mg/ml)/PT(400ng). Suppression: GA (1.2mg/day) i.p started together with disease induction (8 daily injections). Treatment: GA (1.2mg/day) i.p started after the appearance of clinical symptoms (8 daily injections). Mice (n=4 each group) were scored daily for 35 days, and the mean score of each group ±SD is shown.
Mean of the first sign of clinical scores for each animal.
Mean of the sum of scores for each animal over the entire observation period (35 days).
Mean of the highest clinical score exhibited by individual animals within a group during the entire course of EAE.
p< 0.05 by Anova.
A parametric statistical test (Test for Association/Correlation using cor.test {stats} software) was performed between gene expression and disease severity to analyze the correlation and p value in trkB+/+ and trkB+/− mice (Figure 2).
Figure 2. Disease severity correlates with BDNF and NRG1 gene expression in trkB +/+ and +/− mice with less potent immunization conditions.

Brain tissue was collected from individual mice 48 days after the induction of EAE (Figures 1 and Table 1). RNA isolation and qPCR were performed as described in the Methods. Each symbol represents value of gene expression and the cumulative clinical score for an individual mouse. Gene expression = RNA quantity (2−ΔCt) (ΔCt = Ct for targeted gene – Ct for GAPDH). “R” value represents the linear correlation between the gene expression values and the disease severity for expression of each gene in trkB +/+ and +/− mice under less potent (A) and more potent (B) immunization protocols. The samples were considered to show better correlation if the “R” was closer to 1. *: p<0.05 by Test for Association/Correlation using cor.test {stats} software.
Results
Reduced trkB signaling can alter disease severity
Recently, we provided in vivo evidence that endogenous BDNF signaling through axonal trkB receptors promotes a stage-dependent release of type I NRG1 from axons to Schwann cells (Ma et al., 2011). We wanted to know whether BDNF/trkB signaling also plays a role in a pathological condition such as EAE, since previous studies from others reported trkB receptor expression on T cells as well as astrocytes (Colombo et al., 2012; De Santi et al., 2009). We compared EAE induction in heterozygous mice with reduced trkB signaling (trkB+/−) (Klein et al., 1993) to wild type littermates (trkB+/+). We initially used immunization conditions known to produce ‘regular’ EAE in C57BL/6 mice (Figures 3 and 4), but in these particular experiments, wild type littermates developed only mild disease (Figure 1A). Under these conditions, which we have called less potent disease induction, trkB+/− mice had significantly more severe disease than their wild type littermate controls (trkB+/+) (Figures 1A-1B). Because of these unexpected results in the wild type animals we tried to increase disease severity in the wild type littermates by increasing amounts of M. Tbc and pertussis toxin in a second experiment, which we have called potent disease induction (Figures 1C-1D). In this case, the wild type mice had 100% disease incidence and more severe disease that was indistinguishable from the trkB+/− heterozygous mice. These results, summarized in Table 1, suggest that trkB signaling can play a neuroprotective role in EAE, but only under the less potent induction conditions.
Figure 3. BDNF expression increases at EAE onset, while soluble type I NRG1 increases in chronic disease.

C57BL/6 mice were immunized with MOG35-55(200μg)/CFA containing M. Tbc (1mg/ml)/PT(200X2 ng), and were scored daily for disease. The mean score of EAE is shown (A). On the indicated day after immunization (onset day 12-15 or chronic phase day 35), brain tissues were collected from individual mice. RNA isolation and qPCR were performed from individual brain samples as described in the Methods. Each bar represents an individual mouse with indicated clinical score and the fold change in mRNA expression of types I (B) and III NRG1 (C), BDNF (D) and GDNF (E) at disease onset or chronic stage versus the average of the gene expression in naïve mice (n=4). Compared to naive mice (n=4), BDNF increased at disease onset and type I NRG1 expression increased in chronic stages (day 35 post-immunization). The samples were considered to show little change if the fold changes were less than 1.5 or were variable within the same group.
Figure 4. GA treatment reduces disease severity and blunts both BDNF and NRG1 induction.

C57BL/6 mice were immunized with MOG35-55(200 μg)/CFA containing M. Tbc (1mg/ml)/PT(200X2 ng), and were scored daily for EAE with GA treated and untreated groups. GA suppression: GA i.p started together with disease induction (8 daily injections). GA treatment: GA i.p started after the appearance of clinical symptoms (8 daily injections). RNA isolation and qPCR were performed at 35 days after immunization from individual brain samples as described in the Methods. Each bar represents an individual mouse with indicated clinical score and the fold change in mRNA expression of types I (B) and III NRG1 (C), BDNF (D) or GDNF (E) from GA suppression and GA treatment group versus the average of gene expression in untreated group (n=4 mice each group). Compared to untreated mice with EAE, type I NRG1 and BDNF expression was lower in mice treated with GA at disease onset.
Correlation of disease severity with gliotrophic and neurotrophic factor expression is reversed in trkB+/− mice compared to trkB+/+ mice
Given the clear difference in disease severity seen in the trkB heterozygotes with the less potent and immunization conditions (Figures 1A-1B, Table 1), we asked whether there are consistent changes in neurotrophic and gliotrophic factors in EAE, including BDNF that activates trkB. We first examined the expression of type I and III NRG1 together with the neurotrophic factors BDNF and GDNF as functions of disease severity in the brains of individual trkB+/+ and trkB +/− mice at 48 days after EAE induction under less potent and more potent immunization protocols (Figure 2A). Note that two of the trkB +/+ mice did not get disease (Figure 1A), and thus have a cumulative score of 0. While the correlations were not statistically significant, in the trkB+/+ mice, there was a trend toward down-regulation of type I and type III NRG1 (R=0.59 and 0.98) and BDNF (R=0.79), while GDNF was up-regulated (R=0.73) with increasing cumulative disease severity. Conversely, in the trkB +/− mice, there was a trend toward up-regulation of type I and type III NRG1 (R= 0.82 and 0.91) and BDNF (R=0.53), while GDNF was down-regulated (R=0.89). These results indicate that trkB+/− mice differ from wild type trkB+/+ mice in regulation of trophic factor gene expression relative to disease severity at the chronic disease stage. In summary, Figure 2A suggests that disease severity correlates with both NRG1 and BDNF gene expression On the other hand, in Figure 2B, under more severe EAE conditions, there was no clear correlation between NRG1 and BDNF expression with disease severity (Figure 1C-1D). Interestingly, the patterns of GDNF gene expression in EAE in trkB+/+ or +/− mice were not changed between less and more potent immunization protocols. We therefore used the less potent immunization protocol for the rest of this study.
We next looked more closely at the expression of these same four trophic factors at both early and chronic EAE stages in C57BL/6 mice. As shown in Figures 3A, ‘regular’ EAE was induced in C57BL/6 mice under the less potent immunization conditions. Compared to naïve C57BL/6 mice, BDNF expression was increased in the brain during the early stage of disease at disease onset, whereas there was little change in the other three factors. On the other hand, at later stages type I NRG1 mRNA was consistently elevated during the chronic disease phase (35 dpi) (Figure 3B-3E). Together with the trkB+/+ and trkB +/− results in Figures 1 and 2, these data suggest that BDNF expression in the brain may play a neuroprotective role in the early stage of the disease while the subsequent increase in type I NRG1 during the chronic, late stage could play a role in neuroinflammation, glial activation, or myelin repair (Song et al., 2012).
GA treatment reduces disease severity and blunts both BDNF and NRG1 induction in late disease, but has no direct effects in the absence of EAE
A previous study, in which GA was effective in both suppression and treatment of EAE in C57BL/6 mice (Aharoni et al., 2008), showed GA treatment was associated with increased BDNF mRNA expression in EAE using in situ hybridization (Aharoni et al., 2005). Figure 4A and Table 2 show that GA ‘suppression’ (given for the first 8 days) reduced disease incidence, delayed onset, and decreased disease severity, and GA ‘treatment’ (given for 8 days at the time of disease onset) was also effective in reducing disease severity. Compared to EAE mice not receiving GA, mice receiving GA suppression had no consistent change in expression of NRG1, BDNF and GDNF at day 35 post immunization following GA suppression (Figures 4B-4E). These results may be due to marked suppression of EAE by GA in these mice. On the other hand, with GA treatment, there was a consistent decrease in both BDNF and type I NRG1 and also GDNF at 35 days after immunization (27 days after the last GA treatment) (Figures 4B-4E). Taken together, while both GA suppression and treatment inhibit the disease severity, the effects of GA on BDNF and NRG1 expression are greatest in animals that get regular disease and then are treated with GA, suggesting that GA can blunt gliotrophic and neurotrophic effects in late-stage disease.
Given the above result, we asked whether GA had and direct affects on gliotrophic and neurotrophic factor expression in the brain in the absence of EAE. GA, in fact, had no effect on BDNF, GDNF, type I and type III NRG1 expression after an 8 day treatment of C57BL/6 mice (Figure 5), suggesting that GA has no direct effect on the expression of these genes in the brain in the absence of EAE.
Figure 5. GA has no direct effect on brain BDNF and NRG1 expression in untreated mice.
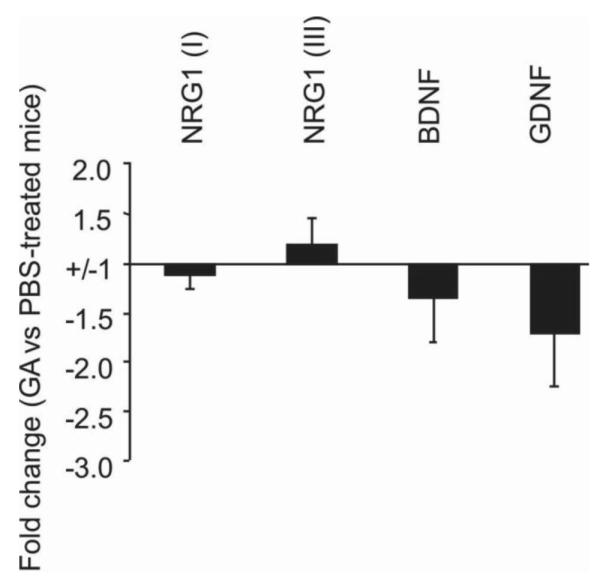
Naïve C57BL/6 mice were treated with or without GA for 8 days. Brain tissues were collected from each group above. RNA isolation and qPCR were performed from individual brain samples as described in the Methods. Each bar represents the mean of fold change ±SD of gene expression from GA treated versus untreated group (n=5 mice each group). Compared to untreated mice, there is no significant change in mRNA expression for types I and III NRG1, BDNF or GDNF in GA treated mice.
Discussion
TrkB regulates disease severity in EAE
To date, it is not clear whether the primary process in MS is neurodegeneration, infiltration of activated inflammatory immune cells into the CNS, or both. Targeted disruption of the trkB receptor gene results in nervous system lesions and neonatal death (Klein et al., 1993) in trkB homozygous mice. Our study using trkB heterozygous mice shows that reduced trkB expression increases disease incidence and severity in EAE using trkB heterozygous mice. This finding is consistent with others (Colombo et al., 2012) that support the hypothesis that trkB receptor activation with ligands such as BDNF could be a possible protective therapeutic strategy. On the other hand, Colombo et al (Colombo et al., 2012) recently reported that trkB is upregulated in human astrocytes in MS lesions, and the absence of trkB expression in mouse astrocytes protected from EAE-induced neurodegeneration in a conditional knock-out for TrkB using the cre/loxp system (Luikart et al., 2005). The increased disease severity we found in trkB+/− mice may arise from the altered regulation of trophic factor expression we observed, along with the possibility that increased BDNF interaction with p75 alone when levels of trkB are reduced may be harmful (Bredesen and Rabizadeh, 1997; Casaccia-Bonnefil et al., 1999). These different findings reinforce the importance of evaluating the trkB receptor as a disease modulator for MS patients. It will also be important to determine whether some of the differences seen could relate to different effects of trkB signaling at different times or in different cell types during the disease course.
As part of our own efforts to reconcile these differences, we repeated EAE in a second group of trkB +/− mice versus wild type littermates under more potent immunization conditions (Figures 1C-1D). Surprisingly, under these conditions we found no clear effects of trkB signaling. This suggests that the effects of trkB are less important with more potent disease induction. An alternative, but not mutually exclusive explanation, is that, in addition to the more potent EAE stimulus, the mice had been further back crossed into C57BL/6 for 7 generations compared to 5 (Figures 1A-1B), which may increase EAE incidence but may not affect disease severity.
Neurotrophic and gliotrophic factors undergo dynamic changes in the different stages of EAE
Based on the inconsistent effects of trkB on various EAE models from our studies and the literature, we explored the dynamics of neurotrophic and gliotrophic ligands and receptors during EAE. One of the reasons we included gliotrophic factors is because the gliotrophic factor NRG1 has been shown to be induced as part of a local BDNF feedback loop that is also important for axoglial communication (Esper and Loeb, 2004; Loeb et al., 2002; Ma et al., 2011) and neuromuscular development (Loeb et al., 2002), and has been implicated as an important activator of microglia in the dorsal horn of the spinal cord in chronic pain models (Calvo et al., 2010). For example, we recently demonstrated that endogenous BDNF signaling through axonal trkB receptors promotes a stage-dependent release of type I NRG1 from axons to Schwann cells critical for normal development (Ma et al., 2011). We have also found that NRG1 may play an important role in microglial activation in the spinal cord in amyotrophic lateral sclerosis (ALS) patients and an ALS-animal model (Song et al., 2012). Here we have found that while BDNF expression is increased at disease onset, type I NRG1 is induced during the chronic phase of the disease during EAE. We also found in the trkB heterozygous mice that both neurotrophin as well as NRG1 expression correlate with disease severity, suggesting that their induction is either a result or the cause of a given stage of the disease.
Therapeutic implications of neurotrophic and gliotrophic expression in EAE
Increased BDNF expression in the brain by GA treatment has been proposed as one possible mechanism for GA protecting mice from EAE (Aharoni et al., 2005), suggesting BDNF might have a neuroprotective role. Our results showing increased BDNF expression at early stages of disease together with increased disease severity in mice with reduced levels of trkB receptor suggest that the BDNF-trkB signaling pathway may play a neuroprotective role at early stages of EAE. In chronic EAE in C57BL/6 mice with normal levels of trkB receptor, we found decreased expression of BDNF in brain 2-3 weeks after GA treatment compared to mice with EAE not receiving GA treatment. Increased production of BDNF in GA-stimulated immune cells has been widely reported (De Santi et al., 2009). As noted above, one study has shown higher expression of BDNF message and protein in neurons and astroglia following GA treatment of EAE mice than in untreated EAE mice, as demonstrated by in situ hybridization and immunocytochemistry (Aharoni et al., 2005). Similar effects on BDNF expression following suppression or treatment of EAE with laquinimod were also reported recently (Aharoni et al., 2012). In our study, we measured total levels of BDNF message in brain tissue, thus averaging changes in all cell types, including immune cells migrating into the brain, which may have low levels of BDNF expression at the time point we investigated, 2-3 weeks after the last dose of GA, compared to 1 week in the other studies. Thus direct comparison of our results with those in the other two studies is not possible. Of note in our study, downregulation of NRG1 in the late stage of disease coincided with GA suppression of EAE severity, suggesting that NRG1, is critical for normal development, could actually promote disease pathogenesis in the chronic phase of EAE. Consistently with this possibility, we recently reported aberrant NRG1 signaling in human ALS spinal cord and in the ALS-SOD1 animal model (Song et al., 2012). Taken together, the results of our study raise the possibility that BDNF expression during disease onset may be protective while increased NRG1 expression during chronic disease could promote further injury, perhaps by microglial activation as we have found recently in chronic pain and in ALS (Calvo et al., 2010; Song et al., 2012).
On the other hand, type II NRG1 (glial growth factor-2) given systemically has been shown to delay EAE disease onset, decrease severity and reduce relapse rate (Cannella et al., 1998). This treatment was also associated with increased Th2 cytokine responses in the immune system and was associated with more remyelination in the CNS lesions of EAE (Cannella et al., 1998; Cannella et al., 1999; Marchionni et al., 1999). However, a follow-up study indicated that effects of systemically delivered type II NRG1 were less likely due to direct effects on remyelination, but to modulation of the immune response that favors remyelination (Penderis et al., 2003). Given that microglial activation is a common pathological change in both ALS and MS, blocking NRG1 signaling could thus be a therapeutic target to slow disease progression in neurodegenerative diseases including MS. We have developed a targeted neuregulin antagonist (Ma et al., 2009) that given intrathecally reduces microglial activation in rat chronic spinal cord pain model (Calvo et al., 2010). Use of this new reagent could help define at which points during the disease process NRG1 activity is beneficial or harmful. More detailed studies will therefore be needed to explore the specific roles of neurotrophic and gliotrophic factors at specific disease stages in order to develop effected targeted therapeutics in patients with MS.
Highlights.
TrkB signaling modulates EAE severity with induction of mild disease.
NRG1-BDNF expression correlates with EAE severity with immunization that induces mild disease.
No correlation between NRG1-BDNF and disease severity in more potent immunization.
A complex interplay between BDNF and NRG1 depends on disease stage and severity.
ACKNOWLEDGEMENTS
This study was supported by a grant from Teva Pharmaceutical Industries (Israel) to R.P.L., F.S, J.A.L., and J.B. (Israel) and by NIH R01 NS059947 (J.A.L. and F. S.). We thank Ms. Samantha Dettloff for her assistance with the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aharoni R, Eilam R, Domev H, Labunskay G, Sela M, Arnon R. The immunomodulator glatiramer acetate augments the expression of neurotrophic factors in brains of experimental autoimmune encephalomyelitis mice. Proc Natl Acad Sci U S A. 2005;102:19045–50. doi: 10.1073/pnas.0509438102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aharoni R, Herschkovitz A, Eilam R, Blumberg-Hazan M, Sela M, Bruck W, Arnon R. Demyelination arrest and remyelination induced by glatiramer acetate treatment of experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105:11358–63. doi: 10.1073/pnas.0804632105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aharoni R, Saada R, Eilam R, Hayardeny L, Sela M, Arnon R. Oral treatment with laquinimod augments regulatory T-cells and brain-derived neurotrophic factor expression and reduces injury in the CNS of mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 2012;251:14–24. doi: 10.1016/j.jneuroim.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–69. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredesen DE, Rabizadeh S. p75NTR and apoptosis: Trk-dependent and Trk-independent effects. Trends Neurosci. 1997;20:287–90. doi: 10.1016/s0166-2236(96)01049-1. [DOI] [PubMed] [Google Scholar]
- Buonanno A. The neuregulin signaling pathway and schizophrenia: from genes to synapses and neural circuits. Brain Res Bull. 2010;83:122–31. doi: 10.1016/j.brainresbull.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo M, Zhu N, Tsantoulas C, Ma Z, Grist J, Loeb JA, Bennett DL. Neuregulin-ErbB signaling promotes microglial proliferation and chemotaxis contributing to microgliosis and pain after peripheral nerve injury. J Neurosci. 2010;30:5437–50. doi: 10.1523/JNEUROSCI.5169-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannella B, Hoban CJ, Gao YL, Garcia-Arenas R, Lawson D, Marchionni M, Gwynne D, Raine CS. The neuregulin, glial growth factor 2, diminishes autoimmune demyelination and enhances remyelination in a chronic relapsing model for multiple sclerosis. Proc Natl Acad Sci U S A. 1998;95:10100–5. doi: 10.1073/pnas.95.17.10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannella B, Pitt D, Marchionni M, Raine CS. Neuregulin and erbB receptor expression in normal and diseased human white matter. J Neuroimmunol. 1999;100:233–42. doi: 10.1016/s0165-5728(99)00201-5. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Gu C, Chao MV. Neurotrophins in cell survival/death decisions. Adv Exp Med Biol. 1999;468:275–82. doi: 10.1007/978-1-4615-4685-6_22. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Colombo E, Cordiglieri C, Melli G, Newcombe J, Krumbholz M, Parada LF, Medico E, Hohlfeld R, Meinl E, Farina C. Stimulation of the neurotrophin receptor TrkB on astrocytes drives nitric oxide production and neurodegeneration. J Exp Med. 2012;209:521–35. doi: 10.1084/jem.20110698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santi L, Annunziata P, Sessa E, Bramanti P. Brain-derived neurotrophic factor and TrkB receptor in experimental autoimmune encephalomyelitis and multiple sclerosis. J Neurol Sci. 2009;287:17–26. doi: 10.1016/j.jns.2009.08.057. [DOI] [PubMed] [Google Scholar]
- De Santi L, Polimeni G, Cuzzocrea S, Esposito E, Sessa E, Annunziata P, Bramanti P. Neuroinflammation and neuroprotection: an update on (future) neurotrophin-related strategies in multiple sclerosis treatment. Curr Med Chem. 2011;18:1775–84. doi: 10.2174/092986711795496881. [DOI] [PubMed] [Google Scholar]
- Derfuss T, Linington C, Hohlfeld R, Meinl E. Axo-glial antigens as targets in multiple sclerosis: implications for axonal and grey matter injury. J Mol Med (Berl) 2010;88:753–61. doi: 10.1007/s00109-010-0632-3. [DOI] [PubMed] [Google Scholar]
- Esper RM, Loeb JA. Rapid axoglial signaling mediated by neuregulin and neurotrophic factors. J Neurosci. 2004;24:6218–27. doi: 10.1523/JNEUROSCI.1692-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esper RM, Pankonin MS, Loeb JA. Neuregulins: versatile growth and differentiation factors in nervous system development and human disease. Brain Res Rev. 2006;51:161–75. doi: 10.1016/j.brainresrev.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Falls DL. Neuregulins and the neuromuscular system: 10 years of answers and questions. J Neurocytol. 2003;32:619–47. doi: 10.1023/B:NEUR.0000020614.83883.be. [DOI] [PubMed] [Google Scholar]
- Hohlfeld R. Neurotrophic cross-talk between the nervous and immune systems: relevance for repair strategies in multiple sclerosis? J Neurol Sci. 2008;265:93–6. doi: 10.1016/j.jns.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. The Journal of cell biology. 2009;187:761–72. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieseier BC, Stuve O. A critical appraisal of treatment decisions in multiple sclerosis--old versus new. Nat Rev Neurol. 2011;7:255–62. doi: 10.1038/nrneurol.2011.41. [DOI] [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–22. [PubMed] [Google Scholar]
- Lalive PH, Neuhaus O, Benkhoucha M, Burger D, Hohlfeld R, Zamvil SS, Weber MS. Glatiramer acetate in the treatment of multiple sclerosis: emerging concepts regarding its mechanism of action. CNS Drugs. 2011;25:401–14. doi: 10.2165/11588120-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Geyer E, Flach AC, Jung K, Gold R, Flugel A, Linker RA, Luhder F. Central nervous system rather than immune cell-derived BDNF mediates axonal protective effects early in autoimmune demyelination. Acta Neuropathol. 2012;123:247–58. doi: 10.1007/s00401-011-0890-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linker R, Gold R, Luhder F. Function of neurotrophic factors beyond the nervous system: inflammation and autoimmune demyelination. Crit Rev Immunol. 2009;29:43–68. doi: 10.1615/critrevimmunol.v29.i1.20. [DOI] [PubMed] [Google Scholar]
- Lisak RP, Behan PO. Experimental autoimmune demyelinating diseases: experimental allergic encephalomyelitis and experimental allergic neuritis. Biomedicine. 1975;22:81–7. [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Loeb JA, Fischbach GD. Neurotrophic factors increase neuregulin expression in embryonic ventral spinal cord neurons. J Neurosci. 1997;17:1416–24. doi: 10.1523/JNEUROSCI.17-04-01416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JA, Khurana TS, Robbins JT, Yee AG, Fischbach GD. Expression patterns of transmembrane and released forms of neuregulin during spinal cord and neuromuscular synapse development. Development. 1999;126:781–91. doi: 10.1242/dev.126.4.781. [DOI] [PubMed] [Google Scholar]
- Loeb JA, Hmadcha A, Fischbach GD, Land SJ, Zakarian VL. Neuregulin expression at neuromuscular synapses is modulated by synaptic activity and neurotrophic factors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;Vol. 22:2206–14. doi: 10.1523/JNEUROSCI.22-06-02206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JA. Neuroprotection and repair by neurotrophic and gliotrophic factors in multiple sclerosis. Neurology. 2007;68:S38–42. doi: 10.1212/01.wnl.0000275231.97764.43. discussion S43-54. [DOI] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Virmani T, Lush ME, Liu Y, Kavalali ET, Parada LF. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci. 2005;25:3774–86. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Li Q, An H, Pankonin MS, Wang J, Loeb JA. Targeting human epidermal growth factor receptor signaling with the neuregulin’s heparin-binding domain. The Journal of biological chemistry. 2009;284:32108–15. doi: 10.1074/jbc.M109.032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Wang J, Song F, Loeb JA. Critical period of axoglial signaling between neuregulin-1 and brain-derived neurotrophic factor required for early Schwann cell survival and differentiation. J Neurosci. 2011;31:9630–40. doi: 10.1523/JNEUROSCI.1659-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchionni MA, Cannella B, Hoban C, Gao YL, Garcia-Arenas R, Lawson D, Happel E, Noel F, Tofilon P, Gwynne D, Raine CS. Neuregulin in neuron/glial interactions in the central nervous system. GGF diminishes autoimmune demyelination, promotes oligodendrocyte progenitor expansion, and enhances remyelination. Adv Exp Med Biol. 1999;468:283–95. [PubMed] [Google Scholar]
- Mei L, Xiong WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nature reviews. Neuroscience. 2008;9:437–52. doi: 10.1038/nrn2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, Yamaai T, Garratt A, Riethmacher-Sonnenberg E, Kane D, Theill LE, Birchmeier C. Isoform-specific expression and function of neuregulin. Development. 1997;124:3575–86. doi: 10.1242/dev.124.18.3575. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10:209–19. doi: 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- O’Neill EJ, Day MJ, Wraith DC. IL-10 is essential for disease protection following intranasal peptide administration in the C57BL/6 model of EAE. J Neuroimmunol. 2006;178:1–8. doi: 10.1016/j.jneuroim.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfuss TL, Rogers CJ, Gienapp I, Yurrita M, McClain M, Damico N, Valo J, Song F, Whitacre CC. Sex differences in experimental autoimmune encephalomyelitis in multiple murine strains. J Neuroimmunol. 2004;150:59–69. doi: 10.1016/j.jneuroim.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Penderis J, Woodruff RH, Lakatos A, Li WW, Dunning MD, Zhao C, Marchionni M, Franklin RJ. Increasing local levels of neuregulin (glial growth factor-2) by direct infusion into areas of demyelination does not alter remyelination in the rat CNS. Eur J Neurosci. 2003;18:2253–64. doi: 10.1046/j.1460-9568.2003.02969.x. [DOI] [PubMed] [Google Scholar]
- Racke MK, Lovett-Racke AE. Glatiramer acetate treatment of multiple sclerosis: an immunological perspective. J Immunol. 2011;186:1887–90. doi: 10.4049/jimmunol.1090138. [DOI] [PubMed] [Google Scholar]
- Ragheb S, Abramczyk S, Lisak D, Lisak R. Long-term therapy with glatiramer acetate in multiple sclerosis: effect on T-cells. Mult Scler. 2001;7:43–7. doi: 10.1177/135245850100700108. [DOI] [PubMed] [Google Scholar]
- Rakhade SN, Yao B, Ahmed S, Asano E, Beaumont TL, Shah AK, Draghici S, Krauss R, Chugani HT, Sood S, Loeb JA. A common pattern of persistent gene activation in human neocortical epileptic foci. Annals of neurology. 2005;58:736–47. doi: 10.1002/ana.20633. [DOI] [PubMed] [Google Scholar]
- Siffrin V, Vogt J, Radbruch H, Nitsch R, Zipp F. Multiple sclerosis - candidate mechanisms underlying CNS atrophy. Trends Neurosci. 2010;33:202–10. doi: 10.1016/j.tins.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Song F, Chiang P, Wang J, Ravits J, Loeb JA. Aberrant neuregulin 1 signaling in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2012;71:104–15. doi: 10.1097/NEN.0b013e3182423c43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadelmann C, Kerschensteiner M, Misgeld T, Bruck W, Hohlfeld R, Lassmann H. BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain. 2002;125:75–85. doi: 10.1093/brain/awf015. [DOI] [PubMed] [Google Scholar]
- Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. 2006;60:12–21. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]