

Abstract
The mechanisms that regulate hematopoietic stem cell (HSC) dormancy and self-renewal are well established and are largely dependent on signals emanating from the HSC niche. Recently, we found that prostate cancer (PCa) cells target the HSC niche in mouse bone marrow (BM) during metastasis. Little is known, however, as to how the HSC niche may regulate dormancy in cancer cells. In this study, we investigated the effects of TANK binding kinase 1 (TBK1) on PCa dormancy in the BM niche. We found that binding with niche osteoblasts induces the expression of TBK1 in PCa cells PC3 and C4-2B. Interestingly, TBK1 interacts with mammalian target of rapamycin (mTOR) and inhibits its function. Rapamycin, an mTOR inhibitor, induces cell cycle arrest of PCa cells and enhances chemotherapeutic resistance of PCa cells. As a result, the knockdown of TBK1 decreases PCa stem-like cells and drug resistance in vitro and in vivo. Taken together, these results strongly indicate that TBK1 plays an important role in the dormancy and drug resistance of PCa.
Introduction
TANK binding kinase 1 (TBK1) is a noncanonical IκB kinase (IKK) family member that mediates the innate immune response. Toll-like receptors (TLR3 and TLR4) and the cytosolic receptors (RIG-I and MDA-5) recognize bacterial lipopolysaccharide or viral dsRNA and induce the phosphorylation of TBK1 [1]. Activated TBK1 then phosphorylates interferon regulatory factors (IRF3 and IRF7), mediating IRF dimerization, nuclear translocation, and transcription of target genes, such as interferon beta (IFN-β) and RANTES [2–5]. In cancer, emerging evidence demonstrates that TBK1 plays an important role in angiogenesis, transformation, and cell survival [6,7]. TBK1 is induced under hypoxic conditions, predominantly increased in solid tumors compared with normal tissue, and promotes secretion of proangiogenic factors, such as RANTES and interleukin 8 (IL-8), resulting in proliferation of endothelial cells [8]. Activation of TBK1 by RalB-mediated Sec5/TBK1 complex assembly is required for oncogenic Ras-induced transformation and tumor cell survival [9]. Systematic RNA interference screening reveals that TBK1 is essential for survival of oncogenic KRAS-driven cancer cells [10]. In addition, recent studies have identified that TBK1 directly phosphorylates AKT to inhibit apoptosis and support oncogenic transformation [11,12].
The mammalian target of rapamycin (mTOR) is the catalytic subunit of two distinct signaling complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [13]. mTORC1, which consists of mTOR, raptor, deptor, mLST8, and PRAS40, activates ribosomal S6 kinase (S6K) and inactivates eukaryotic initiation factor 4E binding protein 1, stimulating protein synthesis, cell proliferation, and cell cycle progression [13–16]. In contrast, mTORC2, which contains mTOR, rictor, deptor, mLST8, and mSIN1, phosphorylates AKT and protein kinase C-α, promoting cell survival and cytoskeleton reorganization [17,18]. Rapamycin directly binds to mTORC1 and inhibits mTORC1 activity [14]. However, mTORC2 is insensitive to rapamycin, although prolonged treatment can inhibit mTORC2 in many cell types [19].
Recent studies have shown that both TBK1 and mTORC2 phosphorylate AKT at S473 [11,12,20] and that mTORC1 inhibition increases AKT phosphorylation at S473 [21]. These studies led us to investigate the relationship between TBK1 and mTOR. In this study, we found that the binding of prostate cancer (PCa) cells to osteoblasts in the hematopoietic stem cell (HSC) niche induces the expression of TBK1 in PCa cells. Interestingly, TBK1 inhibits mTOR signaling pathway. Furthermore, rapamycin, an mTOR inhibitor, induces cell cycle arrest of PCa cells and anticancer drug resistance in PCa cells. Finally, we demonstrate that TBK1 enhances the PCa stem-like cells and drug resistance in vitro and in vivo. Taken together, these results strongly indicate that TBK1 plays an important role in dormancy and drug resistance of PCa.
Materials and Methods
Cell Cultures
Human PCa line PC3 and human breast cancer cell line MDA-MB-231 were obtained from the American Type Culture Collection (Rockville, MD). The metastatic subline LNCaP was originally isolated from a lymph node of a patient with disseminated bony and lymph node involvement and the C4-2B subline established by passage in vivo. PC3Luc cells were constructed by stably transfecting PC3 cells with a luciferase or green fluorescent protein (GFP) construct. PCa cells or MDA-MB-231 cells were grown in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% FBS and 1% penicillin/streptomycin (P/S). The murine bone marrow (BM)-derived stromal cell line ST2 was grown in α-minimum essential medium (Invitrogen) with 10% heat-inactivated FBS and 1% P/S. 293T cells were grown in Dulbecco's modified Eagle's medium (Invitrogen) with 10% FBS and 1% P/S.
RNA Interference
PC3 or PC3Luc cells were transfected with pGIPZ lentiviral shRNA constructs (Vector Core, University of Michigan, Ann Arbor, MI) for scrambled (RHS4346) and human TBK1 (RHS4430-99299280), according to the manufacturer's instructions. Plasmids were co-transfected with psPAX2 and pMD2.G into HEK-293T cells using CaPO4. Supernatants were collected after 60 hours and used to infect cells. Infected cells were selected for 7 days in RPMI 1640 medium containing 1 µg/ml puromycin, and the expression of TBK1 was analyzed by Western blot analysis.
Site-Directed Mutagenesis
TBK1 plasmid was purchased from Addgene (Cambridge, MA). Kinase-dead (K38A) or TOR signaling (TOS) motif mutant (F638A) TBK1 plasmid was generated using the Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions.
In Vivo Isolation of Disseminated Human PCa Cells from Murine Bone Marrow
Intracardiac injections of PCa cells (1 x 106 cells) were performed into male CB.17.SCID mice (4-6 weeks of age; Charles River Laboratories, Wilmington, MA). After 24 hours, BM cells were flushed from the femurs, tibias, and humeri. Cells were incubated with a Lineage Cell Depletion Kit magnetic labeling system with biotinylated anti-Lineage [CD5, CD45R (B220), CD11b, Gr-1 (Ly-6G/C), and Ter-119] antibody cocktail and anti-Biotin MicroBeads (Miltenyi Biotec, Auburn, CA) and then enriched for murine Lineage negative population using an AutoMACS machine (Miltenyi Biotec). Enriched cells were incubated with fluorescein isothiocyanate-HLA-ABC antibody (BioLegend, San Diego, CA), PE-anti-CD133 antibody (Miltenyi Biotec), and APC-anti-CD44 antibody (BD Biosciences, San Diego, CA) for 20 minutes at 4°C. The CD133+/CD44+ and CD133-/CD44- fractions were sorted by gating on HLA-ABC-positive cells (human cells) with a FACSAria High-Speed Cell Sorter (BD Biosciences). All experimental procedures were approved by the University of Michigan Committee for the Use and Care of Animals.
FACS Analysis
For stem-like cell counting, cells were incubated with PE-anti-CD133 antibody (Miltenyi Biotec) and APC-anti-CD44 antibody (BD Biosciences) for 20 minutes at 4°C. For cell cycling analysis, cells were fixed, permeabilized with Perm/Wash Buffer (BD Biosciences), and incubated with polyclonal anti-Ki67 antibody (Abcam, Cambridge, MA) for 30 minutes at 4°C. Subsequently, the cells were incubated with PE-coupled secondary antibodies (Abcam). The CD133+/CD44+ or Ki67- fraction was analyzed with a FACSAria High-Speed Cell Sorter (BD Biosciences).
Quantitative Reverse Transcription-Polymerase Chain Reaction
Total RNA was extracted from cells using the RNeasy mini or micro kit (Qiagen, Valencia, CA) and converted into cDNA using a First-Strand Synthesis Kit (Invitrogen). For isolated CD133+/CD44+ PCa cells from the BM of mice, cDNA was boosted using a PreAmp Master Mix (Applied Biosystems, Foster City, CA). Quantitative polymerase chain reaction (qPCR) was performed on an ABI 7700 sequence detector (Applied Biosystems) using TaqMan Universal PCR Master Mix Kit (Applied Biosystems) according to the manufacturer's instructions. TaqMan MGB probes (Applied Biosystems) were given as follows: human TBK1 (Hs00179410_m1) and human IKKE (Hs01063858_m1). Human β-actin (Hs99999903_m1) was used as an internal control for the normalization of target gene expression.
Western Blot Analysis
Whole-cell lysates were prepared from cells, separated on 10% sodium dodecyl sulfate-polyacrylamide gel, and transferred to polyvinylidene difluoride (PVDF) membrane. The membranes were incubated with 5% milk for 1 hour and incubated with primary antibodies overnight at 4°C. Primary antibodies used were given as follows: polyclonal anti-mTOR (1:1000; Cell Signaling Technology, Danvers, MA), polyclonal anti-raptor (1:1000; Cell Signaling Technology), polyclonal anti-TBK1 (1:1000; Cell Signaling Technology), polyclonal Neoplasia Vol. 15, No. 9, 2013 TBK1 Regulates Prostate Cancer Dormancy Kim et al. 1065 anti-phospho-p70S6K (1:500; Cell Signaling Technology), polyclonal anti-p70S6K (1:1000; Cell Signaling Technology), and monoclonal anti-FLAG M2 (1:2000; Sigma-Aldrich, St Louis, MO). Blots were incubated with peroxidase-coupled secondary antibodies (Promega, Madison, WI) for 1 hour, and protein expression was detected with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL). Membranes were reprobed with polyclonal anti-β-actin antibody (1:1000; Cell Signaling Technology) to control for equal loading.
Immunofluorescence Staining
PCa cells and PCa tissue microarrays (TMA; PR954; US Biomax, Rockville, MD) were used for immunostaining. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 (Sigma-Aldrich) in PBS. TMA sections were unmasked with pepsin solution (Lab Vision, Fremont, CA) at 37°C for 15 minutes and balanced with phosphate-buffered saline (PBS) plus 0.2% Triton X-100 for 5 minutes. Cells and TMA sections were blocked with Image-iT FX signal enhancer for 30 minutes and incubated for 2 hours at room temperature with primary antibodies combined with reagents of Zenon Alexa Fluor 488 (green) or 555 (red) labeling kit (Invitrogen). Polyclonal anti-TBK1 (Cell Signaling Technology), polyclonal anti-mTOR (Cell Signaling Technology), and monoclonal anti-phospho-p70S6K (Millipore, Temecula, CA) were used as primary antibodies. After washing with PBS, cells and TMA sections were mounted with ProLong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). Images were taken with Olympus FV-500 confocal microscope.
Cell Survival Assay
PCa cells (1 x 105) were seeded onto 12-well culture plates and then treated with anticancer drugs and/or rapamycin at 37°C for the indicated time. The final concentration of anticancer drugs used was given as follows: 10 µg/ml cisplatin, 500 nM doxorubicin, 100 nM taxol, 100 µg/ml 5-fluorouracil, and 100 ng/ml taxotere. Viability of the cells was determined by trypan blue assay. Apoptosis was measured by flow cytometry using PE Annexin V Apoptosis Detection Kit I (BD Biosciences) according to the manufacturer's instructions. Assays were performed in triplicate, and the results are representative of three independent experiments.
Co-Immunoprecipitation Assays
Cells were lysed in 0.3% CHAPS buffer (40 mM Hepes, 120 mM NaCl, 0.3% CHAPS, 1 mM PMSF, 1 µg/ml leupeptin, 2 µg/ml aprotinin, and 1 µg/ml pepstatin) on ice for 30 minutes. Lysates containing 1 mg of total protein were precleared with 30 µl of Protein-A/G Plus-Agarose (Santa Cruz Biotechnology, Santa Cruz, CA). Cleared lysates were incubated with 2 µg of monoclonal anti-FLAG M2 antibody (Sigma-Aldrich) or 2 µg of polyclonal anti-TBK1 antibody (Cell Signaling Technology) or 2 µg of normal IgG antibody (Santa Cruz Biotechnology) for 3 hours at 4°C and then incubated with 30 µl of Protein-A/G Plus-Agarose for 1 hour at 4°C. Immunoprecipitates were washed four times in lysis buffer and analyzed by Western blot analysis.
In Vivo Dormancy Assays
All experimental procedures were approved by the University of Michigan Committee for the Use and Care of Animals. Lumbar vertebrae were isolated from 4- to 7-day-old mice. The vertebrae were sectioned into single vertebral bodies (vossicles). Severe combined-immunodeficiency (SCID) mice were used as transplant recipients. Two vossicles per mouse were implanted into subcutaneous pouches. Before implantations, PC3Luc cells containing scrambled or TBK1 shRNA plasmids were introduced into the vossicles (10,000 cells/10 µl of PBS). Mice were treated with taxotere (12 mg/kg, 1x/week) or vehicle for 3 weeks and imaged for 9 weeks by bioluminescent imaging (BLI). BLI was performed through the University of Michigan Small Animal Imaging Resource facility. Briefly, mice were injected with luciferin (40 mg/ml) by intraperitoneal injections, and ventral images were acquired 10 minutes after injection under 1.75% isoflurane/air anesthesia. Total tumor burden of each animal was calculated using regions of interest that encompassed the entire animal. After 9 weeks, tumor volumes were calculated using the following formula: V = the shortest diameter x the longest diameter x height.
In Vivo Tumor Growth and Angiogenesis Assays
PC3 cells (5 x 105) were implanted in the flanks of SCID mice. Mice were treated with rapamycin (1 mg/kg, 2x/week) or vehicle for 3 weeks. After 4 weeks, the tumor volumes were calculated and tumors were fixed. Histologic sections were incubated in antigen retrieval solution for 30 minutes at 90°C, followed by incubation with polyclonal anti-human factor VIII antibody (Lab Vision) overnight at 4°C. The number of microvessels in six random fields were counted under light microscope at x200 magnification.
Statistical Analyses
Results are presented as means ± SD. Significance of the difference between two measurements was determined by unpaired Student's t test, and multiple comparisons were evaluated by the Newman-Keuls multiple comparison test. Values of P < .05 were considered significant.
Results
Expression of TBK1 and IKKE in PCa Cells is Increased after Binding to Osteoblasts and by Anticancer Drug Treatment
Recently, we reported that PCa cells target the HSC niche in mouse BM during metastasis [22]. Because binding to the BM niche induces dormancy and long-term survival of HSCs [23], we hypothesized that targeting the BM niche also induces the dormancy of PCa cells. To determine if binding with niche cells promotes PCa dormancy, PC3 cells were co-cultured with murine BM-derived stromal cell line ST2 cells and then the cell cycle of PCa cells was analyzed by flow cytometry using Ki67 antibody. As expected, PCa cell binding to ST2 cells increased the frequency of the dormant population (Ki67 negative; Figure 1A). In addition, PCa cell binding to ST2 cells significantly prevented cell death induced by the administration of taxotere (Figure 1B).
Figure 1.
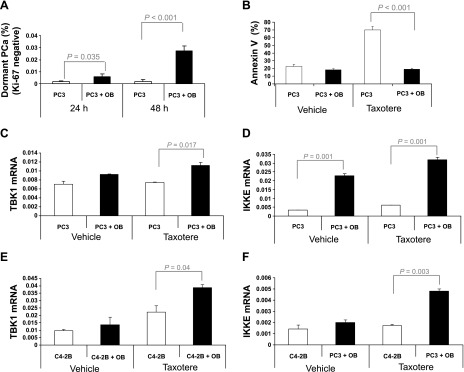
TBK1 and IKKE are increased by osteoblast binding. (A) PC3-GFP cells were cultured for 24 and 48 hours on tissue culture plastic or BM stromal cell ST2 (OB). The dormant population was examined by flow cytometry using Ki67 antibody. (B) PC3-GFP cells were cultured for 72 hours with/without taxotere on tissue culture plastic or ST2 cells. Cell death was measured using flow cytometry with Annexin V staining. (C and D) PC3 cells or (E and F) C42B cells were cultured for 48 hours with/without taxotere on tissue culture plastic or ST2 cells. Expression of TBK1 and IKKE was quantified by qPCR. The results were normalized to β-actin. The results represent mean ± SD values from triplicate assays, and the experiments were performed twice.
Accumulating evidence demonstrates that TBK1 and IKKE play important roles in tumorigenesis and survival [6,9,10,24,25] and are expressed by tumor cells under stressful growth conditions [8] including those that predominate in the HSC niche [26–28]. To evaluate whether the binding of PCa cells to niche osteoblasts induces the expression of TBK1 and IKKE in PCa cells, PCa cells were co-cultured with ST2 cells and then analyzed by qPCR. As shown in Figure 1, C to F, the expression of TBK1 and IKKE in PCa cells was increased following binding to ST2 cells. Interestingly, binding of PCa cells to ST2 cells significantly induced the expression of TBK1 and IKKE when the PCa cells were treated with taxotere (Figure 1, C–F).
TBK1 Inhibits mTOR Signaling
At present, little is known about the function of TBK1 and IKKE in PCa cells. Therefore, we first examined the extent to which TBK1 and IKKE are expressed in PCa cells. Western blot analysis showed that TBK1 is well expressed in PCa cells (PC3 and C42B) compared to breast cancer cells (MDA-MB-231) and that IKKE is poorly expressed in PCa cells (Figure 2A). Therefore, we focused our further studies on TBK1 to understand how PCas become dormant in the BM. As shown in Figure 2, B and C, TBK1 expression was also increased in a tumor stage-dependent manner in human prostate tumor tissues.
Figure 2.
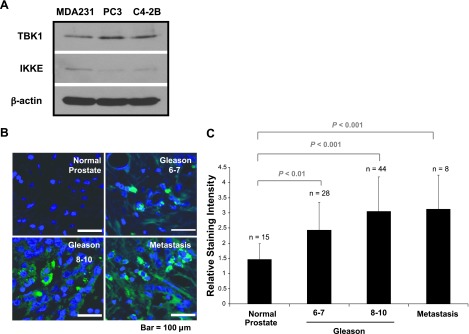
TBK1 is highly expressed in PCa cells. (A) The expression of TBK1 and IKKE was analyzed by Western blot analysis in human PCa cell lines PC3 and C42B and human breast cancer cell line MDA-MB-231 (MDA-231). (B) PCa TMA sections were analyzed by immunofluorescence staining with TBK1 antibody. Representative micrographs of normal prostate and PCa (Gleason score) and bone metastases at x60. (C) Staining intensity was scored blindly as absent (1), weak (2), moderate (3), strong (4), and very strong (5).
To explore the function of TBK1 in PCa cells, TBK1 was stably knocked down using lentiviral shRNA in PC3 cells. Surprisingly, TBK1 knockdown induced the phosphorylation of p70S6K, indicative of mTOR activation (Figure 3A). Moreover, we found that TBK1 is co-localized with mTOR (Figure 3B). To further confirm the interaction, a co-immunoprecipitation assay was performed in TBK1 overexpressing 293T cells (293T/TBK1) and PC3 cells. Indeed, mTOR and raptor interacted with overexpressed TBK1 in 293T cells (Figure 3C) or endogenous TBK1 in PC3 cells (Figure 3D). In C42B cells, overexpression of wild-type TBK1 blocked the phosphorylation of p70S6K, whereas overexpression of kinase-dead (K38A) or TOR signaling (TOS) motif mutant (F638A) TBK1 could not inhibit the phosphorylation (Figure 3E). More importantly, the phosphorylation of p70S6K in human clinical tissues was decreased in a tumor stage-dependent manner (Figure W1, A and B), showing an inverse correlation with TBK1 (Figure 2, B and C). These results suggest that TBK1 expression may be important for regulation of mTOR signaling in PCa cells.
Figure 3.
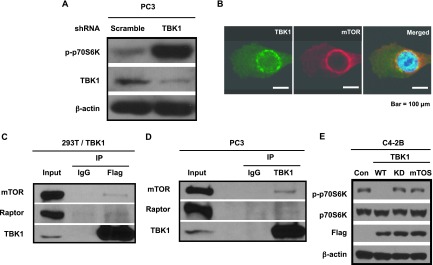
TBK1 inhibits mTOR signaling. (A) PC3 cells were infected with lentivirus containing scrambled or TBK1 shRNA plasmid, selected with puromycin, and analyzed by Western blot analysis using antibodies against p-p70S6K, TBK1, or β-actin. (B) PC3 cells were co-immunostained with TBK1 (green) and mTOR (red) antibodies. Merged image (yellow) shows co-localization of TBK1 and mTOR. (C) 293T cells were transfected with a plasmid encoding Flag-TBK1. Cell lysates were immunoprecipitated with Flag antibodies or normal mouse IgG as negative control and analyzed by Western blot analysis using antibodies against mTOR, raptor, and TBK1. (D) PC3 cell lysates were immunoprecipitated with TBK1 antibodies or normal rabbit IgG as negative control and analyzed by Western blot analysis using antibodies against mTOR, raptor, and TBK1. (E) C4-2B cells were transfected with control empty vector (Con) or a plasmid encoding wild-type (WT) or Kinase-dead (KD) or TOS motif mutant TBK1 (mTOS). Cell lysates were analyzed by Western blot analysis using antibodies against p-p70S6K, p70S6K, Flag, and β-actin.
mTOR Inhibition Induces Dormancy and Drug Resistance in PCa Cells
To investigate whether mTOR signaling is involved in dormancy, we used rapamycin, a specific inhibitor of mTOR. Although rapamycin clearly reduced tumor growth and angiogenesis in PC3 cells implanted in mice (Figure W2, A–C), mTOR inhibition by rapamycin significantly increased the dormant population (Ki67 negative) in a time-dependent manner in both PC3 cells (Figure 4A) and C42B cells (Figure 4B). To determine if the dormant PCa cells are resistant to chemotherapy, PCa cells were pretreated with rapamycin for 3 days and then treated with several anticancer drugs. Indeed, PCa cells pretreated with rapamycin led to the reduction of cell death (Figure 4, C and E) and apoptosis (Figure 4, D and F).
Figure 4.
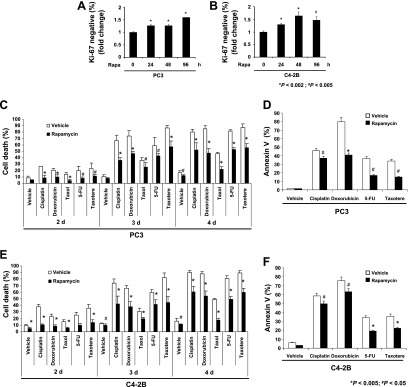
mTOR inhibition induces dormancy and drug resistance in PCa cells. (A) PC3 or (B) C42B cells were treated with 10 nM rapamycin for 0, 24, 48, and 96 hours. The dormant population was examined by flow cytometry using Ki67 antibody. (C) PC3 cells or (E) C42B cells were treated with the indicated anticancer drugs for 2, 3 and 4 days in the presence and absence of rapamycin. Cell death was measured by trypan blue assay. (D) PC3 cells or (F) C42B cells were treated with the indicated anticancer drugs for 72 hours in the presence and absence of rapamycin. Cell death was measured by flow cytometry with Annexin V staining. All results represent average mean ± SD values from triplicate assays, and the experiments were performed twice.
TBK1 Is Important for PCa Stem-Like Cells, Drug Resistance, and Dormancy
We further explored if TBK1 plays an important role in PCa stem-like cells because dormant population may contribute to the stem cell-like properties. PCa stem-like cells were sorted as CD133+/CD44+ cells. Isolated CD133+/CD44+ cells and CD133-/CD44- cells were co-cultured on ST2 cells and then analyzed for the expression of TBK1 by qPCR. As shown in Figure 5, A and B, the expression of TBK1 was increased by binding to ST2 cells in both CD133+/CD44+ cells and CD133-/CD44- cells. However, when CD133+/CD44+ cells and CD133-/CD44- cells were isolated from mouse BM niche (Figure 5C), the expression level of TBK1 in CD133+/CD44+ cells was significantly higher than that in CD133-/CD44- cells (Figure 5, D and E).
Figure 5.
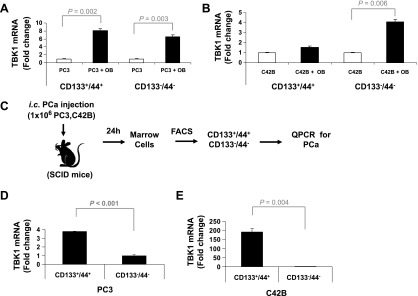
TBK1 is highly expressed in PCa stem-like cells on mouse BM niche. (A) PC3 or (B) C42B cells were sorted with APC-anti-CD44 and PE-anti-CD133 antibodies. The CD133+/CD44+ and CD133-/CD44- cells were cultured for 48 hours on tissue culture plastic or ST2 (OB) cells. Expression of TBK1 was quantified by qPCR. The results were normalized to β-actin. (C) Experimental model of isolation of PCa stem-like cells in vivo. The CD133+/CD44+ and CD133-/CD44- cells were sorted from BM cells of (D) PC3- or (E) C4-2B-injected SCID mice. Expression of TBK1 was quantified by qPCR. The results were normalized to β-actin. All results represent average values from triplicate assays, and the experiments were performed twice.
Next, to investigate the function of TBK1 in PCa stem-like cells, PC3 cells containing TBK1 or scrambled shRNA (Figure 3A) were cultured for 3 days in the presence and absence of rapamycin and analyzed by flow cytometry. Interestingly, TBK1 knockdown decreased the number of CD133+/CD44+ cells in PCa cells (Figure 6, A and B) and increased CD133-/CD44- cell populations (Figure 6, C and D). In contrast, mTOR inhibition by rapamycin increased the population of CD133+/CD44+ cells in PCa-scrambled shRNA cells and also blocked the reduction of stem-like cells in PC3-TBK1 shRNA cells. To examine the effect of TBK1 on drug resistance, these cells were treated for 3 days with taxotere in the presence and absence of rapamycin and analyzed by a cell death assay. As expected, TBK1 knockdown significantly increased cell death by taxotere (Figure 6E). These data suggest that TBK1 is important for PCa stem-like cells and drug resistance.
Figure 6.

TBK1 is important for PCa stem-like cells and drug resistance. PC3 cells were infected with lentivirus containing scrambled or TBK1 shRNA plasmid, selected with puromycin, and then cultured for 3 days in the presence and absence of rapamycin. The CD133+/CD44+ (A and B) and CD133-/CD44- (C and D) cells were analyzed by flow cytometry. (E) PC3 cells containing TBK1 or scramble shRNA were treated for 72 hours with taxotere in the presence and absence of rapamycin. Cell death was measured by the trypan blue assay. All results represent mean ± SD values from triplicate assays, and the experiments were performed twice.
Finally, we examined whether TBK1 is also critical for the dormancy of PCa in vivo. PC3Luc cells containing TBK1 or scrambled shRNA were injected into vertebral bodies (vossicles) derived from mice and then transplanted into subcutaneous pouches of SCID mice. Mice bearing tumors were injected intraperitoneally with taxotere for 3 weeks. Tumor regrowth was induced by stopping treatment. BLI was performed every week to evaluate luciferase activity in the implanted vossicles. After 9 weeks, tumor volume was measured. Taxotere treatment inhibited tumor growth for 4 weeks (Figure 7A). As shown in Figure 7, A to D, TBK1 knockdown inhibited tumor recurrence after 4 weeks, while scramble shRNA failed to prevent PCa from the regrowth. These results suggest that TBK1 is important for the establishment of dormancy by PCa and drug resistance in vivo.
Figure 7.
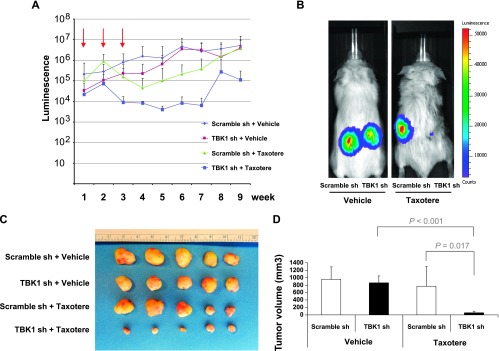
TBK1 is important for tumor dormancy in vivo. (A) PC3Luc cells were infected with lentivirus containing scrambled or TBK1 shRNA plasmid. After selection, cells were injected into mouse vossicles and transplanted into SCID mice. Mice were treated with taxotere or vehicle for 3 weeks. (A) BLI of mice was performed for 9 weeks (n = 9/group). Arrows show taxotere treatment. Representative mice were shown in B. (C and D) After 9 weeks, tumor volume was measured and quantified.
Discussion
In this study, we investigated the mechanisms that regulate dormancy of PCa in the BM. We found that PCa binding to components of the HSC niche increases the dormant population of PCa and their ability to withstand multiple chemotherapeutic insults. As part of the process, it was observed that the expression of TBK1 in PCa cells was increased following binding and in response to the chemotherapeutic agent taxotere.
The disseminated tumor cells (DTCs) from PCa that lodge or home to the marrow appear to be an early feature of “localized” disease. From an evolutionary standpoint, DTCs must have the capacity to not only escape from the primary tumor but also survive in the circulation and engage the marrow microenvironment in such a fashion to establish a foothold in the harsh marrow microenvironment. On the basis of the clinical course of metastatic PCa, two apparent phases of growth were seen in patients who ultimately present with end-stage disease: a latent or dormant phase that may be followed by a proliferative phase. Yet how DTCs switch between these two phases is not clear. One hypothesis is that DTCs are normally metabolically active but may have exited the cell cycle in response to space or resource competition. Proliferative arrest has also been observed in cells in which p38 activity exceeds extracellular signal-regulated kinase (ERK)-kinase expression [29]. An alternative explanation for dormancy is that proliferation and cell death rates are stably coupled. Yet it also remains unclear what regulates the uncoupling of growth from apoptosis. In the context of dormancy in the marrow regulated by the HSC niche, both hypotheses can be accommodated. In the first case, changes in systemic or local host-derived signals or even DTC-secreted paracrine signals that regulate niche activity may tip the balance in favor of proliferative growth. In the second instance, DTC migration or movement away from the niche signals may trigger a slow or even sustained growth by uncoupling apoptosis from proliferation. The role of TBK1 in each of these models of dormancy is possible; however, our data suggest that TBK1 plays a role in DTCs that are likely to have exited the cell cycle induced by niche engagement, although further studies are needed.
Tumor dormancy is defined by the persistence of minimal residual tumor cells over an extended period of time. Experimental and limited clinical data suggest that dormant tumor cells exist in a non-proliferative state having exited the cell cycle. As many conventional anticancer drugs target fast growing cancer cells, dormant cancer cells are thought to be resistant to multiple drugs that ultimately can lead to disease recurrence [30,31]. Like HSCs, the inactivation of oncogenes including MYC induces tumor dormancy even if this results in terminal differentiation, senescence, and apoptosis [32–34]. Moreover, stress signaling resulting in p38 induction regulates cell cycle arrest and dormancy of cancer cells, whereas mitogenic signaling pathway by ERK promotes proliferation and tumorigenicity [29,35]. Yet despite these insights, when compared with HSC quiescence, little is known regarding the molecular mechanism that regulates cancer cell dormancy.
The regulation of dormancy or quiescence is essential for the long-term survival of HSCs. Emerging evidence demonstrates that key cell cycle regulators including p21Cip1, Bmi1, and c-Myc play important roles in HSC maintenance by self-renewal and quiescence [23,36,37]. The absence of cell cycle inhibitors (tumor suppressors), or the increase of cell cycle activators (oncogenes), results in HSC hyperproliferation, terminal differentiation, and ultimately exhaustion. Interestingly, recent mouse studies have shown that deletion of negative regulators of mTOR signaling including TSC1 [38,39], PTEN [40,41], Fbxw7 [42], PML [43], and Lkb1 [44–46] also results in defective HSC quiescence, defects in long-term repopulating abilities, and HSC depletion. Furthermore, Castilho et al. [47] reported that the activation of mTOR by Wnt1 mediates both hair follicle hyperproliferation and epidermal stem cell exhaustion, suggesting that mTOR signaling also plays an important role in self-renewal and quiescence of other stem cell systems.
TBK1 participates in the signaling pathway of TLRs. TLRs are expressed in hematopoietic lineage cells and play a critical role in the activation of innate immunity by recognizing pathogens. TLR signaling also promotes adaptive immune responses by activating immune cells through the secretion of proinflammatory cytokines. Recent studies demonstrate that TLRs are expressed not only in immune cells but also in a variety of cancer cells and that TLR expression induces nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling and stress kinase signaling (p38 and JNK) and contributes to tumorigenesis by cell survival, chemoresistance, immune evasion, and metastasis [48]. Interestingly, González-Reyes et al. [49] found that prostate tumors with high TLR3 or TLR9 expression levels were highly associated with a higher probability of biochemical recurrence, even though Paone et al. [50] showed TLR3-mediated apoptosis and proliferation inhibition in human PCa.
To investigate how TBK1 supports or activates PCa dormancy, TBK1 expression in PCa was downregulated by shRNA. We found that TBK knockdown induces the activation of mTOR signaling and decreased PCa stem-like cells and chemosensitivity in PC3 cells. Moreover, the mTOR inhibitor rapamycin limits the effects of TBK knockdown, suggesting that the inhibition of mTOR signaling by TBK1 is helpful for the PCa stem-like cells and the chemoresistance. Although oncogenic activation of mTOR signaling contributes to tumor growth, survival, and proliferation [51], there is emerging evidence that the inhibition of mTOR signaling is important for tumor dormancy, chemoresistance, and maintenance of cancer stem-like cells. The tumor suppressor ARHI induces autophagy by inhibiting mTOR, and this autophagy contributes to the survival of dormant ovarian cancer cells [52]. In TSC1- and TSC2-deficient mouse embryonic fibroblasts or TSC2 knockdown cancer cell lines, upregulated mTOR activity increases cell death by anticancer drugs or tumor necrosis factor alpha (TNF-α) through the inhibition of NF-κB signaling and the rapamycin-mediated inhibition of mTOR signaling restored NF-κB activation and cell survival [53]. Moreover, rapamycin upregulates the cancer stem cell marker CD133 in gastric, colorectal, and lung cancer cell lines [54]. Yang et al. [55] also showed that rapamycin enhanced the level of CD133 and stem cell-associated genes in human liver tumor cell lines, whereas mTOR activation by Rheb overexpression decreases CD133. Hoshii et al. [56] recently reported that in a mouse model of acute myeloid leukemia, raptor deficiency suppresses mTORC1 and leukemia progression. However, acute myeloid leukemia cells lacking mTORC1 activity still can self renew as stem cells survive for a long time, indicating that the inhibition of mTOR signaling may induce tumor dormancy.
Each year, nearly 30,000 men who had undergone surgery or radiation for their PCa die from incurable metastatic disease that will present as lesions in the bone years after curative treatment. Logically, most, if not all, of the metastases that develop after a protracted period must have disseminated to bone before surgery or radiation. How circulating tumor cells traffic to the BM and how PCa cells become dormant and ultimately overcome dormancy to proliferate remain critical questions. Our work tested the hypothesis that if DTCs are engaged in the HSC niche, then the signals generated by engaging the niche should facilitate the development of dormant disease. We found that TBK1 interacts with mTOR and inhibits its function. mTOR inhibition induces the cell cycle arrest of PCa cells and enhances chemotherapeutic resistance of PCa cells. As a result, the knockdown of TBK1 decreases PCa stem-like cells and drug resistance in vitro and in vivo. Taken together, these results strongly indicate that TBK1 plays an important role in establishing dormancy and maintaining drug resistance of PCa.
Supplementary Material
Acknowledgments
We thank Chris Strayhorn for his assistance with the histology. We also thank the University of Michigan Flow Cytometry Core, the Vector Core and the Imaging Core for their expertise.
Footnotes
This research was supported in part by the National Institutes of Health (NIH) through the University of Michigan Cancer Center support grant (P30 CA046592). This work was directly supported by the National Cancer Institute (CA163124 and CA093900; Y.S., K.J.P., and R.S.T.), the Fund for Cancer Research (R.S.T.), the Department of Defense (K.J.P. and R.S.T.), and the Prostate Cancer Foundation (Y.S., K.J.P., and R.S.T.). K.J.P. receives support as an American Cancer Society Clinical Research Professor and from NIH Specialized Program of Research Excellence (SPORE) in prostate cancer grant P50 CA69568 and the Cancer Center support grant P30 CA46592.
This article refers to supplementary materials, which are designated by Figures W1 and W2 and are available online at www.neoplasia.com.
References
- 1.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 2.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 3.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKɛ and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 4.Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, Akira S. The roles of two IκB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med. 2004;199:1641–1650. doi: 10.1084/jem.20040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med. 2004;199:1651–1658. doi: 10.1084/jem.20040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen RR, Hahn WC. Emerging roles for the non-canonical IKKs in cancer. Oncogene. 2011;30:631–641. doi: 10.1038/onc.2010.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baldwin AS. Regulation of cell death and autophagy by IKK and NF-κB: critical mechanisms in immune function and cancer. Immunol Rev. 2012;246:327–345. doi: 10.1111/j.1600-065X.2012.01095.x. [DOI] [PubMed] [Google Scholar]
- 8.Korherr C, Gille H, Schäfer R, Koenig-Hoffmann K, Dixelius J, Egland KA, Pastan I, Brinkmann U. Identification of proangiogenic genes and pathways by high-throughput functional genomics: TBK1 and the IRF3 pathway. Proc Natl Acad Sci USA. 2006;103:4240–4245. doi: 10.1073/pnas.0511319103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, Balakireva MG, Romeo Y, Kopelovich L, Gale M, Jr, et al. RalB GTPase-mediated activation of the IκB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127:157–170. doi: 10.1016/j.cell.2006.08.034. [DOI] [PubMed] [Google Scholar]
- 10.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T, Brekken R, Wurz R, Tasker A, Polverino T, et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. 2011;41:458–470. doi: 10.1016/j.molcel.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie X, Zhang D, Zhao B, Lu MK, You M, Condorelli G, Wang CY, Guan KL. IκB kinase ɛ and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci USA. 2011;108:6474–6479. doi: 10.1073/pnas.1016132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 15.Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, Hara K, Tanaka N, Avruch J, Yonezawa K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278:15461–15464. doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- 16.Hong F, Larrea MD, Doughty C, Kwiatkowski DJ, Squillace R, Slingerland JM. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol Cell. 2008;30:701–711. doi: 10.1016/j.molcel.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 17.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 18.Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, Sabatini DM. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16:1865–1870. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 20.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 21.Breuleux M, Klopfenstein M, Stephan C, Doughty CA, Barys L, Maira SM, Kwiatkowski D, Lane HA. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol Cancer Ther. 2009;8:742–753. doi: 10.1158/1535-7163.MCT-08-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, Kim JK, Patel LR, Ying C, Ziegler AM, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121:1298–1312. doi: 10.1172/JCI43414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson A, Laurenti E, Trumpp A. Balancing dormant and selfrenewing hematopoietic stem cells. Curr Opin Genet Dev. 2009;19:461–468. doi: 10.1016/j.gde.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, Sjostrom SK, Garraway LA, Weremowicz S, Richardson AL, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 25.Hutti JE, Shen RR, Abbott DW, Zhou AY, Sprott KM, Asara JM, Hahn WC, Cantley LC. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKɛ promotes cell transformation. Mol Cell. 2009;34:461–472. doi: 10.1016/j.molcel.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci USA. 2007;104:5431–5436. doi: 10.1073/pnas.0701152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rehn M, Olsson A, Reckzeh K, Diffner E, Carmeliet P, Landberg G, Cammenga J. Hypoxic induction of vascular endothelial growth factor regulates murine hematopoietic stem cell function in the low-oxygenic niche. Blood. 2011;118:1534–1543. doi: 10.1182/blood-2011-01-332890. [DOI] [PubMed] [Google Scholar]
- 28.Rankin EB, Wu C, Khatri R, Wilson TL, Andersen R, Araldi E, Rankin AL, Yuan J, Kuo CJ, Schipani E, et al. The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell. 2012;149:63–74. doi: 10.1016/j.cell.2012.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ranganathan AC, Adam AP, Zhang L, Aguirre-Ghiso JA. Tumor cell dormancy induced by p38SAPK and ER-stress signaling: an adaptive advantage for metastatic cells? Cancer Biol Ther. 2006;5:729–735. doi: 10.4161/cbt.5.7.2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vessella RL, Pantel K, Mohla S. Tumor cell dormancy: an NCI workshop report. Cancer Biol Ther. 2007;6:1496–1504. doi: 10.4161/cbt.6.9.4828. [DOI] [PubMed] [Google Scholar]
- 31.Quesnel B. Dormant tumor cells as a therapeutic target? Cancer Lett. 2008;267:10–17. doi: 10.1016/j.canlet.2008.02.055. [DOI] [PubMed] [Google Scholar]
- 32.Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 33.Felsher DW. Tumor dormancy and oncogene addiction. APMIS. 2008;116:629–637. doi: 10.1111/j.1600-0463.2008.01037.x. [DOI] [PubMed] [Google Scholar]
- 34.Wikman H, Vessella R, Pantel K. Cancer micrometastasis and tumour dormancy. APMIS. 2008;116:754–770. doi: 10.1111/j.1600-0463.2008.01033.x. [DOI] [PubMed] [Google Scholar]
- 35.Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERKMAPK to p38MAPK activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. 2001;12:863–879. doi: 10.1091/mbc.12.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 37.Gan B, DePinho RA. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle. 2009;8:1003–1006. doi: 10.4161/cc.8.7.8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Liu Y, Zheng P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205:2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gan B, Sahin E, Jiang S, Sanchez-Aguilera A, Scott KL, Chin L, Williams DA, Kwiatkowski DJ, DePinho RA. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci USA. 2008;105:19384–19389. doi: 10.1073/pnas.0810584105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, Haug JS, Rupp D, Porter-Westpfahl KS, Wiedemann LM, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 42.Matsuoka S, Oike Y, Onoyama I, Iwama A, Arai F, Takubo K, Mashimo Y, Oguro H, Nitta E, Ito K, et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev. 2008;22:986–991. doi: 10.1101/gad.1621808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, Rosenblatt J, Avigan DE, Teruya-Feldstein J, Pandolfi PP. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453:1072–1078. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, Fletcher-Sananikone E, Colla S, Wang YA, Chin L, et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468:701–704. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gurumurthy S, Xie SZ, Alagesan B, Kim J, Yusuf RZ, Saez B, Tzatsos A, Ozsolak F, Milos P, Ferrari F, et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2010;468:659–663. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castilho RM, Squarize CH, Chodosh LA, Williams B, Gutkind JS. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell. 2009;5:279–289. doi: 10.1016/j.stem.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basith S, Manavalan B, Yoo TH, Kim SG, Choi S. Roles of toll-like receptors in cancer: a double-edged sword for defense and offense. Arch Pharm Res. 2012;35:1297–1316. doi: 10.1007/s12272-012-0802-7. [DOI] [PubMed] [Google Scholar]
- 49.González-Reyes S, Fernández JM, González LO, Aguirre A, Suárez A, González JM, Escaff S, Vizoso FJ. Study of TLR3, TLR4, and TLR9 in prostate carcinomas and their association with biochemical recurrence. Cancer Immunol Immunother. 2011;60:217–226. doi: 10.1007/s00262-010-0931-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paone A, Starace D, Galli R, Padula F, De Cesaris P, Filippini A, Ziparo E, Riccioli A. Toll-like receptor 3 triggers apoptosis of human prostate cancer cells through a PKC-α-dependent mechanism. Carcinogenesis. 2008;29:1334–1342. doi: 10.1093/carcin/bgn149. [DOI] [PubMed] [Google Scholar]
- 51.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, Kondo S, Kondo Y, Yu Y, Mills GB, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest. 2008;118:3917–3929. doi: 10.1172/JCI35512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghosh S, Tergaonkar V, Rothlin CV, Correa RG, Bottero V, Bist P, Verma IM, Hunter T. Essential role of tuberous sclerosis genes TSC1 and TSC2 in NF-κB activation and cell survival. Cancer Cell. 2006;10:215–226. doi: 10.1016/j.ccr.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 54.Matsumoto K, Arao T, Tanaka K, Kaneda H, Kudo K, Fujita Y, Tamura D, Aomatsu K, Tamura T, Yamada Y, et al. mTOR signal and hypoxia-inducible factor-1α regulate CD133 expression in cancer cells. Cancer Res. 2009;69:7160–7164. doi: 10.1158/0008-5472.CAN-09-1289. [DOI] [PubMed] [Google Scholar]
- 55.Yang Z, Zhang L, Ma A, Liu L, Li J, Gu J, Liu Y. Transient mTOR inhibition facilitates continuous growth of liver tumors by modulating the maintenance of CD133+ cell populations. PLoS One. 2011;6:e28405. doi: 10.1371/journal.pone.0028405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoshii T, Tadokoro Y, Naka K, Ooshio T, Muraguchi T, Sugiyama N, Soga T, Araki K, Yamamura K, Hirao A. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J Clin Invest. 2012;122:2114–2129. doi: 10.1172/JCI62279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.