

Abstract
Poly(ADP-ribose) polymerase inhibitors have gained recent attention due to their highly selective killing of BRCA1/2 mutated and DNA double strand break (DSB) repair deficient tumors. Unfortunately, the majority of sporadic breast cancers carry wild-type BRCA1/2 and are proficient in DSB repair. We and others have shown that BRCA1 is a nuclear/cytoplasm shuttling protein which is transiently exported from the nucleus to the cytosol upon various stimuli. Thus, we hypothesized that depletion of nuclear BRCA1 would compromise DSB repair and subsequently render sporadic tumors susceptible to PARP inhibition. Indeed, in human sporadic breast cancer cells with functional BRCA1 and proficient DSB repair, a transient nuclear depletion of BRCA1 and subsequent HR repair deficit was induced with either truncated BRCA1 or irradiation. This rendered these human sporadic breast cancer cells susceptible to PARP inhibition. These observations were confirmed genetically using mislocated BRCA1 mutants as well as in vivo in mice bearing breast tumor xenografts. These data support the potential strategy of targeting BRCA1 location to convert BRCA1-proficient sporadic tumors to be susceptible to the synthetic lethal combination with PARP inhibitors.
Keywords: breast cancer, PARP inhibitor, radiation, DNA damage, DNA repair
Introduction
Poly(ADP-ribose) polymerase (PARP) inhibitors induce synthetic lethality by targeting homologous recombination (HR)-mediated DNA repair deficient tumors (1-3). However, this approach is only applicable to about 10% of cancers. Thus, much effort has been undertaken to expand the utility of PARP inhibitors beyond the realm of BRCA-associated tumors by combining with agents that alter the DNA damage/repair pathways. Indeed, PARP inhibitors have been reported to enhance cytotoxicity in sporadic tumors when combined with other DNA damaging agents, such as with platinum-based chemotherapy in breast cancer (4). Unfortunately, these combinations have so far failed to be efficacious in clinical trials (5, 6). Thus, novel therapeutic approaches are warranted.
We and others have previously demonstrated that the tumor suppressor BRCA1, a nuclear-cytoplasmic shuttling protein, is critical for DNA damage repair and induction of apoptosis. The function of BRCA1 is regulated by a variety of mechanisms including transcriptional control, phosphorylation, and protein-protein interactions (7). Its subcellular localization is controlled by a nuclear localization signal-mediated nuclear import via the importin receptor pathway and a nuclear export signal-facilitated nuclear export through a CRM1-dependent pathway (7). Importantly, cytoplasmic relocalization of BRCA1 protein is one mechanism whereby BRCA1 function is regulated in response to DNA damage.
We have previously shown that sequestering BRCA1 from the nucleus to the cytoplasm compromises its nuclear function in DNA repair and results in enhanced cytotoxic response to ionizing radiation (IR) and other DNA damaging agents (7-9). Since PARP inhibitors have been reported to induce synthetic lethality in DNA repair deficient cells, we hypothesized that inducing BRCA1 cytoplasmic translocation in sporadic breast cancer, which generates a DSB repair deficiency, will confer enhanced cytotoxicity to the PARP inhibitor ABT-888. Consistent with our hypothesis, ectopic expression of truncated BRCA1 (tr-BRCA1) sequestered endogenous BRCA1 to the cytosol, suppressed HR-mediated DSB repair, and subsequently rendered human sporadic breast cancer cells to become susceptible to the PARP inhibitor ABT-888. These observations were confirmed genetically using mislocated mutants of BRCA1. Furthermore, as a potential therapeutic strategy, we utilized IR to generate a transient depletion of nuclear BRCA1 and a HR repair deficiency. This subsequently conferred breast tumor cytotoxicity to ABT-888. Importantly, these findings were validated in mice bearing breast tumor xenografts. These data support the potential strategy of using radiotherapy to make BRCA1-proficient tumor cells susceptible to systemic DNA damaging agents such as PARP inhibitors. Furthermore, this novel strategy may also be feasible in other tumor types.
Materials and Methods
Cell culture
MCF7 (HTB-22, ATCC), MCF7DRGFP, MCF7 cells stably expressing BRCA1 shRNA (obtained courtesy of Dr. Simon Powell, Memorial Sloan Kettering, NY) were used in this study. Please refer to SI materials and methods for details. The genetic background, including expression and function of key proteins as well as the growth characteristics and their response to genotoxic agents, was tested most recently in December 2011 using western blot analysis, immunohistochemistry, and colony formation assays. The PARP inhibitor ABT-888 (Enzo Life Sciences) was utilized in our study.
Plasmids
Tr-BRCA1 has been described previously (10, 11). Wild-type (WT) BRCA1 YFP and nuclear export signal (NES) mutant BRCA1 YFP were kindly provided by Dr. Beric Hendeson (Westmead Millennium Institute, New South Wales, Australia) and has been previously described (12). Nuclear localization signal (NLS) mutant BRCA1 YFP was created utilizing a site-directed mutagenesis kit (Invitrogen) and had AA503-A506 mutation.
Immunofluorescence staining
BRCA1 location, Rad51 and DNA DSBs were assayed as described previously (8-10, 13, 14).
Clonogenic survival assay
Cell survival was evaluated by the colony formation assay as previously described (15).
Tumor growth delay
Tumor growth delay and immunohistochemical detection of γ-H2AX and BRCA1 were performed as described previously (8, 16). Please refer to SI methods for details. All animal procedures were approved by the Vanderbilt University Institutional Animal Care and Use Committee (protocol M/08/154).
Statistical analysis
The data were analyzed via analysis of variance (ANOVA) followed by a Bonferroni post test using GraphPad Prism version 4.02 (GraphPad Software, San Diego, CA). Data presented as average +/- standard error of mean.
Results
Targeting endogenous wt-BRCA1 to the cytosol using tr-BRCA1 confers susceptibility to ABT-888 in breast cancer cells
BRCA1 shuttles between the nucleus and cytoplasm through the importin and CRM1 pathways, respectively (7, 17). It has been shown that the protein BARD1 prevents BRCA1 export to the cytosol through its binding to the NH2-terminal region of BRCA1. This, in turn, masks the BRCA1 NES and blocks BRCA1 interaction with CRM1/exportin (10, 17). Exogenously expression of the small peptide “tr-BRCA1,” a truncated form (1–301 amino acids) of BRCA1 that contains the BARD1 binding site and competes with endogenous full length BRCA1 in binding with BARD1, has been previously shown to effectively induce BRCA1 cytoplasmic translocation (10). To target BRCA1 to the cytosol, we transiently expressed tr-BRCA1 in MCF7 cells, a well-characterized breast cancer cell line with wt-BRCA1 and wt-p53 (7, 9, 10). Changes in BRCA1 location following tr-BRCA1 was subsequently determined by immunohistochemical staining. Similar to our previous studies (7, 9, 10), cells were categorized into three groups based on BRCA1 staining pattern: strictly nuclear (N), strictly cytoplasmic (C), and mixed nuclear and cytoplasmic staining (NC) (9, 10). As shown in Figure 1A, expression of tr-BRCA1 alone in MCF7 cells effectively decreased endogenous nuclear BRCA1 by 2-fold compared with vector control (49% versus 24%) with a concomitant 3-fold increase in cytosolic fraction of BRCA1 (27% vs 73%). This suggests that tr-BRCA1 is effective in shifting BRCA1 to the cytosol, away from its nuclear repair substrates.
Figure 1. tr-BRCA1 induced BRCA1 cytosolic translocation results in susceptibility to PARP inhibition.
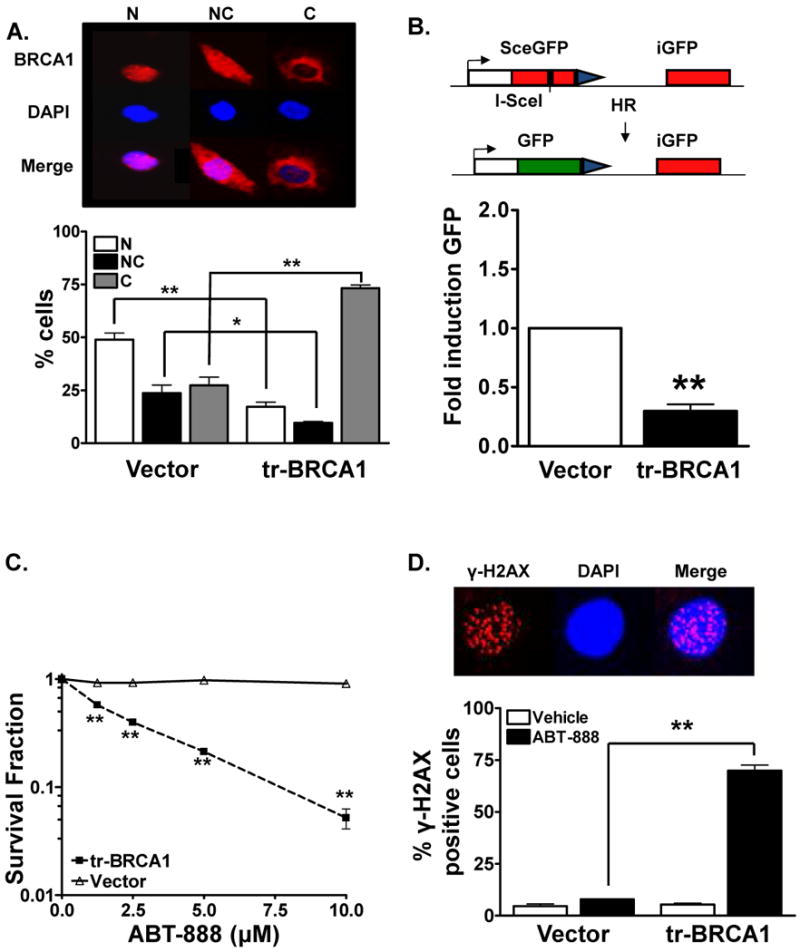
(A). tr-BRCA1 targets endogenous BRCA1 to the cytosol. MCF7 cells were transfected with either YFP or tr-BRCA1 YFP. BRCA1 distribution 24 hours following transfection was analyzed via immunohistochemical staining. A dramatic increase in cytosolic BRCA1 with a concomitant decrease in nuclear BRCA1 was observed in tr-BRCA1 transfected cells compared to vector alone. The top panel is a representative image of MCF7 cells exhibiting nuclear (N), both nuclear and cytosolic (NC), and cytosolic (C) BRCA1. Shown is the representative data of three independent experiments (mean ± SEM, *p<0.05, **p < 0.001). (B). tr-BRCA1 attenuates HR-mediated DSB repair. 8 hours following transfection with tr-BRCA1 or vector, MCF7DRGFP cells were transfected with I-SceI or control vector. 48 hours later cells were harvested for GFP expression analysis via flow cytometry. 4-fold reduction in HR-mediated repair was observed in tr-BRCA1 transfected cells when compared to vector alone. Shown is the representative data of three independent experiments (fold difference ± SEM, **p < 0.001). Inset is a schematic of the DRGFP repair assay. (C). tr-BRCA1 augments breast tumor susceptibility to PARP inhibition. MCF7 cells were transfected with YFP or tr-BRCA1 YFP. 16 hours following transfection, YFP positive cells were sorted via flow cytometry and reseeded for colony formation. 24 and 48 hours following transfection, cells were exposed to various doses of ABT-888. ABT-888 significantly attenuated the colony formation ability of tr-BRCA1 expressing MCF7 breast cancer cells compared to vehicle alone. In contrast, no significant effect on cell viability was observed in vector (YFP) transfected cells. Shown is the representative data of at least three independent experiments (mean ± SEM, **p < 0.001). (D). tr-BRCA1 increase γ-H2AX foci in human breast cancer cells. MCF7 cells were transfected with either YFP or tr-BRCA1 YFP. 24 hours following transfection, cells were exposed to 10μM ABT-888. 24 hours following the treatment period, cells were assessed for γ-H2AX foci. A significant induction in γ-H2AX foci formation, indicative of DSB damage, was observed in tr-BRCA1 expressing cells compared to vector alone. Shown is the representative data of three independent experiments the % of cells (mean ± SEM) with >10 foci (**p < 0.001). Inset, a representative staining of cell exhibiting γ-H2AX foci (red) with the nucleus stained with DAPI (blue).
Having verified that tr-BRCA1 could efficiently drive endogenous BRCA1 to the cytosol in MCF7 cells, we next confirmed its effects on HR-mediated DNA repair. MCF7 DRGFP cells, which carry a single copy of a chromosomally integrated HR repair substrate as described previously (7, 9) and in Figure 1B, were transiently transfected with tr-BRCA1 and subsequently transfected with ISce-1 endonuclease, which generates a DSB within the nonfunctional GFP gene. Only HR-mediated DSB repair restores GFP function. Thus, GFP positive cells, indicative of HR-mediated repair, were sorted for quantification of HR capacity. As shown in Figure 1B, a 4-fold decrease in GFP positive cells was observed in tr-BRCA1 transfected cells compared to vector alone. These HR-mediated repair effects were not due to differences in cell viability or transfection efficiency, which were included as controls in all HR-repair assays. These results suggest that cytosolic translocation of endogenous BRCA1 by tr-BRCA1 indeed induces a DSB repair deficiency in breast cancer cells.
We next assessed cytotoxicity of MCF7 cells expressing tr-BRCA1 in combination with various doses (1-10μM) of ABT-888 (14, 18). In these experiments, MCF7 cells were transfected with either YFP-tr-BRCA1 or YFP and subsequently sorted. 24 hours later, they were exposed to various doses of ABT-888. As shown in Figure 1C, ABT-888 alone failed to have any cytotoxic effect in vector transfected cells while those expressing tr-BRCA1 displayed a dose-dependent cytotoxicity to ABT-888.
We reasoned that compromised DNA repair by the expression of tr-BRCA1 would result in increased levels of persistent DSBs. Indeed, as shown in Figure 1D, increased DSBs as evidenced by persistent γ-H2AX foci were observed in tr-BRCA1 expressing cells treated with ABT-888 compared with vehicle control cells (24 hours post-ABT-888 treatment, 70% versus 5%, P < 0.001). No significant levels of γ-H2AX foci were detected in vector control cells with or without exposure to ABT-888 due to their repair proficiency.
To substantiate that the main determinant of susceptibility to PARP inhibition is the induction of DNA repair deficiency from sequestering BRCA1 to the cytosol, we next utilized a fluorescent YFP tagged NES-BRCA1 genetic mutant [mutation at aa86-aa90 (12, 19) and localized exclusively in the nucleus]. Tr-BRCA1 failed to induce cytosolic translocation (SI Figure 1A) or DNA damage following ABT-888 (SI Figure 1B) in this mutant. These results again substantiate the role of targeting BRCA1 to the cytosol and the subsequent inhibition of HR-mediated DSB repair as a mechanism by which tumor cell susceptibility to PARP inhibition can be achieved.
Genetic exclusion of nuclear BRCA1 confers susceptibility to ABT-888
To further validate if the main determinant of susceptibility to PARP inhibition is the induction of DNA repair deficiency from sequestering BRCA1 to the cytosol, we next utilized YFP tagged wild-type (WT) BRCA1 as well as YFP tagged NES (as above) and NLS mutants. The NLS-BRCA1 mutant (mutation at aa503-aa506) is localized in the cytosol (12, 19). These plasmids were transiently transfected into MCF7 BRCA1 shRNA cells in which the endogenous wt-BRCA1 has been depleted through stably expressing BRCA1 shRNA. BRCA1 location was assessed 48 hours following transfection by staining cells for YFP as a surrogate for BRCA1 since the constructs were YFP tagged. Cells were scored as above. As shown in Figure 2A, compared with cells expressing WT-BRCA1, cells expressing the NLS-BRCA1 mutant had predominantly cytosolic BRCA1 (87%). Conversely, NES-BRCA1 mutant transfected cells had predominantly nuclear BRCA1 (82%). Furthermore the shuttling response of these BRCA1 mutants following IR were also assessed (SI Figure 2A). Similar to our previous observations (7), IR effectively decreased nuclear WT-BRCA1 by 2-fold compared with mock-radiated control (47% versus 19%) while concomitantly increasing cytosolic BRCA1 (24% versus 56%). In contrast, the NES-BRCA1 and NLS-BRCA1 failed to show any significant redistribution following IR.
Figure 2. BRCA1 location determines ABT-888 induced cytotoxicity.
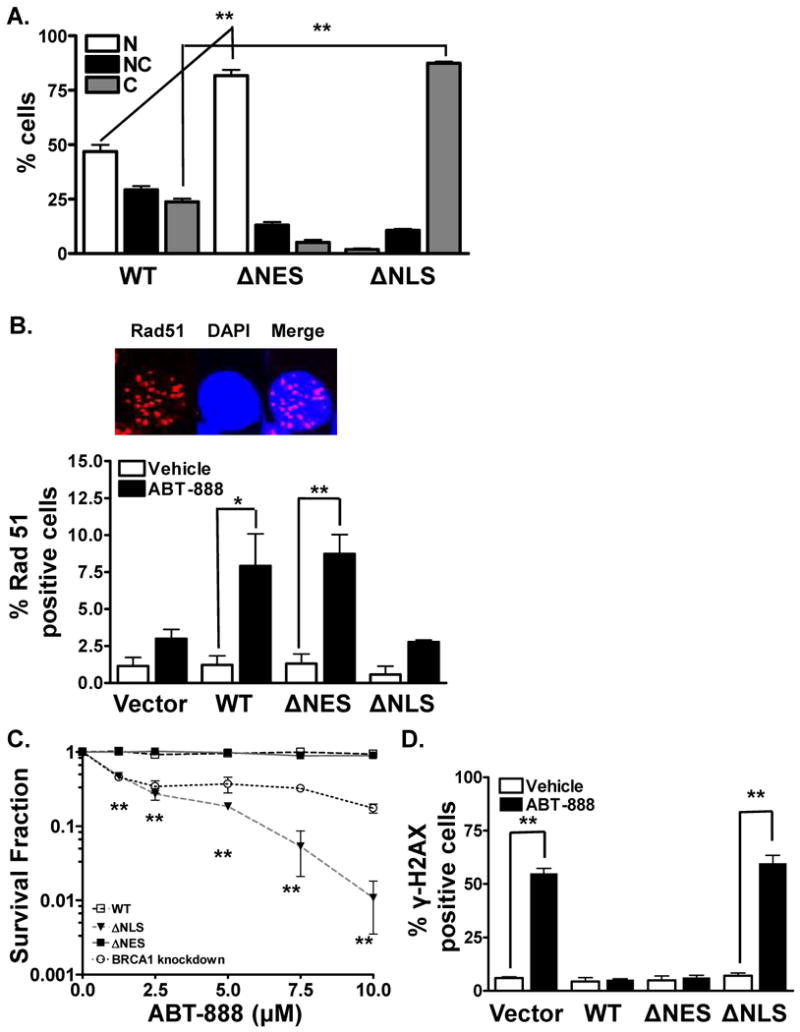
(A). BRCA1 location mutants are located predominantly either in the nucleus or the cytosol. MCF7 BRCA1 shRNA cells were transfected with either WT-BRCA1 YFP, NLS-BRCA1 YFP (cytosolic BRCA1) mutant, or NES-BRCA1 YFP (nuclear BRCA1) mutant. BRCA1 distribution was analyzed via immunohistochemical staining for YFP. NES-BRCA1 mutant was exclusively located in the nucleus whereas NLS-BRCA1 mutant was located in the cytosol. Shown is the representative data of three independent experiments (mean ± SEM, **p < 0.001). (B). Cytosolic BRCA1 mutant abrogates HR-mediated DSB repair. 16 hours following transfection with the various BRCA1 mutants, MCF7 BRCA1 shRNA cells were exposed to 10μM ABT-888. 24 hours following the drug treatment, Rad51 foci levels were analyzed via immunohistochemistry. A robust induction in Rad51 foci was observed in WT-BRCA1 and NES-BRCA1 mutant cells. No significant induction in foci formation was observed in the NLS-BRCA1 mutant or the vector alone, indicative of deficient HR-mediated repair. Shown is the representative data of three independent experiments the percent of cells (mean ± SEM) with Rad51 foci (*p < 0.05, **p < 0.001). Inset, a representative staining of cell exhibiting Rad51 foci (red) with the nucleus stained with DAPI (blue). (C). Cytosolic BRCA1 augments breast tumor response to PARP inhibition. MCF7 BRCA1 shRNA cells were transfected with the various mutants and subsequently sorted for YFP. 24 and 48 hours following transfection, cells were exposed to various doses of ABT-888. ABT-888 significantly attenuated the colony formation ability of NLS-BRCA1 mutant expressing breast cancer cells compared to vehicle alone. In contrast, no significant increase in cytotoxicity was observed in WT-BRCA1 and NES-BRCA1 mutant expressing cells. Shown is the representative data of at least three independent experiments (mean ± SEM, **p < 0.001). (D). Absence of BRCA1 or cytosolic BRCA1 increases γ-H2AX foci in human breast cancer cells. MCF7 BRCA1 shRNA cells were transfected with the various mutants of BRCA1. 40 hours following transfection, cells were treated with vehicle or 10μM ABT-888. 24 hours following the treatment period, cells were assessed for γ-H2AX foci. A robust induction in γ-H2AX foci was observed with ABT-888 treatment in the NLS-BRCA1 mutant or vector expressing cells, indicative of increased DNA damage. On the contrary no significant induction in foci formation was observed with ABT-888 treatment in the NES-BRCA1 mutant or WT-BRCA1 expressing cells, indicative of lack of DNA damage. Shown is the representative data of three independent experiments the % of cells (mean ± SEM) with >10 foci (**p < 0.001).
Based on these results, and given the central role of BRCA1 in promoting the repair of DSBs, we next assessed whether the location mutants of BRCA1 similarly altered HR-mediated DSB repair capacity of cells. We first analyzed Rad51 foci, a well-established functional marker of HR repair activity. It has been demonstrated that accumulation of DNA single strand breaks (SSBs) will be converted to DSBs during DNA replication. Cellular attempts to repair these DSBs lead to induction of HR-mediated repair and Rad51 foci. We thus reasoned that treatment with ABT-888 should induce HR repair in cells expressing the nuclear localized BRCA1 isoforms (NES-BRCA1, WT-BRCA1) but not in cells with cytoplasmic BRCA1 (NLS-BRCA1). Consistent with our hypothesis, no significant levels of Rad51 foci positive cells were detected in the vector or NLS-BRCA1 mutant cells (Figure 2B). Additionally, WT-BRCA1 transfected cells displayed elevated levels of Rad51 foci following exposure to ABT-888, indicative of activated HR-mediated DSB repair (1% versus 8%, p<0.001). Similar results were obtained in NES-BRCA1 mutant expressing cells following exposure to ABT-888 (1% versus 8%, p<0.001). ABT-888-induced Rad51 foci were also potentiated with IR in the WT- and NES-BRCA1 but not the NLS-BRCA1 expressing cells (SI Figure 2B). Furthermore, it was observed that after IR, the enhanced RAD51 foci in response to PARP inhibition was more robustin the NES-BRCA1 compared to WT-BRCA1 expressing cells, which correlates closely with the amount of BRCA1 being retained in the nucleus following IR. These results substantiate the observation that nuclear BRCA1 is required to activate HR-mediated repair, and that targeting BRCA1 location can disrupt HR-mediated DSB repair.
Based on these observations, we hypothesized that expressing the NLS-BRCA1 mutant (cytosolic BRCA1), but not NES-BRCA1 mutant (nuclear BRCA1), in the MCF7 BRCA1 shRNA cells will result in synthetic lethality with ABT-888. To test this hypothesis, we performed colony forming assays with the various mutants in combination with various doses (1-10μM) of ABT-888. IR was used as a positive control with WT-BRCA1 expressing MCF7 cells. As shown in Figure 2C, ABT-888 failed to induce any cytotoxicity in WT-BRCA1 expressing cells. Significantly increased levels of ABT-888-induced dose-dependent cell death were observed in the NLS-BRCA1 mutant expressing cells that express predominantly cytosolic BRCA1. No ABT-888-induced cytotoxicity was observed in the MCF7 NES-BRCA1 mutant cells, which have nuclear accumulation of BRCA1. These results validate genetically that targeting BRCA1 location to the cytosol can augment tumor susceptibility to PARP inhibition via an induced DSB repair deficit.
Because PARP-induced cytotoxicity is mainly attributed to persistent DSBs, we next assessed DNA DSBs in treated cells expressing the various BRCA1 location mutants. As shown in Figure 2D, in the vector alone transfected cells, significantly increased persistent DNA damage was observed 24 hours following ABT-888 treatment as demonstrated by increased percentage of cells with γ-H2AX foci. This is consistent with a DSB-repair deficiency due to absence of functional BRCA1. Similarly, a significantly increased persistent DNA damage was observed in the NLS-BRCA1 mutant expressing cells, which are deficient in HR repair due to cytosolic BRCA1. On the contrary, WT-BRCA1 or NES-BRCA1 mutant expressing cells failed to display increased persistent DNA damage following treatment with ABT-888, likely due to proficient HR repair. However, following IR, ABT-888 induced γ-H2AX foci was potentiated in the NLS-BRCA1 and WT-BRCA1 but not NES-BRCA1 expressing cells (SI Figure 2C). These results indicate that ABT-888-induced cytotoxicity following IR is due to persistent DNA damage from a DNA repair deficiency caused by altered BRCA1 location.
Radiation-induced cytosolic translocation of BRCA1 induces susceptibility to ABT-888 in wt-BRCA1 breast cancer cells
Our results thus far support the notion that targeting BRCA1 to the cytoplasm induces a DNA repair defect and a subsequent susceptibility to ABT-888. As a potential clinical application of this novel finding, we hypothesized that IR, which we have reported to induce BRCA1 nuclear export (10, 17), will induce synthetic lethality with PARP inhibition (7, 10). To test this hypothesis, we first analyzed BRCA1 location via immunohistochemistry at various time points following 4Gy IR in MCF7 breast cancer cells. Indeed, IR-induced BRCA1 cytosolic translocation in MCF7 cells in a time dependent manner, with a 2 fold decrease in nuclear BRCA1 compared with mock-radiated control (40% versus 20%) and a concomitant increase in cytosolic BRCA1 (25% versus 50%). The maximal shift was observed at 24 hours following IR (Figure 3A). Next we verified whether IR-induced decrease in nuclear BRCA1 compromises HR-mediated DSB repair capacity in MCF7DRGFP cells. As shown in Figure 3B, 4Gy IR resulted in a 2-fold decrease in HR-repair efficiency (p<0.001). Importantly, these effects on HR repair were not due to indirect changes in cell cycle distribution (SI Figure 3A-D). Interestingly, NES-BRCA1 mutant rescued IR-induced reduction in HR-mediated repair reaffirming that IR can induce a transient DNA repair deficit by depleting nuclear BRCA1.
Figure 3. Radiation augments cytotoxic response to PARP inhibition.
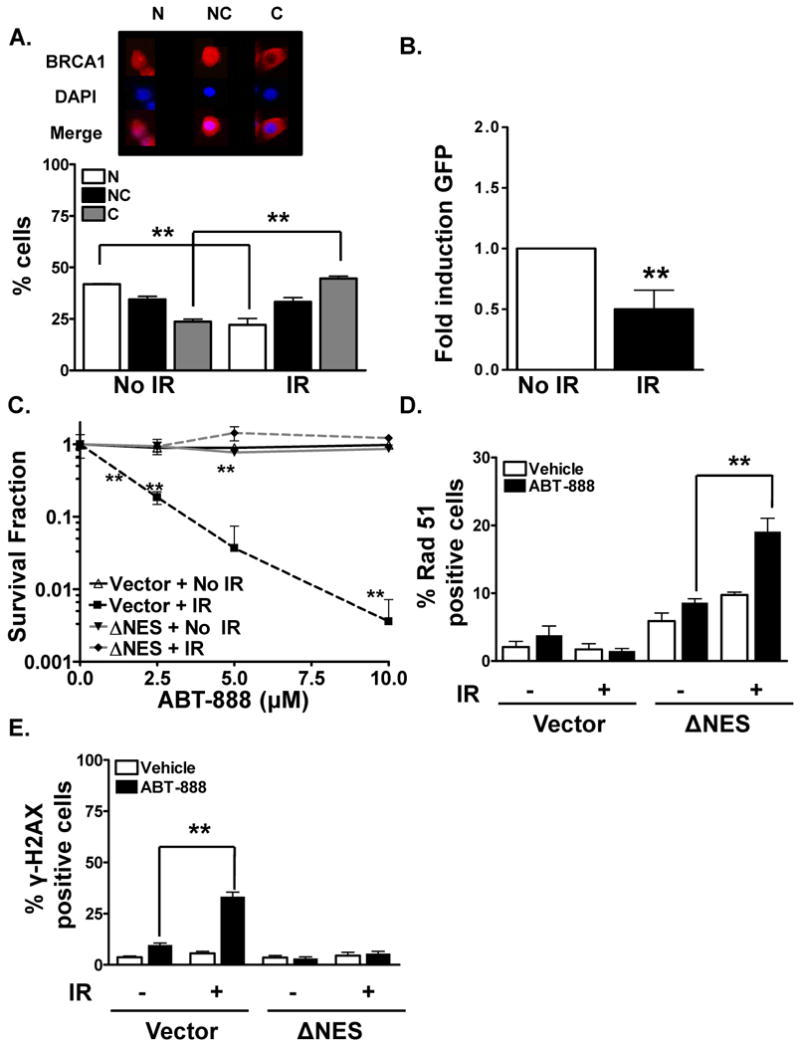
(A). Radiation induces BRCA1 nuclear export. BRCA1 distribution 24 hours following exposure to 4Gy IR was analyzed in MCF7 breast cancer cells via immunohistochemical staining. A dramatic increase in cytosolic BRCA1 with a concomitant decrease in nuclear BRCA1 was observed in irradiated cells compared to mock-IR. The top panel is a representative image of MCF7 cells exhibiting nuclear (N), both nuclear and cytosolic (NC), and cytosolic (C) BRCA1. Shown is the representative data of three independent experiments (mean ± SEM, **p < 0.001). (B). Radiation attenuates BRCA1-dependent and HR-mediated DSB repair. 8 hours following exposure to 4Gy IR, MCF7 DRGFP cells were transfected with control vector or I-SceI endonuclease to induce site-specific chromosomal DSB. 48 hours later cells were harvested for GFP expression analysis via flow cytometry. 2-fold reduction in HR-mediated repair was observed in IR-exposed cells when compared to mock-IR. Shown is the representative data of three independent experiments (fold difference ± SEM, **p < 0.01). (C). Radiation treatment augments breast tumor susceptibility to ABT-888 which is disrupted by the NES-BRCA1 mutant. Combination of IR and ABT-888 reduces the viability of MCF7 breast cancer cells. Cells were transfected with NES-BRCA1 mutant or vector control, sorted for YFP, seeded and exposed to mock or 4Gy IR treatment 24 hours following transfection. 24 and 48 hours following the treatment period, cells were exposed to various doses of ABT-888. 3-fold attenuation in the colony formation ability of IR-exposed MCF7 breast cancer cells was observed following ABT-888 treatment compared to vehicle alone. In contrast, no significant effect on cytotoxicity was observed in the NES-BRCA1 mutant transfected or mock-radiated cells. Shown is the representative data of at least three independent experiments (mean ± SEM, **p < 0.001). (D). Nuclear BRCA1 abrogates radiation-induced HR defect in human breast cancer cells. MCF7 cells were transfected with NES BRCA1 mutant. 24 hours following transfection, cells were exposed to 4Gy radiation. 24 hours later cells were treated with or without ABT-888. 24 hours following the drug treatment, Rad51 foci levels were analyzed via immunohistochemical staining as a surrogate marker for HR-mediated DSB repair. A robust induction in Rad51 foci was observed in NES-BRCA1 mutant cells which demonstrate significant nuclear BRCA1. Shown is the representative data of three independent experiments the percent of cells (mean ± SEM) with Rad51 foci (**p < 0.001). (E). Radiation increases PARP inhibition-induced γ-H2AX foci in human breast cancer cells while the NES-BRCA1 mutant abrogates this effect. MCF7 cells were transfected with NES-BRCA1 mutant, sorted, and treated with either mock or 4Gy IR 24 hours following transfection. 24 hours later, the cells were exposed to 10μM ABT-888. 24 hours following the treatment period cells were assessed for γ-H2AX foci. A robust induction in γ-H2AX foci was observed with the combination treatment of ABT-888 and IR when compared to vehicle alone. On the other hand no significant induction in foci formation was observed with ABT-888, IR treatment alone, or in the NES-BRCA1 mutant transfected cells. Shown is the representative data of three independent experiments the % of cells (mean ± SEM) with >10 foci (**p < 0.001).
Our results thus far suggest that IR induces BRCA1 nuclear export and subsequently decreases HR-mediated DSB repair. We next tested whether IR can induce synthetic lethality with ABT-888 in MCF7 breast cancer cells. As shown in Figure 3C, after normalizing for IR induced toxicity, a differential dose-dependent susceptibility to IR and ABT-888 was observed in MCF7 human breast cancer cells (3-fold reduction in cell viability with combination treatment). As expected, treatment with the inhibitor alone failed to show any cytotoxic effect due to proficient HR repair. Furthermore, the NES-BRCA1 mutant, which is not exported following IR, abrogated synthetic lethality of IR and PARP inhibition.
We next hypothesized that the mechanism of synthetic lethality involved attenuation of HR by IR. Rad51 foci levels were analyzed via immunohistochemical staining as a surrogate marker for HR-mediated DSB repair in NES-BRCA1 mutant cells. As anticipated, a robust induction in Rad51 foci was observed in NES-BRCA1 mutant cells which demonstrate significant nuclear BRCA1 while no significant induction was observed in the WT-BRCA1 cells (Figure 3D).
We also examined whether cells exposed to IR exhibit either increased levels of or persistent ABT-888-induced DSBs by assaying levels of γ-H2AX foci (14, 18, 20). A significant increase in unrepaired DSBs was observed in cells treated with ABT-888 and IR, as demonstrated by increased percentage of cells with γ-H2AX foci (Figure 3E) but, importantly, persistent γ-H2AX foci were not observed in the NES-BRCA1 mutant. These results indicate that IR-induced BRCA1 nuclear export creates a DSB repair deficiency, which results in increased levels of persistent DSBs in treated cells.
Targeting BRCA1 location with radiation delays tumor growth in breast cancer tumor xenografts
We next validated our intriguing observations in vivo in mice bearing MCF7 breast cancer xenografts. Tumors were exposed to 3Gy or mock IR on day 0. Beginning on day 1, tumor-bearing mice were then treated with or without ABT-888 (25 mg/kg daily) for 5 days, at which point treatment ceased, and tumors were followed through day 21. Consistent with results observed in cell culture, tumor growth was modestly inhibited upon treatment with either IR alone or ABT-888 alone as compared to mock-treated mice. However, growth of tumors treated with IR followed by 5 days of PARP inhibition was profoundly reduced as compared to untreated tumors or those treated with either agent alone (Figure 4A).
Figure 4. Cytosolic BRCA1 confers susceptibility to PARP inhibition in vivo.
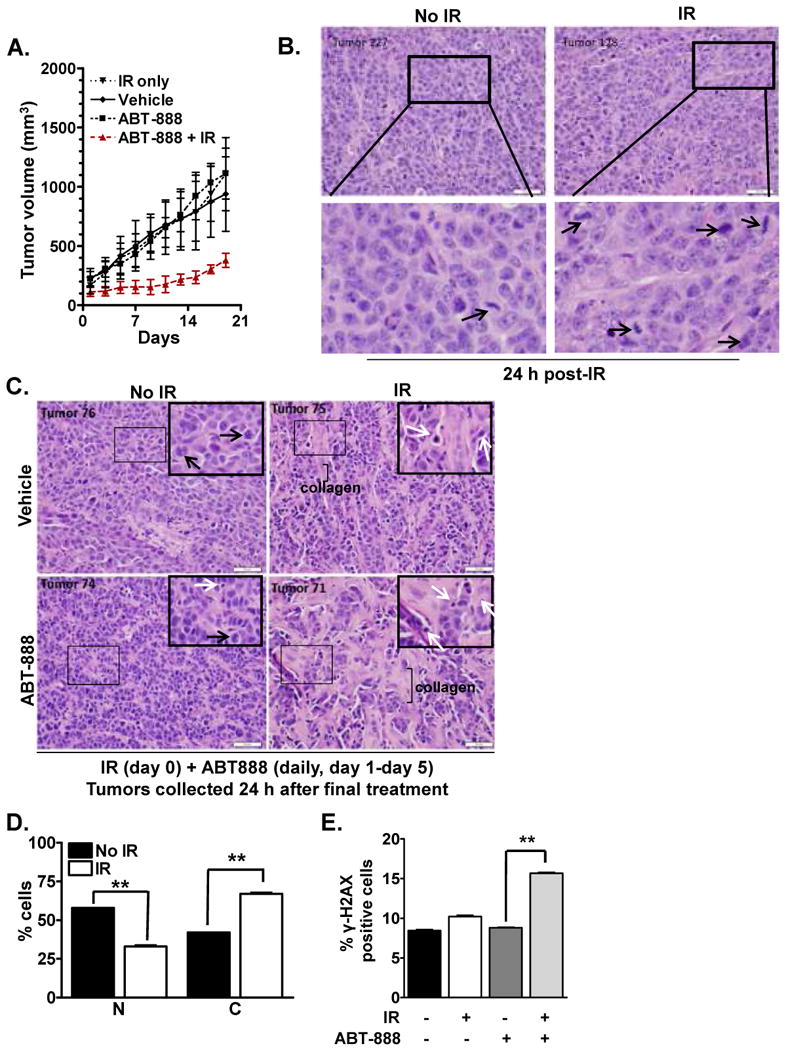
(A). Radiation induces significant tumor growth delay in combination with ABT-888 in vivo. MCF7 tumor-bearing female athymic Foxn1 (nu/nu) mice were randomized into four treatment groups (n=3): control, IR (3Gy, single dose at day 0), ABT-888 (25 mg/kg daily beginning at day 1, for 5 consecutive days), IR+ABT-888. Tumor size was measured for 21 days following which mice were sacrificed and tumor collected. Shown is the tumor volume (mean ± SEM) over time. The combination treatment of ABT-888 and IR induced a significant delay in tumor growth when compared to either agent alone. (B-C). Breast tumor tissues demonstrate increased markers of DNA damage following the combination treatment of ABT-888 and IR. (B.) MCF7 xenografts harvested 24 hours after IR were fixed in 10% formalin and processed for H&E staining. Increased accumulation of mitotic figures (black arrows) was observed in irradiated samples. Boxed areas are shown at higher magnification below each corresponding panel. (C). MCF7 xenografts harvested 24 hours after final treatment were formalin fixed, and sections were stained with H&E. Mitotic figures (black arrows) were evident in untreated samples, and were infrequently seen in ABT-888-treated tumors, but were not found in tumors treated with IR. Apoptotic bodies (white arrows) were found in IR-treated and ABT-888-treated samples, but were more predominant in xenografts treated with combination of IR+ABT-888. Increased collagen scarring was also observed in these tumor environments, consistent with drastically reduced tumor cellularity. Shown are representative images for each treatment group. Inset is magnified image of the boxed tumor field in each panel. (D). Radiation induces BRCA1 cytosolic translocation in MCF7 tumor xenografts. BRCA1 distribution 24 hours following exposure to 4Gy IR was analyzed in MCF7 breast cancer tissues via immunohistochemical staining. A dramatic increase in cytosolic BRCA1 with a concomitant decrease in nuclear BRCA1 was observed in tissue obtained from irradiated mice compared to mock-IR. Shown is the representative data of three independent experiments (mean ± SEM, **p < 0.001). (E). Combined ABT-888 and radiation treatment increases the level of persistent DSBs in tumor xenografts in vivo. γ-H2AX foci levels were analyzed 24 hours following exposure to 4Gy IR in MCF7 breast cancer tissues via immunohistochemical staining. A dramatic increase in γ-H2AX was observed in tissue obtained from irradiated mice treated with ABT-888 compared to either agent alone. Shown is the representative data of three independent experiments (mean ± SEM, **p < 0.001).
Additionally, accumulation of mitotic figures was observed at 24 hours after IR (Figure 4B). These are indicative of DNA damage and typically precede mitotic catastrophe and cell death (21). By 6 days, IR-treated tumors displayed no mitotic figures, but abundant apoptotic bodies (Figure 4C). Interestingly, mitotic figures did not accumulate in tumors treated with ABT-888. Further, apoptotic bodies were more predominant in IR+ABT-888 treated tissues compared to what was seen in response to either agent alone. Increased collagen scarring was also apparent in IR-treated tumors, but was increased in tumors treated with IR+ABT-888, consistent with drastically reduced tumor cellularity.
Similar to our in vitro results, increased cytosolic BRCA1 was observed in tumors obtained from mice irradiated with 3Gy IR (Figure 4D, p<0.001, SI Figure 4A). Furthermore, 2-fold increase in the level of unrepaired residual DNA DSBs was observed in these tumors treated with combination of IR+ABT-888, as measured by the amount of cells with persistent γ-H2AX foci (Figure 4E, p<0.001, SI Figure 4B). Thus these in vivo results validate the role of BRCA1 location as a mechanism by which tumors can be rendered susceptible to PARP inhibition.
Discussion
BRCA1 is a nuclear shuttling protein which is essential in maintaining genomic stability and controlling the cellular response to genotoxic stress. Precise regulation of these BRCA1 functions is vital from an oncologic and cell survival perspective. One emerging target is BRCA1 localization and shuttling, as sequestration of BRCA1 away from the nucleus may switch BRCA1 function from repair in the nucleus to activation of cell death signals in the cytoplasm. When nuclear, BRCA1 controls high fidelity repair of damaged DNA. In contrast, BRCA1 has been shown to enhance p53-independent apoptosis when cytoplasmic (8, 17). Interestingly, we have also previously reported that cytosolic translocation of BRCA1 and subsequent accumulation in the cytosol controls its DNA repair functions and regulates cell death processes following DNA damage (10). These data point to other potential mechanisms in addition to an induced DNA repair deficit that may explain the enhanced cytotoxicity to PARP inhibition following IR-mediated BRCA1 nuclear export, including the potential role of cytosolic BRCA1 in augmenting cell death pathways as well as other cytosolic functions of BRCA1.
Recent studies support the existence of a BRCAness phenotype in sporadic breast cancers, such as the highly aggressive triple negative subtypes, without a BRCA mutation. This has been thought to confer sensitivity of PARP inhibitors (22-25). Whether this BRCAness phenotype involves aberrant localization of BRCA1 is an interesting question and warrants further investigation.
BRCA1 shuttling can be regulated via protein–protein interaction (26, 27). The BRCA1-associated RING domain protein (BARD1) binds and masks the BRCA1 NES located at the N-terminal RING domain, thereby preventing nuclear export of BRCA1 (17, 19). By altering the interaction between BRCA1 and BARD1 with tr-BRCA1, we rendered breast cancer cells susceptible to PARP inhibition by effectively shifting BRCA1 to the cytosol (8-11, 17, 19). Finding the minimal region of tr-BRCA1 that can induce BRCA1 nuclear export could lead to the discovery of novel compounds that could augment tumor cytotoxicity to PARP inhibition in tumors. We and others have also reported the requirement of wild type p53 on IR-, but not tr-BRCA1- induced BRCA1 cytosolic sequestration (7, 8, 10). Thus, with respect to the sensitization of sporadic breast cancer cells to PARP inhibition by targeting BRCA1 location, further preclinical investigation and consideration in clinical studies is needed to determine the role of p53 in this strategy.
In this study, IR was utilized to generate a DNA repair deficit. This is at first glance counterintuitive, as IR is a potent inducer of immediate DNA repair response. It is important to note that the repair deficit generated by IR was measured at a time well after repair of IR-induced DSBs have occurred. Because BRCA1 export is a later DNA damage response event, our repair assays were not performed until 24 hours following IR. These data also indicate that the timing and sequence of treatment are important in order to achieve IR-induced tumor sensitization to PARP inhibition.
Our report suggests that BRCA1 cytoplasmic translocation can be a potential surrogate marker to predict tumor response to PARP inhibitor-based therapy. This is substantiated by our freshly resected human breast cancer specimen ex-vivo results (data not shown) that demonstrate IR-induced BRCA1 cytoplasmic translocation. Further investigation is necessary to validate this method as a potential predictor of PARP inhibitor sensitivity following IR.
Supplementary Material
Acknowledgments
This work was supported by a concept award from the Department of Defense (W81XWH-08-1-0571), Susan G. Komen Breast Cancer Research Award (BCTR0201704)(to FX), a research resident seed grant from the American Society for Therapeutic Radiology and Oncology, the Vanderbilt Institute for Clinical and Translational Research and the CTSA voucher grant, and IMPACT Award from the Department of Radiation Oncology, University of Alabama-Birmingham Comprehensive Cancer Center and University of Alabama-Birmingham School of Medicine (to ESY).
Footnotes
Conflict of interest statement: The authors declare no conflict of interest.
References
- 1.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 2.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 3.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AOH, Zander SAL, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proceedings of the National Academy of Sciences. 2008;105:17079–84. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, et al. ABT-888, an Orally Active Poly(ADP-Ribose) Polymerase Inhibitor that Potentiates DNA-Damaging Agents in Preclinical Tumor Models. Clinical Cancer Research. 2007;13:2728–37. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 5.Guha M. PARP inhibitors stumble in breast cancer. Nature biotechnology. 2011;29:373–4. doi: 10.1038/nbt0511-373. [DOI] [PubMed] [Google Scholar]
- 6.O'Shaughnessy J, Schwartzberg L, Danso M, Rugo H, Miller K, Yardley D, et al. A randomized phase III study of iniparib (BSI-201) in combination with gemcitabine/carboplatin (G/C) in metastatic triple-negative breast cancer (TNBC) J Clin Oncol. 2011;29 doi: 10.1200/JCO.2014.55.2984. [DOI] [PubMed] [Google Scholar]
- 7.Feng Z, Kachnic L, Zhang J, Powell SN, Xia F. DNA damage induces p53-dependent BRCA1 nuclear export. The Journal of biological chemistry. 2004;279:28574–84. doi: 10.1074/jbc.M404137200. [DOI] [PubMed] [Google Scholar]
- 8.Jiang J, Yang ES, Jiang G, Nowsheen S, Wang H, Wang T, et al. p53-dependent BRCA1 nuclear export controls cellular susceptibility to DNA damage. Cancer Res. 2011;71:5546–57. doi: 10.1158/0008-5472.CAN-10-3423. [DOI] [PubMed] [Google Scholar]
- 9.Li L, Wang H, Yang ES, Arteaga CL, Xia F. Erlotinib attenuates homologous recombinational repair of chromosomal breaks in human breast cancer cells. Cancer Res. 2008;68:9141–6. doi: 10.1158/0008-5472.CAN-08-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, Yang ES, Jiang J, Nowsheen S, Xia F. DNA damage-induced cytotoxicity is dissociated from BRCA1's DNA repair function but is dependent on its cytosolic accumulation. Cancer Res. 2010;70:6258–67. doi: 10.1158/0008-5472.CAN-09-4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown MA, Nicolai H, Howe K, Katagiri T, Lalani el N, Simpson KJ, et al. Expression of a truncated Brca1 protein delays lactational mammary development in transgenic mice. Transgenic Res. 2002;11:467–78. doi: 10.1023/a:1020348025139. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez JA, Henderson BR. Identification of a functional nuclear export sequence in BRCA1. The Journal of biological chemistry. 2000;275:38589–96. doi: 10.1074/jbc.M003851200. [DOI] [PubMed] [Google Scholar]
- 13.Nowsheen S, Bonner JA, Yang ES. The poly(ADP-Ribose) polymerase inhibitor ABT-888 reduces radiation-induced nuclear EGFR and augments head and neck tumor response to radiotherapy. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2011 doi: 10.1016/j.radonc.2011.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nowsheen S, Bonner JA, Lobuglio AF, Trummell H, Whitley AC, Dobelbower MC, et al. Cetuximab augments cytotoxicity with poly (adp-ribose) polymerase inhibition in head and neck cancer. PLoS ONE. 2011;6:e24148. doi: 10.1371/journal.pone.0024148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Yang ES, Jiang J, Nowsheen S, Xia F. DNA Damage Induced Cytotoxicity Is Dissociated from BRCA1's DNA Repair Function but Is Dependent on Its Cytosolic Accumulation. Cancer Research. 2010;70:6258–67. doi: 10.1158/0008-5472.CAN-09-4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang ES, Nowsheen S, Wang T, Thotala DK, Xia F. Glycogen synthase kinase 3beta inhibition enhances repair of DNA double-strand breaks in irradiated hippocampal neurons. Neuro Oncol. 2011;13:459–70. doi: 10.1093/neuonc/nor016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fabbro M, Schuechner S, Au WW, Henderson BR. BARD1 regulates BRCA1 apoptotic function by a mechanism involving nuclear retention. Exp Cell Res. 2004;298:661–73. doi: 10.1016/j.yexcr.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 18.Nowsheen S, Bonner JA, Yang ES. The poly(ADP-Ribose) polymerase inhibitor ABT-888 reduces radiation-induced nuclear EGFR and augments head and neck tumor response to radiotherapy. Radiother Oncol. 2011;99:331–8. doi: 10.1016/j.radonc.2011.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fabbro M, Rodriguez JA, Baer R, Henderson BR. BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. The Journal of biological chemistry. 2002;277:21315–24. doi: 10.1074/jbc.M200769200. [DOI] [PubMed] [Google Scholar]
- 20.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, et al. OPINION gamma H2AX and cancer. Nature Reviews Cancer. 2008;8:957–67. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–37. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- 22.Anders CK, Winer EP, Ford JM, Dent R, Silver DP, Sledge GW, et al. Poly(ADP-Ribose) polymerase inhibition: “targeted” therapy for triple-negative breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:4702–10. doi: 10.1158/1078-0432.CCR-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12:852–61. doi: 10.1016/S1470-2045(11)70214-5. [DOI] [PubMed] [Google Scholar]
- 24.Oonk AM, van Rijn C, Smits MM, Mulder L, Laddach N, Savola SP, et al. Clinical correlates of ‘BRCAness’ in triple-negative breast cancer of patients receiving adjuvant chemotherapy. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2012 doi: 10.1093/annonc/mdr621. [DOI] [PubMed] [Google Scholar]
- 25.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–48. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 26.Thompson ME. BRCA1 16 years later: nuclear import and export processes. The FEBS journal. 2010;277:3072–8. doi: 10.1111/j.1742-4658.2010.07733.x. [DOI] [PubMed] [Google Scholar]
- 27.Chen CF, Li S, Chen Y, Chen PL, Sharp ZD, Lee WH. The nuclear localization sequences of the BRCA1 protein interact with the importin-alpha subunit of the nuclear transport signal receptor. The Journal of biological chemistry. 1996;271:32863–8. doi: 10.1074/jbc.271.51.32863. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.