

Abstract
Background
Encapsulating exogenous proteins into a nanosized particulate system for delivery into cells is a great challenge. To address this issue, we developed a novel nanoparticle delivery method that differs from the nanoparticles reported to date because its core was composed of cross-linked dextran glassy nanoparticles which had pH in endosome-responsive environment and the protein was loaded in the core of cross-linked dextran glassy nanoparticles.
Methods
In this study, dextran in a poly(ethylene glycol) aqueous two-phase system created a different chemical environment in which proteins were encapsulated very efficiently (84.3% and 89.6% for enhanced green fluorescent protein and bovine serum albumin, respectively) by thermodynamically favored partition. The structures of the nanoparticles were confirmed by confocal laser scanning microscopy and scanning electron microscopy.
Results
The nanoparticles had a normal size distribution and a mean diameter of 186 nm. MTT assays showed that the nanoparticles were nontoxic up to a concentration of 2000 μg/mL in human hepatocarcinoma cell line SMMC-7721, HeLa, and BRL-3A cells. Of note, confocal laser scanning microscopy studies showed that nanoparticles loaded with fluorescein isothiocyanate-bovine serum albumin were efficiently delivered and released proteins into the cytoplasm of HeLa cells. Flow cytometry and terminal deoxynucleotidyl transferase dUTP nick end labeling assays showed that nanoparticles with a functional protein (apoptin) efficiently induced significant tumor cell apoptosis, which was confirmed by DAPI staining.
Conclusion
Our findings indicate that these nanoparticles meet the high demands for delivering protein medicines and have great potential in protein therapy.
Keywords: cellular uptake, protein delivery, nanoparticles, apoptosis
Introduction
Protein therapy is of great importance in the treatment of protein deficiency disease. Most human diseases are related to the malfunctioning of one or more proteins, and include metabolic disorders, autoimmune diseases, and cancers.1–4 The most effective and direct approach is protein therapy, whereby functional proteins are delivered to the target cell to replace the dysfunctional protein and maintain the balance of the organism. However, clinical application is frequently hampered by various biological barriers to successful delivery. Several problems need to be resolved, including rapid elimination from the circulation, inefficient cell permeability, and endosomal escape.5 For these reasons, we need an effective delivery system to deliver the protein. Any future delivery system must be safe and able to overcome barriers in the organism. In order for protein therapy to be effective, the challenge is to find a safe and effective system that is able to deliver protein to target cells.6
In the past decade, much work has been reported in this field. Kam et al used carbon nanotubes to deliver cytochrome c for induction of apoptosis.7 However, these approaches require use of organic solvents as the transporter which may be difficult for the body to degrade. Polyion complex micelles were also developed for cytoplasmic protein delivery, but the water/oil interfaces that exist when preparing micelles by these methods may lead to protein aggregation and inactivation.8 Specifically, liposomes without aqueous internal spaces have shown potential for lipophilic drug delivery. Kim et al have developed a liposome composed of lipid and apolipoprotein to mediate targeted delivery of intracellular-acting protein to non-small-cell lung tumors.9 Although liposomal systems have been shown to have an immense advantage when entering cells, they have poor stability and degrade rapidly in the circulation in vivo. These characteristics present formidable obstacles to the clinical application of these systems. Therefore, many researchers have focused on development of a new liposomal system utilizing poly(ethylene glycol) (PEG)ylation to increase the half-life in vivo.10 However, if a therapeutic protein is not highly lipophilic, it is difficult to encapsulate it in this system. Nanoparticles have better properties for transporting protein drugs and improved pharmacokinetic profiles in vivo because they can penetrate tissues easily through capillaries and epithelial linings as a result of their nanoscale size.11,12 However, the nanoparticles reported to date lack a thermodynamic driving force to encapsulate protein, which is dependent on volume-based distribution during the formation process. And the low encapsulation efficiency was due to the low volume fraction of nanoparticles interior as compared with that of the continuous PEG aqueous solution phase. For example, Arifin et al reported that the encapsulation efficiency of nanoparticles for bovine serum albumin (BSA) was in the range of 5%–12%.13 Therefore, there is still a lack of a good protein drug delivery system that can not only overcome all biological barriers, but also have high encapsulation efficiency to allow the drug dose to be reduced.
Here, we used a two-phase system, ie, dextran in aqueous PEG, to develop nanoparticles with high encapsulation efficiency. To reduce unwanted side effects and unwarranted interaction with the host, materials in nanoparticles should be compatible with the protein drug as well as the host. Due to preferential partition favoring the dextran phase, nanoparticles were developed using a PEG-dextran two-phase system which worked highly efficiently for encapsulation of exogenous protein (84.3% ± 1.5% and 89.6% ± 3.2% for enhanced green fluorescent protein [EGFP] and BSA, respectively). Further, these nanoparticles may be capable of delivering proteins into cells and releasing them in response to the acidic environment of endosomes.
Materials and methods
Materials
PEG with an average molecular weight of 8000, dextran with an average molecular weight of 60,000–74,000, fluorescein isothiocyanate (FITC)-dextran with an average molecular weight of 10,000, and a micro-bicinchoninic acid assay kit were supplied by Sigma-Aldrich (St Louis, MO, USA). Apoptin and EGFP proteins were purchased from Shanghai Jinan Co, Ltd (Shanghai, People’s Republic of China). PEG-polycaprolactone (PCL), PEG-PCL-maltotriose, and glycidyl methacrylate-derived dextran were synthesized by our laboratory. Nile red was obtained from Alfa Aesar (Tianjin, People’s Republic of China). SMMC-7721, HeLa, and BRL-3A cells were purchased from the Chinese Culture Tissue Collection Center (Shanghai, People’s Republic of China). The cells were cultured in Roswell Park Memorial Institute 1640 complete medium (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum at 37°C and 5% CO2.
Synthesis of glycidyl methacrylate-derived dextran
For solidification of the nanoparticle core by in situ cross-linking, dextran used to form the aqueous two-phase system was grafted with glycidyl methacrylate groups using the procedure described by van Dijk-Wolthuis et al.14 Dextran 50 g was dissolved in 450 mL of dimethyl sulfoxide in a round bottom reaction bulb under a nitrogen atmosphere. A calculated amount of glycidyl methacrylate was added after dissolution of 10 g of 4-dimethylaminopyridine. The solution was stirred at room temperature for 48 hours. The reaction was then stopped by adding an equimolar amount of concentrated HCl to neutralize the 4-dimethylaminopyridine. The reaction mixture was transferred to a dialysis tube and extensively dialyzed for 2 weeks against demineralized water at 4°C. Glycidyl methacrylate-derived dextran was then lyophilized and stored at −20°C before use.
Preparation of nanoparticles
Protein-loaded dextran nanoparticles were prepared using freezing-induced phase separation.14–35 In brief, a cosolution containing protein (0.2%, w/w), glycidyl methacrylate-derived dextran (1%, w/w), and PEG (10%, w/w) was cross-linked by a free radical initiator and a catalyst, ie, ammonium persulfate and N,N,N′,N′-tetramethylethylenediamine (0.2 wt% and 0.4 wt% in concentration, respectively), via grafted methacrylate bearing a C=C double bond. The cosolution was then frozen at −80°C overnight, and lyophilized to powder under 5.25 × 10−3 Pa for 24 hours using a laboratory freeze-drier (Alpha; Martin Christ GmbH, Osterode am Harz, Germany). The lyophilized powders were resuspended in dichloromethane to dissolve the PEG continuous phase, followed by centrifugation at 12,000 rpm for 5 minutes on a centrifuge (Eppendorf 5810R; Eppendorf Inc., Hamburg, Germany) to remove the dissolved PEG. This washing procedure was repeated three times, and the pellets were evaporated at 1.33 Pa for 24 hours using a vacuum drier (DZF-3; Shanghai Fuma Co, Ltd Shanghai, People’s Republic of China).
A 30 mg sample of protein-loaded dextran nanoparticles was suspended in pure water. To solidify the core matrix, glycidyl methacrylate-derived dextran was used to form the dispersed phase of the aqueous two-phase system. After the nanoparticles were formed a free radical initiator and a catalyst, ie, ammonium persulfate and N′,N′,N′,N′-tetramethyl-ethylenediamine (0.2 wt% and 0.4 wt% in concentration, respectively), were added to the system to induce cross-linking of the dextran core through the grafted methacrylate bearing a C=C double bond.14 The reaction was allowed to proceed at room temperature for one hour. The free PEG was then removed by dialysis (molecular weight cutoff 100 kDa) for 24 hours.
In vitro cytotoxicity and bioactivity of biomaterials
HeLa cells were placed in a 96-well plate (5 × 103/well) using Dulbecco’s Modified Eagle’s medium supplemented with 10% fetal bovine serum, 1% L-glutamine, penicillin 100 IU/mL, and streptomycin 100 μg/mL. After 24 hours, prescribed amounts of nanoparticles were added and incubated for 24 hours at 37°C in an atmosphere containing 5% CO2. Next, 10 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium-bromide (MTT) solution in phosphate-buffered saline (5 mg/mL) was added and incubated for another 4 hours. The medium was aspirated the MTT formazan generated by live cells was dissolved in 100 μL of 10% sodium dodecyl sulfate/0.1 M HCl overnight, and the absorbance of each well was measured at a wavelength of 570 nm using a microplate reader (ELX808 IU; Bio-Rad Hercules, CA, USA). Cell viability (%) was determined by comparing absorbance at 570 nm with that in control wells containing only cell culture medium. The experiments were performed four times each.
Scanning electron microscopy, nanoparticle size distribution, and zeta potential
The surface morphology of the nanoparticles was determined using a scanning electron microscope (Sirion 200; FEI Company, Hillsboro, OR USA). All the samples were loaded on double-sided tape that was attached on a metal stub and sprayed with gold vapor for 10 minutes under an argon atmosphere. The images were recorded at 15 kV sputtering energy under high vacuum. Optical microscopic images were taken using a microscope equipped with a digital camera (CX41; Olympus, Tokyo, Japan). To determine mean size and size distribution, the particles were suspended in water and detected using a particle size and shape analyzer (CIS-100; Ankersmid Nijverdal, The Netherlands). All the measurements were carried out at a 90° scattering angle and at 25°C. The morphology of the nanoparticles was imaged using a transmission electron microscope (JEM 2010 system; JEOL, Tokyo, Japan).
Determination of protein encapsulation efficiency in nanoparticles
Dextran particles loaded with BSA were then dissolved in an appropriate volume of phosphate buffer (pH 5.5) and assayed using a high-performance liquid chromatography system (Shimadzu, Tokyo, Japan) equipped with a size exclusion column and a guard column (TSK-GEL G2000SWXL and SWXL, respectively; TSK, Shizuoka, Japan). A phosphate-buffered saline (0.45 mol/L, pH 7.4) was used as the mobile phase running at the flow rate of 0.5 mL per minute. Absorbance of BSA was recorded at 214 nm. The amount of protein was calculated using Chromato-Solution-Light software (Shimadzu).
TUNEL assay
Cells were seeded in 6-well plates. After 6–12 hours, nanoparticles-loaded apoptin or BSA were added and incubated at 37°C. After 48 hours, the cells were washed with phosphate-buffered saline five times, fixed with 4% paraformaldehyde for 10 minutes, and blocked with 3% hydrogen peroxide in absolute methanol for 10 minutes. Tdt-mediated dUTP nick-end labeling (TUNEL) was performed using the In Situ Cell Death Detection Kit, POD (Roche Applied Science, Indianapolis, IN, USA), according to the manufacturer’s instructions, and the cells were stained with hematoxylin (Beyotime Institute of Biotechnology, Jiangsu, People’s Republic of China).
Flow cytometry assay for detection of apoptosis
Estimation of apoptotic HeLa cancer cell subpopulations was determined using Annexin V-fluorescein isothiocyanate and propidium iodide. Briefly, 1 × 105 cells per sample were washed with phosphate-buffered saline and then incubated with 3 μL of Annexin V-fluorescein isothiocyanate 0.5 mg/mL for 5 minutes in darkness. After washing with phosphate-buffered saline, the cells were then incubated with propidium iodide 5 mg/mL and immediately analyzed by flow cytometry. All flow cytometric analyses were performed on a FACSCanto™ flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). A 20 mW blue laser emitting a 488 nm beam served as the excitation source for Annexin V-fluorescein isothiocyanate (FL1) and propidium iodide (FL2). Annexin V (green) and propidium iodide (red) fluorescent signals were collected with 530 nm and 610 nm band pass filters, respectively, for 10,000 cells per sample.
Cell imaging by fluorescence microscopy
EGFP-loaded nanoparticles were added to the cell medium. After incubation at 37°C in 5% CO2 for 4 hours, the medium was removed and the cells on the microscope plates were washed three times with phosphate-buffered saline. Next, HeLa cells were fixed with 4% paraformaldehyde for 20 minutes and subsequently incubated for 30 minutes with LysoTracker(red) (L7528) at 37°C. Nuclei were counter-stained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). Fluorescent images were acquired using a laser fluorescence microscope (TS100; Nikon, Tokyo, Japan).
Data obtained regarding release characteristics, cytotoxicity, and antitumor effects were averaged and are expressed as the mean ± standard deviation. The Student’s t-test was used to determine the significance of the difference. A P-value less than 0.05 (*) or less than 0.01 (**) was considered to be statistically significant.
Results
In vitro cytotoxicity
Toxicity needs to be considered before a delivery system can be considered ideal for clinical application.15 The materials in our nanoparticles were PEG and dextran, which have excellent biocompatibility. The bioactivity of these nanoparticles was compared in SMMC-7721, HeLa, and BRL-3A cells (Figure 1). It was observed that the nanoparticles were nontoxic up to a tested concentration of 2000 μg/mL in all cell lines. Further, the inhibitory concentration (IC50) of the nanoparticles for SMMC-7721, HeLa, and BRL-3A cell growth was 38,562 μg/mL, 46,532 μg/mL, and 31,018 μg/mL, respectively, indicating that they have potential value for administration in vivo.
Figure 1.

Cytotoxicity of nanoparticles determined by MTT ((3-4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) assays using SMMC-7721, HeLa, and BRL-3A cells (n = 6).
Size distribution, zeta potential, and morphology of nanoparticles
It is a significant challenge for drug delivery to many tissues within the body because there are some barriers to systemic delivery in vivo such as egressing from the bloodstream and crossing the vascular endothelial.16 In general, molecules larger than 5 nm in diameter do not readily cross the capillary endothelium and will remain in the circulation until they are cleared from the body17 However, certain tissues, including the liver, spleen, and some tumors allow passage of molecules up to 200 nm in diameter which can accommodate a typical drug delivery nanoparticle.18 The size distribution of the nanoparticles was examined using a particle size and shape analyzer (CIS-100; Ankersmid). Figure 2 shows that the size range of the nanoparticles was 50–500 nm, their mean size was 186 nm, and the polydispersity index was 0.301. It is of benefit for nanoparticles to cross the capillary endothelium. The morphologic images were consistent with those of the scanning electron microscopic images. As shown in Figure 3, the surfaces of the nanoparticles were smooth and rounded, which is beneficial for intravenous administration in future applications.24
Figure 2.
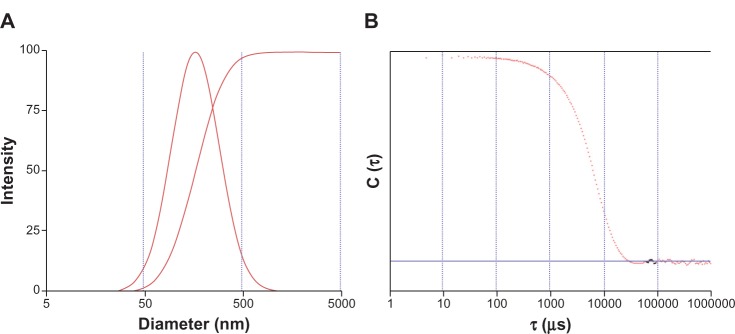
Size distribution of nanoparticles and autocorrelation function curves.
Notes: (A) the size range of the nanoparticles; (B) the polydispersity index of the nanoparticles.
Figure 3.

Scanning electron microscopic images of nanoparticles. Bar, 1 μm.
We found that the size was still 200 ± 12 nm and zeta potential tended toward zero (−2.1 mV). This is possibly because the surfaces of nanoparticles are hydrophilic groups with no adsorbing proteins.
At the same time, the morphology of the nanoparticles was measured using transmission electron microscopy. The diameters of the nanoparticles were around 200 nm (Figure 4), a value consistent with the measurements obtained using dynamic light scattering (Figure 2).
Figure 4.

Transmission electron microscopic images of nanoparticles. Bar, 200 nm.
Loading and releasing of protein in vitro
To study the protein encapsulation efficiency and stabilization effect of nanoparticles, BSA or fluorescein isothiocyanate-BSA is added during the assembly process before they are cross-linked.19 Our nanoparticles were demonstrated to have a remarkably high protein encapsulation efficiency of 84.3% ± 1.5% and 89.6% ± 2.1% for EGFP and BSA, respectively.. It is remarkable that these nanoparticles achieved such a high protein loading efficiency and content, given that low protein loading levels have been an issue for many current protein carriers. High efficiency can reduce the number of times drugs have to be administered and thereby help patients meet their treatment needs. It is dramatically higher than the reported nanoparticles, which encapsulation efficiency of BSA was only 5%.20 The high protein loading level of nanoparticles obtained in this study is likely due to affinity partitioning of the PEG/DEX phase during formation of the nanoparticles. The water-soluble polymers dissolved in water may form two immiscible aqueous phases with increases in their molecular weight and concentration.21 The PEG/DEX two-phase system works efficiently for protein purification due to preferential partition favoring the dextran phase, as well as the protein stabilization effect of the dextran hydroxyls, where KP is the partition coefficient of biomolecules between the two aqueous phases.22,23
The in vitro release of proteins from nanoparticles at pH 7.4 and pH 5.5, simulating body fluid and endosomes, respectively, was investigated. Interestingly, BSA was released from nanoparticles in a sustained manner at pH 5.5, where approximately 90% of the protein was released during 48 hours (Figure 5). On the contrary, only 20% BSA protein was released over 48 hours at pH 7.4 under the same conditions. This comparably rapid protein release at a mildly acidic pH is most probably related to the sensibility of the ester bond in glycidyl methacrylate. The ester bond in the acidic environment can degrade into alcohol and acid rapidly. It has been shown that ester cleavage makes disintegration of nanoparticles and protein release from nanoparticles occur more rapidly.
Figure 5.
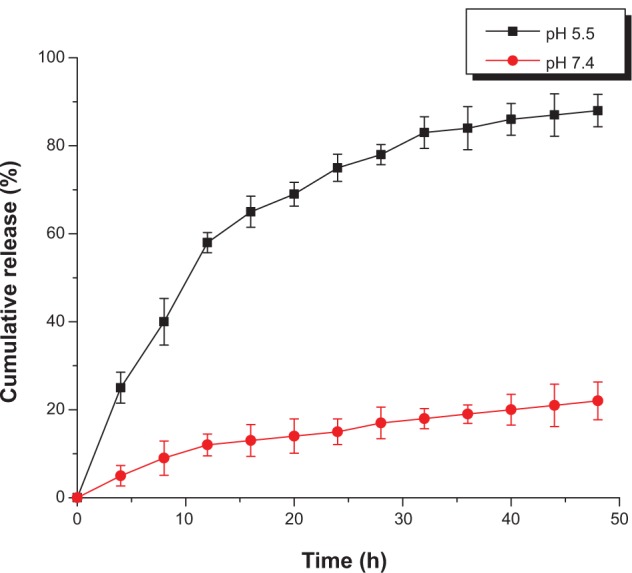
Release profiles for fluorescein isothiocyanate-bovine serum albumin from nanoparticles at ph 7.4 (phosphate-buffered saline, 20 mM) and 5.5 (2-(4-morpholino) ethanesulfonic acid, 20 mM).
Intracellular release of proteins
Intracellular uptake and release of proteins was studied using EGFP protein. Strong fluorescence was observed inside the cells after 4 hours of incubation (Figure 6), indicating that EGFP-loaded nanoparticles were efficiently delivered and released protein into the cells.
Figure 6.
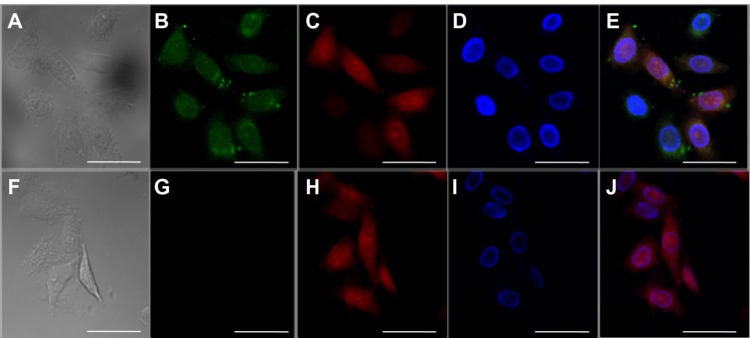
Confocal laser scanning microscopic images of HeLa cells incubated for 4 hours with enhanced green fluorescent protein-loaded nanoparticles. The scale bars correspond to 20 μm in all images. (A and F) Transmittance image, (B and G) fluorescein isothiocyanate-bovine serum albumin (green), (C and H) lysosome stained with lysoTracker® red (red), and (D and I) 4′,6-diamidino-2-phenylindole (DAPI, blue). (E) Overlays of B, C, and D. (J) Overlays of G, H, and I.
To investigate whether protein had escaped from endosomes, the lysosomes were stained by LysoTracker® Red (Invitrogen, Carlsbad, CA, USA) as described by Lee et al.8 Images of HeLa cells following incubation for 4 hours with EGFP-loaded nanoparticles showed strong green fluorescence as well as red fluorescence (Figure 6D), indicating efficient protein release from endosomes. However, for nanoparticles incubated with naked EGFP, green fluorescence could not be detected in the image (Figure 6G). Remarkably, fluorescence studies showed that these carriers can be efficiently loaded and transported into cells.24
Determination of induction of apoptosis
After study of the intracellular uptake of the model protein (EGFP), a functional protein was chosen to test whether the nanoparticles could protect the activity of a protein.25 Apoptin, a chicken anemia virus protein, can induce p53-independent, Bcl-2 insensitive apoptosis in various tumor cells, but not in normal human cells.26 TUNEL assays were performed on HeLa tumor cells treated with apoptin or nanoparticles loading apoptin (Figure 7). They showed that the nanoparticles with apoptin treated tumors contained many apoptotic cells, whereas almost no TUNEL-positive tumor cells could be detected among the controls treated with apoptin.
Figure 7.
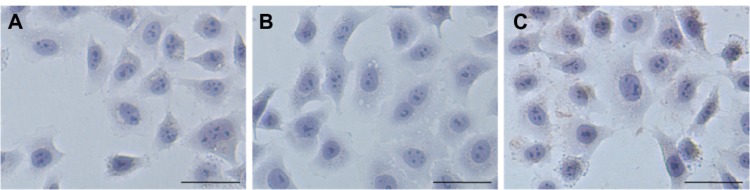
TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling) assays were performed on tumor HeLa cells treated with the different formulations. (A) Naked apoptin, (B) nanoparticles loading bovine serum albumin, and (C) nanoparticles loading apoptin. Bar, 50 μm.
Subsequent DAPI staining assays revealed apoptosis in the nuclei of tumor cells. HeLa cells treated with nanoparticles loading apoptin showed small nuclei containing condensed chromatin characteristics (Figure 8). Cells treated with nanoparticles loading apoptin for 24 hours resulted in fragmentation of the nuclei into apoptotic bodies (indicated by arrow). However, normal morphology of cell nuclei was observed in control cells treated with apoptin.
Figure 8.
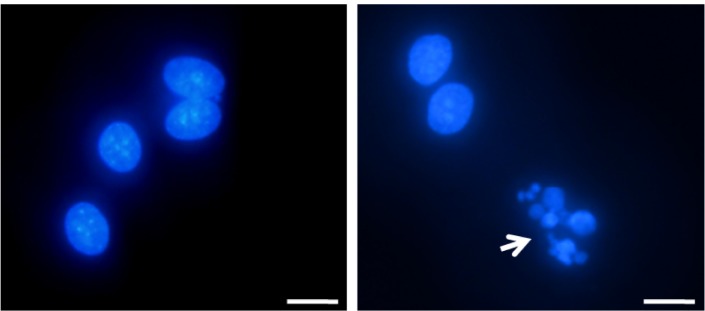
Cells subjected to DAPI (4′,6-diamidino-2-phenylindole) staining, showing condensation of chromatin and nuclear fragmentation (indicated by arrow). Bar, 5 μm. HELA cells treated with naked apoptin (left) and nanoparticles loading apoptin (right).
Next, expression of annexin V and permeation of propidium iodide were measured by flow cytometry in HeLa cells treated for 24 hours. As shown in Figure 9, apoptosis was quantified by the rate of early (upper right) and late (lower right) apoptotic cells. Although there was no significant difference for early apoptosis, 0.58% of control cells treated with empty nanoparticles underwent apoptosis later. In contrast, later apoptosis was induced in 11.79% of cells treated with nanoparticles loaded with apoptin protein. Therefore, one can conclude from the results above that nanoparticles can help protein traverse the cytoplasmic membrane and induce apoptosis in tumor cells.
Figure 9.
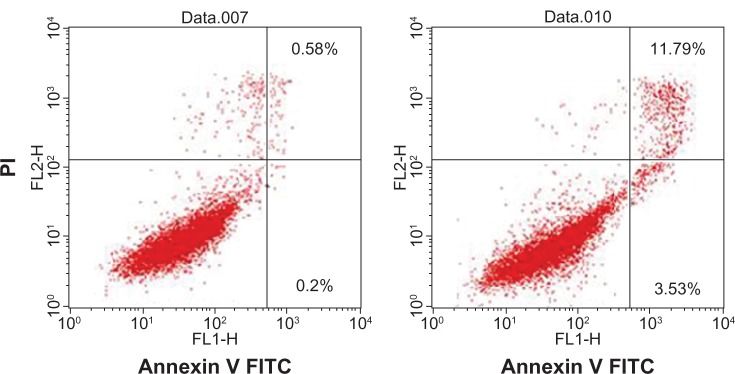
Induction of apoptosis as detected by annexin V (x axis) and propidium iodide (y axis) staining of HELA cells after 24 hours of treatment with naked apoptin (left) or nanoparticles loading apoptin (right). The percentage of apoptotic cells is shown.
Abbreviations: PI, propidium iodide; FITC, fluorescein isothiocyanate.
Discussion
Since Noteborn et al36 discovered the chick anemia virus (CAV)-derived protein, apoptin, in 1991, over 70 analyzed tumor cell lines have been reported by various researchers to be sensitive to apoptin-induced apoptosis.25,26 However, apoptin did not induce apoptosis in normal diploid human cells, because apoptin acts as a multimeric protein complex, and apoptin becomes selectively phosphorylated in tumor cells.27 Based on the above properties, apoptin is expected to be a potential anticancer therapy. However, intracellular delivery is a significant obstacle which needs to be overcome before clinical application. So far, there have been many studies focusing on the apoptin gene therapy to avoid the barrier of the intracellular delivery of drugs. However it also resulted in the other problems in the therapeutic process such as the poor expression efficiency of heterologous gene. Researchers have also used cell-penetrating peptides, such as TAT28 and PTD429, to improve the efficiency of cellular uptake and localization. But the exposed heterologous protein may result in the body’s immune response and be rapidly eliminated by the cells of the reticuloendothelial system (RES) from our body in blood circulation. For all these reasons, we need an effective system to deliver the protein.
To address the issue raised above, the nanoparticulate carrier should have a protein-friendly interior to load the therapeutic protein, a protective and functioning shell to protect and direct the protein into the cell, and a mechanism via which the protein can be encapsulated efficiently without being exposed to hazardous conditions. However, the nanoparticles reported to date lack a thermodynamic driving force to encapsulate protein, and are dependent on volume-based distribution during formation.13,30 They also have low encapsulation efficiency due to the low volume fraction of nanoparticle interior. Our study revealed a PEG-dextran two-phase system which worked efficiently for protein encapsulation due to the preferential partition favoring the dextran phase. This strategy provided not only a thermodynamically favorable partition for the protein with high efficiency (84.3% ± 1.5% and 89.6% ± 2.1% for EGFP and BSA, respectively), but also a friendly environment for protein drugs without water-oil, water-air interfacial tension to protect the protein’s native state and its bioactivity. High encapsulation efficiency of our nanoparticles can reduce the number of times drugs have to be administered and thereby help patients meet their treatment needs. Glycidyl methacrylate-derived dextran was cross-linked to form the nanoparticles in our study which had a relevant mechanical strength for circulation, although nanoparticles may be encountered with serum proteins and other biomolecules in body circulation. Furthermore, we introduced an ester bond into the nanoparticles, which was not sensitive at neutral pH in the blood environment. So nanoparticles can keep their integrality in the delivery system in vivo. In the present study, apoptin, a protein of therapeutic potential, was encapsulated in a polymersome formulation to demonstrate the feasibility of the nanoparticulate carrier system in intracellular protein delivery and therapy.
Conclusion
The protein-loaded nanoparticles used in this study have a good size distribution for endocytosis. Further, acid-sensitive bone among cross-linked glycidyl methacrylate-derived dextran can respond rapidly to release of encapsulated protein when nanoparticles meet the acidic environment of the endosomes after endocytosis. We used this system to encapsulate a functional protein (apoptin) resulting in regression of tumor cells and induction of apoptosis. Based on our results, these characteristics of nanoparticles show tremendous potential for intracellular delivery of protein and peptide drugs and may become a powerful tool for clinical application.
Acknowledgments
This work was supported by the National Science Foundation of China (81373366). The authors thank the technical assistants at the Instrumental Analysis Center of Shanghai Jiao Tong University for their technical support.
Footnotes
Disclosure
The authors declare no competing financial interests in this work.
References
- 1.Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002;2:727–739. doi: 10.1038/nrc905. [DOI] [PubMed] [Google Scholar]
- 2.Harries M, Smith I. The development and clinical use of trastuzumab (Herceptin) Endocr Relat Cancer. 2002;9:75–85. doi: 10.1677/erc.0.0090075. [DOI] [PubMed] [Google Scholar]
- 3.Marshall H. Anti-CD20 antibody therapy is highly effective in the treatment of follicular lymphoma. Trends Immunol. 2001;22:183–184. doi: 10.1016/s1471-4906(01)01909-3. [DOI] [PubMed] [Google Scholar]
- 4.Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006;5:147–159. doi: 10.1038/nrd1957. [DOI] [PubMed] [Google Scholar]
- 5.Torchilin VP, Lukyanov AN. Peptide and protein drug delivery to and into tumors: challenges and solutions. Drug Discov Today. 2003;8:259–266. doi: 10.1016/s1359-6446(03)02623-0. [DOI] [PubMed] [Google Scholar]
- 6.Liu G, Xu D, Jiang M, Yuan W. Preparation of bioactive interferon alpha-loaded polysaccharide nanoparticles using a new approach of temperature-induced water phase/water-phase emulsion. Int J Nanomed. 2012;7:4841–4848. doi: 10.2147/IJN.S35502. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Kam NW, Dai H. Carbon nanotubes as intracellular protein transporters: generality and biological functionality. J Am Chem Soc. 2005;127:6021–6026. [Google Scholar]
- 8.Lee Y, Ishii T, Cabral H, et al. Charge-conversional polyionic complex micelles-efficient nanocarriers for protein delivery into cytoplasm. Angew Chem Int Ed Engl. 2009;48:5309–5312. doi: 10.1002/anie.200900064. [DOI] [PubMed] [Google Scholar]
- 9.Kim SK, Foote MB, Huang L. The targeted intracellular delivery of cytochrome C protein to tumors using lipid-apolipoprotein nanoparticles. Biomaterials. 2012;33:3959–3966. doi: 10.1016/j.biomaterials.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim YH, Chun HY. Effects of the basic-state wind on secondary waves generated by the breaking of gravity waves in the mesosphere. APJAS. 2009;45:91–100. [Google Scholar]
- 11.Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55:329–347. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]
- 12.Yuan W, Liu Z. Controlled-release and preserved bioactivity of proteins from (self-assembled) core-shell double-walled microspheres. Int J Nanomed. 2012;7:257–270. doi: 10.2147/IJN.S27621. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Arifin DR, Palmer AF. Polymersome encapsulated hemoglobin: a novel type of oxygen carrier. Biomacromolecules. 2005;6:2172–2181. doi: 10.1021/bm0501454. [DOI] [PubMed] [Google Scholar]
- 14.Van Dijk-Wolthuis WN, Franssen O, Talsma H, Van Steenbergen MJ, Kettenes-van Den Bosch JJ, Hennink WE. Synthesis, characterization, and polymerization of glycidyl methacrylate derivatized dextran. Macromolecules. 1995;28:6317–6322. [Google Scholar]
- 15.Liu GJ, Ma SB, Li SK, et al. The highly efficient delivery of exogenous proteins into cells mediated by biodegradable chimaeric polymersomes. Biomaterials. 2010;31:7575–7585. doi: 10.1016/j.biomaterials.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 16.Yoo JW, Doshi N, Mitragotri S. Adaptive micro and nanoparticles: temporal control over carrier properties to facilitate drug delivery. Adv Drug Deliv Rev. 2011;63:1247–1256. doi: 10.1016/j.addr.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Zhang JC, Wu LL, Meng FH, et al. pH and reduction dual-bioresponsive polymersomes for efficient intracellular protein delivery. Langmuir. 2012;28:2056–2065. doi: 10.1021/la203843m. [DOI] [PubMed] [Google Scholar]
- 18.Jiang J, Cole D, Westwood N, et al. Crucial roles for protein kinase C isoforms in tumor-specific killing by apoptin. Cancer Res. 2010;70:7242–7252. doi: 10.1158/0008-5472.CAN-10-1204. [DOI] [PubMed] [Google Scholar]
- 19.Yuan W, Geng Y, Wu F, et al. Preparation of polysaccharide glassy microparticles with stabilization of proteins. Int J Pharm. 2009;366:154–159. doi: 10.1016/j.ijpharm.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 20.Kirby C, Gergoriadis G. Dehydration-rehydration vesicles: a simple method for high yield drug entrapment in liposomes. Bio/Technology. 1984;2:979–984. [Google Scholar]
- 21.Benavides J, Rito-Palomares M. Practical experiences from the development of aqueous two-phase processes for the recovery of high value biological products. J Chem Technol Biotechnol. 2008;83:133–142. [Google Scholar]
- 22.Zaslavsky BY. Bioanalytical applications of partitioning in aqueous polymer two-phase systems. Anal Chem. 1992;64:765A–773A. doi: 10.1021/ac00039a001. [DOI] [PubMed] [Google Scholar]
- 23.Jin T, Zhu J, Wu F, Yuan W, Geng LL, Zhu H. Preparing polymer-based sustained-release systems without exposing proteins to water-oil or water-air interfaces and cross-linking reagents. J Control Release. 2008;128:50–59. doi: 10.1016/j.jconrel.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 24.Akagi T, Wang X, Uto T, Baba M, Akashi M. Protein direct delivery to dendritic cells using nanoparticles based on amphiphilic poly(amino acid) derivatives. Biomaterials. 2007;28:3427–3436. doi: 10.1016/j.biomaterials.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 25.Jin JL, Gong J, Yin TJ, et al. PTD4-apoptin protein and dacarbazine show a synergistic antitumor effect on B16-F1 melanoma in vitro and in vivo. Eur J Pharmacol. 2011;654:17–25. doi: 10.1016/j.ejphar.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Danen-Van Oorschot AAAM, Fischer DF, Grimbergen JM, et al. Apoptin induces apoptosis in human transformed and malignant cells but not in normal cells. Proc Natl Acad Sci U S A. 1997;94:5843–5847. doi: 10.1073/pnas.94.11.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rohn JL, Zhang YH, Aalbers RI, et al. A tumor-specific kinase activity regulates the viral death protein apoptin. J Biol Chem. 2002;277:50820–50827. doi: 10.1074/jbc.M208557200. [DOI] [PubMed] [Google Scholar]
- 28.Guelen L, Paterson H, Gaken J, Meyers M, Farzaneh F, Tavassoli M. TAT-apoptin is efficiently delivered and induces apoptosis in cancer cells. Oncogene. 2004;23:1153–1165. doi: 10.1038/sj.onc.1207224. [DOI] [PubMed] [Google Scholar]
- 29.Sun J, Yan Y, Wang X-T, et al. PTD4-apoptin protein therapy inhibits tumor growth in vivo. Int J Cancer. 2009;124:2973–2981. doi: 10.1002/ijc.24279. [DOI] [PubMed] [Google Scholar]
- 30.Liu G, Hong X, Jiang M, Yuan W. Sustained-release G-CSF microspheres using a novel solid-in-oil-in-oil-in-water emulsion method. Int J Nanomed. 2012;7:4559–4569. doi: 10.2147/IJN.S33993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan W, Wu F, Guo M, Jin T. Development of protein delivery microsphere system by a novel S/O/O/W multi-emulsion. Eur J Pharm Sci. 2009;36(2–3):212–218. doi: 10.1016/j.ejps.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 32.Ren T, Yuan W, Zhao H, Jin T. Sustained-release polylactide-co-glycolide microspheres loaded with pre-formulated protein polysaccharide nanoparticles. Micro Nano Lett. 2011;6:70–74. [Google Scholar]
- 33.Yuan W, Zhang Y, Wu F, et al. Preparation of protein-loaded sustained-release microspheres via ‘solid-in-oil-in-hydrophilic oil-in-ethanol (S/O/hO/E)’ emulsification. Colloids Surf B Biointerfaces. 2010;79:326–333. doi: 10.1016/j.colsurfb.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 34.Rong X, Mo X, Ren T, et al. Neuroprotective effect of erythropoietin-loaded composite microspheres on retinal ganglion cells in rats. Eur J Pharm Sci. 2011;43(4):334–342. doi: 10.1016/j.ejps.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 35.Yuan W, Hu Z, Su J, Wu F, Liu Z, Jin T. Preparation and characterization of recombinant human growth hormone-Zn 2+-dextran nanoparticles using aqueous phase-aqueous phase emulsion. Nanomedicine: Nanotechnology, Biology, and Medicine. 2012;8(4):424–427. doi: 10.1016/j.nano.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 36.Noteborn MHM, De Boer GF, Van Roozelaar DJ, Karreman C, Kranenburg O, Vos JG, Jeurissen SHM, Hoeben RC, Zantema A, Koch G, Van Ormondt H, Van der Eb AJ. Characterization of cloned chicken anemia virus DNA that contains all elements for the infections replication cycle. J Virol. 1991;65:3131–3139. doi: 10.1128/jvi.65.6.3131-3139.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]