

Abstract
The Janus kinase 2 (JAK2) mutant V617F and other JAK mutants are found in patients with myeloproliferative neoplasms and leukemias. Due to their involvement in neoplasia and inflammatory disorders, Janus kinases are promising targets for kinase inhibitor therapy. Several small-molecule compounds are evaluated in clinical trials for myelofibrosis, and ruxolitinib (INCB018424, Jakafi®) was the first Janus kinase inhibitor to receive clinical approval. In this review we provide an overview of JAK2V617F signaling and its inhibition by small-molecule kinase inhibitors. In addition, myeloproliferative neoplasms are discussed regarding the role of JAK2V617F and other mutant proteins of possible relevance. We further give an overview about treatment options with special emphasis on possible combination therapies.
Keywords: JAK2V617F, myeloproliferative neoplasms, polycythemia vera, essential thrombocythemia, primary myelofibrosis
Introduction
Soon after their discovery1 the Janus kinases were found to be involved in cytokine signaling.2 The phenotypic analysis of knock-out mice for all four JAKs revealed that the lack of each JAK protein is linked to deficiencies in the signaling of specific cytokines using these JAKs in their receptor complexes3-8 (reviewed in refs. 9 and 10). Janus kinase 2 is essential in the signaling of cytokines using homodimeric receptors (Epo, Tpo, prolactin, leptin, and growth hormone). It has been shown that JAK2 plays a crucial role in hematopoiesis as JAK2 knockout mice die at day 13 of gestation due to failure of the development of definite hematopoiesis.4,5 JAK2 also plays a central role in the signaling of cytokines employing the common β chain receptor (IL3, IL5, and GM-CSF), of certain members of the IL10 type cytokine family (IFNγ, IL19, IL20, and IL24), of the IL12 type family members (IL12 and IL23) and in TSLP signaling.11
Many detailed studies have shown how the four members of the Janus kinase family mediate cytokine-induced signal transduction through cytokine receptors and regulate proliferation, differentiation, survival, and cell migration and thereby play a major role in hematopoiesis and the immune system. Due to this immunomodulatory role it is evident that Janus kinases are major regulators of inflammatory disorders (e.g., rheumatoid arthritis and psoriasis12) and cytokine-dependent cancers (e.g., multiple myeloma13) and, thus, have long been identified as druggable targets. Mutations in JAKs have first been described for JAK3 and have been found to elicit severe combined immunodeficiency (SCID).14 Fusion of JAK2 with certain proteins (e.g., Tel, Bcr, or PCM1) resulting in constitutively active signaling molecules has been described in a variety of hematopoietic malignancies as CML, AML, or ALL.15-18
Additionally, a point mutation in JAK2—JAK2V617F—was discovered in the majority of Philadelphia chromosome-negative myeloproliferative neoplasm (MPN) patients in 2005.19-23 JAK2V617F is found with high incidence in patients with polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). In different murine models, it has been shown that the expression of JAK2V617F is sufficient to induce a MPN-like phenotype.24-29 JAK2V617F is also, albeit rarely, found in other hematologic malignancies like the hypereosinophilic syndrome (HES), chronic or juvenile myelomonocytic leukemia (CMML or JMML), acute myeloid leukemia (AML), and refractory anemia with ringed sideroblasts (with thrombocytosis) (RARS or RARS-T) (reviewed in ref. 11). The JAK2V617F mutation is an acquired somatic event of the hematopoietic compartment, where it has been identified in hematopoietic stem cells (CD34+CD38−CD90+lin−) and multi-potent progenitor cells22,30 as well as in differentiated cells like granulocytes.20 It was also found in cells from the lymphoid lineage (e.g., natural killer cells) in a considerable amount of MPN patients31,32 suggesting that JAK2V617F occurs in multi-potent hematopoietic progenitor cells, although the phenotype of MPN is related to a selective proliferative advantage of the myeloid lineages. In the last years, many more genetic alterations affecting all members of the Janus kinase family have been discovered in leukemias and other hematopoietic neoplasia.11
JAK-STAT Signaling and the JAK2V617F Mutant
Structural organization of JAKs
The size of Janus kinases ranges from 120 to 140 kDa. All JAK family members share a similar sequence consisting of seven JAK homology (JH) domains,33 which only partially match the JAK domain structure. The JH1 and JH2 domains represent the adjacent kinase and pseudokinase domain, a feature only found in five kinases (in the four JAKs and in GCN2). The domains JH3 to JH7 correspond to the SH2 and FERM domains33,34 and are involved in cytokine receptor binding. Structural aspects of receptor binding have been reviewed recently11,35,36 and will not be covered here. Since the discovery of JAK2V617F, a great number of mutations (~70) have been described throughout all the structural domains of the JAKs and many (~30) have been biochemically validated to lead to constitutively active proteins.37 Mutations in the kinase domain can have direct consequences on kinase domain conformation and activation, but the molecular consequences of mutations in other domains of the JAKs are not as easily understood. The pseudokinase domain mutations (e.g., V617F) are thought to relieve the negative regulatory interaction between the pseudokinase domain and the kinase domain36,38 and result in constitutive activation of the kinase. Recently, the pseudokinase domain has been described to have residual kinase activity and to phosphorylate inhibitory amino acid residues within JAK2 (serine 523 and tyrosine 570).39 This might imply that mutations in the pseudokinase domain could alternatively represent loss-of-function mutations regarding the pseudokinase domain’s remaining kinase activity. Still, the pseudokinase domain mutations are not fully understood, while the consequences of the mutations within the FERM and SH2 domains are not understood at all. This is due to the lack of detailed structural information concerning the full-length JAK proteins. Structural models of JAK240,41 have been used to explain the molecular details of processes involved in JAK2V617F activation.42-44 However, 3D reconstructions of isolated JAK1 from an electron microscopy imaging approach45 have shown that the pseudokinase and kinase domain form a closely associated cluster, the conformation of which does not correspond to the molecular model described above. The isolated JAK1 showed great flexibility and could adopt different conformations from an “open” conformation (relatively linear with contacts between the adjacent domains in the polypeptide chain) to a “closed” conformation (in addition to contacts between adjacent domains, the FERM, SH2 domains are in contact with the kinase and pseudokinase domains). Although mutational studies have already suggested these contacts between the FERM and kinase domains,46-48 there is no certainty that the conformation of the JAKs bound to a cytokine receptor is entirely comparable to these conformational states. Unfortunately, the conformation of JAK1 bound to gp130 could not be resolved in this study. This might show that even when bound to a cytokine receptor the JAKs have great conformational flexibility.
JAK activation at the receptor
Janus kinases are tightly associated to the intracellular parts of cytokine receptors mediated by their FERM and SH2 domains and are maintained in an inactive state, when no cytokine is bound to the receptor.35 Binding of a cytokine to a cytokine receptor leads to conformational changes in the receptor which are transmitted to the cytoplasmically associated JAKs, leading to their activation and phosphorylation (reviewed in refs. 11 and 35). Recently, a study using kinase-inactive and constitutively active mutants of JAK1 and JAK3 in the context of IL-2 receptor signaling suggested that the conformational and phosphorylation events of JAK activation are independent of one another (Fig. 1), and that both events are necessary to induce full activation of the JAKs.37 However, the exact molecular details of JAK activation upon binding of a cytokine to the receptor remains elusive, because of lacking structural information of the full-length protein bound to a receptor. The transformation potential of JAK2V617F is also dependent on binding to a cytokine receptor (EpoR, thrombopoietin receptor [TpoR], or G-CSF receptor)49 and it has been demonstrated that a functional FERM domain as well as an intact SH2 domain are required for the JAK2V617F-mediated transformation.50,51

Figure 1. Conformational and phosphorylation events leading to JAK activation. IL2 signaling is used as an example. For clarity only the signal transducing receptor chains (IL2Rβ [β] and IL2Rγ [γ]) are shown. The scheme on the left shows the inactive state of a receptor/JAK complex. The binding of the cytokine (1a) impinges conformational changes in the receptor complex. JAKs are sensitive to these changes since they bind to the membrane-proximal region of cytokine receptors. This results in a conformational, phosphorylation-independent activation of the two JAKs (1b). Activation of downstream signaling is already promoted at this stage (2), although at a non-maximal level. The now activated JAKs phosphorylate each other “in trans” (3a and 3b). This leads to a full-fledged activation of the JAKs and maximal downstream signaling (4) (here: STAT5 phosphorylation). The different steps of the activation process are derived from a study using kinase-inactive, constitutively active and analog-sensitive mutants of JAK1 and JAK3 in the context of IL2 signaling.37
JAK2V617F-mediated activation of diverse signaling pathways
The activated JAKs phosphorylate tyrosine residues in the cytoplasmic part of the receptor, thereby providing docking sites for SH2 domain-containing signaling molecules (Fig. 2). JAK2V617F leads to constitutive activation of downstream signaling through the JAK-STAT (STAT5 and STAT3), the MAPK, and the PI3K/Akt pathways,23,49,52,53 which lead to the expression of the mitotic serine/threonine-protein kinases Pim, anti-apoptotic genes (BclxL and Bcl2), and cell cycle regulatory proteins (cyclin D1 and Cdc25A).54-58 This results in a proliferative advantage of the affected cells.23 It has recently been shown that STAT5 is absolutely essential for the cellular transformation mediated by JAK2V617F,59-61 whereas activation of Akt might also play a role in the process of transformation.62 JAK2V617F has been implicated in promoting transition from G1 to S phase of the cell cycle which could be reverted by the inhibition of JAK2V617F with a small molecule JAK inhibitor.63 The inhibition of JAK2V617F correlated with a decreased expression of cyclin D2 and an increased expression of the cyclin-dependent kinase (Cdk) inhibitor 1B (p27Kip1) (Fig. 2). p27 expression could also be blocked by Akt- or Erk1/2-mediated phosphorylation and subsequent degradation of FOXO transcription factors.64,65 JAK2 has also been reported to phosphorylate p27Kip1, thereby impairing its function and stability, which then leads to partial activation of Cdk and cell cycle progression.66 Pim kinases, which are upregulated by JAK2V617F-mediated signaling,50,57 have been described to inactivate Bad by phosphorylation, thereby activating the anti-apoptotic BclxL.57 Akt can also display its anti-apoptotic role via phosphorylation of BH3-only proteins resulting in a recruitment of Bcl2 and BclxL to the mitochondrial membrane.64 Furthermore Akt can inactivate Gsk3 by phosphorylation, thus impairing normal downstream Gsk3 functions such as inhibition of the cell cycle (e.g., by phosphorylation of cyclin/cyclin-dependent kinase complexes or by inhibition of mitotic transcription factors) or promotion of apoptosis (e.g., by increasing BH3-only protein [Bax/Bak]-mediated apoptosis or by inhibition of Bcl2 family member expression).64,67,68 Inhibition of FOXO by Akt is also known to lead to a downregulation of pro-apoptotic BH3-only proteins. Interestingly, the activation of Gsk3 by DNA damage stress was shown to synergize with JAK inhibitors in inducing apoptosis in cells expressing JAK2V617F.69

Figure 2. Schematic representation of pathways related to signaling of JAK2V617F and regulation of its expression levels. A number of possible pharmacological approaches have been described that target proteins in the scheme (e.g., mTOR, MEK, HSP90, Aurora A/B, …). Other kinases involved in these pathways (e.g., PI3K, Akt, Pim, Erk1/2, …) might also be promising targets for combination treatments. In addition to Aurora kinases, further kinases influencing cell cycle progression also represent interesting targets (cyclin-dependent kinases [Cdk] and polo-like kinases [Plk]). Inhibition of the Bcl-2 family members might counteract anti-apoptosis. Interference with JAK expression levels has been shown to suppress JAK-STAT signaling either by inhibiting chaperone functions (HSP90) or by using deubiquitinase inhibitors.216 Future approaches could also involve the targeting of adaptor proteins such as GAB1/2 which orchestrate the activation of the different signaling pathways in the signalosome at the receptor.
Additionally, it has also been described that JAK2V617F phosphorylates a histone arginine methyltransferase (PRMT5) and thus inhibits its activity resulting in altered chromatin modifications and gene expression.70 This contributes then to myeloproliferation and erythroid differentiation in JAK2V617F-positive cells. JAK2 has been described to phosphorylate histone H3 at tyrosine 41 resulting in the displacement of heterochromatin protein (HP) 1α71 leading to expression of leukemogenic oncogenes like LMO2. However, the direct implication of JAK2V617F in this process remains controversial,72 and it cannot be excluded that a kinase downstream of JAK2V617F may be involved in promoting this nuclear function. An active JAK homolog, HOP, in Drosophila has also been implicated in changes of chromatin condensation and STAT-independent gene transcription.73
Negative Regulatory Mechanisms of JAK Activity
To prevent a permanent and/or excessive activation of JAK-STAT signaling a number of negative regulatory mechanisms that modulate the pathway at different levels have been reported.
Phosphatases and PIAS proteins
Negative regulatory mechanisms include the dephosphorylation of cytokine receptors, JAKs or STATs by protein tyrosine phosphatases (PTP)74 or the prevention of STAT factors to bind DNA by protein inhibitors of activated STAT (PIAS).75 No specific regulations of JAK-STAT phosphatases or PIAS family members have been reported for JAK2V617F to our knowledge.
SH2B protein family members
LNK (SH2B3), an adaptor protein comprising a dimerization domain, proline-rich regions, a PH domain, and an SH2 domain, negatively regulates activated JAK2 by directly binding to the phosphorylated tyrosine residue 813 via its SH2 domain.76,77 LNK has been reported to negatively regulate TpoR and EpoR signaling.78,79 LNK mutations have been detected in JAK2V617F-positive and -negative myeloproliferative neoplasms80-83 and LNK mRNA in MPN patients was reported to positively correlate with JAK2V617F allele burden.84 Interestingly, other family members, SH2B1 (SH2Bβ) and SH2B2 (APS), have been described to associate with Janus kinases and to positively85-87 or negatively88-90 regulate their kinase activity. Concerning EpoR signaling, however, all three family members have been reported to act as negative regulators (SH2B190). Moreover, SH2B2 was reported to cooperate with CBL (see below) in doing so.88
Regulation of JAK and receptor protein expression (internalization, SOCS, and CBL)
On the cellular52 and the organism level as well as in patients (see sections below) it is well established that the levels of mutant JAK2V617F protein influence the signaling intensity and its pathological consequences. This underscores the importance of understanding the regulation of the cytokine receptor/JAK complexes at the protein level.
Cytokine signaling can be regulated on the level of plasma membrane localization of receptor/JAK complexes. Cytokine receptor/complexes can be internalized and processed either for recycling back to the plasma membrane or be targeted for degradation of their components via the lysosome or proteasome91-93 (reviewed in ref. 94). JAK2V617F has been described to lead to the internalization, ubiquitination, and degradation of TpoR.95
Downregulation by ubiquitination in the JAK-STAT pathway has been described to be mediated by two families of proteins, SOCS proteins and CBL proteins. Both types of proteins possess E3 ubiquitin ligase activity. Among the two types of ubiquitin ligases, SOCS and CBL proteins are both part of the RING finger E3 family, but they belong to different subgroups. While CBL proteins are single subunit E3s (having the RING finger and the substrate recruiting subunit on the same polypeptide chain), the SOCS proteins are part of the multi-subunit E3s (including a small RING finger protein, a member of the Cullin family, and multiple other subunits among which there is the substrate recruiting domain).96
The suppressor of cytokine signaling (SOCS) protein97 family (all having a central SH2 domain and a C-terminal SOCS box) comprises eight family members (SOCS1–7 and CIS) that can suppress JAK-STAT signaling by inhibiting JAK kinase activity, by competing with STAT factors for docking sites on the cytokine receptor and/or by facilitating the proteasomal degradation of signaling proteins. Constitutively active JAK2 mutants are susceptible to negative regulation by SOCS proteins, show decreased stability, increased ubiquitination, and are degraded via the proteasome.52 Thus, mechanisms interfering with this negative regulation could considerably contribute to the development and progression of MPNs by increasing the levels of constitutively active JAK2 mutants, although this is still under debate.98 Mechanisms that were reported to interfere with SOCS function are methylation,99-101 mutations,102 and deletions103 of SOCS genes. Importantly, epigenetic silencing of SOCS3 and SOCS1 was recently reported in about 40% of patients with Philadelphia chromosome-negative chronic myeloid disorders.104,105 The Casitas B-cell lymphoma (CBL) family consists of 3 mammalian members, CBL, CBL-b, and CBL-c. All CBL proteins have a conserved N-terminal tyrosine kinase binding domain (TKB) (itself comprised of a 4-helix bundle [4H], an EF-hand [EF] and an atypical SH2 domain) connected by an α-helical linker to a RING finger (RF) domain. C-terminally to the RF, CBL proteins contain proline-rich sequences, tyrosine residues and an ubiquitin-associated domain (UBA). CBL proteins can function as ubiquitin ligases but are also adaptor proteins which can mediate signal transduction events by offering binding sites for SH3 and SH2 domain-containing proteins.106 CBL proteins are known to mediate ubiquitination and degradation of kinases and were described to interact with many receptor tyrosine kinases, cytokine receptors, and cytoplasmic kinases (including the JAKs) and oncogenic mutants of CBL have been reported to uncouple kinases from degradation.107-109 CBL mutations are also found in myeloproliferative neoplasms110-113 and have been associated with a poor prognosis.
Myeloproliferative Neoplasms and JAK2 Mutations
Myeloproliferative neoplasms
Myeloproliferative neoplasms are characterized by a dysregulated enhanced proliferation of one or more of the myeloid lineages (i.e., the erythroid, granulocytic, megakaryocytic, and monocytic lineages), which is considered to result from genetic abnormalities at the level of hematopoietic stem/progenitor cells. Myeloproliferative neoplasms comprise chronic myeloid leukemia (Bcr-Abl-positive) (CML), polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF), chronic neutrophilic leukemia (CNL), chronic eosinophilic leukemia (CEL-NOS), mast cell disease, and unclassified myeloproliferative neoplasms (MPN-U). CML, PV, ET, and PMF were known since long to be clonal stem cell disorders.114-117 Patients suffering from MPN usually show an increased amount of functional and terminally differentiated myeloid cells (i.e., erythrocytes, granulocytes, monocytes, and/or platelets) in their peripheral blood. However, the diseases can progress to ineffective hematopoiesis and failure of the bone marrow due to myelofibrosis and/or transformation to acute leukemia.
In addition to CML (for which the fusion protein kinase Bcr-Abl was already identified as the disease-causing mutation118), three other MPNs (PV, ET, and PMF) were shown to harbor a mutated kinase—JAK2V617F,19,20,22,23,119 which can result from a heterozygous or homozygous mutation. Cells homozygous for JAK2V617F can be found in most of the PV patients but only rarely in ET patients.120 The homozygous mutation was demonstrated to result from a duplication of the mutant allele by mitotic recombination.20-23
Polycythemia vera
Polycythemia vera (PV) is the only acquired primary polycythemia. It has an incidence of 1–3 per 100 000 people per year and is most frequently diagnosed in people aged between 60 and 70 y. The vast majority of PV patients is positive for the JAK2V617F mutation and most of them bear cells which are homozygous for the mutation.120 PV patients, who do not carry the JAK2V617F mutant, mostly display other activating mutations in exon 12 of JAK2 (see Fig. 3).121
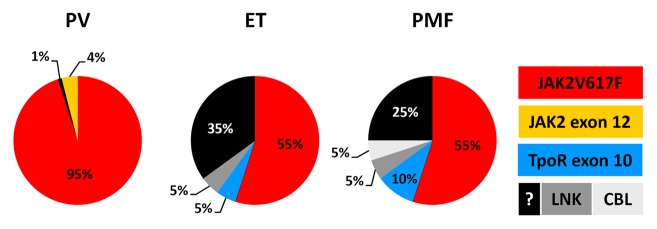
Figure 3. Proportion of patients with PV, ET, or PMF carrying different genetic abnormalities related to JAK-STAT signaling.
Polycythemia vera is characterized by the dysregulated proliferation of the erythroid, granulocytic, and/or megakaryocytic lineages. This leads to the hypercellularity of the bone marrow (i.e., panmyelosis) and an increase of the red cell mass in the peripheral blood as well as leukocytosis and thrombocytosis. However, patients with mutations in JAK2 exon 12 mainly demonstrate an isolated erythrocytosis without associated increase of platelet number or white blood count.122,123 In contrast to PMF and ET, the megakaryocytes in PV show mainly a normal phenotype and size.
The course of PV can be divided into three phases:124 (1) the pre-polycythemic phase characterized by a borderline or mild erythrocytosis often in combination with significant thrombocytosis (sometimes associated with thrombotic events), (2) the apparent polycythemic phase, and (3) the post-polycythemic phase defined by cytopenia (including anemia), bone marrow fibrosis, and extramedullary hematopoiesis (post-polycythemia myelofibrosis). Almost all patients are diagnosed when they are in the polycythemic phase and the first symptoms appear. These include e.g., headache, dizziness, paresthesia, aquagenic pruritus, and erythromelalgia mainly due to thrombotic events in the microvasculature. However, a thrombosis of major blood vessels (e.g., splanchnic vein thrombosis) can occur as well. Additionally, many patients suffer from splenomegaly and/or hepatomegaly. Upon appropriate treatment the survival time of PV is very much prolonged, but life expectancy of PV patients is nevertheless reduced when compared with that of the general population.125
The probability of PV patients to develop a post-polycythemic myelofibrosis is ~15% at 10 y and ~35% at 15 y after the initial diagnosis.126 A major risk factor to progress to myelofibrosis seems to be the JAK2V617F allele load since the incidence is much higher in patients with a high JAK2V617F allele burden compared with those with a low allele load.126,127 On the other hand, the incidence of progression to myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML) is very low, but is increased with higher age at diagnosis or due to treatment with certain cytotoxic agents (e.g., busulfan or pipobroman128).
Essential thrombocythemia
ET has an annual incidence of 0.5–2.5 per 100 000 people.129 It can occur at any age (including children), but the disease is mostly diagnosed in patients who are in their sixties or around 30 y old.130 Approximately half of the ET patients carry the JAK2V617F mutation; these patients mainly bear cells that are heterozygous for the mutation.120 About 5% of the ET patients are positive for a mutation in exon 10 of the Tpo receptor and additional 5% bear a mutation in the adaptor protein LNK. The remaining ET patients (~1/3) do not display any known mutation affecting the JAK-STAT signaling pathway (see Fig. 3).
Essential thrombocythemia is mainly characterized by an enhanced proliferation of the megakaryocytic lineage leading to sustained thrombocytosis (with a platelet count ranging from more than 450 to more than 2000 × 109/l). The platelets are not equal in size ranging from small to giant and display abnormal functions (e.g., spontaneous aggregation and activation) resulting in an increased risk of thrombosis and/or bleeding.131 The bone marrow of ET patients is typically normal or slightly hypercellular apart from the megakaryocytic lineage. The number of megakaryocytes is elevated and megakaryocytes in ET patients have extremely lobulated nuclei and their size is increased varying from large to giant.
In general, ET is a rather indolent disorder with long symptom-free periods and only occasional events of thrombosis or bleeding. Up to 50% of the patients are asymptomatic at diagnosis; the disease is then mostly detected by a routine examination. The other patients demonstrate symptoms related to thrombotic events in the microvasculature. However, the thrombosis of major blood vessels (leading to, e.g., seizures, stroke, myocardial infarct, and deep-vein thrombosis) can occur as well. The life expectancy of the majority of ET patients is near normal132 and only a minority of patients either progress to post-ET myelofibrosis or to AML.133
Primary myelofibrosis
Myelofibrosis is defined as an increase in quantity and density of extracellular matrix proteins, which normally provide a scaffold for the hematopoietic (stem and progenitor) cells in the bone marrow. Myelofibrosis can occur secondary to, e.g., infections and inflammatory or neoplastic disorders.
Primary myelofibrosis (PMF) occurs with an incidence of 0.5–1.5 per 100 000 people per year. The median age at diagnosis is usually >70 y.134 Importantly, the clinical characteristics of post-polycythemic or post-ET myelofibrosis are the same as for PMF in the fibrotic phase and can only be distinguished when the initial disease was well diagnosed. Approximately half of the patients with PMF carry the JAK2V617F mutant, whereas approximately 10% are positive for a mutation in exon 10 of the Tpo receptor. Additionally, mutations in the adaptor proteins LNK or CBL can be found in PMF patients as well (each ~5%). The remaining PMF patients (~25%) do not display any known mutation affecting the JAK-STAT signaling pathway (see Fig. 3).
Primary myelofibrosis is characterized by enhanced proliferation mainly of the megakaryocytic lineage and the alteration of the bone marrow structure including progressive myelofibrosis and hyperactive angiogenesis, which is often accompanied by extramedullary hematopoiesis. The disease course can be divided in two phases:124 The prefibrotic or early phase is characterized by a hypercellular bone marrow (due to an increase of the megakaryocytic and the granulocytic lineages; erythropoiesis is often decreased) with no or slight reticulin fibrosis and an increased platelet count in the peripheral blood. The fibrotic phase displays a hypocellular bone marrow with marked reticulin and/or collagen fibrosis and also osteosclerosis. Megakaryocytes and platelets for instance produce PDGF, TGFβ, or OSM,135,136 which stimulate fibroblast proliferation and activity. The peripheral blood of PMF patients in the fibrotic phase demonstrates decreased erythrocyte levels up to anemia, low levels of large abnormal platelets, and also leukopenia. Moreover, the plasma levels of inflammatory cytokines (e.g., IL1β, IL6, IL8, IFNγ, and TNFα) are highly increased.137,138 In the advanced stages, bone marrow failure results in relocation of the hematopoiesis to other organs. Most common sites of extramedullary hematopoiesis are the spleen and the liver, but any other organ (e.g., kidney, lung, or the gastrointestinal tract) can be affected. Bone marrow failure also leads to high levels of CD34+ cells in the peripheral blood, which normally reside in the bone marrow.
The median overall survival of PMF patients who have been diagnosed in the fibrotic phase is approximately five years. However, the survival times can be much longer if the disease has been diagnosed in the prefibrotic stage.132,139 The main causes of death for PMF patients include the progression to acute leukemia (observed in 20% of the patients at 10 y of diagnosis), infection, and bleeding secondary to bone marrow failure, and portal hypertension or hepatic failure caused by hepatic vein thrombosis or extramedullary hematopoiesis.125
JAK2 mutations and other mechanisms contributing to PV, ET, and PMF
The discovery of an activating mutation downstream of cytokine receptors playing an essential role in myeloid hematopoiesis was a major breakthrough in understanding the development of the Philadelphia chromosome-negative MPNs. However, this raised the question of how a single mutation can lead to the development of three distinct diseases (PV, ET, and PMF). Subsequently, it was demonstrated in a murine bone marrow transplantation model introducing JAK2V617F-positive cells that the MPN phenotype was influenced by the genetic background of the respective mouse strain.140 Furthermore, the amount of JAK2V617F seems to play a role in the pathogenesis as well, given that cells that are homozygous for JAK2V617F are more often found in PV patients than in ET patients.120 This could be recapitulated in a JAK2V617F transgenic mouse model, which allowed the expression of varying ratios of JAK2 wild-type to JAK2V617F.25 Low expression of JAK2V617F resulted in a MPN phenotype resembling human ET, while higher expression of JAK2V617F led to a PV-like phenotype.
Since 2005, many more mutations affecting proteins important in JAK-STAT signaling have been identified in JAK2V617F-negative MPN patients (for a review see ref. 11). Scott and colleagues discovered several additional mutations in exon 12 of JAK2 including K539L by sequencing JAK2 in JAK2V617F-negative PV patients. Several more point mutations, deletions, and insertions affecting JAK2 exon 12 have been identified in PV patients since then.11,122 Somatic gain of function mutations often affecting the amino acid residues W515 and S505 have been found in the Tpo receptor gene (MPL) of patients with ET and PMF.141-145 Both JAK2 exon 12 (K539L) and Tpo receptor (W515K/L) mutants have been shown to lead to the transformation of BA/F3 cells and induce a MPN-like phenotype in murine bone marrow transplantation models.122,142 Furthermore, mutations in adaptor proteins involved in the negative regulation of cytokine signaling, i.e., LNK and CBL, have been described in ET and PMF patients.80,112
A great variety of new data could contribute to understand the development of the three diseases with differing phenotypes. The high proportion of patients with ET and PMF not displaying any known mutation affecting JAK-STAT signaling shows that for these two diseases at least other players can be sufficient to induce the disease state. Indeed, further recurrent somatic mutants of different proteins (e.g., ASXL1, EZH2, IKZF1, IDH1/2, RUNX1, and TET2) have been found with variable frequency in PV, ET, and PMF.111,146 Some of the affected proteins are implicated in the epigenetic regulation (e.g., ASXL1 and TET2), whereas IKZF1 and RUNX1 are transcription factors. The different mutations are not specific for any of the MPN subtypes and can occur concomitantly with JAK2V617F or the other mutations. However, these mutations and/or their accumulation might partially explain the clinical differences among PV, ET, and PMF.147 Furthermore, some of the mutations are associated to disease progression and are more frequently found in post-MPN acute leukemia (e.g., mutants of IDH1/2, IKFZ1, and also LNK146).
Additional mechanisms like epigenetic silencing, post-transcriptional regulation, or post-translational modifications could account for the development of different phenotypes. For instance, it has been reported that the SOCS1, SOCS2, and SOCS3 genes are hypermethylated in MPN.104,105,148-150 Furthermore, the comparison of microRNA expression in MPN patients and healthy controls identified among others miRNA-150 to be differentially expressed.151-153 Interestingly, miRNA-150 has been reported to regulate the lineage fate in megakaryocyte–erythrocyte progenitor cells.154 Furthermore, Pardanani and colleagues found several germline single nucleotide polymorphisms (SNPs) in the region of the JAK2 gene that are different in PV and ET patients and could contribute to the differences in MPN phenotype.155 Subsequently, several groups reported that a common haplotype (referred to as “46/1”) in the JAK2 locus is associated with the acquisition of JAK2V617F as well as the development of MPN.156-158 They demonstrated that patients who were heterozygous for this haplotype were significantly more likely to acquire JAK2V617F. The same haplotype also predisposes to mutations in JAK2 exon 12 as well as in the Tpo receptor.159,160 However, the mechanism by which a germline SNP in the 46/1 haplotype increases the risk to develop MPN or acquire JAK2V617F or other mutations is not known. In general, the 46/1 haplotype seems to be a major germline factor involved in MPN development and to date no other common SNP associated with MPN has been reported.147 The newly discovered genetic abnormalities also played a central role in the revision of the WHO classification for MPN in 2008161 as they can be used as diagnostic parameters. The new classification includes CML, the “classic” Philadelphia chromosome-negative MPN (PV, ET, and PMF) and several other rare diseases that demonstrate many features of MPN.
Inflammation and an aberrant activation of the JAK-STAT signaling pathway are also hallmarks of MPN162-165 irrespective of mutations influencing the JAK-STAT pathway. The JAK-STAT pathway not only drives myeloproliferation but also mediates the activity of inflammatory cytokines, whose levels are commonly increased in myelofibrosis patients.137,138 Since an initiating event in MPN is not known, inflammation has also been discussed to be an incipient event. It has been reviewed recently166 that inflammation can induce epigenetic changes and genomic mutations. High levels of inflammatory cytokines and chemokines are found in the plasma of MPN patients and in supernatants of cells expressing JAK2V617F136-138,167-170 and a number of cytokines, e.g., IL6, IL11, TNFα, and HGF have been reported to promote survival of cells carrying JAK2V617F.171-173 Cytokines are involved in the development of fibrosis, e.g., megakaryocytes and platelets produce PDGF, TGFβ, or OSM,135,136 which stimulate fibroblast proliferation and activity. On the other hand, the stroma also secretes cytokines, which regulate the behavior of JAK2V617F mutated cells.171-173
Janus Kinase Inhibitors in the Treatment of MPN
Classic treatment of MPNs
For PV and ET the treatment rationale is mainly the prevention of thrombotic complications which is the major reason for morbidity and mortality in these patients.174 Low-risk patients with PV are normally treated with phlebotomy and low-dose aspirin. High-risk PV patients additionally receive hydroxyurea or pegylated IFN-α as first-line treatment. ET patients at low thrombotic risk are either monitored without therapeutic intervention or they receive low-dose aspirin as well. High-risk patients with ET are usually treated with hydroxyurea, pegylated IFN-α, or anagrelide.
There are several treatment approaches for patients with myelofibrosis that are primarily aimed at relieving the diverse disease symptoms and improve the patient’s quality of life. The only curative treatment of myelofibrosis is allogeneic hematopoietic stem cell transplantation (HSCT). However, the mortality and morbidity of this procedure is still very high and it is questionable if it leads to substantial increase in overall survival for eligible patients.174 The main issues that are targeted by conventional treatment strategies are anemia and splenomegaly/extramedullary hematopoiesis. Blood transfusion or treatment with corticosteroids, androgens, or erythropoiesis-stimulating agents is used to treat the anemia. Anemia as well as splenomegaly can be treated with immunomodulatory agents like thalidomide or lenalidomide. Furthermore, cytoreductive drugs as hydroxyurea or pegylated IFNα or chemotherapeutic agents (e.g., cladribine or decitabine) are used to reduce the spleen size. Alternative treatment options for splenomegaly/extramedullary hematopoiesis are radiation therapy or splenectomy, either of which is rare and only performed if no other treatment option is feasible. However, there is no evidence that any conventional treatment approach improves the constitutional symptoms.175 In addition, none of the conventional treatment strategies except allogeneic stem cell transplantation shows durable effects/benefits and they also demonstrate significant toxicities.176-179
Treatment of MPN with JAK inhibitors
The discovery of the JAK2V617F mutant defined JAK2 as “druggable” target for Philadelphia chromosome-negative MPNs. Although JAK2V617F is not found in all patients with ET and PMF, an aberrant activation of the JAK-STAT signaling pathway plays a central role in the pathogenesis of most PV, ET, and PMF patients.162 The JAK-STAT pathway not only drives myeloproliferation but also mediates the activity of inflammatory cytokines, whose levels are commonly increased in myelofibrosis patients.137,138 Since 2005, many inhibitors of JAK(2) have been developed; several of those are currently evaluated in clinical trials (see Table 1).
Table 1. JAK inhibitors in clinical trials for myelofibrosis.
| Compound | Targets | Clinical trial | Responses observed so far |
|---|---|---|---|
| AZD1418 |
JAK1/2217 |
I/II |
No information available |
| BMS911543 |
JAK2218 |
I/II |
No information available |
| CEP701 |
JAK2/3219 |
II |
Splenomegaly ↓220 |
| CYT387 |
JAK1/2, Tyk2168 |
II |
Splenomegaly ↓ Improvement of constitutional symptoms Improvement of anemia221 |
| LY2784544 |
JAK2222 |
II |
Splenomegaly ↓ Improvement of constitutional symptoms (improvement of bone marrow fibrosis)223 |
| NS-018 |
JAK2224 |
I/II |
No information available |
| SB1518 (Pacritinib) |
JAK2, Tyk2225 |
I/II |
Splenomegaly ↓ Improvement of constitutional symptoms226 |
| TG101348 (SAR302503) |
JAK2227 |
III |
Splenomegaly ↓ Improvement of constitutional symptoms Normalization of leukocytosis and thrombocytosis (JAK2V617f allele burden ↓)189 |
| XL019 | JAK2228 | I/terminated | Neuronal toxicity229 |
INCB018424 (ruxolitinib, Jakafi®)
To date, only ruxolitinib (INCB018424) has received approval by the FDA (in November 2011) and the European Commission (in August 2012) for the treatment of intermediate- and high-risk myelofibrosis (primary and post-PV/ET). Ruxolitinib is a JAK1 and JAK2 inhibitor.180 The basis of its approval were two phase III clinical studies for myelofibrosis (COMFORT I and II) which provided evidence that application of ruxolitinib leads to the reduction of spleen size and an improvement of symptoms.181,182 In addition, ruxolitinib decreases leukocytosis and thrombocytosis as well as inflammatory cytokine levels and thereby enhances the patients’ quality of life. Recently, long-term results from the before mentioned studies have shown that ruxolitinib-treated patients have a survival advantage over the control groups (placebo in COMFORT I, best available therapy [BAT] in COMFORT II) and that the JAK2V617F allele burden was reduced (>20% in 13% of the patients).181,183-186 Interestingly, also the requirement of blood transfusions (due to the side effects of anemia and thrombocytopenia) observed in the early phases for patients receiving ruxolitinib decreased to rates similar to the control groups.
It will be interesting to determine to what extent the relief of symptoms in myelofibrosis patients by ruxolitinib is in fact due to the inhibition of inflammatory cytokine action (via its role as JAK1 and JAK2 inhibitor). This will probably only be recognized when data from studies with more JAK2-specific inhibitors advance to the same stage in clinical studies. As mentioned before, inflammatory cytokines are a hallmark of myelofibrosis (even in those cases without apparent mutations affecting the JAK-STAT pathway).
Also for the treatment of PV it will be interesting to follow the performance of specific JAK2 vs. multi-JAK inhibitors since PV patients do not generally demonstrate elevated serum levels of inflammatory cytokines. In fact, the phenotype of PV is mainly characterized by myeloproliferation resulting in the increase of red blood cell count often accompanied by leukocytosis and/or thrombocytosis. However some studies have shown that inflammatory cytokines are also detectable in PV and contribute to the growth of clonal erythroblast independently of JAK2V617F.169,173 Additionally, the underlying mechanism of PV is more closely connected to hyperactivated JAK2, since almost all PV patients either bear the JAK2V617F mutant or a mutation in exon 12 of JAK2. Thus, one might speculate that in the treatment of PV a JAK2-specific inhibitor (e.g., TG101348) might be more efficient; however, this remains to be shown. Ruxolitinib has been assessed in a phase II clinical trial in PV and ET patients intolerant or resistant to treatment with hydroxyurea.187 Application of ruxolitinib led to a decrease of hematocrit levels, platelet count, and JAK2V617F allele burden.188 The most common side effect was anemia for both patient cohorts, which was clinically well manageable. Two clinical studies on PV patients (http://www.clinicaltrials.gov/, NCT01243944 [RESPONSE] and NCT01632904 [RELIEF]) are currently being conducted.
TG101348 (SAR302503)
TG101348, an inhibitor described to be specific for JAK2, is also evaluated in a phase II clinical trial in patients with PV and ET (http://www.clinicaltrials.gov/, NCT01420783). When tested in a phase I/II clinical trial in myelofibrosis patients, it led to the normalization of leukocytosis and thrombocytosis, while a decrease in inflammatory cytokine levels could not be observed for this compound.189 This suggests that TG101348 acts rather anti-proliferative than anti-inflammatory. So it will be very interesting, how this inhibitor with a stronger preference for JAK2 in in vitro kinase assays will perform in myelofibrosis, PV, and ET patients in comparison to ruxolitinib.
Other JAK inhibitors
Many potent JAK inhibitors (showing nanomolar activities in intact cell assays) have been developed in the last years and several are evaluated in clinical trials.177,190-193Table 1 shows promising JAK(2) inhibitors in clinical trials for MPN. More comparative studies of these inhibitors are needed to show possible differences of potency and to uncover potential additional activities of these compounds (see ref. 194). For instance CEP701, a JAK2 inhibitor, was recently shown to also target Aurora kinases in the sub-micromolar concentration range in intact cells.194
However, most of the JAK inhibitors demonstrate inhibitory activity toward more than one JAK family member (or other kinases), which, on the other hand, might be beneficial in the setting of inflammatory disorders. In line with this, tofacitinib (CP-690550, a pan-JAK inhibitor) has been successfully applied in patients with rheumatoid arthritis195 and has recently been approved by the FDA (Xeljanz®) for the treatment of patients with moderately to severely active rheumatoid arthritis.
The majority of ATP-competitive kinase inhibitors bind the kinase domain of their respective targets in the active state (also known as type I inhibitors); the clinically approved drugs gefitinib, erlotinib, and sunitinib are prominent examples of this inhibitor class.196 Most inhibitors developed against Janus kinases are type I inhibitors.197 Since kinase domains in their active conformation are highly similar to each other it is especially difficult to accomplish high selectivity by using type I inhibitors. A strategy to gain selectivity would be the targeting of the inactive conformation of a kinase domain. This class of compounds (type II inhibitors) also acts ATP-competitively but targets an extended ATP-binding site by spreading into the hydrophobic deep pocket which is only accessible in the inactive conformation of the kinase.196 Recently, NVP-BBT594 (originally designed to target Bcr-Abl) was described as first compound to bind JAK2 in its inactive conformation.197
Some of the JAK targeting compounds (including some that are not tested in clinical trials) are also very valuable tools for research: some by their pan-JAK activity and some by their specificity for individual JAKs. Table 2 shows some of these potent inhibitors of Janus kinases that are commercially available.
Table 2. A selection of potent commercially available JAK inhibitors.
| Compound | JAK1 | JAK2 | JAK3 | Tyk2 | References |
|---|---|---|---|---|---|
| AZ960 |
- |
3 |
9 |
- |
57 |
| BMS911543 |
356 |
1 |
73 |
66 |
218 |
|
CEP33779 |
~72 |
1.8 |
85 |
~1440 |
230 |
| JAK inhibitor 1 (JI1, pyridone 6, CMP6) |
15 |
1 |
5 |
1 |
231 |
| NVP-BSK805 |
~32 |
0.5 |
~19 |
~11 |
232 |
| TG101209 | - | 6 | 169 | - | 233 |
IC50 values in nM obtained in in vitro kinase assays
Combination treatment with JAK2 inhibitors
Combinations of different kinase inhibitors have been shown to have beneficial effects on growth inhibition of JAK2V617F-expressing cells. The combination of an Aurora kinase inhibitor with a JAK2 inhibitor has recently been shown to synergistically reduce the proliferation of JAK2V617F-positive cells.194 Also the use of a JAK2 inhibitor in combination with the suppression of the PI3K/Akt/mTOR pathway synergistically reduces the proliferation of JAK2V617F-positive cells.198,199 Moreover, a combined application of an inhibitor of the dual specificity mitogen-activated protein kinase kinase (MEK)—selumetinib (AZD6244)—and a JAK2 inhibitor has been demonstrated to act synergistically on the proliferation of JAK2V617F-positive cells.200
Additionally, compounds modifying the epigenome have been tested for their potential therapeutic activity in MPN. However, it is not clear if there is a therapeutic indication for DNA demethylation in MPN since the reports on alterations in DNA methylation patterns are controversial. Demethylating agents as azacitidine and decitabine are tested as single drug or in combination with JAK2 inhibitors in MPN patients.177 Barrio and colleagues found a homogeneous and very similar methylation pattern in MPN compared with healthy control populations.201 On the other hand, it was described that PV and ET are characterized by an aberrant hypermethylation while PMF is characterized by both aberrant hyper and hypomethylation.202 Histone deacetylases (HDACs) are also known to epigenetically regulate gene expression by removing acetyl groups from lysine residues on histone proteins and also non-histone proteins like transcription factors.203,204 It has been shown that both the level and activity of HDACs are elevated in primary myelofibrosis patients.205 Therefore the potent pan-HDAC inhibitor panobinostat (LBH589) has been evaluated in vitro in JAK2V617F-positive cells.206 The treatment with panobinostat decreased JAK2V617F expression levels and its downstream signaling probably by mediating hyperacetylation of heat shock protein (HSP) 90 and thereby disrupting the association between JAK2 and the chaperone, leading to its proteasomal degradation. Myelofibrosis patients treated with panobinostat as a single agent experienced an improvement of constitutional symptoms and a reduction of spleen size.205,207 Moreover, when applying a JAK2 inhibitor and panobinostat in combination, the proliferation of JAK2V617F-positive cells was synergistically suppressed206 and demonstrated enhanced efficacy in comparison to each single agent in murine MPN models.208 Based on these findings a phase I clinical trial was initiated to test the combination of ruxolitinib and panobinostat in myelofibrosis patients (http://www.clinicaltrials.gov/, NCT01433445). As mentioned, the disturbance of the association between JAK2V617F and its chaperone HSP90 can lead to lower JAK2V617F expression levels. This can also be achieved by inhibiting HSP90. It has been shown that the inhibition of HSP90 chaperone function by e.g., PU-H71 or AUY922 leads to the loss of binding to JAK2 resulting in attenuated expression of JAK2 (V617F) and inhibition of JAK-STAT signaling. The combination of a JAK2 inhibitor and a HSP90 inhibitor showed enhanced efficacy in the proliferation of JAK2V617F-positive cells in comparison to each single compound.209,210 Furthermore, AUY922 was demonstrated to overcome resistance to JAK2 inhibitor treatment in cells expressing JAK2V617F.209,211 Taken together, inhibition of HSP90 and/or the combination with JAK2 inhibitors might be a valuable treatment approach to test in MPN patients, especially in those who do not respond to JAK2 inhibitory treatment. However, it has to be considered that HSP90 has many other client proteins besides JAK2 that are prone to degradation upon inhibition of HSP90 as well. This might lead to additional side effects compared with a more specific treatment.
In conclusion, a combination of JAK2 inhibitors with other agents that have demonstrated a clinical benefit in MPN patients might help to further improve the treatment outcome in comparison to JAK2 inhibitors as single drug. Thereby, the efficacy of the treatment can be enhanced while possibly decreasing the drug dosage resulting in reduced toxicity. In addition, combining two compounds with different mechanisms of action would decrease the probability of developing resistance to either of the drug.
Perspectives
The clinical development of ruxolitinib and other JAK inhibitors appears to be a breakthrough in the treatment of myelofibrosis patients. These drugs significantly improve the patients’ quality of life, which is remarkable progress over conventional treatment strategies. In addition to the reduction of symptoms, the recent data indicate that ruxolitinib treatment leads to a reduction of the JAK2V617F allele load and presents a survival advantage. It will be interesting to follow up to what extent the ruxolitinib-induced relief of symptoms and decrease of JAK2V617F allele load in myelofibrosis and PV is due to the inhibition of inflammatory cytokine action (since ruxolitinib targets both JAK1 and JAK2). This will probably only be recognized when data from studies with more JAK2-specific inhibitors (e.g., TG101348 [SAR302503] or BMS911543) will have reached comparable stages in clinical studies. It is conceivable that a JAK2-specific inhibitor might actually perform less well in comparison to ruxolitinib, due to a lack of activity against JAK1. It could also be possible, that a specific JAK2 inhibitor might be more adequate for the treatment of PV, as almost all PV patients carry a mutant of JAK2 (V617F or exon 12 mutations) and the inflammatory cytokine levels are much lower in PV patients than in myelofibrosis patients. For PV and JAK2V617F-positive ET patients a JAK1-targeting inhibitor might also have more undesired side effects.
No JAK2-specific compound has yet been approved for clinical application and the development of specific JAK inhibitors also for other indications besides MPN is still required. Additionally, the generation of a JAK2-specific inhibitor targeting the inactive state of the kinase (type II inhibitor)197 is especially interesting. If type II inhibitors are more efficient in inhibiting JAK2 (V617F) activity and reducing the JAK2V617F allele burden compared with a type I compound remains to be elucidated. The occurrence of JAK2 mutations in MPN patients conferring resistance to JAK2 inhibition has not been described so far. However, the acquisition of secondary mutations to evade therapeutic targeting is a common mechanism in cancer.212 Nevertheless, several mutations in the JAK2 kinase domain that evade JAK2 inhibition have been identified in in vitro studies.211,213-215 These mutations may also emerge in patients under prolonged JAK2 inhibitory treatment. More specific JAK inhibitors are necessary to investigate the above mentioned issues and will provide more insight in understanding the perspective of JAK inhibitors in the treatment of MPN.
Furthermore, the disease-driving mechanisms in the three MPNs with high JAK2V617F levels have not been fully elucidated. It is not well understood how the various genetic aberrations interact and contribute to the pathogenesis of MPN. Thus, the elucidation of underlying molecular mechanisms including the interplay between the JAK-STAT signaling pathway (with JAK2 as central node), other signaling pathways and epigenetic abnormalities remains a major subject of research in the field of MPN. Better therapies for MPN patients are sought, which provide better treatment of symptoms, can efficiently change the course of these disorders and increase the patients’ survival time. The development of combination treatment approaches affecting key cellular regulators (including JAK2 inhibitors as well as other drugs) might contribute to reach this goal.
Acknowledgments
This work was also supported by the grant “FSC-PUL09-MyeloJAK” of the University of Luxembourg. KG was funded by the grant “Aides à la Formation-Recherche” of the Fonds National de la Recherche Luxembourg (FNR, AFR PHD-08-030).
Glossary
Abbreviations:
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- Bad
Bcl2-associated agonist of cell death
- Bax
Bcl2-associated X protein
- Bcl-2
B-cell CLL/Lymphoma 2
- Bcl-xL
B-cell lymphoma-extra large
- Bcr
breakpoint cluster region protein
- Bcr-Abl
Bcr/Abl fusion protein
- Bim
Bcl-2 interacting mediator of cell death
- c-Abl
Abelson tyrosine protein kinase 1
- cA/cD
cyclin A/cyclin D
- CBL
Casitas B-cell lymphoma
- CD
cluster of differentiation
- Cdc25A
cell division cycle 25 homolog A
- Cdk
cyclin-dependent kinase
- CML
chronic myeloid leukemia
- CMML
chronic myelomonocytic leukemia
- EGF
epidermal growth factor
- eIF2α
eukaryotic translation initiation factor 2-alpha
- Epo
erythropoietin
- ET
essential thrombocythemia
- FDA
US Food and Drug Administration
- FERM
four point one protein, ezrin, radixin, moesin
- FOXO
forkhead box protein O
- GCN2, general control non-derepressible 2
eIF2α kinase 4
- G-CSF
granulocyte colony stimulating factor
- GM-CSF
granulocyte macrophage colony stimulating factor
- Grb2
growth factor receptor-bound protein 2
- Gsk3
glycogen synthase kinase 3
- HES
hypereosinophilic syndrome
- HSP90
heat shock protein 90
- IL
interleukin
- IFN
interferon
- KAK
Janus kinase
- JMML
juvenile myelomonocytic leukemia
- KD
kinase domain
- LNK
lymphocyte linker protein
- MCL1
myeloid cell leukemia sequence 1
- MDS
myelodysplastic syndrome
- mdm2
mouse double minute 2 homolog
- MEK1
MAP kinase/Erk kinase 1
- MPN
myeloproliferative neoplasm
- mTOR
mammalian target of rapamycin
- mTORC
mammalian target of rapamycin complex
- PDGF
platelet-derived growth factor
- PI3K
phosphoinositide 3 kinase
- Pim
proviral integration site for Moloney murine leukemia virus
- PTEN
phosphatase and tensin homolog
- PV
polycythemia vera
- PDK1
3-phosphoinositide-dependant protein kinase-1
- PCM1
pericentriolar material 1
- PIAS
protein inhibitor of activated STAT
- PKD
pseudokinase domain
- PMF
primary myelofibrosis
- PPT
protein phosphatase
- RARS (-T)
refractory anemia with ringed sideroblasts (-with thrombocytosis)
- SCID
severe combined immunodeficiency
- SH2
scr homology 2
- SNP
single nucleotide polymorphism
- SOCS
suppressor of cytokine signaling
- Sos
son of sevenless
- STAT
signal transducers and activators of transcription
- Tpo
thrombopoietin
- TSC
tuberous sclerosis
- TSLP
thymic stromal lymphopoietin
- Tyk2
tyrosine kinase 2
- VEGF
vascular endothelial growth factor
- XIAP
X-linked inhibitor of apoptosis
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/25025
References
- 1.Wilks AF. Two putative protein-tyrosine kinases identified by application of the polymerase chain reaction. Proc Natl Acad Sci U S A. 1989;86:1603–7. doi: 10.1073/pnas.86.5.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Velazquez L, Fellous M, Stark GR, Pellegrini S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell. 1992;70:313–22. doi: 10.1016/0092-8674(92)90105-L. [DOI] [PubMed] [Google Scholar]
- 3.Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93:373–83. doi: 10.1016/S0092-8674(00)81166-6. [DOI] [PubMed] [Google Scholar]
- 4.Neubauer H, Cumano A, Müller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93:397–409. doi: 10.1016/S0092-8674(00)81168-X. [DOI] [PubMed] [Google Scholar]
- 5.Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93:385–95. doi: 10.1016/S0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 6.Park SY, Saijo K, Takahashi T, Osawa M, Arase H, Hirayama N, et al. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity. 1995;3:771–82. doi: 10.1016/1074-7613(95)90066-7. [DOI] [PubMed] [Google Scholar]
- 7.Karaghiosoff M, Neubauer H, Lassnig C, Kovarik P, Schindler H, Pircher H, et al. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity. 2000;13:549–60. doi: 10.1016/S1074-7613(00)00054-6. [DOI] [PubMed] [Google Scholar]
- 8.Seto Y, Nakajima H, Suto A, Shimoda K, Saito Y, Nakayama KI, et al. Enhanced Th2 cell-mediated allergic inflammation in Tyk2-deficient mice. J Immunol. 2003;170:1077–83. doi: 10.4049/jimmunol.170.2.1077. [DOI] [PubMed] [Google Scholar]
- 9.O’Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(Suppl):S121–31. doi: 10.1016/S0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- 10.Strobl B, Stoiber D, Sexl V, Mueller M. Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front Biosci. 2011;16:3214–32. doi: 10.2741/3908. [DOI] [PubMed] [Google Scholar]
- 11.Haan C, Behrmann I, Haan S. Perspectives for the use of structural information and chemical genetics to develop inhibitors of Janus kinases. J Cell Mol Med. 2010;14:504–27. doi: 10.1111/j.1582-4934.2010.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riese RJ, Krishnaswami S, Kremer J. Inhibition of JAK kinases in patients with rheumatoid arthritis: scientific rationale and clinical outcomes. Best Pract Res Clin Rheumatol. 2010;24:513–26. doi: 10.1016/j.berh.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Mahindra A, Cirstea D, Raje N. Novel therapeutic targets for multiple myeloma. Future Oncol. 2010;6:407–18. doi: 10.2217/fon.10.2. [DOI] [PubMed] [Google Scholar]
- 14.O’Shea JJ, Husa M, Li D, Hofmann SR, Watford W, Roberts JL, et al. Jak3 and the pathogenesis of severe combined immunodeficiency. Mol Immunol. 2004;41:727–37. doi: 10.1016/j.molimm.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 15.Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffé M, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–12. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 16.Peeters P, Raynaud SD, Cools J, Wlodarska I, Grosgeorge J, Philip P, et al. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood. 1997;90:2535–40. [PubMed] [Google Scholar]
- 17.Reiter A, Walz C, Watmore A, Schoch C, Blau I, Schlegelberger B, et al. The t(8;9)(p22;p24) is a recurrent abnormality in chronic and acute leukemia that fuses PCM1 to JAK2. Cancer Res. 2005;65:2662–7. doi: 10.1158/0008-5472.CAN-04-4263. [DOI] [PubMed] [Google Scholar]
- 18.Griesinger F, Hennig H, Hillmer F, Podleschny M, Steffens R, Pies A, et al. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11.2) translocation in a patient with a clinically typical chronic myeloid leukemia. Genes Chromosomes Cancer. 2005;44:329–33. doi: 10.1002/gcc.20235. [DOI] [PubMed] [Google Scholar]
- 19.Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280:22788–92. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 21.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 22.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Cancer Genome Project Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 23.James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 24.Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108:1652–60. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 25.Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111:3931–40. doi: 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 26.Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107:4274–81. doi: 10.1182/blood-2005-12-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akada H, Yan D, Zou H, Fiering S, Hutchison RE, Mohi MG. Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood. 2010;115:3589–97. doi: 10.1182/blood-2009-04-215848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marty C, Lacout C, Martin A, Hasan S, Jacquot S, Birling MC, et al. Myeloproliferative neoplasm induced by constitutive expression of JAK2V617F in knock-in mice. Blood. 2010;116:783–7. doi: 10.1182/blood-2009-12-257063. [DOI] [PubMed] [Google Scholar]
- 29.Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010;17:584–96. doi: 10.1016/j.ccr.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jamieson CH, Gotlib J, Durocher JA, Chao MP, Mariappan MR, Lay M, et al. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci U S A. 2006;103:6224–9. doi: 10.1073/pnas.0601462103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bellanné-Chantelot C, Chaumarel I, Labopin M, Bellanger F, Barbu V, De Toma C, et al. Genetic and clinical implications of the Val617Phe JAK2 mutation in 72 families with myeloproliferative disorders. Blood. 2006;108:346–52. doi: 10.1182/blood-2005-12-4852. [DOI] [PubMed] [Google Scholar]
- 32.Delhommeau F, Dupont S, Tonetti C, Massé A, Godin I, Le Couedic JP, et al. Evidence that the JAK2 G1849T (V617F) mutation occurs in a lymphomyeloid progenitor in polycythemia vera and idiopathic myelofibrosis. Blood. 2007;109:71–7. doi: 10.1182/blood-2006-03-007146. [DOI] [PubMed] [Google Scholar]
- 33.Wilks AF, Harpur AG, Kurban RR, Ralph SJ, Zürcher G, Ziemiecki A. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol. 1991;11:2057–65. doi: 10.1128/mcb.11.4.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Girault JA, Labesse G, Mornon JP, Callebaut I. Janus kinases and focal adhesion kinases play in the 4.1 band: a superfamily of band 4.1 domains important for cell structure and signal transduction. Mol Med. 1998;4:751–69. [PMC free article] [PubMed] [Google Scholar]
- 35.Haan C, Kreis S, Margue C, Behrmann I. Jaks and cytokine receptors--an intimate relationship. Biochem Pharmacol. 2006;72:1538–46. doi: 10.1016/j.bcp.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 36.Haan C, Ungureanu D, Pekkala T, Silvennoinen O, Haan S. Regulation of JAKs: Insights Gleaned from the Functional Protein Domains. In: Decker T, Müller M, eds. JAK-Stat Signaling: From Basics to Disease. Vienna: Springer Verlag, 2012:5-25. [Google Scholar]
- 37.Haan C, Rolvering C, Raulf F, Kapp M, Drückes P, Thoma G, et al. Jak1 has a dominant role over Jak3 in signal transduction through γc-containing cytokine receptors. Chem Biol. 2011;18:314–23. doi: 10.1016/j.chembiol.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 38.Saharinen P, Takaluoma K, Silvennoinen O. Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol Cell Biol. 2000;20:3387–95. doi: 10.1128/MCB.20.10.3387-3395.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ungureanu D, Wu J, Pekkala T, Niranjan Y, Young C, Jensen ON, et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat Struct Mol Biol. 2011;18:971–6. doi: 10.1038/nsmb.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindauer K, Loerting T, Liedl KR, Kroemer RT. Prediction of the structure of human Janus kinase 2 (JAK2) comprising the two carboxy-terminal domains reveals a mechanism for autoregulation. Protein Eng. 2001;14:27–37. doi: 10.1093/protein/14.1.27. [DOI] [PubMed] [Google Scholar]
- 41.Giordanetto F, Kroemer RT. Prediction of the structure of human Janus kinase 2 (JAK2) comprising JAK homology domains 1 through 7. Protein Eng. 2002;15:727–37. doi: 10.1093/protein/15.9.727. [DOI] [PubMed] [Google Scholar]
- 42.Lee TS, Ma W, Zhang X, Giles F, Kantarjian H, Albitar M. Mechanisms of constitutive activation of Janus kinase 2-V617F revealed at the atomic level through molecular dynamics simulations. Cancer. 2009;115:1692–700. doi: 10.1002/cncr.24183. [DOI] [PubMed] [Google Scholar]
- 43.Dusa A, Mouton C, Pecquet C, Herman M, Constantinescu SN. JAK2 V617F constitutive activation requires JH2 residue F595: a pseudokinase domain target for specific inhibitors. PLoS One. 2010;5:e11157. doi: 10.1371/journal.pone.0011157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008;372:1484–92. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- 45.Lupardus PJ, Skiniotis G, Rice AJ, Thomas C, Fischer S, Walz T, et al. Structural snapshots of full-length Jak1, a transmembrane gp130/IL-6/IL-6Rα cytokine receptor complex, and the receptor-Jak1 holocomplex. Structure. 2011;19:45–55. doi: 10.1016/j.str.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou YJ, Chen M, Cusack NA, Kimmel LH, Magnuson KS, Boyd JG, et al. Unexpected effects of FERM domain mutations on catalytic activity of Jak3: structural implication for Janus kinases. Mol Cell. 2001;8:959–69. doi: 10.1016/S1097-2765(01)00398-7. [DOI] [PubMed] [Google Scholar]
- 47.Funakoshi-Tago M, Pelletier S, Moritake H, Parganas E, Ihle JN. Jak2 FERM domain interaction with the erythropoietin receptor regulates Jak2 kinase activity. Mol Cell Biol. 2008;28:1792–801. doi: 10.1128/MCB.01447-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haan S, Margue C, Engrand A, Rolvering C, Schmitz-Van de Leur H, Heinrich PC, et al. Dual role of the Jak1 FERM and kinase domains in cytokine receptor binding and in stimulation-dependent Jak activation. J Immunol. 2008;180:998–1007. doi: 10.4049/jimmunol.180.2.998. [DOI] [PubMed] [Google Scholar]
- 49.Lu X, Levine R, Tong W, Wernig G, Pikman Y, Zarnegar S, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci U S A. 2005;102:18962–7. doi: 10.1073/pnas.0509714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wernig G, Gonneville JR, Crowley BJ, Rodrigues MS, Reddy MM, Hudon HE, et al. The Jak2V617F oncogene associated with myeloproliferative diseases requires a functional FERM domain for transformation and for expression of the Myc and Pim proto-oncogenes. Blood. 2008;111:3751–9. doi: 10.1182/blood-2007-07-102186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorantla SP, Dechow TN, Grundler R, Illert AL, Zum Büschenfelde CM, Kremer M, et al. Oncogenic JAK2V617F requires an intact SH2-like domain for constitutive activation and induction of a myeloproliferative disease in mice. Blood. 2010;116:4600–11. doi: 10.1182/blood-2009-07-236133. [DOI] [PubMed] [Google Scholar]
- 52.Haan S, Wüller S, Kaczor J, Rolvering C, Nöcker T, Behrmann I, et al. SOCS-mediated downregulation of mutant Jak2 (V617F, T875N and K539L) counteracts cytokine-independent signaling. Oncogene. 2009;28:3069–80. doi: 10.1038/onc.2009.155. [DOI] [PubMed] [Google Scholar]
- 53.Röder S, Steimle C, Meinhardt G, Pahl HL. STAT3 is constitutively active in some patients with Polycythemia rubra vera. Exp Hematol. 2001;29:694–702. doi: 10.1016/S0301-472X(01)00637-3. [DOI] [PubMed] [Google Scholar]
- 54.Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673–83. doi: 10.1038/nrc2210. [DOI] [PubMed] [Google Scholar]
- 55.Oku S, Takenaka K, Kuriyama T, Shide K, Kumano T, Kikushige Y, et al. JAK2 V617F uses distinct signalling pathways to induce cell proliferation and neutrophil activation. Br J Haematol. 2010;150:334–44. doi: 10.1111/j.1365-2141.2010.08249.x. [DOI] [PubMed] [Google Scholar]
- 56.Tognon R, Gasparotto EP, Neves RP, Nunes NS, Ferreira AF, Palma PV, et al. Deregulation of apoptosis-related genes is associated with PRV1 overexpression and JAK2 V617F allele burden in Essential Thrombocythemia and Myelofibrosis. J Hematol Oncol. 2012;5:2. doi: 10.1186/1756-8722-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gozgit JM, Bebernitz G, Patil P, Ye M, Parmentier J, Wu J, et al. Effects of the JAK2 inhibitor, AZ960, on Pim/BAD/BCL-xL survival signaling in the human JAK2 V617F cell line SET-2. J Biol Chem. 2008;283:32334–43. doi: 10.1074/jbc.M803813200. [DOI] [PubMed] [Google Scholar]
- 58.Gautier EF, Picard M, Laurent C, Marty C, Villeval JL, Demur C, et al. The cell cycle regulator CDC25A is a target for JAK2V617F oncogene. Blood. 2012;119:1190–9. doi: 10.1182/blood-2011-01-327742. [DOI] [PubMed] [Google Scholar]
- 59.Walz C, Ahmed W, Lazarides K, Betancur M, Patel N, Hennighausen L, et al. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in mice. Blood. 2012;119:3550–60. doi: 10.1182/blood-2011-12-397554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yan D, Hutchison RE, Mohi G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood. 2012;119:3539–49. doi: 10.1182/blood-2011-03-345215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Funakoshi-Tago M, Tago K, Abe M, Sonoda Y, Kasahara T. STAT5 activation is critical for the transformation mediated by myeloproliferative disorder-associated JAK2 V617F mutant. J Biol Chem. 2010;285:5296–307. doi: 10.1074/jbc.M109.040733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kamishimoto J, Tago K, Kasahara T, Funakoshi-Tago M. Akt activation through the phosphorylation of erythropoietin receptor at tyrosine 479 is required for myeloproliferative disorder-associated JAK2 V617F mutant-induced cellular transformation. Cell Signal. 2011;23:849–56. doi: 10.1016/j.cellsig.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 63.Walz C, Crowley BJ, Hudon HE, Gramlich JL, Neuberg DS, Podar K, et al. Activated Jak2 with the V617F point mutation promotes G1/S phase transition. J Biol Chem. 2006;281:18177–83. doi: 10.1074/jbc.M600064200. [DOI] [PubMed] [Google Scholar]
- 64.Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal. 2011;23:1515–27. doi: 10.1016/j.cellsig.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 65.Yang JY, Hung MC. A new fork for clinical application: targeting forkhead transcription factors in cancer. Clin Cancer Res. 2009;15:752–7. doi: 10.1158/1078-0432.CCR-08-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jäkel H, Weinl C, Hengst L. Phosphorylation of p27Kip1 by JAK2 directly links cytokine receptor signaling to cell cycle control. Oncogene. 2011;30:3502–12. doi: 10.1038/onc.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–86. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gómez-Sintes R, Hernández F, Lucas JJ, Avila J. GSK-3 Mouse Models to Study Neuronal Apoptosis and Neurodegeneration. Front Mol Neurosci. 2011;4:45. doi: 10.3389/fnmol.2011.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nagao T, Oshikawa G, Wu N, Kurosu T, Miura O. DNA damage stress and inhibition of Jak2-V617F cause its degradation and synergistically induce apoptosis through activation of GSK3β. PLoS One. 2011;6:e27397. doi: 10.1371/journal.pone.0027397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu F, Zhao X, Perna F, Wang L, Koppikar P, Abdel-Wahab O, et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell. 2011;19:283–94. doi: 10.1016/j.ccr.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dawson MA, Bannister AJ, Göttgens B, Foster SD, Bartke T, Green AR, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461:819–22. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Girodon F, Steinkamp MP, Cleyrat C, Hermouet S, Wilson BS. Confocal imaging studies cast doubt on nuclear localization of JAK2V617F. Blood. 2011;118:2633–4. doi: 10.1182/blood-2011-02-336479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi S, Calhoun HC, Xia F, Li J, Le L, Li WX. JAK signaling globally counteracts heterochromatic gene silencing. Nat Genet. 2006;38:1071–6. doi: 10.1038/ng1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bourdeau A, Dubé N, Tremblay ML. Cytoplasmic protein tyrosine phosphatases, regulation and function: the roles of PTP1B and TC-PTP. Curr Opin Cell Biol. 2005;17:203–9. doi: 10.1016/j.ceb.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 75.Shuai K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 2006;16:196–202. doi: 10.1038/sj.cr.7310027. [DOI] [PubMed] [Google Scholar]
- 76.Gery S, Cao Q, Gueller S, Xing H, Tefferi A, Koeffler HP. Lnk inhibits myeloproliferative disorder-associated JAK2 mutant, JAK2V617F. J Leukoc Biol. 2009;85:957–65. doi: 10.1189/jlb.0908575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Devallière J, Charreau B. The adaptor Lnk (SH2B3): an emerging regulator in vascular cells and a link between immune and inflammatory signaling. Biochem Pharmacol. 2011;82:1391–402. doi: 10.1016/j.bcp.2011.06.023. [DOI] [PubMed] [Google Scholar]
- 78.Seita J, Ema H, Ooehara J, Yamazaki S, Tadokoro Y, Yamasaki A, et al. Lnk negatively regulates self-renewal of hematopoietic stem cells by modifying thrombopoietin-mediated signal transduction. Proc Natl Acad Sci U S A. 2007;104:2349–54. doi: 10.1073/pnas.0606238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tong W, Zhang J, Lodish HF. Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways. Blood. 2005;105:4604–12. doi: 10.1182/blood-2004-10-4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oh ST, Simonds EF, Jones C, Hale MB, Goltsev Y, Gibbs KD, Jr., et al. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood. 2010;116:988–92. doi: 10.1182/blood-2010-02-270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lasho TL, Pardanani A, Tefferi A. LNK mutations in JAK2 mutation-negative erythrocytosis. N Engl J Med. 2010;363:1189–90. doi: 10.1056/NEJMc1006966. [DOI] [PubMed] [Google Scholar]
- 82.Lasho TL, Tefferi A, Finke C, Pardanani A. Clonal hierarchy and allelic mutation segregation in a myelofibrosis patient with two distinct LNK mutations. Leukemia. 2011;25:1056–8. doi: 10.1038/leu.2011.45. [DOI] [PubMed] [Google Scholar]
- 83.Hurtado C, Erquiaga I, Aranaz P, Miguéliz I, García-Delgado M, Novo FJ, et al. LNK can also be mutated outside PH and SH2 domains in myeloproliferative neoplasms with and without V617FJAK2 mutation. Leuk Res. 2011;35:1537–9. doi: 10.1016/j.leukres.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 84.Baran-Marszak F, Magdoud H, Desterke C, Alvarado A, Roger C, Harel S, et al. Expression level and differential JAK2-V617F-binding of the adaptor protein Lnk regulates JAK2-mediated signals in myeloproliferative neoplasms. Blood. 2010;116:5961–71. doi: 10.1182/blood-2009-12-256768. [DOI] [PubMed] [Google Scholar]
- 85.Maures TJ, Kurzer JH, Carter-Su C. SH2B1 (SH2-B) and JAK2: a multifunctional adaptor protein and kinase made for each other. Trends Endocrinol Metab. 2007;18:38–45. doi: 10.1016/j.tem.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 86.O’Brien KB, O’Shea JJ, Carter-Su C. SH2-B family members differentially regulate JAK family tyrosine kinases. J Biol Chem. 2002;277:8673–81. doi: 10.1074/jbc.M109165200. [DOI] [PubMed] [Google Scholar]
- 87.Kurzer JH, Saharinen P, Silvennoinen O, Carter-Su C. Binding of SH2-B family members within a potential negative regulatory region maintains JAK2 in an active state. Mol Cell Biol. 2006;26:6381–94. doi: 10.1128/MCB.00570-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wakioka T, Sasaki A, Mitsui K, Yokouchi M, Inoue A, Komiya S, et al. APS, an adaptor protein containing Pleckstrin homology (PH) and Src homology-2 (SH2) domains inhibits the JAK-STAT pathway in collaboration with c-Cbl. Leukemia. 1999;13:760–7. doi: 10.1038/sj.leu.2401397. [DOI] [PubMed] [Google Scholar]
- 89.Nishi M, Werner ED, Oh BC, Frantz JD, Dhe-Paganon S, Hansen L, et al. Kinase activation through dimerization by human SH2-B. Mol Cell Biol. 2005;25:2607–21. doi: 10.1128/MCB.25.7.2607-2621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Javadi M, Hofstätter E, Stickle N, Beattie BK, Jaster R, Carter-Su C, et al. The SH2B1 adaptor protein associates with a proximal region of the erythropoietin receptor. J Biol Chem. 2012;287:26223–34. doi: 10.1074/jbc.M112.382721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hitchcock IS, Chen MM, King JR, Kaushansky K. YRRL motifs in the cytoplasmic domain of the thrombopoietin receptor regulate receptor internalization and degradation. Blood. 2008;112:2222–31. doi: 10.1182/blood-2008-01-134049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Walrafen P, Verdier F, Kadri Z, Chrétien S, Lacombe C, Mayeux P. Both proteasomes and lysosomes degrade the activated erythropoietin receptor. Blood. 2005;105:600–8. doi: 10.1182/blood-2004-03-1216. [DOI] [PubMed] [Google Scholar]
- 93.Yen CH, Yang YC, Ruscetti SK, Kirken RA, Dai RM, Li CC. Involvement of the ubiquitin-proteasome pathway in the degradation of nontyrosine kinase-type cytokine receptors of IL-9, IL-2, and erythropoietin. J Immunol. 2000;165:6372–80. doi: 10.4049/jimmunol.165.11.6372. [DOI] [PubMed] [Google Scholar]
- 94.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pecquet C, Diaconu CC, Staerk J, Girardot M, Marty C, Royer Y, et al. Thrombopoietin receptor down-modulation by JAK2 V617F: restoration of receptor levels by inhibitors of pathologic JAK2 signaling and of proteasomes. Blood. 2012;119:4625–35. doi: 10.1182/blood-2011-08-372524. [DOI] [PubMed] [Google Scholar]
- 96.Weissman AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol. 2001;2:169–78. doi: 10.1038/35056563. [DOI] [PubMed] [Google Scholar]
- 97.Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19:414–22. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hookham MB, Elliott J, Suessmuth Y, Staerk J, Ward AC, Vainchenker W, et al. The myeloproliferative disorder-associated JAK2 V617F mutant escapes negative regulation by suppressor of cytokine signaling 3. Blood. 2007;109:4924–9. doi: 10.1182/blood-2006-08-039735. [DOI] [PubMed] [Google Scholar]
- 99.Yoshikawa H, Matsubara K, Qian GS, Jackson P, Groopman JD, Manning JE, et al. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet. 2001;28:29–35. doi: 10.1038/ng0501-29. [DOI] [PubMed] [Google Scholar]
- 100.Galm O, Yoshikawa H, Esteller M, Osieka R, Herman JG. SOCS-1, a negative regulator of cytokine signaling, is frequently silenced by methylation in multiple myeloma. Blood. 2003;101:2784–8. doi: 10.1182/blood-2002-06-1735. [DOI] [PubMed] [Google Scholar]
- 101.Ekmekci CG, Gutiérrez MI, Siraj AK, Ozbek U, Bhatia K. Aberrant methylation of multiple tumor suppressor genes in acute myeloid leukemia. Am J Hematol. 2004;77:233–40. doi: 10.1002/ajh.20186. [DOI] [PubMed] [Google Scholar]
- 102.Melzner I, Bucur AJ, Brüderlein S, Dorsch K, Hasel C, Barth TF, et al. Biallelic mutation of SOCS-1 impairs JAK2 degradation and sustains phospho-JAK2 action in the MedB-1 mediastinal lymphoma line. Blood. 2005;105:2535–42. doi: 10.1182/blood-2004-09-3701. [DOI] [PubMed] [Google Scholar]
- 103.Melzner I, Weniger MA, Bucur AJ, Brüderlein S, Dorsch K, Hasel C, et al. Biallelic deletion within 16p13.13 including SOCS-1 in Karpas1106P mediastinal B-cell lymphoma line is associated with delayed degradation of JAK2 protein. Int J Cancer. 2006;118:1941–4. doi: 10.1002/ijc.21485. [DOI] [PubMed] [Google Scholar]
- 104.Capello D, Deambrogi C, Rossi D, Lischetti T, Piranda D, Cerri M, et al. Epigenetic inactivation of suppressors of cytokine signalling in Philadelphia-negative chronic myeloproliferative disorders. Br J Haematol. 2008;141:504–11. doi: 10.1111/j.1365-2141.2008.07072.x. [DOI] [PubMed] [Google Scholar]
- 105.Teofili L, Martini M, Cenci T, Guidi F, Torti L, Giona F, et al. Epigenetic alteration of SOCS family members is a possible pathogenetic mechanism in JAK2 wild type myeloproliferative diseases. Int J Cancer. 2008;123:1586–92. doi: 10.1002/ijc.23694. [DOI] [PubMed] [Google Scholar]
- 106.Lipkowitz S, Weissman AM. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat Rev Cancer. 2011;11:629–43. doi: 10.1038/nrc3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Thien CB, Langdon WY. Negative regulation of PTK signalling by Cbl proteins. Growth Factors. 2005;23:161–7. doi: 10.1080/08977190500153763. [DOI] [PubMed] [Google Scholar]
- 108.Schmidt MH, Dikic I. The Cbl interactome and its functions. Nat Rev Mol Cell Biol. 2005;6:907–18. doi: 10.1038/nrm1762. [DOI] [PubMed] [Google Scholar]
- 109.Dikic I, Schmidt MH. Malfunctions within the Cbl interactome uncouple receptor tyrosine kinases from destructive transport. Eur J Cell Biol. 2007;86:505–12. doi: 10.1016/j.ejcb.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 110.Aranaz P, Hurtado C, Erquiaga I, Miguéliz I, Ormazábal C, Cristobal I, et al. CBL mutations in myeloproliferative neoplasms are also found in the gene’s proline-rich domain and in patients with the V617FJAK2. Haematologica. 2012;97:1234–41. doi: 10.3324/haematol.2011.052605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010;24:1128–38. doi: 10.1038/leu.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C, et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood. 2009;113:6182–92. doi: 10.1182/blood-2008-12-194548. [DOI] [PubMed] [Google Scholar]
- 113.Schwaab J, Ernst T, Erben P, Rinke J, Schnittger S, Ströbel P, et al. Activating CBL mutations are associated with a distinct MDS/MPN phenotype. Ann Hematol. 2012;91:1713–20. doi: 10.1007/s00277-012-1521-3. [DOI] [PubMed] [Google Scholar]
- 114.Fialkow PJ, Gartler SM, Yoshida A. Clonal origin of chronic myelocytic leukemia in man. Proc Natl Acad Sci U S A. 1967;58:1468–71. doi: 10.1073/pnas.58.4.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Adamson JW, Fialkow PJ, Murphy S, Prchal JF, Steinmann L. Polycythemia vera: stem-cell and probable clonal origin of the disease. N Engl J Med. 1976;295:913–6. doi: 10.1056/NEJM197610212951702. [DOI] [PubMed] [Google Scholar]
- 116.Fialkow PJ, Faguet GB, Jacobson RJ, Vaidya K, Murphy S. Evidence that essential thrombocythemia is a clonal disorder with origin in a multipotent stem cell. Blood. 1981;58:916–9. [PubMed] [Google Scholar]
- 117.Jacobson RJ, Salo A, Fialkow PJ. Agnogenic myeloid metaplasia: a clonal proliferation of hematopoietic stem cells with secondary myelofibrosis. Blood. 1978;51:189–94. [PubMed] [Google Scholar]
- 118.Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–30. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 119.Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp Hematol. 2002;30:229–36. doi: 10.1016/S0301-472X(01)00789-5. [DOI] [PubMed] [Google Scholar]
- 120.Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood. 2006;108:2435–7. doi: 10.1182/blood-2006-04-018259. [DOI] [PubMed] [Google Scholar]
- 121.Scott LM. The JAK2 exon 12 mutations: a comprehensive review. Am J Hematol. 2011;86:668–76. doi: 10.1002/ajh.22063. [DOI] [PubMed] [Google Scholar]
- 122.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–68. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bernardi M, Ruggeri M, Albiero E, Madeo D, Rodeghiero F. Isolated erythrocytosis in V617F negative patients with JAK2 exon 12 mutations: report of a new mutation. Am J Hematol. 2009;84:258–60. doi: 10.1002/ajh.21357. [DOI] [PubMed] [Google Scholar]
- 124.Bueso-Ramos CE, Vardiman JW. Diagnosis and Classification of the BCR-ABL1-Negative Myeloproliferative Neoplasms. In: Verstovsek S, Tefferi A, eds. Myeloproliferative Neoplasms - Biology and Therapy. New York: Humana Press, Springer Science+Business Media, LLC, 2011:1-37. [Google Scholar]
- 125.Cervantes F, Hernández-Boluda J-C. Prognostic Factors in Classic Myeloproliferative Neoplasms. In: Verstovsek S, Tefferi A, eds. Myeloproliferative Neoplasms - Biology and Therapy. New York: Humana Press, Springer Science+Business Media, LLC, 2011:85-96. [Google Scholar]
- 126.Alvarez-Larrán A, Bellosillo B, Martínez-Avilés L, Saumell S, Salar A, Abella E, et al. Postpolycythaemic myelofibrosis: frequency and risk factors for this complication in 116 patients. Br J Haematol. 2009;146:504–9. doi: 10.1111/j.1365-2141.2009.07804.x. [DOI] [PubMed] [Google Scholar]
- 127.Vannucchi AM, Antonioli E, Guglielmelli P, Rambaldi A, Barosi G, Marchioli R, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110:840–6. doi: 10.1182/blood-2006-12-064287. [DOI] [PubMed] [Google Scholar]
- 128.Finazzi G, Caruso V, Marchioli R, Capnist G, Chisesi T, Finelli C, et al. ECLAP Investigators Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood. 2005;105:2664–70. doi: 10.1182/blood-2004-09-3426. [DOI] [PubMed] [Google Scholar]
- 129.Jensen MK, de Nully Brown P, Nielsen OJ, Hasselbalch HC. Incidence, clinical features and outcome of essential thrombocythaemia in a well defined geographical area. Eur J Haematol. 2000;65:132–9. doi: 10.1034/j.1600-0609.2000.90236.x. [DOI] [PubMed] [Google Scholar]
- 130.Finazzi G, Harrison C. Essential thrombocythemia. Semin Hematol. 2005;42:230–8. doi: 10.1053/j.seminhematol.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 131.Elliott MA, Tefferi A. Thrombosis and haemorrhage in polycythaemia vera and essential thrombocythaemia. Br J Haematol. 2005;128:275–90. doi: 10.1111/j.1365-2141.2004.05277.x. [DOI] [PubMed] [Google Scholar]
- 132.Kvasnicka HM, Thiele J. The impact of clinicopathological studies on staging and survival in essential thrombocythemia, chronic idiopathic myelofibrosis, and polycythemia rubra vera. Semin Thromb Hemost. 2006;32:362–71. doi: 10.1055/s-2006-942757. [DOI] [PubMed] [Google Scholar]
- 133.Cervantes F, Alvarez-Larrán A, Talarn C, Gómez M, Montserrat E. Myelofibrosis with myeloid metaplasia following essential thrombocythaemia: actuarial probability, presenting characteristics and evolution in a series of 195 patients. Br J Haematol. 2002;118:786–90. doi: 10.1046/j.1365-2141.2002.03688.x. [DOI] [PubMed] [Google Scholar]
- 134.Passamonti F, Cervantes F, Vannucchi AM, Morra E, Rumi E, Pereira A, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment) Blood. 2010;115:1703–8. doi: 10.1182/blood-2009-09-245837. [DOI] [PubMed] [Google Scholar]
- 135.Lataillade JJ, Pierre-Louis O, Hasselbalch HC, Uzan G, Jasmin C, Martyré MC, et al. French INSERM and the European EUMNET Networks on Myelofibrosis Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence. Blood. 2008;112:3026–35. doi: 10.1182/blood-2008-06-158386. [DOI] [PubMed] [Google Scholar]
- 136.Hoermann G, Cerny-Reiterer S, Herrmann H, Blatt K, Bilban M, Gisslinger H, et al. Identification of oncostatin M as a JAK2 V617F-dependent amplifier of cytokine production and bone marrow remodeling in myeloproliferative neoplasms. FASEB J. 2012;26:894–906. doi: 10.1096/fj.11-193078. [DOI] [PubMed] [Google Scholar]
- 137.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117–27. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011;29:1356–63. doi: 10.1200/JCO.2010.32.9490. [DOI] [PubMed] [Google Scholar]
- 139.Barosi G, Rosti V, Bonetti E, Campanelli R, Carolei A, Catarsi P, et al. Evidence that prefibrotic myelofibrosis is aligned along a clinical and biological continuum featuring primary myelofibrosis. PLoS One. 2012;7:e35631. doi: 10.1371/journal.pone.0035631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zaleskas VM, Krause DS, Lazarides K, Patel N, Hu Y, Li S, et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS One. 2006;1:e18. doi: 10.1371/journal.pone.0000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472–6. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- 142.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Boyd EM, Bench AJ, Goday-Fernández A, Anand S, Vaghela KJ, Beer P, et al. Clinical utility of routine MPL exon 10 analysis in the diagnosis of essential thrombocythaemia and primary myelofibrosis. Br J Haematol. 2010;149:250–7. doi: 10.1111/j.1365-2141.2010.08083.x. [DOI] [PubMed] [Google Scholar]
- 144.Ma W, Zhang X, Wang X, Zhang Z, Yeh CH, Uyeji J, et al. MPL mutation profile in JAK2 mutation-negative patients with myeloproliferative disorders. Diagn Mol Pathol. 2011;20:34–9. doi: 10.1097/PDM.0b013e3181ecd261. [DOI] [PubMed] [Google Scholar]
- 145.Chaligné R, Tonetti C, Besancenot R, Roy L, Marty C, Mossuz P, et al. New mutations of MPL in primitive myelofibrosis: only the MPL W515 mutations promote a G1/S-phase transition. Leukemia. 2008;22:1557–66. doi: 10.1038/leu.2008.137. [DOI] [PubMed] [Google Scholar]
- 146.Campregher PV, Santos FP, Perini GF, Hamerschlak N. Molecular biology of Philadelphia-negative myeloproliferative neoplasms. Rev Bras Hematol Hemoter. 2012;34:150–5. doi: 10.5581/1516-8484.20120035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Harutyunyan AS, Kralovics R. Role of germline genetic factors in MPN pathogenesis. Hematol Oncol Clin North Am. 2012;26:1037–51. doi: 10.1016/j.hoc.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 148.Fourouclas N, Li J, Gilby DC, Campbell PJ, Beer PA, Boyd EM, et al. Methylation of the suppressor of cytokine signaling 3 gene (SOCS3) in myeloproliferative disorders. Haematologica. 2008;93:1635–44. doi: 10.3324/haematol.13043. [DOI] [PubMed] [Google Scholar]
- 149.Jost E, do O N, Dahl E, Maintz CE, Jousten P, Habets L, et al. Epigenetic alterations complement mutation of JAK2 tyrosine kinase in patients with BCR/ABL-negative myeloproliferative disorders. Leukemia. 2007;21:505–10. doi: 10.1038/sj.leu.2404513. [DOI] [PubMed] [Google Scholar]
- 150.Quentmeier H, Geffers R, Jost E, Macleod RA, Nagel S, Röhrs S, et al. SOCS2: inhibitor of JAK2V617F-mediated signal transduction. Leukemia. 2008;22:2169–75. doi: 10.1038/leu.2008.226. [DOI] [PubMed] [Google Scholar]
- 151.Bruchova H, Yoon D, Agarwal AM, Mendell J, Prchal JT. Regulated expression of microRNAs in normal and polycythemia vera erythropoiesis. Exp Hematol. 2007;35:1657–67. doi: 10.1016/j.exphem.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bruchova H, Merkerova M, Prchal JT. Aberrant expression of microRNA in polycythemia vera. Haematologica. 2008;93:1009–16. doi: 10.3324/haematol.12706. [DOI] [PubMed] [Google Scholar]
- 153.Guglielmelli P, Tozzi L, Pancrazzi A, Bogani C, Antonioli E, Ponziani V, et al. MPD Research Consortium MicroRNA expression profile in granulocytes from primary myelofibrosis patients. Exp Hematol. 2007;35:1708–18. doi: 10.1016/j.exphem.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 154.Lu J, Guo S, Ebert BL, Zhang H, Peng X, Bosco J, et al. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev Cell. 2008;14:843–53. doi: 10.1016/j.devcel.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pardanani A, Fridley BL, Lasho TL, Gilliland DG, Tefferi A. Host genetic variation contributes to phenotypic diversity in myeloproliferative disorders. Blood. 2008;111:2785–9. doi: 10.1182/blood-2007-06-095703. [DOI] [PubMed] [Google Scholar]
- 156.Jones AV, Chase A, Silver RT, Oscier D, Zoi K, Wang YL, et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet. 2009;41:446–9. doi: 10.1038/ng.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kilpivaara O, Mukherjee S, Schram AM, Wadleigh M, Mullally A, Ebert BL, et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat Genet. 2009;41:455–9. doi: 10.1038/ng.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Olcaydu D, Harutyunyan A, Jäger R, Berg T, Gisslinger B, Pabinger I, et al. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet. 2009;41:450–4. doi: 10.1038/ng.341. [DOI] [PubMed] [Google Scholar]
- 159.Jones AV, Campbell PJ, Beer PA, Schnittger S, Vannucchi AM, Zoi K, et al. The JAK2 46/1 haplotype predisposes to MPL-mutated myeloproliferative neoplasms. Blood. 2010;115:4517–23. doi: 10.1182/blood-2009-08-236448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Olcaydu D, Skoda RC, Looser R, Li S, Cazzola M, Pietra D, et al. The ‘GGCC’ haplotype of JAK2 confers susceptibility to JAK2 exon 12 mutation-positive polycythemia vera. Leukemia. 2009;23:1924–6. doi: 10.1038/leu.2009.110. [DOI] [PubMed] [Google Scholar]
- 161.Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22:14–22. doi: 10.1038/sj.leu.2404955. [DOI] [PubMed] [Google Scholar]
- 162.Anand S, Stedham F, Gudgin E, Campbell P, Beer P, Green AR, et al. Increased basal intracellular signaling patterns do not correlate with JAK2 genotype in human myeloproliferative neoplasms. Blood. 2011;118:1610–21. doi: 10.1182/blood-2011-02-335042. [DOI] [PubMed] [Google Scholar]
- 163.Hasselbalch HC. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk Res. 2013;37:214–20. doi: 10.1016/j.leukres.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 164.Vannucchi AM. How do JAK2-inhibitors work in myelofibrosis: an alternative hypothesis. Leuk Res. 2009;33:1581–3. doi: 10.1016/j.leukres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 165.Barosi G, Gale RP. Bone marrow fibrosis in myeloproliferative neoplasms-associated myelofibrosis: deconstructing a myth? Leuk Res. 2011;35:563–5. doi: 10.1016/j.leukres.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 166.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 167.Skov V, Larsen TS, Thomassen M, Riley CH, Jensen MK, Bjerrum OW, et al. Molecular profiling of peripheral blood cells from patients with polycythemia vera and related neoplasms: identification of deregulated genes of significance for inflammation and immune surveillance. Leuk Res. 2012;36:1387–92. doi: 10.1016/j.leukres.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 168.Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, et al. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood. 2010;115:5232–40. doi: 10.1182/blood-2009-05-223727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gangemi S, Allegra A, Pace E, Alonci A, Ferraro M, Petrungaro A, et al. Evaluation of interleukin-23 plasma levels in patients with polycythemia vera and essential thrombocythemia. Cell Immunol. 2012;278:91–4. doi: 10.1016/j.cellimm.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 170.Hintzen C, Haan C, Tuckermann JP, Heinrich PC, Hermanns HM. Oncostatin M-induced and constitutive activation of the JAK2/STAT5/CIS pathway suppresses CCL1, but not CCL7 and CCL8, chemokine expression. J Immunol. 2008;181:7341–9. doi: 10.4049/jimmunol.181.10.7341. [DOI] [PubMed] [Google Scholar]
- 171.Manshouri T, Estrov Z, Quintás-Cardama A, Burger J, Zhang Y, Livun A, et al. Bone marrow stroma-secreted cytokines protect JAK2(V617F)-mutated cells from the effects of a JAK2 inhibitor. Cancer Res. 2011;71:3831–40. doi: 10.1158/0008-5472.CAN-10-4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Fleischman AG, Aichberger KJ, Luty SB, Bumm TG, Petersen CL, Doratotaj S, et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood. 2011;118:6392–8. doi: 10.1182/blood-2011-04-348144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Boissinot M, Cleyrat C, Vilaine M, Jacques Y, Corre I, Hermouet S. Anti-inflammatory cytokines hepatocyte growth factor and interleukin-11 are over-expressed in Polycythemia vera and contribute to the growth of clonal erythroblasts independently of JAK2V617F. Oncogene. 2011;30:990–1001. doi: 10.1038/onc.2010.479. [DOI] [PubMed] [Google Scholar]
- 174.Vannucchi AM, Pieri L, Susini MC, Guglielmelli P. BCR-ABL1-negative chronic myeloid neoplasms: an update on management techniques. Future Oncol. 2012;8:575–93. doi: 10.2217/fon.12.50. [DOI] [PubMed] [Google Scholar]
- 175.Reilly JT, McMullin MF, Beer PA, Butt N, Conneally E, Duncombe A, et al. Writing group: British Committee for Standards in Haematology Guideline for the diagnosis and management of myelofibrosis. Br J Haematol. 2012;158:453–71. doi: 10.1111/j.1365-2141.2012.09179.x. [DOI] [PubMed] [Google Scholar]
- 176.Randhawa J, Ostojic A, Vrhovac R, Atallah E, Verstovsek S. Splenomegaly in myelofibrosis--new options for therapy and the therapeutic potential of Janus kinase 2 inhibitors. J Hematol Oncol. 2012;5:43. doi: 10.1186/1756-8722-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Harrison C, Verstovsek S, McMullin MF, Mesa R. Janus kinase inhibition and its effect upon the therapeutic landscape for myelofibrosis: from palliation to cure? Br J Haematol. 2012;157:426–37. doi: 10.1111/j.1365-2141.2012.09108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jabbour E, Thomas D, Kantarjian H, Zhou L, Pierce S, Cortes J, et al. Comparison of thalidomide and lenalidomide as therapy for myelofibrosis. Blood. 2011;118:899–902. doi: 10.1182/blood-2010-12-325589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Abdel-Wahab O, Pardanani A, Bernard OA, Finazzi G, Crispino JD, Gisslinger H, et al. Unraveling the genetic underpinnings of myeloproliferative neoplasms and understanding their effect on disease course and response to therapy: proceedings from the 6th International Post-ASH Symposium. Am J Hematol. 2012;87:562–8. doi: 10.1002/ajh.23169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Quintás-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115:3109–17. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–98. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 183.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, Dipersio JF, et al. The clinical benefit of ruxolitinib across patient subgroups: analysis of a placebo-controlled, Phase III study in patients with myelofibrosis. Br J Haematol. 2013;161:508–16. doi: 10.1111/bjh.12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Vannucchi AM, Passamonti F, Al-Ali HK, Barosi G, Harrison CN, Sirulnik A, et al. Reductions in JAK2 V617F Allele Burden with Ruxolitinib Treatment in Comfort-II, a Phase 3 Study Comparing the Safety and Efficacy of Ruxolitinib with Best Available Therapy (BAT) Blood. 2012;120:A802. [Google Scholar]
- 185.Mesa RA, Gotlib J, Gupta V, Catalano JV, Deininger MW, Shields AL, et al. Effect of ruxolitinib therapy on myelofibrosis-related symptoms and other patient-reported outcomes in COMFORT-I: a randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2013;31:1285–92. doi: 10.1200/JCO.2012.44.4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cervantes F, Mesa R, Harrison C. JAK inhibitors: beyond spleen and symptoms? Haematologica. 2013;98:160–2. doi: 10.3324/haematol.2012.083543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Verstovsek S, Passamonti F, Rambaldi A, Barosi G, Rosen PJ, Levy R, et al. Durable Responses with the JAK1/JAK2 Inhibitor, INCB018424, In Patients with Polycythemia Vera (PV) and Essential Thrombocythemia (ET) Refractory or Intolerant to Hydroxyurea (HU) Blood. 2010;116:A313. [Google Scholar]
- 188.Verstovsek S, Passamonti F, Rambaldi A, Barosi G, Rosen PJ, He S, et al. Long-Term Efficacy and Safety Results From a Phase II Study of Ruxolitinib in Patients with Polycythemia Vera. Blood. 2012;120:A804. [Google Scholar]
- 189.Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29:789–96. doi: 10.1200/JCO.2010.32.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tefferi A. JAK inhibitors for myeloproliferative neoplasms: clarifying facts from myths. Blood. 2012;119:2721–30. doi: 10.1182/blood-2011-11-395228. [DOI] [PubMed] [Google Scholar]
- 191.Santos FP, Verstovsek S. Therapy with JAK2 inhibitors for myeloproliferative neoplasms. Hematol Oncol Clin North Am. 2012;26:1083–99. doi: 10.1016/j.hoc.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Laurence A, Pesu M, Silvennoinen O, O’Shea J. JAK Kinases in Health and Disease: An Update. Open Rheumatol J. 2012;6:232–44. doi: 10.2174/1874312901206010232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Furqan M, Mukhi N, Lee B, Liu D. Dysregulation of JAK-STAT pathway in hematological malignancies and JAK inhibitors for clinical application. Biomarker Res. 2013;1:5. doi: 10.1186/2050-7771-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Gäbler K, Rolvering C, Kaczor J, Eulenfeld R, Méndez SA, Berchem G, et al. Cooperative effects of Janus and Aurora kinase inhibition by CEP701 in cells expressing Jak2V617F. J Cell Mol Med. 2013;17:265–76. doi: 10.1111/jcmm.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Leah E. Clinical trials: Phase III trial results for tofacitinib bring new oral DMARD therapy a step closer for patients with rheumatoid arthritis. Nat Rev Rheumatol. 2012;8:561. doi: 10.1038/nrrheum.2012.145. [DOI] [PubMed] [Google Scholar]
- 196.Backes AC, Müller G, Sennhenn PC. Design Principles of Deep Pocket-Targeting Protein Kinase Inhibitors. In: Klebl B, Müller G, Hamacher M, eds. Protein Kinases as Drug Targets. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA, 2011:145-93. [Google Scholar]
- 197.Andraos R, Qian Z, Bonenfant D, Rubert J, Vangrevelinghe E, Scheufler C, et al. Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov. 2012;2:512–23. doi: 10.1158/2159-8290.CD-11-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Vannucchi AM, Bogani C, Bartalucci N, Tozzi L, Martinelli S, Guglielmelli P, et al. Inhibitors of PI3K/Akt and/or mTOR Inhibit the Growth of Cells of Myeloproliferative Neoplasms and Synergize with JAK2 Inhibitor and Interferon. Blood. 2011;118:A3835. [Google Scholar]
- 199.Bogani C, Bartalucci N, Martinelli S, Tozzi L, Guglielmelli P, Bosi A, et al. Associazione Italiana per la Ricerca sul Cancro AGIMM Gruppo Italiano Malattie Mieloproliferative mTOR inhibitors alone and in combination with JAK2 inhibitors effectively inhibit cells of myeloproliferative neoplasms. PLoS One. 2013;8:e54826. doi: 10.1371/journal.pone.0054826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Fiskus W, Manepalli RR, Balusu R, Bhalla KN. Synergistic Activity of Combinations of JAK2 Kinase Inhibitor with PI3K/mTOR, MEK or PIM Kinase Inhibitor Against Human Myeloproliferative Neoplasm Cells Expressing JAK2V617F. Blood. 2010;116:A798. [Google Scholar]
- 201.Barrio S, Gallardo M, Albizua E, Jimenez A, Rapado I, Ayala R, et al. Epigenomic profiling in polycythaemia vera and essential thrombocythaemia shows low levels of aberrant DNA methylation. J Clin Pathol. 2011;64:1010–3. doi: 10.1136/jclinpath-2011-200175. [DOI] [PubMed] [Google Scholar]
- 202.Nischal S, Bhattacharyya S, Christopeit M, Yu Y, Zhou L, Bhagat T, et al. Methylome profiling reveals distinct alterations in phenotypic and mutational subgroups of myeloproliferative neoplasms. Cancer Res. 2013;73:1076–85. doi: 10.1158/0008-5472.CAN-12-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 204.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 205.Wang JC, Chen C, Dumlao T, Naik S, Chang T, Xiao YY, et al. Enhanced histone deacetylase enzyme activity in primary myelofibrosis. Leuk Lymphoma. 2008;49:2321–7. doi: 10.1080/10428190802527699. [DOI] [PubMed] [Google Scholar]
- 206.Wang Y, Fiskus W, Chong DG, Buckley KM, Natarajan K, Rao R, et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood. 2009;114:5024–33. doi: 10.1182/blood-2009-05-222133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.DeAngelo DJ, Tefferi A, Fiskus W, Mesa RA, Paley CS, Wadleigh M, et al. A Phase II Trial of Panobinostat, an Orally Available Deacetylase Inhibitor (DACi), in Patients with Primary Myelofibrosis (PMF), Post Essential Thrombocythemia (ET), and Post Polycythemia Vera (PV) Myelofibrosis. Blood. 2010;116:A630. doi: 10.1111/bjh.12384. [DOI] [PubMed] [Google Scholar]
- 208.Baffert F, Evrot E, Ebel N, Roelli C, Andraos R, Qian ZY, et al. Improved Efficacy Upon Combined JAK1/2 and Pan-Deacetylase Inhibition Using Ruxolitinib (INC424) and Panobinostat (LBH589) in Preclinical Mouse Models of JAK2V617F-Driven Disease. Blood. 2011;118:A798. [Google Scholar]
- 209.Fiskus W, Verstovsek S, Manshouri T, Rao R, Balusu R, Venkannagari S, et al. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin Cancer Res. 2011;17:7347–58. doi: 10.1158/1078-0432.CCR-11-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Marubayashi S, Koppikar P, Taldone T, Abdel-Wahab O, West N, Bhagwat N, et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Invest. 2010;120:3578–93. doi: 10.1172/JCI42442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Weigert O, Lane AA, Bird L, Kopp N, Chapuy B, van Bodegom D, et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J Exp Med. 2012;209:259–73. doi: 10.1084/jem.20111694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev. 2008;18:73–9. doi: 10.1016/j.gde.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 213.Deshpande A, Reddy MM, Schade GO, Ray A, Chowdary TK, Griffin JD, et al. Kinase domain mutations confer resistance to novel inhibitors targeting JAK2V617F in myeloproliferative neoplasms. Leukemia. 2012;26:708–15. doi: 10.1038/leu.2011.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Marit MR, Chohan M, Matthew N, Huang K, Kuntz DA, Rose DR, et al. Random mutagenesis reveals residues of JAK2 critical in evading inhibition by a tyrosine kinase inhibitor. PLoS One. 2012;7:e43437. doi: 10.1371/journal.pone.0043437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hornakova T, Springuel L, Devreux J, Dusa A, Constantinescu SN, Knoops L, et al. Oncogenic JAK1 and JAK2-activating mutations resistant to ATP-competitive inhibitors. Haematologica. 2011;96:845–53. doi: 10.3324/haematol.2010.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kapuria V, Levitzki A, Bornmann WG, Maxwell D, Priebe W, Sorenson RJ, et al. A novel small molecule deubiquitinase inhibitor blocks Jak2 signaling through Jak2 ubiquitination. Cell Signal. 2011;23:2076–85. doi: 10.1016/j.cellsig.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 217.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–97. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Purandare AV, McDevitt TM, Wan H, You D, Penhallow B, Han X, et al. Characterization of BMS-911543, a functionally selective small-molecule inhibitor of JAK2. Leukemia. 2012;26:280–8. doi: 10.1038/leu.2011.292. [DOI] [PubMed] [Google Scholar]
- 219.Hexner EO, Serdikoff C, Jan M, Swider CR, Robinson C, Yang S, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2008;111:5663–71. doi: 10.1182/blood-2007-04-083402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Santos FP, Kantarjian HM, Jain N, Manshouri T, Thomas DA, Garcia-Manero G, et al. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood. 2010;115:1131–6. doi: 10.1182/blood-2009-10-246363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Pardanani A, Gotlib J, Gupta V, Roberts AW, Wadleigh M, Sirhan S, et al. An Expanded Multicenter Phase I/II Study of CYT387, a JAK-1/2 Inhibitor for the Treatment of Myelofibrosis. Blood. 2011;118:A3849. [Google Scholar]
- 222.Ma LD, Zhao BH, Walgren R, Clayton JA, Blosser WD, Burkholder TP, et al. Efficacy of LY2784544, a Small Molecule Inhibitor Selective for Mutant JAK2 Kinase, In JAK2 V617F-Induced Hematologic Malignancy Models. Blood. 2010;116:A4087. [Google Scholar]
- 223.Verstovsek S, Mesa RA, Rhoades SK, Giles JLK, Pitou C, Jones E, et al. Phase I Study of the JAK2 V617F Inhibitor, LY2784544, in Patients with Myelofibrosis (MF), Polycythemia Vera (PV), and Essential Thrombocythemia (ET) Blood. 2011;118:A2814. [Google Scholar]
- 224.Nakaya Y, Shide K, Niwa T, Homan J, Sugahara S, Horio T, et al. Efficacy of NS-018, a potent and selective JAK2/Src inhibitor, in primary cells and mouse models of myeloproliferative neoplasms. Blood Cancer J. 2011;1:e29. doi: 10.1038/bcj.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Hart S, Goh KC, Novotny-Diermayr V, Hu CY, Hentze H, Tan YC, et al. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia. 2011;25:1751–9. doi: 10.1038/leu.2011.148. [DOI] [PubMed] [Google Scholar]
- 226.Komrokji RS, Wadleigh M, Seymour JF, Roberts AW, To LB, Zhu HJ, et al. Results of a Phase 2 Study of Pacritinib (SB1518), a Novel Oral JAK2 Inhibitor, In Patients with Primary, Post-Polycythemia Vera, and Post-Essential Thrombocythemia Myelofibrosis. Blood. 2011;118:A282. [Google Scholar]
- 227.Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13:311–20. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 228.Paquette R, Sokol L, Shah NP, Silver RT, List AF, Clary DO, et al. A Phase I Study of XL019, a Selective JAK2 Inhibitor, in Patients with Polycythemia Vera. Blood. 2008;112:A2810. [Google Scholar]
- 229.Shah NP, Olszynski P, Sokol L, Verstovsek S, Hoffman R, List AF, et al. A Phase I Study of XL019, a Selective JAK2 Inhibitor, in Patients with Primary Myelofibrosis, Post-Polycythemia Vera, or Post-Essential Thrombocythemia Myelofibrosis. Blood. 2008;112:A98. [Google Scholar]
- 230.Stump KL, Lu LD, Dobrzanski P, Serdikoff C, Gingrich DE, Dugan BJ, et al. A highly selective, orally active inhibitor of Janus kinase 2, CEP-33779, ablates disease in two mouse models of rheumatoid arthritis. Arthritis Res Ther. 2011;13:R68. doi: 10.1186/ar3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Thompson JE, Cubbon RM, Cummings RT, Wicker LS, Frankshun R, Cunningham BR, et al. Photochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitor. Bioorg Med Chem Lett. 2002;12:1219–23. doi: 10.1016/S0960-894X(02)00106-3. [DOI] [PubMed] [Google Scholar]
- 232.Baffert F, Régnier CH, De Pover A, Pissot-Soldermann C, Tavares GA, Blasco F, et al. Potent and selective inhibition of polycythemia by the quinoxaline JAK2 inhibitor NVP-BSK805. Mol Cancer Ther. 2010;9:1945–55. doi: 10.1158/1535-7163.MCT-10-0053. [DOI] [PubMed] [Google Scholar]
- 233.Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G, et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia. 2007;21:1658–68. doi: 10.1038/sj.leu.2404750. [DOI] [PubMed] [Google Scholar]