

Background: Increased PP2A levels have been linked to autoimmunity in SLE patients and transgenic mice.
Results: In T cells, PP2A overexpression increases the transcription of proinflammatory genes and facilitates chromatin accessibility at the Il17 locus.
Conclusion: Increased levels of PP2A promote the inflammatory capacity of T cells.
Significance: PP2A dysregulation may contribute to SLE by directly affecting lymphocyte gene expression.
Keywords: Autoimmunity, Chromatin Histone Modification, Inflammation, Interleukin, PP2A, T Cell
Abstract
Protein phosphatase 2A (PP2A) is a heterotrimeric serine/threonine phosphatase involved in essential cellular functions. T cells from patients with systemic lupus erythematosus (SLE) express high levels of the catalytic subunit of PP2A (PP2Ac). A mouse overexpressing PP2Ac in T cells develops glomerulonephritis in an IL-17-dependent manner. Here, using microarray analyses, we demonstrate that increased expression of PP2Ac grants T cells the capacity to produce an array of proinflammatory effector molecules. Because IL-17 is important in the expression of glomerulonephritis, we studied the mechanism through which PP2Ac dysregulation facilitates its production. We report that PP2Ac is involved in the regulation of the Il17 locus by enhancing histone 3 acetylation through a mechanism that involves activation of interferon regulatory factor 4. Increased histone 3 acetylation of the Il17 locus is shared between T cells of PP2Ac transgenic mice and patients with SLE. We propose that, by promoting the inflammatory capacity of T cells, PP2Ac dysregulation contributes to the pathogenesis of SLE.
Introduction
Protein phosphatase 2A (PP2A)4 is an evolutionarily conserved and ubiquitously expressed serine/threonine phosphatase (1). Its assembly requires a scaffold, a catalytic subunit, and a regulatory subunit (2, 3). The scaffold (subunit A) and catalytic (subunit C) proteins form a heterodimeric core that can associate with various regulatory (B) subunits thought to determine the substrate specificity of the holoenzyme (4). PP2A regulates a large number of cellular processes, including cell cycle and apoptosis (5–7), and modulates several signaling cascades, including the PI3K-AKT-mammalian target of rapamycin (8, 9), MAP kinase (10), and NF-κB (11) pathways. Defects in PP2A expression and/or function have been linked to cancer (12), neurodegenerative diseases (13, 14), and systemic lupus erythematosus (SLE) (15, 7).
Patients with SLE develop a chronic autoimmune response that leads to multiorgan inflammatory damage (16). The immune response in SLE is affected at several levels, but evidence from human patients and lupus-prone mice implicate T cells as a key element in the development of disease and in the instigation of inflammation (17). T cells from patients with SLE exhibit a number of phenotypic alterations. However, it has been difficult to attribute a pathogenic role to these defects. Levels of the catalytic subunit of PP2A (PP2Ac) are higher in T cells from SLE patients than in T cells from healthy controls (15), and this has been linked to T cell defects that include abnormal cytokine production (15). We demonstrated previously that a mouse overexpressing PP2Ac in a T cell-specific manner developed florid glomerulonephritis in response to antiglomerular basement membrane antibodies, a process that was dependent on IL-17 (18). The clinical relevance of these findings is further signified by the fact that T cells from patients with SLE produce large amounts of IL-17 and infiltrate the kidneys (19, 20).
To understand how dysregulation of PP2A grants T cells an increased capacity to amplify autoimmune pathology, we performed unbiased gene expression profile analyses. We present evidence that increased levels of PP2Ac induce a broad proinflammatory program that includes, but is not limited to, unrestrained IL-17 production. In addition, we reveal that aberrant chromatin remodeling, associated with increased activity and DNA binding of interferon regulatory factor 4 (IRF4), underlies the heightened capacity of PP2Ac transgenic T cells to produce IL-17 in response to TCR stimulation in a manner that resembles observations made previously in T cells from patients with SLE (21, 22).
EXPERIMENTAL PROCEDURES
Mice
The PP2Ac transgenic mice were generated as described previously (18). Mice were housed in specific pathogen-free conditions in accordance with the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee and bred with C57BL/6J (The Jackson Laboratory) mice. Non-transgenic littermates were used as controls. Mice used in experiments were between 8 and 12 weeks old.
T Cell Isolation and Stimulation
TCR-αβ+ CD4+ CD25− CD62L+ naïve T cells were isolated from spleens and peripheral lymph nodes (axillary and inguinal) of transgenic and control mice by magnetic cell sorting (CD4+ CD62L+ T cell isolation kit II, Miltenyi Biotec). Post-sorting cell purity was > 95%. Cells were cultured in RPMI 1640 supplemented with 10% FCS and antibiotics. When indicated, serum-free medium was used (X-vivo 10, Lonza). T cells were stimulated with plate-bound goat anti-hamster antibodies (MP Biomedicals) and soluble anti-CD3 (0.25 μg/ml, clone no. 145-2C11, Biolegend) and anti-CD28 (0.5 μg/ml, clone no. 37.51, Biolegend) antibodies. For T cell differentiation studies, cells were cultured during 4 days in the presence of the following cytokines that were replenished every 48 h: Th0, IL-2 (100 units/ml); Th1, IL-12 (10 ng/ml), and IL-2 (100 units/ml); and Th17, IL-6 (25 ng/ml), TGF-β (2.5 ng/ml), and IL-23 (10 ng/ml).
Microarray Analyses
Naïve CD4 T cells (CD3+CD4+CD25−CD62L+) from six transgenic and six wild-type mice were sorted in a FACSAria II (BD Biosciences) (> 98% purity). Cells were lysed directly or after stimulation with anti-CD3 and anti-CD28 for 6 or 24 h. RNA was labeled and hybridized to Affymetrix Mouse Gene 1.0 ST microarrays. Raw data were background-corrected and normalized using RMAExpress. Data were analyzed with R (23). Functional annotation clustering was performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID). False discovery rate control was performed using the Benjamin correction (24, 25). p < 0.001 was considered significant in this analysis.
RNA Isolation and Real-time PCR
RNA was isolated using TRIzol (Invitrogen). cDNA was produced from 500 ng of RNA (reverse transcription system, Promega). Real-time PCR was performed using SYBR Green (LightCycler 480 SYBR Green I Master, Roche). Primer sequences and amplification conditions are available upon request.
Antibodies and Reagents
The following antibodies were used for Western blot and/or ChIP experiments: anti-PP2A C (clone no. 1D6, Millipore), anti-Stat3 (clone no. 124H6, Cell Signaling Technology), anti-phospho-Stat3 (Tyr-705, catalog no. 9131, Cell Signaling Technology), anti-H3K4me3 (catalog no. ab8580, Abcam), anti-H3K9me3 (catalog no. ab8898, Abcam), anti-H3K27me3 (catalog no. 07-449, Millipore), anti-acetyl-H3 (catalog no. 06-599, Millipore), and anti-IRF4 (catalog no. sc-6059 X, Santa Cruz Biotechnology).
Western Blot Analysis
Naïve CD4 T cells were lysed in radioimmune precipitation assay buffer. Cell lysates were separated in either conventional acrylamide gels or in SuperSep Phos-tag (Wako Pure Chemical Industries) acrylamide gels to retard the migration of phosphorylated proteins. Proteins were transferred to a PVDF membrane and blotted with the indicated antibodies.
ROCK Activity Quantification
Activated naïve CD4 T cells were lysed in radioimmune precipitation assay buffer. ROCK kinase activity was measured in cell lysates using Rho kinase (ROCK) activity assay according to the instructions of the manufacturer (Millipore).
Chromatin Immunoprecipitation
The MAGnify ChIP system (Invitrogen) was used following the instructions of the manufacturer. Briefly, between 1 and 3 million cells were cross-linked with 1% formaldehyde for 10 min at 37 °C. The reaction was stopped with glycine for 5 min, and the samples were lysed and sonicated to obtain 200- to 500-bp fragments. Immunoprecipitation was performed with the indicated antibodies and protein A/G Dynabeads (Invitrogen). Cross-linking was reversed, and DNA was eluted and purified using DNA purification magnetic beads (Invitrogen). Enrichment of specific DNA sequences was quantified by real-time PCR and normalized against the input.
Statistical Analyses
Student's two-tailed t tests and Mann-Whitney U tests were used. p < 0.05 was considered significant. Results are expressed as the mean ± S.E. unless noted otherwise.
RESULTS
Increased PP2Ac Levels Skew T Cell Gene Expression toward Inflammation
To investigate whether PP2Ac overexpression modifies the transcriptional profile of CD4 T cells, we isolated naïve CD4 cells from PP2Ac transgenic mice and WT littermates and performed microarray analyses in untreated and stimulated cells for 6 and 24 h (Fig. 1). PP2Ac overexpression affected a relatively small number of genes (n = 130), a great majority of which were up-regulated in the transgenic compared with the WT CD4 T cells (Fig. 1A). To determine whether the set of genes up-regulated by PP2Ac was enriched in molecules associated with particular biological functions, we performed a functional gene clustering analysis that yielded 89 categories of biological processes. After the p value was adjusted by the Benjamin correction, only seven biological processes remained associated with the PP2Ac gene set in a statistically significant manner (p < 0.001) (Fig. 1B). The three most significant associations were with immune response (p = 4.9 × 10−17), defense response (p = 3.7 × 10−13), and inflammatory response (p = 1.2 × 10−11). Of the 124 genes up-regulated in the PP2Ac transgenic CD4 T cells, 25 (∼1 of 5) were found higher in all conditions and 58 (∼1 of 2) only in activated cells (data not shown). As predicted by the functional clustering analysis, the latter encoded primarily for immune response effector molecules, including cytokines and chemokines (Fig. 1C).
FIGURE 1.

PP2Ac dysregulation induces a proinflammatory transcriptional profile in CD4 T cells. A, comparison of microarray expression values in naïve CD4 T cells from PP2Ac transgenic and wild-type mice 24 h after activation with anti-CD3 and anti-CD28. Red dots indicate genes up-regulated ≥ 2-fold in transgenic mice. Blue dots indicate genes down-regulated ≥ 2-fold. Some genes involved in the inflammatory response are highlighted. B, functional gene clustering analysis showing that the set of genes up-regulated in the PP2Ac transgenic mice is enriched in genes involved in cell movement and inflammation. The dotted gray line indicates statistical significance after p value correction (p ≤ 0.001). C, microarray expression values were normalized to unstimulated cells, and the magnitude of their increase upon 6 and 24 h of T cell activation (Δ 6 h and Δ 24 h, respectively) was compared between transgenic (Tg) and wild-type mice. Transcripts whose activation-induced regulation differed ≥ 2-fold in transgenic and wild-type CD4 T cells were included in the heat maps.
These results reveal that even though PP2Ac is known to control a large variety of fundamental cellular functions, such as cell cycle and apoptosis (1), its overexpression in T cells facilitates the transcription of proinflammatory genes.
PP2Ac Allows Rapid IL-17 Transcription Independently of Known Th17-related Factors
In a previous report, we showed that the presence of increased PP2Ac levels in T cells exacerbates autoimmune glomerulonephritis in an IL-17-dependent manner (18). Because the gene expression data indicated that transcription of molecules functionally related to IL-17 was increased in the transgenic T cells, including Il17a, Il17f, and Il1a (Fig. 1), we chose to analyze the mechanisms through which increased PP2Ac facilitated Il17 transcription.
For this purpose, we performed a time course experiment to determine the kinetics of transcription of Il17a in the PP2Ac transgenic mice. As shown in Fig. 2A, ll17a mRNA was detected in transgenic mice as soon as 3 h after CD3/CD28 stimulation, and IL-17 levels reached a plateau at 6 h. In sharp contrast, T cells from WT mice produced no detectable amounts of IL-17 during the first 24 h after activation.
FIGURE 2.

PP2Ac does not induce Th17-associated transcription factors. A, naïve CD4 T cells from WT or PP2Ac transgenic mice were stimulated for the indicated time periods. Il17a mRNA was normalized to Actb levels (mean ± S.E.). *, p ≤ 0.01. B, microarray expression values of the indicated genes are shown. C, naïve CD4 T cells were stimulated overnight. Expression of the indicated genes was normalized to Actb. Shown is the fold expression relative to WT cells (mean ± S.E.). D, naïve CD4 T cells were stimulated during 48 h, expanded with IL-2 during 5 days, rested 48 h, washed and incubated with IL-6 for 3 h. Cells were lysed in radioimmune precipitation assay buffer in the presence of protease and phosphatase inhibitors. E, naïve CD4 T cells were stimulated with anti-CD3 and anti-CD28 and lysed at the indicated time points. Data are representative of at least four experiments, each with ≥ 3 mice/group (A and C), or ≥ 2 experiments, each performed with cells pooled from ≥ 3 mice (D and E).
Transcription of IL-17 in CD4 T cells is restricted to activated Th17 cells (26). This specificity is achieved by epigenetic control of the Il17 locus that is normally inaccessible to transcription factors in naïve cells (27). Remodeling of chromatin at the Il17 locus is driven by the combined action of TGF-β and inflammatory cytokines, including IL-1β, IL-6, and IL-23 (28–30), and depends on the presence of certain lineage-determining transcription factors, such as retinoic acid receptor-related orphan receptor γ (ROR-γt) (31) and signal transducer and activator of transcription 3 (STAT3) (32–35). In addition, other transcription factors regulate the Th17 program. These include IRF4 (36, 37), aryl-hydrocarbon receptor (Ahr) (38, 39), runt-related transcription factor 1 (Runx1) (40), B-cell-activating transcription factor (Batf) (41), and IκBζ (42).
We found no differences in the expression values of these Th17-related transcription factors between transgenic and control mice in the microarray analyses. In fact, the abundance of their transcripts was not modified at the early time points analyzed (6 and 24 h), with the exception of Irf4 whose transcription was strongly induced by activation in both WT and transgenic mice (Fig. 2B). Levels of these factors were also analyzed with quantitative PCR, and the absence of significant differences was confirmed (Fig. 2C).
The ability of STAT3 to act as a Th17-inducing factor is controlled by the phosphorylation of its tyrosine residue 705 (35). To rule out the possibility that differential phosphorylation of STAT3 facilitated IL-17 production in naïve CD4 T cells from transgenic mice, we analyzed Tyr-705 STAT3 phosphorylation induced by IL-6 and by TCR activation. IL-6-induced pSTAT3 was not different in transgenic and WT mice, suggesting that high levels of cellular PP2Ac do not facilitate cytokine-induced STAT3 phosphorylation (Fig. 2D). In time course experiments of TCR activation, STAT3 phosphorylation was only detected at late time points and was not different in transgenic and WT cells (Fig. 2E), suggesting that pSTAT3 was not involved in the early IL-17 production observed in transgenic mice. Taken together, these results indicate that the overexpression of PP2Ac allows rapid transcription of Il17a upon TCR stimulation without affecting the induction of Th17-associated transcription factors.
Increased PP2Ac Levels Allow Non-Th17 Cells to Produce IL-17
To determine whether PP2Ac overexpression facilitated the differentiation of CD4 T cells into the Th17 helper subset, we stimulated naïve CD4 T cells from WT and transgenic mice in non-polarizing (Th0) and Th17-polarizing conditions for 5 days. At the end of the Th0 stimulation period, cells from transgenic mice produced ∼30-fold more Il17a (p = 0.03) and ∼15-fold more Il17f (p = 0.05) than WT cells. Transcription of Rorc was modestly increased in transgenic cells (∼3-fold, p = 0.03), whereas abundance of Il21 did not differ (Fig. 3A). When Th17 differentiation was induced, the differences in expression of the analyzed genes were minimized as WT cells acquired the Th17 genetic profile.
FIGURE 3.
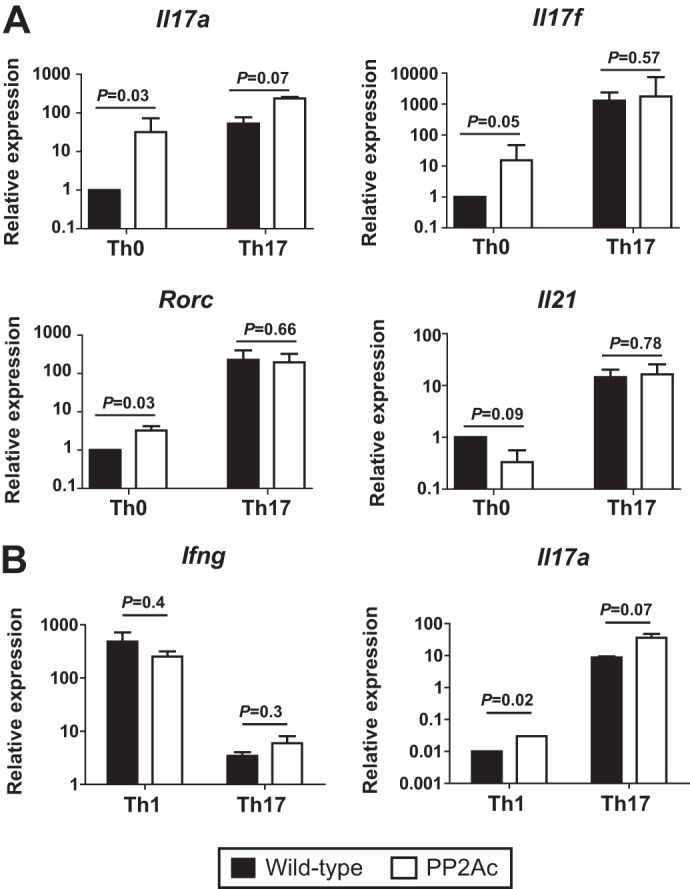
The PP2Ac effect is independent of T cell effector differentiation. A, naïve CD4 T cells were stimulated in neutral (Th0) or Th17-inducing conditions (TGF-β1 and IL-6). At day 5, expression of the indicated genes was analyzed by real-time PCR. Results were normalized to Actb. Shown is the fold expression relative to WT Th0 cells (mean ± S.E.). B, naïve CD4 T cells were stimulated in Th1- (IL-12) or Th17-polarizing conditions. After 5 days, expression of Ifng and Il17a was analyzed by real-time PCR. Results are expressed as fold expression relative to Actb. Data are representative of four (A) or three (B) experiments, each with three mice per group.
To determine whether high levels of PP2Ac affected other effector T cell subsets, we stimulated naïve CD4 T cells under Th1- or Th17-polarizing conditions. After 5 days, the production of the signature cytokines IFN-γ and IL-17A was analyzed. As shown in Fig. 3B, production of IFN-γ was not different in cells from WT and transgenic mice, indicating that under Th1 or Th17 polarization, the transcriptional regulation of this cytokine is not altered. In contrast, similar to what we observed in non-polarized cells (Th0), transcription of Il17a was significantly increased (p = 0.02) in the Th1-differentiated transgenic cells, indicating that the inhibitory effect mediated by Th1 cytokines (i.e. IFN-γ) on IL-17 production was incomplete in the presence of high levels of PP2Ac.
PP2Ac Increases Chromatin Accessibility at the Il17 Locus
T cells acquire the capacity to produce signature effector cytokines during helper subset differentiation by epigenetic modifications of chromatin at the corresponding cytokine loci (27). This, along with the fact that Il17a and Il17f, both affected by high PP2Ac levels, are encoded in the same locus and share common regulatory elements (43), suggested that the effect of PP2Ac could be exerted by chromatin remodeling. Histones undergo posttranslational modifications that determine the accessibility and, thus, the transcriptional activity of neighboring genes (44). For this reason, we determined the levels of permissive and repressive histone modifications known to be important for the regulation of the Il17 locus (43, 45). To this end, ChIP was performed in resting and stimulated naïve CD4 T cells using antibodies that bind specifically to different histone 3 (H3) posttranslational modifications. H3 configuration was analyzed at several conserved non-coding sequences throughout the Il17 locus (43) and also within the Il17a and Il17f genes (Fig. 4). As shown in Fig. 4B, an increase in H3 acetylation (a permissive mark) was observed throughout the Il17 locus, particularly in the proximal promoter region (PrPr) of both Il17a and Il17f. This effect was already present in unstimulated cells and increased after stimulation. In sharp contrast, other H3 modifications, including trimethylation of lysines 9 (H3K9, repressive), 4 (H3K4, permissive), and 27 (H3K27, repressive) were not different in WT and transgenic cells regardless of the stimulation status of the cells (Fig. 4B).
FIGURE 4.

PP2Ac increases chromatin accessibility at the Il17 locus. A, the mouse Il17 locus is depicted. The conserved non-coding sequences as well as the sites within the Il17a and Il17f genes that were amplified during the ChIP experiments are indicated. B, naïve CD4 T cells were stimulated and fixed, and ChIP was performed for acetylated H3, H3K9me3, H3K4me3, and H3K27me3. Shown is relative binding (PP2Ac:WT ratio) in resting (blue) or stimulated (red) cells. Data were normalized to the corresponding input DNA and are representative of three or four independent experiments, each with ≥ 3 mice/group. PrPr, proximal promoter.
Together, these results indicate that high levels of PP2Ac are associated with increased accessibility to the Il17 locus enabled by constitutively high local H3 acetylation. This is similar to what has been observed in T cells from patients with SLE that have an increased abundance of PP2Ac (15), produce high amounts of IL-17 upon activation, and have higher levels of H3 acetylation at the IL17 locus (21).
H3 Acetylation at the Il17 Locus in PP2Ac Transgenic Mice Does Not Require Th17-inducing Cytokines
The differentiation of Th17 cells requires chromatin remodeling at the loci of specific effector genes, such as Il17a and Il17f. These changes eliminate epigenetic restrictions and allow rapid transcription of key cytokines upon T cell activation by increasing the accessibility of the loci to transcription factors (46–48). During the initial period of Th17 differentiation, permissive chromatin changes occur at the Rorc locus that will facilitate the production of this signature transcription factor (31). At later time points, opening of the Il17 locus occurs. Because naïve CD4 T cells from PP2Ac transgenic mice were able to produce high levels of IL-17A and IL-17F during the first 24 h following TCR activation alone (Fig. 2A) (18), we hypothesized that increased levels of PP2Ac could induce epigenetic changes similar to those observed during Th17 differentiation. For this purpose, we stimulated naïve CD4 T cells in the absence or presence of Th17-polarizing cytokines (TGF-β and IL-6) and, after 18 h, analyzed the acetylation and Lys-4 trimethylation of H3 in the Rorc and Il17 loci. In WT mice, addition of TGF-β and IL-6 was associated with a significant increase in acetylated and Lys-4 trimethylated H3 at the Rorc promoter region (Fig. 5A). As predicted by the normal ROR-γt levels (Fig. 2), PP2Ac mice showed a normal remodeling pattern at the Rorc locus (Fig. 5A). As expected in this early time point, no H3 modification was observed in the Il17 locus of WT mice even when the cells were stimulated in the presence of TGF-β and IL-6 (Fig. 5B). In concordance with our previous findings, overexpression of PP2Ac was associated with a robust enrichment of acetylated H3 in the absence of Th17-inducing cytokines. Moreover, addition of TGF-β and IL-6 to transgenic cells had a negligible effect on the already acetylated H3 of the Il17a promoter region (Fig. 5B).
FIGURE 5.
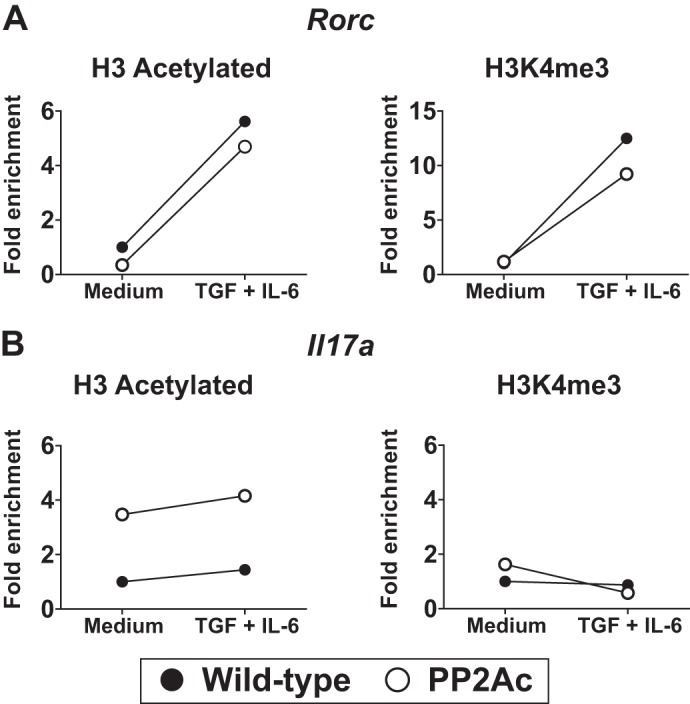
Increased PP2Ac levels induce H3 acetylation in the Il17 locus in a constitutive manner. A and B, naïve CD4 T cells were stimulated in serum-free medium in the absence or presence of TGF-β and IL-6. After 18 h, the promoter regions of the Rorc and Il17a genes were analyzed by ChIP assays using anti-H3 acetylated or anti-H3K4me3 antibodies. Results were normalized against the input and are expressed as fold change over the WT stimulated in culture medium alone.
These results indicate that increased PP2Ac levels cause H3 acetylation in a manner independent of Th17-inducing cytokines. Moreover, they suggest that high levels of PP2Ac are specifically associated with increased H3 acetylation.
PP2Ac Transgenic CD4 T Cells Exhibit Increased IRF4 Recruitment to the Il17 Locus
IRF4 is one of the earliest transcription factors recruited to the Il17 locus during Th17 cell differentiation and has been shown to mediate the local initial chromatin remodeling events (46, 47). IRF4 activity is posttranslationally regulated by Rho kinases (ROCK) through serine phosphorylation (49). Abnormally high activity of ROCK has been linked to aberrant T cell phenotypes in patients with SLE (22, 50). To determine whether IRF4 facilitated IL-17 production in PP2Ac transgenic mice, we performed ChIP assays in unstimulated and anti-CD3/CD28-stimulated naïve CD4 T cells. As shown in Fig. 6A, IRF4 recruitment at the Il17a locus was enriched in PP2Ac transgenic T cells both before and after TCR stimulation. This was associated with increased levels of phosphorylated IRF4 in PP2Ac transgenic T cells, as shown in Fig. 6B using an acrylamide gel where phosphorylated proteins migrate more slowly. As a control, we show that PP2Ac phosphorylation did not differ between WT and transgenic T cells and was unaffected by CD3/CD28 stimulation. Finally, ROCK kinase activity was significantly higher in activated PP2Ac transgenic T cells compared with their WT counterparts (Fig. 6C). Taken together, our results indicate that increased levels of PP2Ac in T cells are associated with enhanced ROCK activity and subsequent IRF4 activation that binds to the Il17a and promotes its transcription.
FIGURE 6.
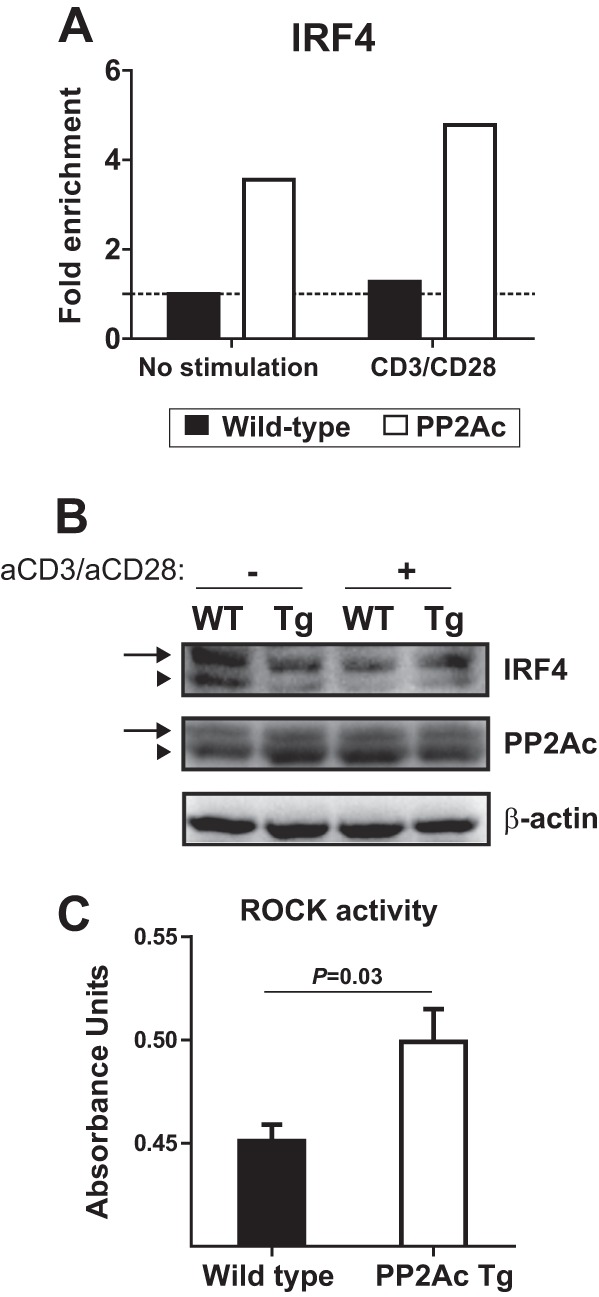
PP2Ac promotes IRF4 activation and recruitment to the Il17a locus. A, ChIP experiments were performed to analyze IRF4 occupancy of the Il17a promoter region in naïve CD4 T cells before and after stimulation with anti-CD3 and anti-CD28. Results were normalized against the input and are expressed as fold change over the unstimulated WT cells. B, naïve CD4 T cells were isolated from PP2Ac Tg and WT mice and were lysed before or after activation with anti-CD3 and anti-CD28 (1 h). Lysates were separated in a SuperSep Phos-tag acrylamide gel that retards the migration of phosphorylated proteins. Proteins were transferred to a PVDF membrane that was blotted with anti-IRF4, anti-PP2Ac, and anti-β-actin. Arrows indicate the phosphorylated proteins. Arrowheads indicate unphosphorylated proteins. C, cell lysates of stimulated naïve CD4 T cells were probed for Rho kinase activity.
DISCUSSION
In this report, we show that dysregulation of the serine/threonine phosphatase PP2A induces the expression of a proinflammatory genetic profile in T cells characterized by increased transcription of chemokines and cytokines upon TCR activation. We also demonstrate that increased levels of PP2Ac modify the epigenetic landscape of the Il17 locus, allowing unrestricted transcription of Il17a and Il17f. This phenomenon is explained by heightened IRF4 activity that is associated with increased H3 acetylation of the Il17 locus that is poised to undergo rapid transcription following T cell activation.
SLE is a complex inflammatory disease that develops in genetically predisposed individuals. Multiple immune and non-immune factors synergize to promote a chronic autoimmune response directed toward a multitude of self-antigens. This process manifests as target organ disease when products of the autoimmune response, such as activated T cells and autoantibodies, cause inflammation. Several molecules have been linked to SLE either by genetic associations or functional studies (16). The complexity of the disease has hindered the efforts to identify which molecules are causal and how they impact pathology. We have established previously that PP2Ac dysregulation has an independent role in the expression of lupus-related pathology. By studying a mouse that has increased levels of this phosphatase in T cells in the absence of other autoimmunity-associated abnormalities, we were able to demonstrate in vivo that high levels of PP2Ac in T cells facilitate autoimmune inflammation in an IL-17-dependent manner (18). Here we have shown that PP2Ac dysregulation shifts the T cell gene expression pattern toward a proinflammatory phenotype. The study of PP2Ac expression solely in T cells reveals a mechanism through which a single molecular abnormality contributes to autoimmune pathology. It also defines in a reductionist manner the relative contribution of PP2Ac overexpression to the development of SLE.
Healthy T cell function relies on the capacity of the T cell to interpret external cues and mount adequate responses. These are shaped through the modulation of gene expression and are imprinted through epigenetic changes. This concept is illustrated by the inability of naïve T cells to produce effector cytokines, such as IFN-γ, IL-4, or IL-17, that is overcome when access to the respective loci is granted during activation and differentiation in the presence of lineage-determining cytokines (51, 52). Alterations in epigenetic regulatory mechanisms are well described in T cells from patients with SLE and have been attributed a pathological role (53, 54). The fact that the aberrant epigenetic changes associated with high PP2Ac levels are present in a constitutive manner could affect, importantly, the response of the T cell to antigen or other external cues. Here we have associated the rapid production of inflammatory cytokines by naïve T cells as well as the incomplete suppression of IL-17 production during Th1 differentiation with high levels of PP2Ac. This phenotypic anomaly may be relevant in the setting of SLE, where PP2Ac is overexpressed in T cells (15) and production of large amounts of IL-17 is observed upon activation in the absence of classical IL-17-inducing factors (19, 22, 55–57). Moreover, abnormally high levels of H3 acetylation in the IL17 locus have been associated with increased IL-17 production in T cells from patients with SLE (21).
Rho kinases have been associated to lupus (22) through their capacity to regulate cytoskeletal proteins (50) and to activate the transcription factor IRF4 (49). The importance of IRF4 in the pathogenesis of lupus is supported by work performed in animal models where IRF4 deficiency (58) or its pharmacological inhibition (59) abrogate lupus pathology, whereas increased function of IRF4 causes autoimmunity (60, 49). Our results suggest that PP2Ac dysregulation may contribute to IRF4 increased activity by promoting ROCK activation. IRF4 colocalizes with histone acetyltransferases such as p300 to modulate chromatin remodeling at the Il17 locus (46). We believe that activated IRF4 may be responsible for the recruitment of histone acetyltransferases to the Il17 locus in the PP2Ac transgenic mice (Fig. 7).
FIGURE 7.
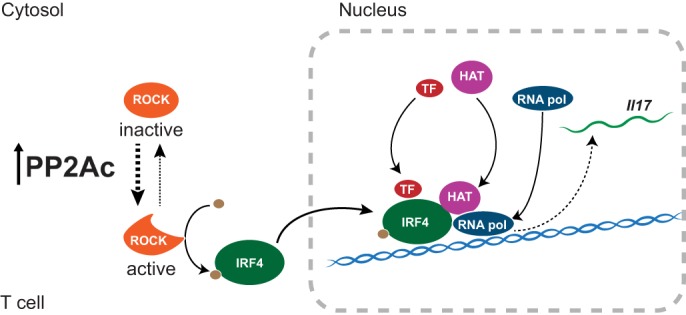
PP2Ac promotes IL-17 production by facilitating IRF4 activity. Increased levels of PP2Ac promote Rho kinase (ROCK) activation. Activated ROCK phosphorylates IRF4, which then localizes to the Il17 locus. There, IRF4 recruits histone acetyltransferases (HAT), as well as other transcription factors (TF) and RNA polymerase (RNA pol) to initiate transcription of Il17.
PP2A regulates a large number of cellular processes (1). However, dysregulation of PP2Ac mainly affected immune response genes, as shown in the functional clustering analysis. This may reflect the fact that in T cells, the regulation of basic cellular processes, including cell cycle and metabolism, are intimately involved in determining T cell effector functions (61–64). PP2A has been implicated in the regulation of gene expression through various mechanisms, including direct association with chromatin remodeling complexes (65–67). However, we could not detect the presence of PP2Ac on the Il17 locus (data not shown). We demonstrate, though, a novel pathway by which PP2Ac promotes the transcription of Il17 that depends on IRF4. This mechanism is probably not responsible for the whole spectrum of gene transcriptional effects because most of the genes affected by PP2Ac are not known to be regulated by IRF4 (47, 68). Whether PP2Ac overexpression induces the rest of the observed proinflammatory changes by modifying fundamental evolutionary conserved cellular processes or directly by its association to molecular complexes that regulate gene expression will be the focus of future work.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 AI068787 (to G. C. T.). This work was also supported by an Alliance for Lupus Research grant (to J. C. C.).
The amino acid sequence of this protein can be accessed through the NCBI Protein Database under NCBI accession number GSE 49466.
- PP2A
- protein phosphatase 2A
- SLE
- systemic lupus erythematosus
- PP2Ac
- protein phosphatase 2A catalytic subunit
- IRF
- interferon regulatory factor
- H3
- histone
- TCR
- T cell receptor
- ROCK
- Rho-associated, coiled-coil–containing protein kinase.
REFERENCES
- 1. Janssens V., Goris J. (2001) Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 353, 417–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xu Y., Xing Y., Chen Y., Chao Y., Lin Z., Fan E., Yu J. W., Strack S., Jeffrey P. D., Shi Y. (2006) Structure of the protein phosphatase 2A holoenzyme. Cell 127, 1239–1251 [DOI] [PubMed] [Google Scholar]
- 3. Cho U. S., Xu W. (2007) Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 445, 53–57 [DOI] [PubMed] [Google Scholar]
- 4. Janssens V., Longin S., Goris J. (2008) PP2A holoenzyme assembly. In cauda venenum (the sting is in the tail). Trends Biochem. Sci. 33, 113–121 [DOI] [PubMed] [Google Scholar]
- 5. Lee T. Y., Lai T. Y., Lin S. C., Wu C. W., Ni I. F., Yang Y. S., Hung L. Y., Law B. K., Chiang C. W. (2010) The B56γ3 regulatory subunit of protein phosphatase 2A (PP2A) regulates S phase-specific nuclear accumulation of PP2A and the G1 to S transition. J. Biol. Chem. 285, 21567–21580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kurimchak A., Graña X. (2012) PP2A holoenzymes negatively and positively regulate cell cycle progression by dephosphorylating pocket proteins and multiple CDK substrates. Gene 499, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crispin J. C., Apostolidis S. A., Finnell M. I., Tsokos G. C. (2011) Induction of PP2A Bβ, a regulator of IL-2 deprivation-induced T-cell apoptosis, is deficient in systemic lupus erythematosus. Proc. Natl. Acad. Sci. U.S.A., 108, 12443–12448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peterson R. T., Desai B. N., Hardwick J. S., Schreiber S. L. (1999) Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycin-associated protein. Proc. Natl. Acad. Sci. U.S.A. 96, 4438–4442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hahn K., Miranda M., Francis V. A., Vendrell J., Zorzano A., Teleman A. A. (2010) PP2A regulatory subunit PP2A-B′ counteracts S6K phosphorylation. Cell Metab. 11, 438–444 [DOI] [PubMed] [Google Scholar]
- 10. Chung H., Brautigan D. L. (1999) Protein phosphatase 2A suppresses MAP kinase signalling and ectopic protein expression. Cell Signal. 11, 575–580 [DOI] [PubMed] [Google Scholar]
- 11. Eitelhuber A. C., Warth S., Schimmack G., Düwel M., Hadian K., Demski K., Beisker W., Shinohara H., Kurosaki T., Heissmeyer V., Krappmann D. (2011) Dephosphorylation of Carma1 by PP2A negatively regulates T-cell activation. EMBO J. 30, 594–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mumby M. (2007) PP2A. Unveiling a reluctant tumor suppressor. Cell 130, 21–24 [DOI] [PubMed] [Google Scholar]
- 13. Sontag E., Nunbhakdi-Craig V., Lee G., Brandt R., Kamibayashi C., Kuret J., White C. L., 3rd, Mumby M. C., Bloom G. S. (1999) Molecular interactions among protein phosphatase 2A, Tau, and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J. Biol. Chem. 274, 25490–25498 [DOI] [PubMed] [Google Scholar]
- 14. Landrieu I., Smet-Nocca C., Amniai L., Louis J. V., Wieruszeski J. M., Goris J., Janssens V., Lippens G. (2011) Molecular implication of PP2A and Pin1 in the Alzheimer's disease specific hyperphosphorylation of Tau. PLoS ONE 6, e21521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Katsiari C. G., Kyttaris V. C., Juang Y. T., Tsokos G. C. (2005) Protein phosphatase 2A is a negative regulator of IL-2 production in patients with systemic lupus erythematosus. J. Clin. Invest. 115, 3193–3204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsokos G. C. (2011) Systemic lupus erythematosus. N. Engl. J. Med. 365, 2110–2121 [DOI] [PubMed] [Google Scholar]
- 17. Crispín J. C., Kyttaris V. C., Terhorst C., Tsokos G. C. (2010) T cells as therapeutic targets in SLE. Nat. Rev. Rheumatol. 6, 317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crispín J. C., Apostolidis S. A., Rosetti F., Keszei M., Wang N., Terhorst C., Mayadas T. N., Tsokos G. C. (2012) Cutting edge. Protein phosphatase 2A confers susceptibility to autoimmune disease through an IL-17-dependent mechanism. J. Immunol. 188, 3567–3571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crispín J. C., Oukka M., Bayliss G., Cohen R. A., Van Beek C. A., Stillman I. E., Kyttaris V. C., Juang Y. T., Tsokos G. C. (2008) Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J. Immunol. 181, 8761–8766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Y., Ito S., Chino Y., Goto D., Matsumoto I., Murata H., Tsutsumi A., Hayashi T., Uchida K., Usui J., Yamagata K., Sumida T. (2010) Laser microdissection-based analysis of cytokine balance in the kidneys of patients with lupus nephritis. Clin. Exp. Immunol. 159, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rauen T., Hedrich C. M., Juang Y. T., Tenbrock K., Tsokos G. C. (2011) cAMP-responsive element modulator (CREM)α protein induces interleukin 17A expression and mediates epigenetic alterations at the interleukin-17A gene locus in patients with systemic lupus erythematosus. J. Biol. Chem. 286, 43437–43446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Isgro J., Gupta S., Jacek E., Pavri T., Duculan R., Kim M., Kirou K. A., Salmon J. E., Pernis A. B. (2013) Enhanced Rho-associated protein kinase activation in patients with systemic lupus erythematosus. Arthritis Rheum. 65, 1592–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. R Core Team (2013) R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 24. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 25. Huang da W., Sherman B. T., Lempicki R. A. (2009) Bioinformatics enrichment tools. Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Korn T., Bettelli E., Oukka M., Kuchroo V. K. (2009) IL-17 and Th17 Cells. Annu. Rev. Immunol. 27, 485–517 [DOI] [PubMed] [Google Scholar]
- 27. O'Shea J. J., Paul W. E. (2010) Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327, 1098–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mangan P. R., Harrington L. E., O'Quinn D. B., Helms W. S., Bullard D. C., Elson C. O., Hatton R. D., Wahl S. M., Schoeb T. R., Weaver C. T. (2006) Transforming growth factor-β induces development of the T(H)17 lineage. Nature 441, 231–234 [DOI] [PubMed] [Google Scholar]
- 29. Bettelli E., Carrier Y., Gao W., Korn T., Strom T. B., Oukka M., Weiner H. L., Kuchroo V. K. (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238 [DOI] [PubMed] [Google Scholar]
- 30. Ghoreschi K., Laurence A., Yang X. P., Tato C. M., McGeachy M. J., Konkel J. E., Ramos H. L., Wei L., Davidson T. S., Bouladoux N., Grainger J. R., Chen Q., Kanno Y., Watford W. T., Sun H. W., Eberl G., Shevach E. M., Belkaid Y., Cua D. J., Chen W., O'Shea J. J. (2010) Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature 467, 967–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ivanov I. I., McKenzie B. S., Zhou L., Tadokoro C. E., Lepelley A., Lafaille J. J., Cua D. J., Littman D. R. (2006) The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 [DOI] [PubMed] [Google Scholar]
- 32. Chen Z., Laurence A., Kanno Y., Pacher-Zavisin M., Zhu B. M., Tato C., Yoshimura A., Hennighausen L., O'Shea J. J. (2006) Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl. Acad. Sci. U.S.A. 103, 8137–8142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Beaucoudrey L., Puel A., Filipe-Santos O., Cobat A., Ghandil P., Chrabieh M., Feinberg J., von Bernuth H., Samarina A., Jannière L., Fieschi C., Stéphan J. L., Boileau C., Lyonnet S., Jondeau G., Cormier-Daire V., Le Merrer M., Hoarau C., Lebranchu Y., Lortholary O., Chandesris M. O., Tron F., Gambineri E., Bianchi L., Rodriguez-Gallego C., Zitnik S. E., Vasconcelos J., Guedes M., Vitor A. B., Marodi L., Chapel H., Reid B., Roifman C., Nadal D., Reichenbach J., Caragol I., Garty B. Z., Dogu F., Camcioglu Y., Gülle S., Sanal O., Fischer A., Abel L., Stockinger B., Picard C., Casanova J. L. (2008) Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J. Exp. Med. 205, 1543–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harris T. J., Grosso J. F., Yen H. R., Xin H., Kortylewski M., Albesiano E., Hipkiss E. L., Getnet D., Goldberg M. V., Maris C. H., Housseau F., Yu H., Pardoll D. M., Drake C. G. (2007) Cutting edge. An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J. Immunol. 179, 4313–4317 [DOI] [PubMed] [Google Scholar]
- 35. Yang X. O., Panopoulos A. D., Nurieva R., Chang S. H., Wang D., Watowich S. S., Dong C. (2007) STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 282, 9358–9363 [DOI] [PubMed] [Google Scholar]
- 36. Brüstle A., Heink S., Huber M., Rosenplänter C., Stadelmann C., Yu P., Arpaia E., Mak T. W., Kamradt T., Lohoff M. (2007) The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 8, 958–966 [DOI] [PubMed] [Google Scholar]
- 37. Chen Q., Yang W., Gupta S., Biswas P., Smith P., Bhagat G., Pernis A. B. (2008) IRF-4-binding protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity 29, 899–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Veldhoen M., Hirota K., Westendorf A. M., Buer J., Dumoutier L., Renauld J. C., Stockinger B. (2008) The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109 [DOI] [PubMed] [Google Scholar]
- 39. Veldhoen M., Hirota K., Christensen J., O'Garra A., Stockinger B. (2009) Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J. Exp. Med. 206, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang F., Meng G., Strober W. (2008) Interactions among the transcription factors Runx1, RORγt and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat. Immunol. 9, 1297–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schraml B. U., Hildner K., Ise W., Lee W. L., Smith W. A., Solomon B., Sahota G., Sim J., Mukasa R., Cemerski S., Hatton R. D., Stormo G. D., Weaver C. T., Russell J. H., Murphy T. L., Murphy K. M. (2009) The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature 460, 405–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Okamoto K., Iwai Y., Oh-Hora M., Yamamoto M., Morio T., Aoki K., Ohya K., Jetten A. M., Akira S., Muta T., Takayanagi H. (2010) IκBζ regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature 464, 1381–1385 [DOI] [PubMed] [Google Scholar]
- 43. Akimzhanov A. M., Yang X. O., Dong C. (2007) Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J. Biol. Chem. 282, 5969–5972 [DOI] [PubMed] [Google Scholar]
- 44. Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 45. Wei G., Wei L., Zhu J., Zang C., Hu-Li J., Yao Z., Cui K., Kanno Y., Roh T. Y., Watford W. T., Schones D. E., Peng W., Sun H. W., Paul W. E., O'Shea J. J., Zhao K. (2009) Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ciofani M., Madar A., Galan C., Sellars M., Mace K., Pauli F., Agarwal A., Huang W., Parkurst C. N., Muratet M., Newberry K. M., Meadows S., Greenfield A., Yang Y., Jain P., Kirigin F. K., Birchmeier C., Wagner E. F., Murphy K. M., Myers R. M., Bonneau R., Littman D. R. (2012) A validated regulatory network for Th17 cell specification. Cell 151, 289–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Glasmacher E., Agrawal S., Chang A. B., Murphy T. L., Zeng W., Vander Lugt B., Khan A. A., Ciofani M., Spooner C. J., Rutz S., Hackney J., Nurieva R., Escalante C. R., Ouyang W., Littman D. R., Murphy K. M., Singh H. (2012) A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science 338, 975–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yosef N., Shalek A. K., Gaublomme J. T., Jin H., Lee Y., Awasthi A., Wu C., Karwacz K., Xiao S., Jorgolli M., Gennert D., Satija R., Shakya A., Lu D. Y., Trombetta J. J., Pillai M. R., Ratcliffe P. J., Coleman M. L., Bix M., Tantin D., Park H., Kuchroo V. K., Regev A. (2013) Dynamic regulatory network controlling T17 cell differentiation. Nature 496, 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Biswas P. S., Gupta S., Chang E., Song L., Stirzaker R. A., Liao J. K., Bhagat G., Pernis A. B. (2010) Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J. Clin. Invest. 120, 3280–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li Y., Harada T., Juang Y. T., Kyttaris V. C., Wang Y., Zidanic M., Tung K., Tsokos G. C. (2007) Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. J. Immunol. 178, 1938–1947 [DOI] [PubMed] [Google Scholar]
- 51. Ansel K. M., Lee D. U., Rao A. (2003) An epigenetic view of helper T cell differentiation. Nat. Immunol. 4, 616–623 [DOI] [PubMed] [Google Scholar]
- 52. Zhu J., Yamane H., Paul W. E. (2010) Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 28, 445–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ballestar E., Esteller M., Richardson B. C. (2006) The epigenetic face of systemic lupus erythematosus. J. Immunol., 176, 7143–7147 [DOI] [PubMed] [Google Scholar]
- 54. Hedrich C. M., Tsokos G. C. (2011) Epigenetic mechanisms in systemic lupus erythematosus and other autoimmune diseases. Trends Mol. Med. 17, 714–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pisitkun P., Ha H. L., Wang H., Claudio E., Tivy C. C., Zhou H., Mayadas T. N., Illei G. G., Siebenlist U. (2012) Interleukin-17 cytokines are critical in development of fatal lupus glomerulonephritis. Immunity 37, 1104–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang J., Chu Y., Yang X., Gao D., Zhu L., Yang X., Wan L., Li M. (2009) Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum. 60, 1472–1483 [DOI] [PubMed] [Google Scholar]
- 57. Doreau A., Belot A., Bastid J., Riche B., Trescol-Biemont M. C., Ranchin B., Fabien N., Cochat P., Pouteil-Noble C., Trolliet P., Durieu I., Tebib J., Kassai B., Ansieau S., Puisieux A., Eliaou J. F., Bonnefoy-Bérard N. (2009) Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. 10, 778–785 [DOI] [PubMed] [Google Scholar]
- 58. Lech M., Weidenbusch M., Kulkarni O. P., Ryu M., Darisipudi M. N., Susanti H. E., Mittruecker H. W., Mak T. W., Anders H. J. (2011) IRF4 deficiency abrogates lupus nephritis despite enhancing systemic cytokine production. J. Am. Soc. Nephrol. 22, 1443–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stirzaker R. A., Biswas P. S., Gupta S., Song L., Bhagat G., Pernis A. B. (2012) Administration of fasudil, a ROCK inhibitor, attenuates disease in lupus-prone NZB/W F1 female mice. Lupus 21, 656–661 [DOI] [PubMed] [Google Scholar]
- 60. Biswas P. S., Gupta S., Stirzaker R. A., Kumar V., Jessberger R., Lu T. T., Bhagat G., Pernis A. B. (2012) Dual regulation of IRF4 function in T and B cells is required for the coordination of T-B cell interactions and the prevention of autoimmunity. J. Exp. Med. 209, 581–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. MacIver N. J., Michalek R. D., Rathmell J. C. (2013) Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 31, 259–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sena L. A., Li S., Jairaman A., Prakriya M., Ezponda T., Hildeman D. A., Wang C. R., Schumacker P. T., Licht J. D., Perlman H., Bryce P. J., Chandel N. S. (2013) Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 38, 225–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sinclair L. V., Rolf J., Emslie E., Shi Y. B., Taylor P. M., Cantrell D. A. (2013) Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 14, 500–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chang C. H., Curtis J. D., Maggi L. B., Jr., Faubert B., Villarino A. V., O'Sullivan D., Huang S. C., van der Windt G. J., Blagih J., Qiu J., Weber J. D., Pearce E. J., Jones R. G., Pearce E. L. (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nowak S. J., Pai C. Y., Corces V. G. (2003) Protein phosphatase 2A activity affects histone H3 phosphorylation and transcription in Drosophila melanogaster. Mol. Cell. Biol. 23, 6129–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Martin M., Potente M., Janssens V., Vertommen D., Twizere J. C., Rider M. H., Goris J., Dimmeler S., Kettmann R., Dequiedt F. (2008) Protein phosphatase 2A controls the activity of histone deacetylase 7 during T cell apoptosis and angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 4727–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Paroni G., Cernotta N., Dello Russo C., Gallinari P., Pallaoro M., Foti C., Talamo F., Orsatti L., Steinkühler C., Brancolini C. (2008) PP2A regulates HDAC4 nuclear import. Mol. Biol. Cell 19, 655–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fu W., Ergun A., Lu T., Hill J. A., Haxhinasto S., Fassett M. S., Gazit R., Adoro S., Glimcher L., Chan S., Kastner P., Rossi D., Collins J. J., Mathis D., Benoist C. (2012) A multiply redundant genetic switch “locks in” the transcriptional signature of regulatory T cells. Nat. Immunol. 13, 972–980 [DOI] [PMC free article] [PubMed] [Google Scholar]