

Abstract
We recently reported the systematic ligand-based rational design and synthesis of monovalent Smac mimetics that bind preferentially to the BIR2 domain of the anti-apoptotic protein XIAP. Expanded structure-activity relationship (SAR) studies around these peptidomimetics led to compounds with significantly improved selectivity (> 60-fold) for the BIR2 domain vs. the BIR3 domain of XIAP. The potent and highly selective IAP antagonist 8q (ML183) sensitized TRAIL-resistant prostate cancer cells to apoptotic cell death, highlighting the value of this probe compound as a valuable tool to investigate the biology of XIAP.
Keywords: Apoptosis, XIAP, Monovalent Smac mimetics, Probe compounds
With an estimated 12.1 million new cases of malignant disease diagnosed every year resulting in approximately 7.6 million deaths worldwide, cancer represents a significant public health concern and a substantial burden on society.1 Despite progress in the last several years towards the development of improved cancer medications, current therapeutic agents remain prone to intrinsic or acquired resistance, which is a major barrier to effective treatment.2 The evasion of apoptosis (programmed cell death) by tumorigenic cells is one of the defining hallmarks of cancer and is an underlying cause of therapeutic resistance.3 The development of therapeutic agents that correct defective apoptotic signaling and allow cell death to proceed selectively in malignant cells is therefore an approach with great promise for the treatment of cancer.
The inhibitor of apoptosis proteins (IAPs) have attracted attention recently as potential targets for the development of new cancer therapeutics.4 Distinct members of the IAP family of antiapoptotic proteins inhibit caspases, the group of intracellular cysteine proteases which act as executioner enzymes by performing apoptosis in cells.5 Thus, inhibition of specific IAPs de-represses caspases, allowing apoptosis to occur in malignant cells but not normal cells. Human X-linked inhibitor of apoptosis protein (XIAP) is the most potent caspase inhibitor in the IAP family.6 XIAP contains three ~70 amino acid motifs termed baculovirus IAP repeat (BIR) domains (designated 1, 2, and 3), with two of these BIRs exhibiting specificity for different caspases. The BIR2 domain and an adjacent linker peptide between BIR1 and BIR2 of XIAP combine to mediate the two-site interaction with the apoptosis effector proteases caspases-3 and -7, whereas the BIR3 domain of XIAP targets the apoptosis initiator protease caspase-9.7
In the intrinsic cell death pathway, apoptotic signaling is regulated by the mitochondrial protein Smac, an endogenous dimeric proapoptotic antagonist of XIAP. The release of Smac from the inter-membrane space of the mitochondria into the cytosol perpetuates the apoptotic signal by competing with caspases for binding to the BIR domains of XIAP.8 The design of peptidomimetics that bind to the BIR3 domain of XIAP and in part mimic the activity of Smac has been investigated by a number of research groups and companies that have developed antagonists of XIAP.4,9 For example, the Genentech group recently reported the discovery and characterization of the clinical candidate peptidomimetic GDC-0152 (1, Figure 1).10 While some success has been achieved using this approach, it has become clear that other strategies for alleviating the repression of caspases by XIAP should also be viable therapeutically. Since all apoptotic signaling, triggered via either the intrinsic or the extrinsic pathway, converges on caspases-3 or -7, it would seem logical to develop XIAP inhibitors with high affinity for the BIR2 domain of XIAP. However, somewhat surprisingly there have been few reports on the design and synthesis of compounds that effect inhibition by binding to the BIR2 domain of XIAP.11
Figure 1.
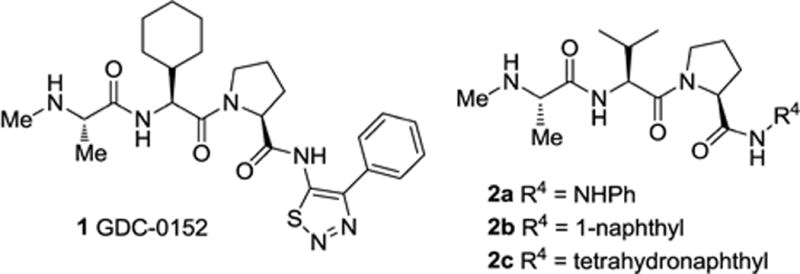
Tripeptide IAP antagonists.
We recently described the systematic rational design and synthesis of new monovalent IAP antagonists that bind preferentially to the BIR2 domain of XIAP.12 Using the AVPI motif found at the N-terminus of Smac as an initial template, we developed the tripeptide model shown in Figure 2. We then synthesized a series of analogues and determined their binding affinity for the BIR2 or BIR3 domains of XIAP. In this initial study we held constant the N-methyl alanine moiety at the P1 position and the proline residue at P3, while investigating the effects of varying the P2 position (R2) and the C-terminal substituent (R4). This led to the discovery of compounds such as 2a (R4 = NHPh) and 2b (R4 = 1-naphthyl) with preferential affinity for XIAP BIR2 vs. BIR3 and promising cell killing activity. We also confirmed that a tetrahydronaphthyl C-terminal capping group (R4) confers selectivity for BIR3 and potent cell killing activity, as observed for peptidomimetic 2c. Herein we report our further investigation of the SAR around this series of tripeptides leading to the discovery of novel compounds with unique selectivity for the BIR2 domain of XIAP.
Figure 2.
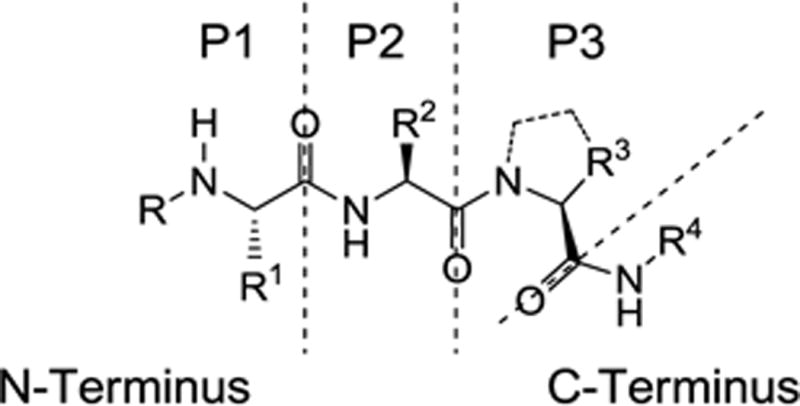
Tripeptide binding model.
The present study is a continuation of work performed within the Molecular Libraries Probe Production Centers Network (MLPCN; http://mli.nih.gov/mli/mlpcn/) with the goal of developing novel XIAP BIR2-selective probes. In this phase of the study we synthesized analogues that retained the P2 substituent (R2) as valine and the optimized C-terminal capping groups while varying the substituents at P3 (Figure 2). Modifications were also made to the P1 substituents in some of the optimized compounds. The general synthesis of the tripeptide analogues 8 is illustrated in Scheme 1. Condensation of Boc-amino acid derivative 3 with amino acid derivative 4, followed by acidic cleavage of the N-terminal Boc protecting group afforded dipeptide 5. Condensation of dipeptide 5 with the Boc-protected derivative 6 followed by hydrogenolysis of the benzyl ester afforded the tripeptide acid 7. Finally, condensation of 7 with the appropriate amine followed by removal of the Boc group provided the target IAP inhibitors 8.13
Scheme 1.
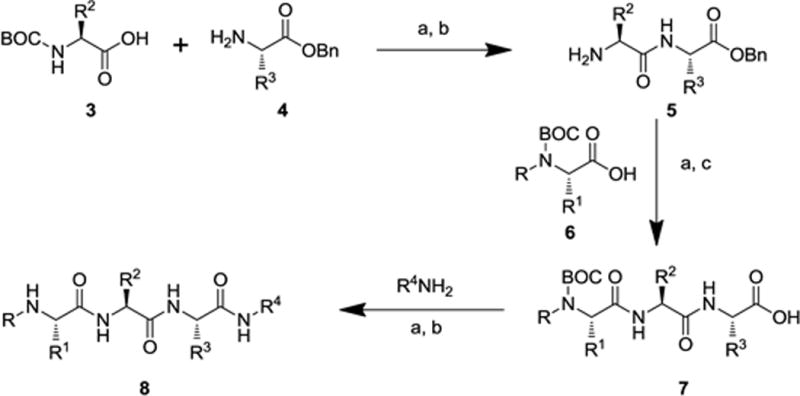
General synthetic sequence for the synthesis of small molecule IAP antagonists. Reagents and conditions: (a) EDC, HOBt, N-methylmorpholine, DMF; (b) TFA, CH2Cl2; (c) H2, Pd/C, MeOH.
All compounds synthesized were evaluated for their ability to bind the BIR2 or BIR3 domains of XIAP by employing fluorescence-polarization (FP) competition assays.14 The results are expressed as competitive inhibition constants (Ki), derived from the corresponding IC50 values by application of a mathematical equation developed by Wang and coworkers.15
We first synthesized and tested a series of analogues where P1 = NMeAla, P2 = Val and R4 = 1,2,3,4-tetrahydronaphthyl, while varying the P3 residue. The results are shown in Table 1. The specific amino acid residues that were incorporated into the tripeptide scaffold were selected to maximize diversity and to probe the size of the binding pocket in the P3 region of the protein. Thus, several of the amino acids chosen for P3 contained long chain substituents, some of which were hydrophilic and others with hydrophobic characteristics. Compared with the lead structure 2c (P3 = Pro) none of the new analogues had improved affinity for the BIR3 domain of XIAP. For example, replacing Pro with Ala at P3 as in 8a led to a reduction in potency of two-fold for BIR2 and fourteen-fold for BIR3 compared with 2c. However, it is noteworthy that some analogues with long chain residues at P3 (e.g. 8b, 8d, 8g) bound with submicromolar affinity to the BIR3 domain, including the ArgNO2 analogue 8b, with a Ki value of 0.29 μM at XIAP BIR3 and 12:1 selectivity for BIR3 vs. BIR2. Interestingly, the BIR3 affinity for 8b (0.29 μM) is comparable with the clinical compound 1 (GDC-0152) (0.26 μM). These results suggest that the BIR3 domain of XIAP tolerates relatively bulky substituents in the P3 region.
Table 1.
Binding affinities to BIR2 or BIR3 domains of XIAP for compound 1, and for analogues where R4 = 1,2,3,4-tetrahydronaphthyl (2c, 8a–g).
| ||||
|---|---|---|---|---|
| Cmpds. | P3 | BIR2 Ki, μMa | BIR3 Ki, μMa | BIR2 vs BIR3 Selectivity |
| 1 | — | 29.80 (± 4.60) | 0.26 (± 0.015) | 1 : 115 |
| 2c | Pro | 1.37 (± 0.11) | 0.06 (± 0.011) | 1 : 25 |
| 8a | Ala | 2.65 (± 0.15) | 0.78 (± 0.11) | 1 : 3.4 |
| 8b | ArgNO2 | 3.51 (± 0.40) | 0.29 (± 0.03) | 1 : 12 |
| 8c | Gln | 10.77 (± 1.70) | 2.02 (± 0.01) | 1 : 5.3 |
| 8d | LysCBZ | 4.30 (± 0.18) | 0.67 (± 0.04) | 1 : 6.4 |
| 8e | Orn | 4.36 (± 0.18) | 1.13 (± 0.06) | 1 : 3.9 |
| 8f | Ser | 4.57 (± 0.38) | 2.05 (± 0.11) | 1 : 2.2 |
| 8g | OrnCBZ | 2.75 (± 0.07) | 0.53 (± 0.01) | 1 : 5.2 |
Values are determined in duplicate and reported ± standard deviation which is given in parentheses.
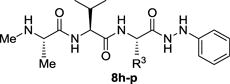
We next investigated a set of analogues that retained P1 = NMeAla, P2 = Val and introduced phenylhydrazine into the R4 position. The results of varying the P3 residue among the analogues are shown in Table 2. For compound 2a, where P3 = Pro, we had previously found that a phenylhydrazine C-terminal capping group (R4) confers selectivity for BIR2 of XIAP.12 In the new series this trend proved to hold true regardless of the P3 residue. In fact, the BIR2 vs. BIR3 selectivity ratio of potencies for all the new compounds was equal to or better than the 3 : 1 ratio exhibited by the potent XIAP antagonist 2a. Furthermore, the BIR3 potency of the compounds was poor, with none of them exhibiting Ki values better than 8 μM. Although the BIR2 Ki value of 0.39 μM for 2a was not improved upon, the selectivity of several compounds for BIR2 was significantly better. For example, the next most potent compounds in this series were 8j (BIR2 Ki = 1.18 μM) and 8p (BIR2 Ki = 1.88 μM) with ArgNO2 and Lys residues at P3, respectively. The selectivity of these compounds was encouraging, with BIR2 : BIR3 ratios of 7.5 and 11.4 to 1, respectively. Of particular note were the data for 8h (P3 = Ala) and 8i (P3 = Val) with BIR2 Ki values of 2.29 μM and 3.04 μM, respectively, and an impressive BIR2 : BIR3 selectivity ratio of more than 20 : 1. Taken together, these data suggest that analogues in which P3 = Ala or Val exhibit favorable selectivity for XIAP BIR2 versus BIR3.
Table 2.
Binding affinities to BIR2 or BIR3 domains of XIAP for tripeptide analogues where R4 = NHPh (2a, 8h–p).
| ||||
|---|---|---|---|---|
| Cmpds. | P3 | BIR2 Ki, μMa | BIR3 Ki, μMa | BIR2 vs BIR3 Selectivity |
| 2a | Pro | 0.39 (± 0.17) | 1.08 (± 0.06) | 3 : 1 |
| 8h | Ala | 2.29 (± 0.37) | 51.79 (± 2.16) | 22.6 : 1 |
| 8i | Val | 3.04 (± 0.28) | 65.62 (± 6.22) | 21.6 : 1 |
| 8j | ArgNO2 | 1.18 (± 0.11) | 8.79 (± 1.11) | 7.5 : 1 |
| 8k | Arg | 3.65 (± 0.23) | 21.15 (± 1.77) | 5.8 : 1 |
| 8l | Thr | 4.67 (± 0.37) | 30.07 (± 0.55) | 6.4 : 1 |
| 8m | OrnCBZ | 1 70 (± 01 3) | 9.82 (± 1.17) | 5.8 : 1 |
| 8n | Orn | 13.14 (± 1.19) | ~39 | 3 : 1 |
| 8o | Ser | 5.46 (± 0.56) | > 39 | > 7.1 : 1 |
| 8p | Lys | 1.88 (± 0.13) | 21.48 (± 1.52) | 11.4 : 1 |
Values are determined in duplicate and reported ± standard deviation which is given in parentheses.
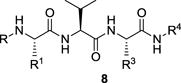
The data for our final iteration of analogues are shown in Table 3. In this series we initially synthesized analogues of the potent XIAP antagonist 2b, in which R4 = 1-naphthyl, by varying the P3 substituent. As noted above, we previously found that compound 2b (with P3 = Pro) is a potent inhibitor of XIAP BIR2 but the BIR2 : BIR3 selectivity ratio is only 3.4 : 1. Using the knowledge gained from the series with R4 = NHPh (Table 2), we prepared analogues with P3 = Ala (8q), Val (8r) or OrnCBZ (8s). Gratifyingly, we found that all three analogues were selective for BIR2, and essentially inactive against BIR3 with Ki values > 39 μM. In particular, compound 8q16 exhibited very respectable potency against XIAP BIR2 with Ki = 1.74 μM. Remarkably, the analogue 8t in which P1 = Ala (R = H) rather than NMeAla is highly potent against BIR2 (Ki = 0.64 μM) and more than sixty-fold selective for BIR2 over BIR3. Interestingly, the corresponding analogue in the R4 = N-phenylhydrazine series (8x, Table 3) did not quite exhibit the same BIR2 potency enhancement compared with the parent compound (8h, Table 2). Analogues of 8t in which R = CH2OH (8u) or R1 = Et (8v) were also highly selective for BIR2, although less potent. An analogue of 8q in which R4 = naphthalen-1-ylmethanamine (8w) retained the BIR2 selectivity of the parent compound but also with reduced BIR2 potency (Ki = 3.00 μM). Overall, the combination of P3 = Ala and R4 = 1-naphthyl provides XIAP antagonists with excellent BIR2 selectivity and good in vitro potency. Despite the excellent potency and selectivity of 8t, unfortunately when tested in a cellular assay 8t did not show any activity, presumably due to a lack of cell permeability (see Figure S1 in the Supplementary Material). We therefore proceeded with additional characterization of 8q as the optimal probe for studies in cells.
Table 3.
Binding affinities to BIR2 or BIR3 domains of XIAP for tripeptide analogues 2b and 8q–8y.
| |||||||
|---|---|---|---|---|---|---|---|
| Cmpds. | R | R1 | P3 | R4 | BIR2 Ki, μMa | BIR3 Ki, μMa | BIR2 vs BIR3 Selectivity |
| 2b | CH3 | CH3 | Pro | 1-naphthyl | 0.44 (± 0.02) | 1.51 (± 0.13) | 3.4 : 1 |
| 8q | CH3 | CH3 | Ala | 1-naphthyl | 1.74 (± 0.30) | > 39 | > 22.4 : 1 |
| 8r | CH3 | CH3 | Val | 1-naphthyl | 4.54 (± 0.23) | > 39 | > 8.6 : 1 |
| 8s | CH3 | CH3 | OrnCBZ | 1-naphthyl | 1.94 (± 0.11) | > 39 | > 20.1 : 1 |
| 8t | H | CH3 | Ala | 1-naphthyl | 0.64 (± 0.01) | 40.15 (± 2.69) | 62.7 : 1 |
| 8u | H | CH2OH | Ala | 1-naphthyl | 2.43 (± 0.25) | > 39 | > 16 : 1 |
| 8v | H | CH2CH3 | Ala | 1-naphthyl | 3.78 (± 0.37) | > 39 | > 10.3 : 1 |
| 8w | CH3 | CH3 | Ala | CH2-1-naphthyl | 3.00 (± 0.40) | 34.18 (± 2.15) | 11.4 : 1 |
| 8x | H | CH3 | Ala | NHPh | 1.83 (± 0.19) | 41.32 (± 0.86) | 22.6 : 1 |
| 8y | CH3 | CH3 | Val | NH-3-FPh | 2.38 (± 0.28) | > 39 | > 16.4 : 1 |
Values are determined in duplicate and reported ± standard deviation which is given in parentheses.
Based on these encouraging results we then sought to explore whether the selectivity of compound 8q for the BIR2 domain of XIAP might be explained by the relative structural properties of the BIR2 and BIR3 domains. Thus we modeled the binding of 8q onto the Smac binding groove of a BIR2 domain. We chose the structure of XIAP BIR2 from our earlier structure of the caspase-3–BIR2 complex.7 In addition to being the only available crystal structure of a XIAP/cIAP BIR2 domain, this structure also depicts “Smac-like” binding by the N-terminus of the caspase-3 small subunit to XIAP BIR2 (Figure 3a).7 Using this interaction we overlaid the structure of 8q into the Smac-binding groove, conserving the backbone geometry of residues 310–312 from the N-terminus of the caspase-3 small subunit. Conformational freedom of the 1-naphthyl ring permitted by the surrounding protein was explored followed by energetic minimization of the compound, and the lowest energy result is displayed in Figure 3b.
Figure 3.
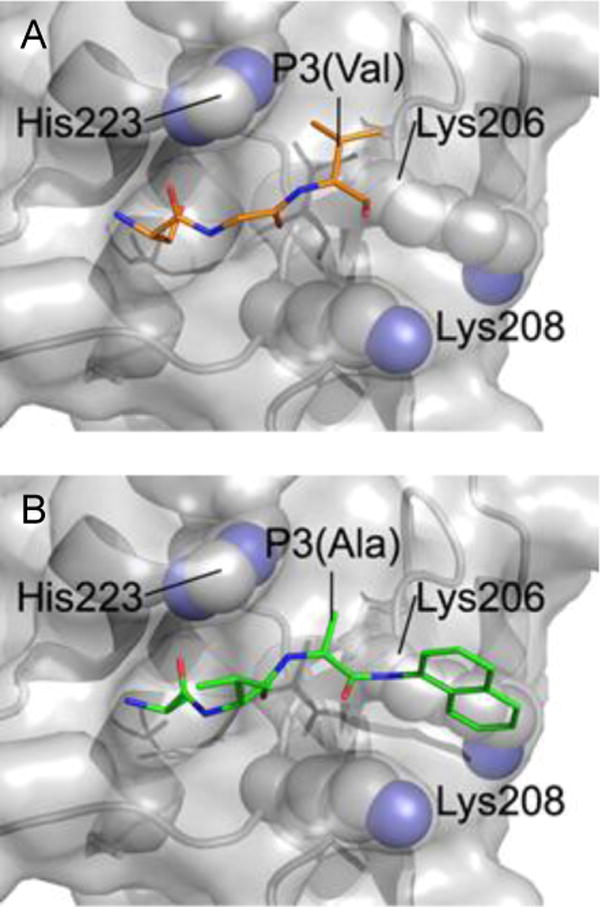
Model of compound 8q binding to XIAP BIR2. (A) Smac-like binding to BIR2 of XIAP (according to PMID 11257232; PDB 1i3o). Residues 310–312 from a neighboring Caspase-3 molecule are presented in stick form (orange) binding to the Smac-binding groove of BIR2. (B) An equivalent view of the model of 8q (green) bound to XIAP BIR2. Residues that potentially contribute to the selectivity of 8q are represented as spheres (C = grey, N = blue).
This model suggests several factors that could contribute to the BIR2 selectivity of 8q. Firstly, as already visualized in the contact from the caspase-3 BIR2 structure, a small non-proline entity is highly compatible with P3-binding adjacent to His223 of BIR2 (Figure 3). In the template caspase-3/BIR2 contact structure (Figure 3a) this residue is valine, and recent publications suggest an alanine in this position to be favorable for BIR2 selectivity.17,18 The alanine moiety of 8q allows a similar backbone conformation in P3 (Figure 3b). Secondly, the model (Figure 3b) visualizes that the 1-naphthyl ring is positioned between the aliphatic stems of Lys206 and Lys208 with a plausible positive effect on the interaction of 8q with the BIR2 domain. These residues correspond to glycine and threonine residues respectively in the BIR3 domain of XIAP, thus negating a potentially favorable interaction.
We next determined the activity of the new inhibitors in a disease-relevant cellular assay. In particular, the ability to sensitize TRAIL-resistant cells to apoptosis was investigated. Thus, PPC-1 prostate cancer cells were exposed to each compound (5 μM) for 4 h, followed by the addition of various concentrations of TRAIL, and incubated for 20 h.19 Cell viability was then assessed using ATP content as a readout (ATPlite). As shown in Figure 4, the cell penetrant IAP antagonists 2a–c or 8q all act synergistically with TRAIL to reduce the number of viable cells to much lower levels than those observed with TRAIL alone. The LD50 values for TRAIL in the presence of the compounds 2b, 2c, or 8q (5 μM) are in the 0.073–0.089 nM range. Compound 2b (5 μM) also acts synergistically with various concentrations of TNF to induce cell death in MDA-MB-231 cells (see Figure S2 in the Supplementary Material). To evaluate the effects in normal cells, compounds 2b and 8q were tested against human foreskin fibroblasts (HFF) (Figure 5). No toxicity was observed, even in the presence of TRAIL (100 ng/mL). Furthermore, 8q showed no toxicity against Fa2N-4 immortalized human hepatocytes (LC50 > 50μM).20 Taken together, these data suggest that our new compounds are cell penetrant and exhibit effects on prostate cancer cells consistent with their observed in vitro activity as IAP antagonists.
Figure 4.
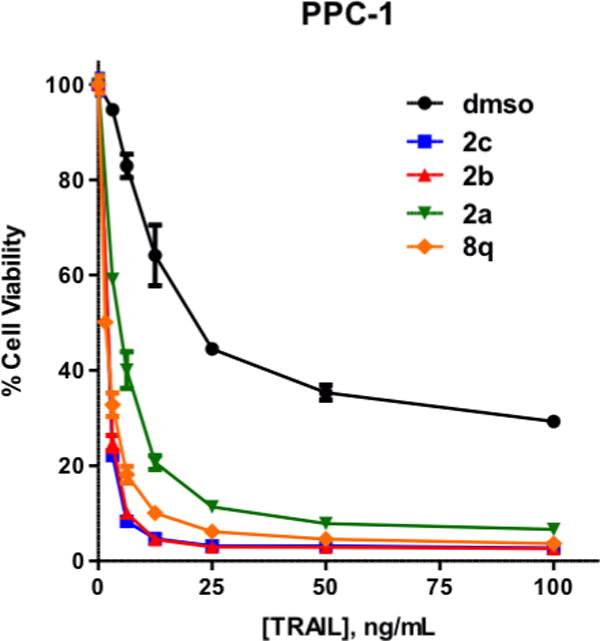
TRAIL-resistant PPC-1 cells challenged with TRAIL, or compounds 2a–c or 8q (5 μM) alone or in the presence of TRAIL.
Figure 5.
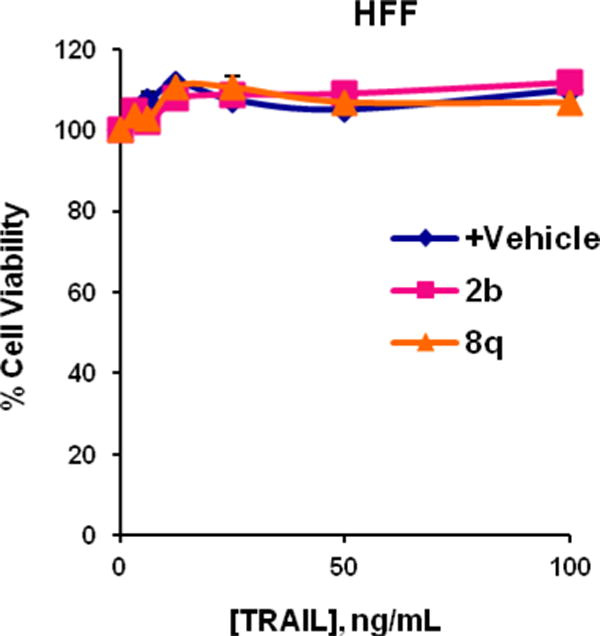
Normal human foreskin fibroblasts (HFF) challenged with TRAIL, or compounds 2b or 8q (5 μM) alone or in the presence of TRAIL.
In conclusion, by using a ligand-based rational design approach we have systematically optimized small molecule monovalent IAP antagonists that inhibit XIAP by binding selectively to the BIR2 domain without interacting with the BIR3 domain at detectable levels. The in vitro probe compound 8t binds to the BIR2 domain with submicromolar affinity and > 60-fold selectivity for BIR2 over BIR3. In addition, the XIAP inhibitory potency observed in vitro translates into cell killing activity in prostate cancer cells for the cell penetrant probe 8q. Consistent with our previous observations with IAP antagonists,21 the highly BIR2 selective, cell penetrant analogue 8q showed no toxicity as a single agent (Figures 4, 5 and S1) but rather sensitized the cells to TRAIL-induced apoptosis. Our results suggest that these compounds are potentially useful tools for probing apoptosis signaling pathways in cells. Further optimization of these compounds towards the discovery of potent and selective drug-like analogues is currently in progress.
Supplementary Material
Acknowledgments
This work was supported by NIH grants HG005033 and R01CA163743 to J.C.R., and R01AA017238 to S.J.R The authors thank Andrey Bobkov for expert technical assistance. M.G.L. acknowledges Fundación Ramón Areces for a postdoctoral fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Figures are provided showing the effects of compounds on cells (alone or in combination with TRAIL or TNF) and NMR spectra for 8q.
References and notes
- 1.http://www.cancerresearchuk.org/cancer-info/cancerstats/world/cancer-worldwide-the-global-picture
- 2.Gottesman MM. Annu Rev Med. 2002;53:615. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Cell. 2000;100:57. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 4.(a) Fulda S, Vucic D. Nat Rev Drug Discovery. 2012;11:109. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]; (b) Condon S. Ann Rep Med Chem. 2011;46:211. [Google Scholar]
- 5.Pop C, Salvesen GS. J Biol Chem. 2009;284:21777. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schimmer AD, Dalili S, Batey RA, Riedl SJ. Cell Death Differ. 2006;13:179. doi: 10.1038/sj.cdd.4401826. [DOI] [PubMed] [Google Scholar]
- 7.Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS. Cell. 2001;104:791. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 8.Shiozaki EN, Shi Y. Trends Biochem Sci. 2004;39:486. doi: 10.1016/j.tibs.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 9.(a) Sun H, Nikolovska-Coleska Z, Yang C-Y, Qian D, Lu J, Qiu S, Bai L, Peng Y, Cai Q, Wang S. Acc Chem Res. 2008;41:1264. doi: 10.1021/ar8000553. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mannhold R, Fulda S, Carosati E. Drug Discov Today. 2010;15:210. doi: 10.1016/j.drudis.2010.01.003. [DOI] [PubMed] [Google Scholar]; (c) Flygare JA, Fairbrother WJ. Expert Opin Ther Pat. 2010;20:251. doi: 10.1517/13543770903567077. [DOI] [PubMed] [Google Scholar]
- 10.(a) Flygare JA, Beresini M, Budha N, Chan H, Chan IT, Cheeti S, Cohen F, Deshayes K, Doerner K, Eckhardt SG, Elliott LO, Feng B, Franklin MC, Reisner SF, Gazzard L, Halladay J, Hymowitz SG, La H, LoRusso P, Maurer B, Murray L, Plise E, Quan C, Stephan J-P, Young SG, Tom J, Tsui V, Um J, Varfolomeev E, Vucic D, Wagner AJ, Wallweber HJA, Wang L, Ware J, Wen Z, Wong H, Wong JM, Wong M, Wong S, Yu R, Zobel K, Fairbrother WJ. J Med Chem. 2012;55:4101. doi: 10.1021/jm300060k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Erickson RI, Tarrant J, Cain G, Lewin-Koh SC, Dybdal N, Wong H, Blackwood E, West K, Steigerwalt R, Mamounas M, Flygare JA, Amemiya K, Dambach D, Fairbrother WJ, Diaz D. Toxicol Sci. 2013;131:247. doi: 10.1093/toxsci/kfs265. [DOI] [PubMed] [Google Scholar]
- 11.(a) Sun H, Liu L, Lu J, Bai L, Li X, Nikolovska-Coleska Z, McEachern D, Yang CY, Qiu S, Yi H, Sun D, Wang S. J Med Chem. 2011;54:3306. doi: 10.1021/jm101651b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sun H, Liu L, Lu J, Qiu S, Yang CY, Yi H, Wang S. Bioorg Med Chem Lett. 2010;20:3043. doi: 10.1016/j.bmcl.2010.03.114. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schimmer AD, Welsh K, Pinilla C, Wang Z, Krajewska M, Bonneau MJ, Pedersen IM, Kitada S, Scott FL, Bailly-Maitre B, Glinsky G, Scudiero D, Sausville E, Salvesen G, Nefzi A, Ostresh JM, Houghten RA, Reed JC. Cancer Cell. 2004;5:25. doi: 10.1016/s1535-6108(03)00332-5. [DOI] [PubMed] [Google Scholar]; (d) Orzaez M, Gortat A, Sancho M, Carbajo RJ, Pineda-Lucena A, Palacios-Rodriguez Y, Perez-Paya E. Apoptosis. 2011;16:460. doi: 10.1007/s10495-011-0582-4. [DOI] [PubMed] [Google Scholar]
- 12.González-Lopez M, Welsh K, Finlay D, Ardecky RJ, Ganji SR, Su Y, Yuan H, Teriete P, Mace PD, Riedl SJ, Vuori K, Reed JC, Cosford NDP. Bioorg Med Chem Lett. 2011;21:4332–4336. doi: 10.1016/j.bmcl.2011.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.All synthesized compounds were purified by either medium pressure silica gel chromatography or preparative HPLC using a XBridge Prep C18 column (19 × 100 mm) eluted with 0.05 % formic acid in a methanol-water gradient of 10:90 to 100:0 over 10 to 15 min. with a flow rate of 25 mL/min. Compound purity was determined using analytical HPLC/MS and NMR (1H and 13C). The yields for the first two steps ranged from 70 – 90% while the yields for the last step ranged from 50 – 90%. The purity of each final product was greater than 95% as determined by HPLC/MS analysis.
- 14.Fluorescence polarization assays were run in 20 μL volume in 384 well format with black plates. The assay solution was 25 mM Hepes at pH 7.5/1 mM tris(2-carboxyethyl)phosphine/0.005% Tween 20 and 20 nM AVPIAQK-rhodamine. XIAP BIR2 was present at 1 μM or XIAP BIR3 at 0.2 μM. Compound was present within a range of 100 μM to 6.1 nM using two fold dilutions starting at 100 μM generating a 16 point curve. Plates were read on an Analyst HT in fluorescence polarization mode with excitation at 530 nm, emission at 580 nm and a dichroic mirror at 565 nm. Resulting data in mP was fit in GraphPad Prism 5 with a non-linear regression using a sigmodial curve with variable slope.
- 15.Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito N, Stuckey J, Wang S. Anal Biochem. 2004332:261. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 16.Spectroscopic data for compound 8q: 1H NMR (400 MHz, DMSO-d6) δ 9.99 (1H, s), 8.80 (1H, b), 8.60 (1H, d, J = 8.5 Hz), 8.38 (1H, d, J = 8.5 Hz), 7.75 (1H, m), 7.59 (1H, m), 7.52 (1H, d, J = 5.9Hz), 7.51 (1H, m), 7.50 (4H, bm), 4.71 (1H, m), 4.34 (1H, m) 3.74 (1H, m), 3.64 (1H, b), 2.05 (1H, m), 1.40 (3H, d, J = 5.9 Hz), 1.35 (3H, d, J = 5.9 Hz), 0.88 (6H, m), 13C NMR (100 MHz, DMSO-d6) δ 171.8, 170.2, 168.5, 133.7, 133.3, 128.1, 127.9, 126.0, 125.8, 122.7, 121.6, 57.5, 56.0, 48.9, 30.7, 19.1, 17.9, 15.8. Exact mass for chemical formula: C22H30N4O3398.23 [M], HR-MS (ESI) 399.30 [M + H].
- 17.Speer KF, Cosimini CL, Splan KE. Biopolymers. 2012;98:122. doi: 10.1002/bip.21732. [DOI] [PubMed] [Google Scholar]
- 18.Eckelman BP, Drag M, Snipas SJ, Salvesen GS. Cell Death Differ. 2008;15:920. doi: 10.1038/cdd.2008.6. [DOI] [PubMed] [Google Scholar]
- 19.Cell viability was assessed using the ATPlite 1step Luminescence Assay System (PerkinElmer Inc., Waltham, MA). Briefly, cells were seeded at 2000 cells/well in 38 μL medium with 5% FBS and allowed to attach overnight. Test compound (1 μL) was added to a final concentration of 5 μM and the cells were incubated at 37oC for 4 h. TRAIL (1 μL) was then added to the desired final concentration and the cells again incubated at 37oC for 20 h. The plates were removed to room temperature for 30 min. before addition of one half volume (20 μL) of ATPlite reagent. The plates were shaken at 1,100 rpm for 2 min. to ensure proper mixing before luminescence measurement on a BMG POLARstar Omega plate reader. PPC-1 cells were maintained in RPMI 1640 (Cellgro, Manassas, VA) with 10% (v/v) FBS (HyClone, Logan, UT), and penicillin/streptomycin/L-Glutamine (Life Technologies, Carlsbad, CA).
- 20.Lopez M, Welsh K, Yuan H, Stonich D, Su Y, Garcia X, Cuddy M, Houghten R, Sergienko E, Reed JC, Ardecky R, Reddy S, Finlay D, Vuori K, Dad S, Chung TDY, Cosford NDP. Antagonists of IAP-family anti-apoptotic proteins – Probe 2 Probe Reports from the NIH Molecular Libraries Program [Internet] Bethesda (MD): National Center for Biotechnology Information (US); 2010–2009. [PubMed] [Google Scholar]
- 21.Vamos M, Welsh K, Finlay D, San Lee P, Mace PD, Snipas SJ, Gonzalez ML, Reddy Ganji S, Ardecky RJ, Riedl SJ, Salvesen GS, Vuori K, Reed JC, Cosford NDP. ACS Chem Biol. 2013 doi: 10.1021/cb3005512. in press. http://pubs.acs.org/doi/abs/10.1021/cb3005512. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.