

Summary
Mitochondrial dysfunction plays a key pathogenic role in aging skeletal muscle resulting in significant healthcare costs in the developed world. However, there is no pharmacologic treatment to rapidly reverse mitochondrial deficits in the elderly. Here we demonstrate that a single treatment with the mitochondrial targeted peptide SS-31 restores in vivo mitochondrial energetics to young levels in aged mice after only one hour. Young (5 month old) and old (27 month old) mice were injected intraperitoneally with either saline or 3 mg/kg of SS-31. Skeletal muscle mitochondrial energetics were measured in vivo one hour after injection using a unique combination of optical and 31P magnetic resonance spectroscopy. Age related declines in resting and maximal mitochondrial ATP production, coupling of oxidative phosphorylation (P/O), and cell energy state (PCr/ATP) were rapidly reversed after SS-31 treatment, while SS-31 had no observable effect on young muscle. These effects of SS-31 on mitochondrial energetics in aged muscle were also associated with a more reduced glutathione redox status and lower mitochondrial H2O2 emission. Skeletal muscle of aged mice was more fatigue resistant in situ one hour after SS-31 treatment and eight days of SS-31 treatment led to increased whole animal endurance capacity. These data demonstrate that SS-31 represents a new strategy for reversing age-related deficits in skeletal muscle with potential for translation into human use.
Introduction
Impaired mitochondrial function is associated with exercise intolerance, fatigue, and muscle atrophy (Williams et al. 2007; Min et al. 2011), which contribute to poor quality of life and loss of independence in the elderly. The resulting increased rates of nursing home placement and hospitalization make the loss of skeletal muscle function with age (sarcopenia) a growing public health crisis in terms of both quality of life and economic costs to society, which were estimated to be $18 billion in the U.S. in 2000 (Janssen et al. 2004). Despite these societal costs, there is no known pharmacologic treatment to reverse age-related mitochondrial deficits and improve skeletal muscle function in vivo.
Mitochondrial deficits resulting from a buildup of oxidative stress is one mechanism that may link advanced age to skeletal muscle dysfunction. Mitochondria produce reactive O2 species (ROS) at the electron transport chain (ETC) as a by-product of oxidative phosphorylation (St-Pierre et al. 2002). As organisms age the rate of mitochondrial ROS production increases, resulting in a more oxidized intracellular environment (Mansouri et al. 2006). This increased oxidative state may modify cell function by 1) damaging proteins, lipids, and DNA, 2) activating cellular stress response pathways, and 3) modifying protein function through reversible redox sensitive post-translational modification such as glutathionylation (Applegate et al. 2008; Sohal & Orr 2012). The traditional view is that the accumulation of oxidative damage to proteins, lipids, and mitochondrial DNA resulting from exposure to increased levels of ROS results in mitochondrial dysfunction with age. However, a growing body of evidence describes more subtle interactions between the intracellular redox environment and mitochondrial energetics (Mailloux et al. 2011; McLain et al. 2011; Siegel et al. 2011), which may contribute to age-related changes in mitochondrial function.
We have previously demonstrated that reduced mitochondrial efficiency and maximal ATP production is accompanied by a loss of energy homeostasis in aged skeletal muscle in both humans (Amara et al. 2007) and mice (Marcinek et al. 2005) (Siegel et al. 2012). Increasing oxidative stress in young skeletal muscle leads to reduced mitochondrial efficiency and altered energy homeostasis in vivo, similar to aging muscle, without a change in mitochondrial function ex vivo (Siegel et al. 2011). These data suggest that mild oxidative stress results in a “regulatory” change in mitochondrial function that is dependent on the cell environment without leading to “structural” changes that are intrinsic to the mitochondria. The difference between the effects of oxidative stress on in vivo and ex vivo mitochondrial function supports the importance of considering the interaction between the cell environment and mitochondria in age-related mitochondrial dysfunction. This idea is also consistent with data suggesting that previous studies may have overestimated the extent of mitochondrial damage in aging muscle (Rasmussen et al. 2003; Picard et al. 2010).
If age-related energy deficits are due to regulation of mitochondrial function by oxidative stress, it should be possible to rapidly reverse mitochondrial dysfunction by altering the oxidative environment. We directly test the role of regulatory vs. structural changes (Sohal & Orr 2012) in mitochondrial dysfunction in aged skeletal muscle by manipulating mitochondrial oxidative stress with an acute treatment of the mitochondrial targeted peptide SS-31. SS-31, like other Szeto-Schiller (SS) peptides, has an alternating aromatic-cationic structure that allows it to freely cross the cell membrane and concentrate greater than 1000 fold in the mitochondrial inner membrane independently of mitochondrial membrane potential. SS-31 associates with mitochondrial cardiolipin (Birk et al.), improves ATP production, reduces mitochondrial ROS production, and lowers oxidative damage (Szeto et al. 2011). These effects are associated with protection against ischemia-reperfusion injury (Szeto et al. 2011), cardiac hypertrophy and failure (Dai et al. 2011), disuse muscle atrophy (Min et al. 2011), high fat diet-induced insulin resistance (Anderson et al. 2009), and MPTP-induced dopaminergic neuron cell death, a model of Parkinson’s disease (Yang et al. 2009) in animal models. Here we report that treatment with SS-31 rapidly reverses in vivo deficits in mitochondrial energetics in aged skeletal muscle and improves skeletal muscle performance. These data support the hypothesis that altered regulation of mitochondria by an oxidative shift in the redox environment plays a significant role in the age-related decline in skeletal muscle function.
Results
To determine whether SS-31 improves mitochondrial energetics in old mice we administered 3mg of SS-31 per kg of body weight by intraperitoneal injection and used simultaneous in vivo optical and 31P magnetic resonance (MR) spectroscopy (Siegel et al. 2012) to measure mitochondrial function approximately one hour after treatment. Figure S1 shows that SS-31 is rapidly taken up by skeletal muscle, with maximal levels observed as early at 30 min after administration. Representative optical and MR spectra and the associated analysis of in vivo mitochondrial function are provided in Fig. 1. As controls we administered volume-matched doses of saline (C) to both young and old mice and the same dose of SS-31 to young mice. Measurement of in vivo mitochondrial function 1 hr after a single acute treatment precludes an increase in mitochondrial content as an explanation for the improved energetics as demonstrated by the lack of effect of SS-31 on protein expression of ETC in old mice (Fig. S2).
Figure 1.

(A) Representative in vivo 31P MR spectra acquired during dynamic experiment following 20 Hz line-broadening and summing 3 consecutive FIDs. (B) Representative in vivo NIR optical spectra acquired during dynamic experiment. (C) Analysis of dynamic in vivo spectra in conjunction with biochemical measures of hemoglobin, myoglobin, and ATP yields a noninvasive assessment of mitochondrial O2 consumption, resting ATP demand (ATPase) rate, and oxidative phosphorylation capacity (ATPmax). Total O2 content on the left axis and PCr concentration on the right.
Aging was associated with a reduction in free reduced glutathione (GSH) levels (Fig. 2A) with no significant change in oxidized glutathione (GSSG) (Fig. 2B). One hour after treatment with SS-31 GSH increased in the aged mice. The increase in GSH was associated with a reduction of the glutathione redox potential in the old mice (more negative potential in Fig. 2C). SS-31 in the young mice or age alone had no significant effect on GSH redox status in the young mice (Fig. 2C). Aging was also associated with an increased capacity for H2O2 production by permeabilized extensor digitorum longus (EDL) muscle (Fig. 2D). The addition of 500nM SS-31 to the assay reduced H2O2 production in permeabilized EDL from aged mice (Fig. 2D).
Figure 2.

In gastrocnemius muscle, (A) GSH was depressed in old vs. young and was restored to near young levels one hour after a single SS-31 injection, while (B) neither age nor SS-31 treatment affected GSSG content. (C) The glutathione redox potential was not significantly higher (more oxidized) in old vs. young, but was significantly reduced by in vivo SS-31 treatment in old mice. (D) H2O2 emission in permeabilized EDL was elevated in old vs. young and was restored to near young levels in the presence of exogenous SS-31. Means ± SEM. For glutathione data n = 8 for young controls and 4 for other groups per group for glutathione data. For H2O2 data n = 3 for young and n = 9 for old data. *p<0.05 relative to old control. #p<0.05, ###p<0.001 relative to young control.
SS-31 Rapidly Reverses Age-Related Energetic Deficits
In vivo P/O ratio and mitochondrial phosphorylation capacity (ATPmax) were significantly depressed in old vs. young adult mice. One hour after treatment with SS-31, both measures of mitochondrial function returned to young levels (Figs. 3A and 3E). The changes in in vivo P/O parallel the changes in resting mitochondrial ATP production with age and SS-31 treatment (Fig. 3B), while neither age nor SS-31 affected the resting rate of mitochondrial O2 consumption (Fig. 3C). Figure 3D illustrates the improved recovery of PCr following ischemia, indicative of improved ATPmax (Fig. 3E)(Blei et al. 1993).
Figure 3.
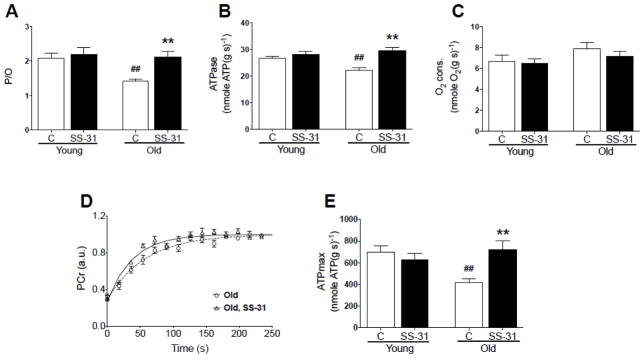
(A) In vivo P/O ratio was depressed with age and fully restored one hour after SS-31 injection. (B) Resting ATPase rate was significantly lower in old mice and recovered one hour following SS-31 treatment, while (C) O2 consumption was unchanged. (D) After ischemia, PCr recovered to resting concentration at a faster rate in old mice following SS-31 treatment, revealing (E) a significant increase in ATPmax. SS-31 had no observed affect on these measures of in vivo mitochondrial function in young mice. Means ± SEM. n = 5–7 per group. **p<0.01 relative to age-matched control. ##p<0.01 relative to young control.
Improvements in cell energetics were associated with a reduction in energy stress in the old animals. Cell energy state, as measured by the PCr/ATP ratio, was also significantly depressed with age and recovered to young levels one hour after SS-31 treatment (Table 1). Figure S3 illustrates the greater relative peak area of PCr after SS-31 treatment in representative fully-relaxed 31P MR spectra from old control and old SS-31 treated mice. Metabolite concentrations used for metabolic analyses are provided in Table 1.
Table 1.
Metabolites from young and old mouse skeletal muscle with and without SS-31 treatment.
| YOUNG | OLD | |||
|---|---|---|---|---|
|
| ||||
| C | SS31 | C | SS31 | |
| Body wt (g) | 25.20±0.92 | 24.81±0.48 | 29.30±0.87* | 28.67±0.54 |
| GA wt (mg) | 124.4±3.6 | 123.8±1.1 | 109.5±3.6* | 106.1±4.0 |
| ATP(mM) | 10.26±0.29 | 9.20±0.14 | 7.98±0.22** | 7.69±0.15 |
| PCr(mM) | 36.83±1.31 | 33.36±1.20 | 24.43±0.75*** | 29.29±1.89# |
| Pi(mM) | 3.55±0.37 | 2.31±0.31 | 2.40±0.23* | 2.18±0.22 |
| Cr(mM) | 43.19±1.40 | 41.48±0.61 | 38.11±1.16* | 38.06±0.90 |
| ADP(μM) | 15.56±2.45 | 19.82±3.13 | 29.22±2.13* | 19.45±3.00 |
| AMP(nM) | 25.41±7.12 | 42.02±12.46 | 97.03±11.77*** | 51.07±18.33 |
| pHRest | 7.07±0.01 | 7.10±0.05 | 7.02±0.02 | 7.06±0.03 |
| pHIschemia | 6.96±0.01 | 6.95±0.01 | 6.90±0.03 | 6.88±0.02 |
| Mb (nmol/g) | 0.034±0.002 | 0.028±0.005 | 0.031±0.002 | 0.035±0.002 |
| Hb (nmol/g) | 0.031±0.002 | 0.032±0.003 | 0.037±0.002 | 0.048±0.009 |
Data for ATP and total creatine (Cr) are from HPLC, Mb and Hb are from SDS-PAGE analysis, and all other values were calculated from MR spectra. Mean ± SEM. n = 3–9.
p<0.05,
p<0.01,
p<0.001 relative to young control;
p<0.05,
p<0.001 relative to age-matched control.
In contrast with our in vivo results, there was no effect of age or SS-31 treatment on mitochondrial respiration in permeabilized muscles. Neither proton leak driven respiration (state 4, Fig. 4A) nor maximal ADP-stimulated respiration (state 3, Fig. 4B) were significantly different in permeabilized edl muscles from adult and old mice treated in vivo with saline or SS-31.
Figure 4.

Mitochondrial respiration was measured in permeabilized edl muscles from young and old mice treated in vivo with either saline or SS-31. (A) There was no effect of age or SS-31 on state 4 respiration (complex I substrates, no ADP) or (B) state 3 respiration (complex I + II substrates, saturating ADP). Means ± SEM. n = 3–4 per group.
SS-31 Improves Fatigue Resistance in Old Mouse Muscle
We hypothesized that improved mitochondrial energetics would lead to improved muscle endurance and resistance to fatigue. To test muscle function we measured in situ muscle fatigue in the tibialis anterior (TA) of old mice using tetanic stimulation (200 Hz) every 2 sec 1 hour after treatment. This preparation maintains the in vivo circulatory and nervous inputs into the muscle. The rate of force decay was slower (Fig. 5A) and the percent of original force at the end of the fatigue protocol (Fig. 5B) was significantly greater in SS-31 treated mice relative to control. Maximum specific force (Fig. 5C) and the force-frequency response (Fig. 5D), two parameters that should be independent of acute changes in skeletal muscle mitochondrial function, were not affected by acute SS-31 treatment in aged TA.
Figure 5.

(A) Plots of force vs. time during repetitive tetanic stimuli were distinctly different in old control and old SS-31-treated mice, with SS-31 treatment slowing the rate and magnitude of force decay. (B) Net force maintained by TA muscle at the end of fatigue experiments was significantly higher in SS-31-treated mice. (C) Maximum specific force and (D) the force-frequency response in old muscle was the same regardless of SS-31 treatment. Means ± SEM. n = 6–7 per group. *p<0.05.
SS-31 Improves Whole Body Endurance in Old Mice
Reversal of mitochondrial dysfunction also led to improvements in whole body exercise capacity in the old mice. In the young adults SS-31 treatment did not significantly improve endurance capacity. However, SS-31 treatment did lead to a trend toward greater running capacity in the top performing young adults. As illustrated in Fig. 6B, old mice treated daily for 8 days with SS-31 were able to run at a rate of 30m/min and an upward incline of 10° for a significantly longer period of time than untreated old controls. However, both groups of old animals performed worse than either young group Fig. 6A. Despite the improved fatigue resistance and endurance capacity in the 27 mo animals, SS-31 treatment over a two week period did not lead to an increase in voluntary wheel running (Fig S4).
Figure 6.

SS-31 improves exercise tolerance in old mice. (A) 8 days of SS-31 treatment had no significant effect on treadmill endurance in young mice. (B) After SS-31 treatment old mice had increased exercise tolerance. Not the different time axes in A and B. n = 7–8 per group for old and n=5 for young. *p<0.05.
Discussion
We tested whether in vivo mitochondrial energy deficits in aged skeletal muscle could be rapidly reversed with a single treatment of the mitochondrial targeted peptide SS-31. Using a combined MR and optical spectroscopy approach we identified declines in mitochondrial coupling efficiency (P/O), phosphorylation capacity (ATPmax), and energy state (PCr/ATP) in aged muscle that were restored to young levels 1 hour after a single treatment with SS-31. The improvements in mitochondrial energetics were associated with a reduction in the cell redox state and improved performance in aged skeletal muscle.
Oxidative stress leads to reduced in vivo P/O and PCr/ATP in the absence of intrinsic mitochondrial changes in young mouse skeletal muscles (Siegel et al. 2011) and a further decline of ATPmax in aged muscles (Siegel et al. 2012). The effects of increasing oxidative stress with paraquat treatment on in vivo mitochondrial function parallel those associated with normal aging in mouse and human skeletal muscles (Amara et al. 2007; Siegel et al. 2012). This suggests that oxidation of the cell environment with age may reversibly inhibit mitochondrial energetics and underlie energy deficits in aged skeletal muscle. We tested this hypothesis by targeting mitochondrial ROS production and cell redox state with the mitochondrial targeted peptide SS-31. The more reduced environment was associated with improvements in in vivo P/O, ATPmax, and PCr/ATP. This rapid reversal of depressed energetic function was not due to increased mitochondrial content or intrinsic functional capacity as measured by state 3 respiration in permeabilized muscle fibers from young and aged animals. The short time between treatment with SS-31 and measurement of in vivo energetics also indicates that the improvements are not due to repairing or replacing damaged mitochondria (Hood 2001). Therefore these results support the hypothesis that the interaction between mitochondria and cell environment in aged skeletal muscle contributes significantly to mitochondrial deficits. This insight suggests that manipulation of the oxidative state of the cell may be a therapeutic target for reversal of mitochondrial deficits in aged skeletal muscle.
In efforts to ameliorate age-related changes by targeting mitochondrial oxidative stress other groups have used both long-term pharmacologic (Skulachev et al. 2009; McManus et al. 2011) and transgenic (Schriner et al. 2005; Jang et al. 2009) strategies to deliver antioxidants to mitochondria. However, the long-term nature of these interventions prevents distinction between “regulatory” (reversible inhibition by oxidative stress) from the “structural” (repair or prevention of oxidative damage by protein turnover or compensation by mitochondrial biogenesis). A recent study also demonstrated improved mitochondrial coupling in human muscle following nitrate supplementation (Larsen et al, 2011). However, this effect followed a 3 day nitrate supplementation resulting in changes intrinsic to the mitochondria that were reflected both in vivo and in permeabilized muscle fibers and in decreased ANT protein expression. In contrast, the reversal of in vivo mitochondrial deficits by SS-31 on this short time scale indicate that the shift in redox state and improved function are independent of adaptive or compensatory changes to mitochondrial protein expression. Previous work has suggested that mitochondrial function can be altered in vivo by acute manipulation of redox signaling. Acute inhibition of nitric oxide synthase in rat skeletal muscle by L-NAME led to reduced O2 cost for muscle contraction (Baker et al. 2006), which is consistent with an acute in increase in mitochondrial coupling efficiency. Thus, we provide strong evidence that reversible regulatory inhibition by oxidative stress contribute to functional deficits in old mitochondria independently of permanent structural modification by oxidative damage.
One potential mechanism by which the mitochondrial redox environment can control mitochondrial energetics without inducing permanent damage is through post-translational modification of mitochondrial proteins. Reversible glutathionylation is one such example. Increased oxidative stress has been found to lead to increased glutathionylation and inhibition of the activity of ETC proteins, F1F0 ATPase (Garcia et al. 2010) and complex I (Hurd et al. 2008), and of TCA cycle proteins, succinyl-CoA-transferase (Garcia et al. 2010) and α-ketoglutarate dehydrogenase (Applegate et al. 2007). GSH bound to proteins would be lost during our extraction procedure and would result in a reduction in the measured free GSH as we found in the aged gastrocnemius. The rapid increase in the total GSH pool in the SS-31 treated aged muscles is consistent with a release of bound GSH from proteins (Garcia et al. 2010). Further experiments are required to determine the biochemical mechanisms that underlie the reversible inhibition of in vivo mitochondrial function in aged skeletal muscle demonstrated here.
Exercise capacity declines dramatically with age and SS-31 treatment improved skeletal muscle performance and exercise capacity in aged mice. The improved fatigue resistance in the aged TA in situ suggests that SS-31 treatment results in increased performance due to parameters that are intrinsic to the skeletal muscle. The TA was stimulated to contract by direct stimulation of the peroneal nerve, which remains intact in this preparation. Therefore, the absence of an effect of SS-31 on maximal tetanic force or the force-frequency relationship in situ indicates that the fatigue resistance was not due to improved function of the neuromuscular junctions or the contractile apparatus (Percival et al. 2008).
Previous studies have demonstrated improved skeletal muscle fatigue resistance with the antioxidant N-acetylcysteine (NAC). Multiple studies have demonstrated that acute NAC treatment increases fatigue resistance to submaximal (low frequency) fatigue, but has no effect on muscle fatigue during high frequency (tetanic) stimulation in limb muscles (Reid et al. 1994; Matuszczak et al. 2005). The effects of oxidative stress on low frequency fatigue have been suggested to work through redox modification EC coupling, or myofibrillar proteins (reviewed by (Reid 2008)). In order to more directly test the link between improved muscle energetics and fatigue our stimulation protocol used tetanic stimulations to induce high frequency fatigue, a condition in which general antioxidant treatment has been shown to have no effect. Thus, these data are consistent with the conclusion that the fatigue resistance of aged muscle 1 hr after SS-31 treatment is due to the reversal of mitochondrial deficits in aged muscle and not the direct result of increased antioxidant activity. However, we cannot rule out the possibility that the improved fatigue resistance is due to effects of the reduced oxidative state that are independent of mitochondrial energetics.
In addition to the resistance to muscle fatigue observed in situ, we found that aged mice had improved exercise tolerance following SS-31 treatment, which was particularly evident in the top performers in both age groups. This improvement further supports a role for oxidative stress dependent inhibition of mitochondrial function in the loss of exercise capacity with age. SS-31 has been shown to protect both the cardiovascular and nervous system from stress induced dysfunction (Yang et al. 2009; Dai et al. 2011) so it is possible that improvements in both systems in aged mice contribute to the improved exercise tolerance observed in this study. Targeting catalase to skeletal muscle and heart mitochondria also increased exercise tolerance in young mice after three months (Li et al. 2009), but had no effect on ex vivo fatigue resistance in the extensor digitorum longus. However, catalase expression was mosaic in the skeletal muscle and mitochondrial energetics or oxidative stress were not measured so it is not clear if catalase expression and SS-31 treatment in this study are acting through the same mechanisms to improve exercise tolerance.
A causal link between improved skeletal muscle energetics and improved muscle function has been demonstrated previously. Quercetin administration for seven days increased mitochondrial biogenesis and resulted in a 25% increase in cytochrome c expression in skeletal muscle and brain, leading to significantly improved exercise capacity in young mice (Davis et al. 2009). Increased mitochondrial content in skeletal muscle following administration of (−)-epicatechin, an extract found in dark chocolate, also led to improved high frequency fatigue resistance in isolated skeletal muscles and increased treadmill performance in young mice (Nogueira et al. 2011). However, the improvements with SS-31 in this study are unique in that they show an increase in ATPmax and muscle performance in the absence of increased mitochondrial content. This indicates that there is extra capacity for mitochondrial ATP production that is inhibited under conditions of normal aging in mouse skeletal muscle.
Interestingly, SS-31-treated mice did not run farther than untreated mice when given voluntary access to running wheels. Comparison of voluntary wheel running with treadmill test separates exercise capacity from motivation to exercise. Unlike forced treadmill running to exhaustion, voluntary wheel running tests an animal’s motivation more than the limits of its endurance.
In conclusion, this study demonstrates the recovery of depressed mitochondrial energetics and restoration of the cell redox environment in aged mouse skeletal muscle by a single injection of the mitochondrial targeted peptide SS-31. Interestingly, this acute treatment had no effect on skeletal muscle energetics in young healthy mice indicating that SS-31 is not simply enhancing mitochondrial function. Rather, SS-31 appears to reverse age-related inhibition of mitochondrial energetics. The capacity for rapid functional recovery supports the hypothesis that age-related loss of mitochondrial function results from regulatory modifications associated with increased capacity for mitochondrial ROS production rather than permanent structural changes. The energetic improvements that we observed translated into enhanced fatigue resistance and exercise capacity, supporting the notion that SS-31 may have therapeutic benefits for elderly humans.
Experimental Procedures
Animals and SS-31
This study was approved by the Institutional Animal Care and Use Committee of the University of Washington. Five month (young) and 27 month (old) old female C57BL/6 mice were purchased from the NIA aged mouse colony. All mice were exposed to a 12 hour light/dark cycle in a fixed-temperature environment with free access to water and standard mouse chow until immediately prior to experimentation. Treatments were administered by intraperitoneal injection of isotonic saline or 3 mg/kg SS-31 dissolved in isotonic saline at a concentration of 0.3 mg/mL. Mouse body temperatures were maintained at 36° ± 1° throughout in vivo and in situ experiments.
In Vivo Metabolic Spectroscopy
Mice were anesthetized by intraperitoneal injection of 0.01 ml/g of 2.5% tribromoethanol (“Avertin”, Sigma), the distal hindlimb was shaved, and the mouse was suspended by flexible straps within a custom built combined MR/optics probe for use with a 7T vertical bore spectrometer (Varian, Palo Alto, CA) (Siegel et al. 2012). The distal hindlimb was centered within a horizontal MR solenoid coil tunable to both 1H and 31P with fiber optic bundles positioned on either side to simultaneously collect MR and optical spectra from intact skeletal muscle. After positioning the mouse, MR signal was optimized by shimming the 1H of tissue water and optical signal was optimized by adjusting acquisition time. Next, a high signal to noise 31P spectrum was acquired under fully relaxed conditions (32 transients, 4096 complex points, 10 kHz sweep width, 25 sec interpulse delay). Finally, dynamic optical (0.5 sec delay) and MR (45° flip angle, 4 transients, 4096 complex points, 10 kHz sweep width, 1.5 sec interpulse delay) spectra were acquired continuously through periods of rest (2 min), ischemia (11 min), and recovery (7 min). After the first minute of rest mice breathed 100% O2 for the remainder of each dynamic experiment. In vivo spectroscopy data were acquired approximately one hour after a single injection with either saline or SS-31.
Tissue Preparation
Immediately following in vivo spectroscopy the skeletal muscles of the distal hindlimb were dissected and flash-frozen in liquid nitrogen. From the left leg, extensor digitorum longus, gastrocnemius, soleus, and tibialis anterior muscles were pooled and pulverized over liquid nitrogen for measurement of mixed muscle metabolite, hemoglobin, and myoglobin concentrations. From the right leg, gastrocnemius was pulverized over liquid nitrogen and prepared for western blotting. All muscle samples were stored at −80°C until the day of assay.
Metabolite, Hemoglobin, and Myoglobin Concentrations
Concentrations of ATP, PCr, and total creatine were determined in mixed muscle by HPLC (Waters, Milford, MA) using protocols described previously (Marcinek et al. 2004b). Hemoglobin and myoglobin concentrations were measured in mixed muscles after separating proteins with SDS-PAGE staining with Coomassie Brilliant Blue stain (Bio-Rad) according to (Marcinek et al. 2004a).
In vivo Spectroscopy Data Analysis
31P MR spectra were exponentially multiplied, Fourier transformed, and manually phase corrected using Varian VNMR (7T) software. The resulting spectra were taken to custom written MATLAB software (Mathworks, Natick, MA) for the remainder of analysis. Raw optical spectra files collected using WinSpec (Princeton Instruments) were taken directly to custom written MATLAB software for analysis. The method used for analyzing MR spectra is described in detail elsewhere (Marcinek et al. 2004a). Relative peak integrals from fully relaxed 31P MR spectra were used to calculate the resting inorganic phosphate (Pi)/ATP and PCr/ATP ratios. Three consecutive dynamic spectra were summed to improve signal-to-noise ratio and then the Fit-to-Standard algorithm (Heineman et al. 1990) was used to determine PCr and Pi peak magnitudes throughout dynamic acquisition. After correcting for variable relaxation, the ATP concentration from HPLC analysis of mixed muscle was used as an internal reference to calculate absolute PCr and Pi concentrations over time. pH was determined using the chemical shift between Pi and PCr peaks, and ADP and AMP concentrations were calculated using the known kinetics of the creatine kinase and adenylate kinase reactions, assuming equilibrium conditions and a Mg2+ concentration of 0.6 mM (Golding et al. 1995; Kushmerick 1997).
Optical spectra were analyzed using a partial-least squares routine to determine the O2 saturations of Hb and Mb throughout dynamic spectral acquisition (Marcinek et al. 2004a). Second derivatives of optical spectra were used to minimize the influence of tissue scattering (Arakaki et al. 2007). The concentrations and known O2 binding kinetics of Hb and Mb were then used to calculate net O2 flux in the closed system of the ischemic hindlimb.
The resting rates of mitochondrial ATP production (ATPase) and O2 consumption were calculated during ischemia from least-squares linear approximations of the decline in PCr and O2, respectively, during the initial phase of ischemia (Fig. 1C) (Marcinek et al. 2004a). The maximum rate of oxidative phosphorylation (ATPmax) was calculated using a least-squares monoexponential approximation of PCr recovery during recovery from ischemia (Blei et al. 1993).
Glutathione Redox
The quantities of reduced and oxidized glutathione were determined in gastrocnemius using a modified version of the fluorescent HPLC method described in detail by White, et al. (White et al. 2001). One hour after injection with either 3 mg/kg SS-31 or volume-matched saline, muscle was dissected from live, Avertin-anesthetized mice, weighed quickly, submerged in 0.5 mL of cold 5% salicylic acid (SSA), and homogenized over ice. After incubating on ice for 15 min to allow proteins to precipitate, homogenates were centrifuged at 4°C for 15 min at 10,000 X G, and the resulting supernatant taken for analysis. For GSSG: 100 μL of sample was combined with 400 μL phosphate buffered saline (pH 7.4), 50 μL of 10% triethanolamine, and 4 μL of 2-vinylpyridine (to derivatize GSH). Chloroform was used to phase-separate GSH-2-vinylpyridine conjugate from GSSG. After centrifugation the aqueous phase was combined with 200 μL 100 mM NaH2PO4, 1 mM EDTA and 10 μL tris(2-carboxyethy)phosphine to reduce GSSG in solution. After incubating for 20min at room temperature, 20 μL of 6.25mM monobromobimane (MBB) was added and incubated for 30 min to derivatize the GSH yielded by GSSG reduction. For GSH: the original sample was diluted 1:9 with SSA and then 100 μL was combined with 400μL phosphate buffered saline (pH 7.4), 50 μL of 10% triethanolamine. 100 μL of this mixture was combined with 200 μL 100mM NaH2PO4, 1mM EDTA and 10 μL H2O and then allowed 30min to incubate with 20 μL of 6.25mM MBB. The fluorescent derivative of GSH and MBB (excitation/emission of 375/475nm) was then quantified by HPLC (Shimadzu SCL-10AVP Columbia, MD) for all samples. The GSH redox potential was calculated using the Nernst equation (Nicholls & Ferguson 2002) with a standard midpoint potential of −246 mV at a cell pH of 7.1 (Table 1).
Ex vivo Mitochondrial Respiration
Freshly dissected, gently separated and permeabilized (50 μg/ml saponin, 4°C, 40 min) EDL and soleus muscle fibers were stirred at 25°C in a 2 ml chamber of an O2K Respirometer (Oroboros Instruments, Innsbruck, Austria). Corresponding muscle from contralateral legs were run simultaneously, with one chamber receiving 5 μL of 40 mM SS-31 (final concentration of 100 μM SS-31) and the other receiving 5 μL of water for volume control. Leak driven (“state 4”) and ADP stimulated respiration was measured as described (Siegel et al. 2011).
H2O2 Emission in Permeabilized Muscle Fibers
H2O2 production was measured using Amplex Red, which reacts with H2O2 to produce the fluorescent molecule resorufin (excitation/emission of 590/530–560nm). EDL muscles were dissected, mechanically separated, and permeabilized following the method used for ex vivo mitochondrial respiration measurements described below. Muscles were then combined with 12.5 μM Amplex Red, 2U/mL horseradish peroxidase, 10 μM succinate and either SS-31 or volume-matched water in a total reaction volume of 200 μL. SS-31 concentrations of either 500 nM or 100 μM were used, with no difference in H2O2 production between the two concentrations. Succinate was used as substrate to measure the capacity for H2O2 production by reverse electron flow through complex I. Fluorescence was measured on a standard plate-reader and calibrated using standards of known H2O2 concentration.
In situ Muscle Force and Fatigue
In situ analysis of TA muscle function was performed following a protocol described elsewhere (Percival et al. 2008). Following injection with either 3mg/kg SS-31 or volume-matched saline, mice were anesthetized with Avertin and the distal TA tendon was surgically isolated. Mice were then positioned on a heated platform with the limb restrained at the knee joint and the distal tendon attached by silk sutures to the lever arm of a servomotor (Model 305B-LR, Aurora Scientific, Aurora, ON, Canada). The exposed surface of the muscle was kept moist with isotonic saline and muscle stimulation was delivered at a voltage of 4.5V through the peroneal nerve using two needle electrodes. Custom-written software in LabVIEW (National Instruments, Austin, TX) was used to control electrical stimulation and to acquire force vs. time data. After adjusting muscle length to optimize tetanic force output (tetanic stimulus: 200 Hz stimulation frequency for 300msec), force-frequency measurements were made by delivering stimuli once per minute at frequencies ranging from 10 to 250 Hz to test muscle contractile response to submaximal and maximal stimulation frequencies. Muscle fatigue was measured by delivering tetanic stimuli (200 Hz) at 2 sec intervals for 4 min and then 1 min and 5 min after the final stimulus to assess recovery from fatigue. If tetanic muscle force failed to reach >90% of pre-fatigue levels after 5 min of recovery muscle damage was likely to have occurred and data were discarded. After the completion of testing, muscle length and mass used to calculate specific tetanic force (Percival et al. 2008). Following in situ experiments, raw data were taken to custom-written MATLAB software for analysis. For twitch force and force-frequency measurements, peak force during contraction was recorded. For tetanic stimuli, the (force)x(time) integral (FTI) was calculated for the entire duration of contraction.
Treadmill Test
Following 8 days of daily injections with 3 mg/kg SS-31 or volume-matched saline, endurance capacity of mice was assessed by measuring time to failure during forced running on a treadmill (Eco 3/6, Columbus Instruments, Columbus, OH). Mice first underwent two days of acclimation (on injection days 6 and 7), during which they were allowed to explore motionless treadmill lanes for 1 min, become familiar with the motivational shocking grid for 1 minute, and walk at a rate of 20 m/min at a 0° incline for 2 min. On the following day (commencing one hour after injection on the 8th day), mice ran at a rate of 30m/min at a 10° incline until failure. Time to failure was recorded manually and failure was determined when mice were unable to maintain position on the treadmill despite electrical shock and light prodding for 10 sec. Studies were carried out between 6 pm and 8 pm to observe mice during their more active dark cycle.
Statistics
Statistical analysis was carried out using Prism 5 software (GraphPad, La Jolla, CA). For treadmill data, a survival analysis was carried out using a Weibull model to fit times to exhaustion. For all other data sets, two-tailed student’s t-tests were used to compare groups. The levels of statistical significance are indicated in table and figure legends.
Supplementary Material
Figure S1. SS-31 is rapidly elevated in skeletal muscle and declines to 10% of its maximum value after 16 hr.
Figure S2. ETC complex protein expression was not different one hour after injection of 3mg/kg SS-31 in old mice. Means ± SEM, normalized to control averages, with n = 5 per group.
Figure S3. Representative fully relaxed spectra from SS-31-treated and control old mice illustrate an increase in relative peak area of PCr to γATP.
Figure S4. Daily SS-31 treatment did not affect voluntary running distance in old mice. Mean ± SEM. n = 6–7 per group. Day 1 is the first injection day when wheels were unlocked, so no distance data were recorded. Due to a software malfunction wheel data from days 4 and 5 were lost.
Acknowledgments
We thank Dr. Mary Emond for her statistical consultation and Rudy Stuppard for technical assistance. This work was supported by NIH grants AG028455, AG042637, AG036606, AG001751, ES007033 and the Ellison Medical Foundation and the University of Washington Nathan Shock Center of Excellence in the Basic Biology of Aging.
Footnotes
Disclosure
The SS peptides described in this article are licensed for commercial research and development to Stealth Peptides Inc, a clinical stage biopharmaceutical company, in which Hazel H. Szeto and the Cornell Research Foundation have financial interests.
Author Contributions
MPS and DJM designed and conducted experiments, analyzed data, and wrote the paper; SEK designed and conducted experiments and analyzed data; JMP, JG, HCH, CCW conducted experiments; TJK and PSR contributed to experimental design and writing; HHS contributed to experimental design and conducted experiments.
References
- Amara C, Shankland E, Jubrias S, Marcinek D, Kushmerick M, Conley K. Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proceedings of the National Academy of Science. 2007;104:1057–1062. doi: 10.1073/pnas.0610131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E, Lustig M, Boyle K, Woodlief T, Kane D, Lin C, Price JI, Kang L, Rabinovitch P, Szeto H, Houmard J, Cortright R, Wasserman D, Neufer P. Mitochondrial h2o2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. The Journal of Clinical Investigation. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Applegate MA, Humphries KM, Szweda LI. Reversible inhibition of alpha-ketoglutarate dehydrogenase by hydrogen peroxide: Glutathionylation and protection of lipoic acid. Biochemistry. 2008;47:473–478. doi: 10.1021/bi7017464. [DOI] [PubMed] [Google Scholar]
- Applegate MAB, Humphries KM, Szweda LI. Reversible inhibition of α-ketoglutarate dehydrogenase by hydrogen peroxide: Glutathionylation and protection of lipoic acid†. Biochemistry. 2007;47:473–478. doi: 10.1021/bi7017464. [DOI] [PubMed] [Google Scholar]
- Arakaki L, Burns D, Kushmerick M. Accurate myoglobin oxygen saturation by optical spectroscopy measured in blood-perfused rat muscle. Applied Spectroscopy. 2007;61:978–985. doi: 10.1366/000370207781745928. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Krause DJ, Howlett RA, Hepple RT. Nitric oxide synthase inhibition reduces o2 cost of force development and spares high-energy phosphates following contractions in pump-perfused rat hindlimb muscles. Exp Physiol. 2006;91:581–589. doi: 10.1113/expphysiol.2005.032698. [DOI] [PubMed] [Google Scholar]
- Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, Seshan SV, Pardee JD, Szeto HH. Cardiolipin as a novel target to re-energize ischemic mitochondria. J Am Soc Nephrol. doi: 10.1681/ASN.2012121216. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blei ML, Conley KE, Kushmerick MJ. Separate measures of atp utilization and recovery in human skeletal muscle. The Journal of Physiology. 1993;465:203–222. doi: 10.1113/jphysiol.1993.sp019673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai D-F, Chen T, Szeto H, Nieves-Cintrón M, Kutyavin V, Santana LF, Rabinovitch PS. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. Journal of the American College of Cardiology. 2011;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JM, Murphy EA, Carmichael MD, Davis B. Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2009;296:R1071–R1077. doi: 10.1152/ajpregu.90925.2008. [DOI] [PubMed] [Google Scholar]
- Garcia J, Han D, Sancheti H, Yap LP, Kaplowitz N, Cadenas E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J Biol Chem. 2010;285:39646–39654. doi: 10.1074/jbc.M110.164160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding EM, Teague WE, Jr, Dobson GP. Adjustment of k′ to varying ph and pmg for the creatine kinase, adenylate kinase and atp hydrolysis equilibria permitting quantitative bioenergetic assessment. J Exp Biol. 1995;198:1775–1782. doi: 10.1242/jeb.198.8.1775. [DOI] [PubMed] [Google Scholar]
- Heineman FW, Eng J, Berkowitz BA, Balaban RS. Nmr spectral analysis of kinetic data using natural lineshapes. Magn Reson Med. 1990;13:490–497. doi: 10.1002/mrm.1910130316. [DOI] [PubMed] [Google Scholar]
- Hood DA. Invited review: Contractile activity-induced mitochondrial biogenesis in skeletal muscle. Journal of Applied Physiology. 2001;90:1137–1157. doi: 10.1152/jappl.2001.90.3.1137. [DOI] [PubMed] [Google Scholar]
- Hurd TR, Requejo R, Filipovska A, Brown S, Prime TA, Robinson AJ, Fearnley IM, Murphy MP. Complex i within oxidatively stressed bovine heart mitochondria is glutathionylated on cys-531 and cys-704 of the 75-kda subunit. Journal of Biological Chemistry. 2008;283:24801–24815. doi: 10.1074/jbc.M803432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC, Pérez VI, Song W, Lustgarten MS, Salmon AB, Mele J, Qi W, Liu Y, Liang H, Chaudhuri A, Ikeno Y, Epstein CJ, Van Remmen H, Richardson A. Overexpression of mn superoxide dismutase does not increase life span in mice. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2009;64A:1114–1125. doi: 10.1093/gerona/glp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen I, Shepard DS, Katzmarzyk PT, Roubenoff R. The healthcare costs of sarcopenia in the united states. Journal of the American Geriatrics Society. 2004;52:80–85. doi: 10.1111/j.1532-5415.2004.52014.x. [DOI] [PubMed] [Google Scholar]
- Kushmerick M. Multiple equilibria of cations with metabolites in muscle bioenergetics. American Journal of Physiology Cell Physiology. 1997;272:C1739–C1747. doi: 10.1152/ajpcell.1997.272.5.C1739. [DOI] [PubMed] [Google Scholar]
- Li D, Lai Y, Yue Y, Rabinovitch PS, Hakim C, Duan D. Ectopic catalase expression in mitochondria by adeno-associated virus enhances exercise performance in mice. PLOS ONE. 2009;4:e6673. doi: 10.1371/journal.pone.0006673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailloux RJ, Seifert EL, Bouillaud F, Aguer C, Collins S, Harper M-E. Glutathionylation acts as a control switch for uncoupling proteins ucp2 and ucp3. Journal of Biological Chemistry. 2011;286:21865–21875. doi: 10.1074/jbc.M111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri A, Muller FL, Liu Y, Ng R, Faulkner J, Hamilton M, Richardson A, Huang TT, Epstein CJ, Van Remmen H. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mech Ageing Dev. 2006;127:298–306. doi: 10.1016/j.mad.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Marcinek D, Schenkman K, Ciesielski W, Conley K. Mitochondrial coupling in vivo in mouse skeletal muscle. The American Journal of Cell Physiology. 2004a;286:C457–C463. doi: 10.1152/ajpcell.00237.2003. [DOI] [PubMed] [Google Scholar]
- Marcinek D, Schenkman K, Ciesielski W, Lee D, Conley K. Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. Journal of Physiology. 2005;569:467–473. doi: 10.1113/jphysiol.2005.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinek DJ, Schenkman KA, Ciesielski WA, Conley KE. Mitochondrial coupling in vivo in mouse skeletal muscle. Am J Physiol Cell Physiol. 2004b;286:C457–463. doi: 10.1152/ajpcell.00237.2003. [DOI] [PubMed] [Google Scholar]
- Matuszczak Y, Farid M, Jones J, Lansdowne S, Smith MA, Taylor AA, Reid MB. Effects of n-acetylcysteine on glutathione oxidation and fatigue during handgrip exercise. Muscle Nerve. 2005;32:633–638. doi: 10.1002/mus.20385. [DOI] [PubMed] [Google Scholar]
- McLain AL, Szweda PA, Szweda LI. A-ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Radical Research. 2011;45:29–36. doi: 10.3109/10715762.2010.534163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus MJ, Murphy MP, Franklin JL. The mitochondria-targeted antioxidant mitoq prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of alzheimer’s disease. The Journal of Neuroscience. 2011;31:15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min K, Smuder AJ, Kwon O-s, Kavazis AN, Szeto HH, Powers SK. Mitochondrial-targeted antioxidants protect the skeletal muscle against immobilization-induced muscle atrophy. Journal of Applied Physiology. 2011 doi: 10.1152/japplphysiol.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Ferguson SJ. Bioenergetics 3. London, UK: Academic Press; 2002. [Google Scholar]
- Nogueira L, Ramirez-Sanchez I, Perkins GA, Murphy A, Taub PR, Ceballos G, Villarreal FJ, Hogan MC, Malek MH. (−)-epicatechin enhances fatigue resistance and oxidative capacity in mouse muscle. The Journal of Physiology. 2011 doi: 10.1113/jphysiol.2011.209924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival JM, Anderson KNE, Gregorevic P, Chamberlain JS, Froehner SC. Functional deficits in nnosμ-deficient skeletal muscle: Myopathy in nnos knockout mice. PLOS ONE. 2008;3:e3387. doi: 10.1371/journal.pone.0003387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Ritchie D, Wright KJ, Romestaing C, Thomas MM, Rowan SL, Taivassalo T, Hepple RT. Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibers. Aging Cell. 2010;9:1032–1046. doi: 10.1111/j.1474-9726.2010.00628.x. [DOI] [PubMed] [Google Scholar]
- Rasmussen UF, Krustrup P, Kjær M, Rasmussen HN. Experimental evidence against the mitochondrial theory of aging a study of isolated human skeletal muscle mitochondria. Experimental Gerontology. 2003;38:877–886. doi: 10.1016/s0531-5565(03)00092-5. [DOI] [PubMed] [Google Scholar]
- Reid MB. Free radicals and muscle fatigue: Of ros, canaries, and the ioc. Free Radic Biol Med. 2008;44:169–179. doi: 10.1016/j.freeradbiomed.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Reid MB, Stokic DS, Koch SM, Khawli FA, Leis AA. N-acetylcysteine inhibits muscle fatigue in humans. J Clin Invest. 1994;94:2468–2474. doi: 10.1172/JCI117615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- Siegel MP, Kruse SE, Knowels G, Salmon A, Beyer R, Xie H, Van Remmen H, Smith SR, Marcinek DJ. Reduced coupling of oxidative phosphorylation in vivo precedes electron transport chain defects due to mild oxidative stress in mice. PLOS ONE. 2011;6:e26963. doi: 10.1371/journal.pone.0026963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel MP, Wilbur T, Mathis M, Shankland EG, Trieu A, Harper ME, Marcinek DJ. Impaired adaptability of in vivo mitochondrial energetics to acute oxidative insult in aged skeletal muscle. Mech Ageing Dev. 2012;133:620–628. doi: 10.1016/j.mad.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulachev VP, Anisimov VN, Antonenko YN, Bakeeva LE, Chernyak BV, Erichev VP, Filenko OF, Kalinina NI, Kapelko VI, Kolosova NG, Kopnin BP, Korshunova GA, Lichinitser MR, Obukhova LA, Pasyukova EG, Pisarenko OI, Roginsky VA, Ruuge EK, Senin II, Severina II, Skulachev MV, Spivak IM, Tashlitsky VN, Tkachuk VA, Vyssokikh MY, Yaguzhinsky LS, Zorov DB. An attempt to prevent senescence: A mitochondrial approach. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2009;1787:437–461. doi: 10.1016/j.bbabio.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Orr WC. The redox stress hypothesis of aging. Free Radic Biol Med. 2012;52:539–555. doi: 10.1016/j.freeradbiomed.2011.10.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Buckingham J, Roebuck S, Brand M. Topology of superoxide production from different sites in the mitochondrial electron transport chain. The Journal of Biological Chemistry. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng F-Y, Zhao Z, Ganger M, Tow CY, Seshan SV. Mitochodria-targeted peptide accelerates atp recovery and reduces ischemic kidney injury. Journal of the American Society of Nephrology. 2011 doi: 10.1681/ASN.2010080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CC, Krejsa CJ, Eaton DL, Kavanagh TJ. Current protocols in toxicology. John Wiley & Sons, Inc; 2001. Hplc-based assays for enzymes of glutathione biosynthesis. [DOI] [PubMed] [Google Scholar]
- Williams AD, Carey MF, Selig S, Hayes A, Krum H, Patterson J, Toia D, Hare DL. Circuit resistance training in chronic heart failure improves skeletal muscle mitochondrial atp production rate—a randomized controlled trial. Journal of Cardiac Failure. 2007;13:79–85. doi: 10.1016/j.cardfail.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Yang L, Zhao K, Calingasan NY, Luo G, Szeto HH, Beal MF. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxidants & Redox Signaling. 2009;11:2095–2104. doi: 10.1089/ars.2009.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. SS-31 is rapidly elevated in skeletal muscle and declines to 10% of its maximum value after 16 hr.
Figure S2. ETC complex protein expression was not different one hour after injection of 3mg/kg SS-31 in old mice. Means ± SEM, normalized to control averages, with n = 5 per group.
Figure S3. Representative fully relaxed spectra from SS-31-treated and control old mice illustrate an increase in relative peak area of PCr to γATP.
Figure S4. Daily SS-31 treatment did not affect voluntary running distance in old mice. Mean ± SEM. n = 6–7 per group. Day 1 is the first injection day when wheels were unlocked, so no distance data were recorded. Due to a software malfunction wheel data from days 4 and 5 were lost.