

Abstract
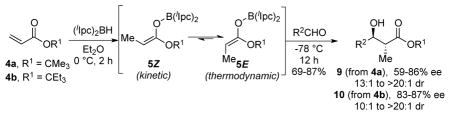
The (diisopinocampheyl)borane promoted reductive aldol reaction of acrylate esters 4 is described. Isomerization of the kinetically formed Z(O)-enolborinate 5Z to the thermodynamic E(O)-enolborinate 5E via 1,3-boratropic shifts, followed by treatment with representative achiral aldehydes, leads to anti-α-methyl-β-hydroxy esters 9 or 10 with excellent diastereo- (up to ≥20:1 dr) and enantioselectivity (up to 87% ee). Results of double asymmetric reactions of 5E with several chiral aldehydes are also presented.
The aldol reaction is a powerful method for the stereocontrolled construction of carbon-carbon bonds.1,2 Although the formation of syn-aldols with exceptional stereoselectivtity is well established, efficient means to access anti-aldols with synthetically useful diastereo- and enantioselectivity remains a significant challenge.1 Noteworthy contributions towards the enantioselective anti-aldol reaction have emerged utilizing chiral auxiliary-based,3 metal-promoted4 and organocatalytic procedures.5 In 2005, Nishiyama reported an efficient rhodium-catalyzed anti-selective reductive aldol reaction of acrylates predominantly with aromatic aldehydes.6 To the best of our knowledge, this work represents the only reductive anti-aldol reaction originating from acyclic precursors.7
We recently reported the highly enantio- and diastereoselective reductive syn-aldol reaction8 of N-acryloylmorpholine (1) with (diisopinocampheyl)borane [(Ipc)2BH] as the reducing agent (Scheme 1(a)).9 Isomerization of 2Z to the corresponding E(O)-enol borinate did not occur evidently due to A1,3 strain that develops between the morpholine unit and the enolborinate methyl substituent. Hence the reductive aldol reactions of N-acryloylmorpholine (1) were highly selective for the syn-aldol 3.9 We reasoned that replacing the morpholine amide of 1 with an ester unit in 4 would eliminate this interaction, and that enolborinate 5Z obtained from 1,4-reduction8 of acrylate 4 would undergo a 1,3-boratropic shift to give the presumably more stable enolate 5E,8c thereby providing access to anti-aldols 6 (Scheme 1(b)).
Scheme 1.

Reductive aldol reactions of 1 and 4.
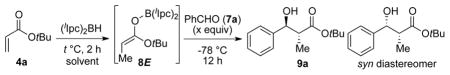
We selected the inexpensive, commercially available tert-butyl acrylate 4a as the initial substrate for this study.4d,10 The reductive aldol reaction of 4a, with (lIpc)2BH11 and benzaldehyde (7a) was used to optimize reaction conditions (Table 1).
Table 1.
Optimization of reaction parameters.a
| ||||||
|---|---|---|---|---|---|---|
| entry | solvent | t (°C) | x | yieldb | dr (9a:syn)c | ee (9a)d |
| 1 | toluene | 0 | 1.1 | 61 | 15:1 | 85 |
| 2 | THF | 0 | 1.1 | 92 | 11:1 | 76 |
| 3 | CH2Cl2 | 0 | 1.1 | 84 | 11:1 | 80 |
| 4 | Et2O | 0 | 1.1 | 76 | 16:1 | 85 |
| 5 | toluene | 0 | 0.85 | 81 | 16:1 | 85 |
| 6 | toluene | −30 | 0.85 | 29 | 13:1 | ND |
| 7 | Et2O | 0 | 0.85 | 79 | 18:1 | 86 |
Reactions were performed by treating 4a (0.275 mmol, 1.1 equiv) with (lIpc)2BH (0.25 mmol, 1 equiv) in solvent (1 mL) at the indicated temperature for 2 h, followed by addition of 7a at −78 °C. After being stirred for 12 h at −78 °C, the reaction was subjected to oxidative hydrolysis (buffer/MeOH/H2O2) followed by product isolation.
Isolated yield of aldols following silica gel chromatography.
Diastereomer ratio (dr) determined by 1H NMR analysis of crude reaction mixtures.
Enantiomeric excess (% ee) and absolute configuration were determined by using the Mosher ester analysis.12
Treatment of acrylate 4a with (lIpc)2BH (1.1 equiv) in toluene at 0° C for 2 h followed by addition of benzaldehyde at −78° C provided a 15:1 mixture of 9a and the syn diastereomer in 61% yield (entry 1). As indicated by the formation of anti-aldol 9a as the major product, this initial experiment suggested that enolborinate 8E is indeed the dominant species in this reaction. Reactions performed in toluene (entry 1) and Et2O (entry 4) exhibited greater diastereo- and enantioselectivity than those in THF and CH2Cl2 (Entries 2 and 3). Decreasing the amount of aldehyde to 0.85 equiv led to improved product yields (calculated based on aldehyde as the limiting reagent; entries 5, 7). Lowering the temperature of the hydroboration reaction had a dramatic effect on yield (entry 6), presumably due to incomplete reaction under these conditions. Ultimately, the best compromise between product yield, diastereo- and enantioselectivity was achieved by performing the hydroboration reaction at 0 °C in Et2O (entry 7).
These conditions were applied to the reductive anti-aldol reactions of acrylate 4a with a series of achiral aldehydes 7a–f (Scheme 2). Anti-α-methyl-β-hydroxy tert-butyl esters 9a–f were obtained in 69–87% yield with excellent diastereoselectivity (dr 13:1 to ≥20:1), and with moderate to good enantioselectivity (59–86% ee).13 Interestingly, the sense of absolute stereochemical induction by the (diisopinocampheyl)boryl unit in these anti-selective aldol reactions is opposite to that determined in our studies of the syn aldol reactions of acrylamide 1.9,14 This leads us to speculate that the major anti-aldol in each of the reactions summarized in Scheme 2 may possibly arise by way of the boat-like transition state TS-I. It is known that anti-selective boron-mediated aldol reactions proceed preferentially through boat-like transition states.15 Indeed, ab initio calculations for the boron-mediated aldol reaction of ethyl methyl ketone with acetaldehyde showed that the lowest energy transition state for the anti-aldol reaction of the E-enolborinate is boat-like (analogous to TS-I), but also that a competitive chair-like and a second boat-like transition state are only 0.55 and 0.67 kcal/mmol higher in energy than the predominant boat-like transition structure.15 Boat-like transition states also appear to dominate in the (diisopinocampheyl)borane-mediated aldol reactions of methyl ketones.16 Thus, that small structural changes in the substrates impact the overall reaction enantioselectivity may not be surprising.
Scheme 2.

Scope of the anti-reductive aldol reaction of 4a with achiral aldehydes.
a) Isolated yield after purification on silica gel. b) Diastereomer ratio (dr) determined by 1H NMR analysis of crude reaction mixture. c) Enantiomeric excess (% ee) and absolute configuration determined by using the Mosher ester analysis.12
At present, we rationalize the good to excellent enantioselectivity data presented in Scheme 2 by a competition between the boat-like TS-I and the chair-like TS-II (Scheme 3). In an effort to improve the enantioselectivity of these reactions, especially with aliphatic aldehydes, we anticipated that increasing the size of the ester alkyl group might further destabilize chair-like TS-II relative to the major boat-like TS-I.
Scheme 3.

Postulated TS for the formation of anti-aldols 9.
Based on this analysis we examined the more hindered acrylate 4b as the substrate for the anti-selective aldol reactions.17 Gratifyingly, markedly enhanced levels of enantioselectivity (83–87% ee) were obtained for anti-aldols 10a–e, in comparison to the results summarized in Scheme 2 for aldols 9a–e.
In order to investigate the potential for application of this methodology to the synthesis of more complex polyketide structures, we turned our attention to double asymmetric19 reductive aldol reactions (Scheme 5).
Scheme 5.

Double asymmetric aldol reactions of chiral aldehydes and the chiral E-enolborinate generated from 4a,b.a–d
a) Isolated yield of the indicated aldol products (major product in all cases except 9k from 7i) after purification by silica gel chromatography. b) Diastereomer ratio (dr) determined by 1H NMR analysis of crude reaction mixture. c) Absolute and relative configuration of 9g–9n determined using Mosher ester analysis12 and the Rychnovsky acetonide method.18 (see Supporting Information). d) Relative configuration of 10k,l determined by analogy with 9k,l.
Four chiral aldehydes 7g–j were used in aldol reactions with the E(O)-enolborinates generated by reduction of from 4a with both (lIpc)2BH and (dIpc)2BH. Reductive aldol reactions of β-alkoxy aldehydes 7g, 7h20 and 7j9 furnished anti-aldols 9g–j,m,n (50–74% isolated yield of major aldol isomer) with moderate to good diastereoselectivity (dr 3:1 to 8:1, as determined by analysis of crude product mixtures). However, when the double stereodifferentiating reactions were carried out with syn-a-methyl-β-alkoxy aldehyde 7i,21 it was not possible to achieve the synthesis of anti, anti stereotriad 9k with acceptable mismatched stereoselectivity (when acrylate 4a (via 8E) was used as the starting material). However, when these reactions were performed by using the more sterically demanding acrylate 4b (via enolborinate 11E), the anti, anti stereotriad 10k was obtained with 2:1 dr in the mismatched case, and the diastereomer 10l was obtained with 13:1 dr in the matched double aymmetric reaction using 11E generated from the hydroboration of 4b with (dIpc)2BH. These results confirm the conclusion from Scheme 4 that the enolborinate 11E generated from hindered acrylate 4b exhibits a higher level of enantioselectivity than 8E deriving from 4a, and that 11E should be used in the most stereochemically demanding applications of this methodology.
Scheme 4.
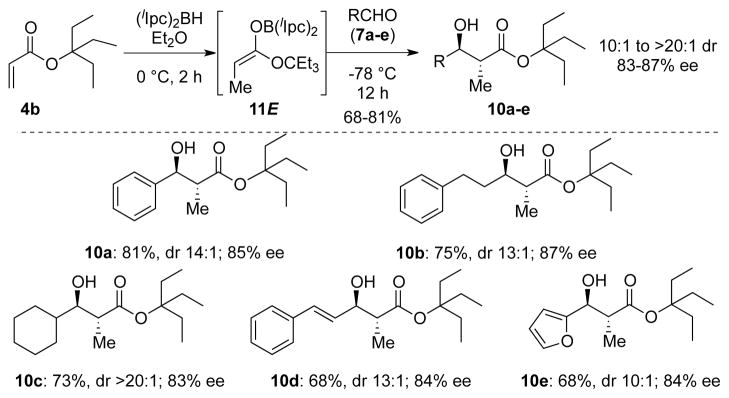
Reductive anti-aldol reactions of acrylate 4b.a,b.c
a) Isolated yield after purification on silica gel. b) Diastereomer ratio (dr) determined by 1H NMR analysis of crude reaction mixture. c)Enantiomeric excess (% ee) and absolute configuration determined using Mosher ester analysis.12
In summary, we have developed an enantio- and diastereoselective synthesis of anti-a-methyl-β-hydroxy propionate esters from achiral and chiral aldehydes, via the hydroboration of tert-butyl acrylate 4a or 4b with (diisopinocampheyl)borane. This highly cost-effective9 methodology takes advantage of the in situ formation of enolborinates 8E (from 4a) or 11E (from 4b) under neutral reaction conditions that is compatible with various protecting groups. As an example, the highly acid sensitive dimethoxytrityl -ODMTr ether 9f (Scheme 2) is well tolerated under standard reaction conditions. Hydroboration of acrylate 4a directly produces the (diisopinocampheyl)enolborinate 8Z which presumably isomerizes to 8E via 1,3-boratropic shifts. The latter then undergoes aldol reactions with achiral aldehydes (dr 13:1 to >20:1; 59–86% ee, Scheme 2). Higher levels of enantioselectivity were reached when the reaction was performed with bulkier acrylate 4b (Scheme 4). The study of double asymmetric reactions with chiral aldehydes demonstrated that this methodology can be applied to the synthesis of polyketide fragments of natural products (Scheme 5). Synthetic applications of this methodology are in progress and will be reported in due course.
Supplementary Material
Acknowledgments
We thank the National Institute of Health (GM038436) for supporting this research. We also thank Professor Glenn Micalizio and Dr. Daniel Canterbury, both at Scripps Florida when this work was being performed, for helpful discussions.
Footnotes
Supporting Information Available. Experimental procedures and spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Selected reviews of enantioselective aldol reactions: Heathcock CH. In: Comprehensive Organic Synthesis. Heathcock CH, editor. Vol. 2. Pergamon Press; New York: 1991. p. 181.Kim BM, Williams SF, Masamune S. In: Comprehensive Organic Synthesis. Heathcock CH, editor. Vol. 2. Pergamon Press; New York: 1991. p. 239.Cowden CJ, Paterson I. Org React. 1997;51:1.Arya P, Qin H. Tetrahedron. 2000;56:917–947.Mahrwald R. Modern Aldol Reactions. Wiley-VCH; Weinheim, Germany: 2004. Denmark SE, Fiujimori S. In: Modern Aldol Reactions. Mahrwald R, editor. Vol. 2. Wiley-VCH; 2004. pp. 229–326.Shibasaki M, Matsunaga S, Kumagai N. In: Modern Aldol Reactions. Mahrwald R, editor. Vol. 2. Wiley-VCH; 2004. pp. 197–227.Johnson JS, Nicewicz DA. In: Modern Aldol Reactions. Rainer M, editor. Vol. 2. Wiley VCH; 2006. pp. 69–103.Geary LM, Hultin PG. Tetrahedron: Asymmetry. 2009;20:131–173.Bisai V, Bisai A, Singh VK. Tetrahedron. 2012;68:4541–4580.
- 2.(a) Guo HC, Ma JA. Angew Chem Int Ed. 2006;45:354–366. doi: 10.1002/anie.200500195. [DOI] [PubMed] [Google Scholar]; (b) Nishiyama H, Shiomi T. Top Curr Chem. 2007;279:105–137. [Google Scholar]; (c) Han SB, Hassan A, Krische MJ. Synthesis. 2008;17:2669–2679. doi: 10.1055/s-2008-1067220. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Garner SA, Han SB, Krische MJ. In: In Modern Reduction Methods. Andersson PG, Munslow IJ, editors. 2008. pp. 387–417. [Google Scholar]
- 3.(a) Danda H, Hansen MM, Heathcock CH. J Org Chem. 1990;55:173–181. [Google Scholar]; (b) Walker MA, Heathcock CH. J Org Chem. 1991;56:5747–5750. [Google Scholar]; (c) Oppolzer W, Starkemann C, Rodriguez I, Bernardinelli G. Tetrahedron Lett. 1991;32:61–64. [Google Scholar]; (d) Oppolzer W, Lienard P. Tetrahedron Lett. 1993;34:4321–4324. [Google Scholar]; (e) Abiko A, Liu JF, Masamune S. J Am Chem Soc. 1997;119:2586–2587. [Google Scholar]; (f) Gabriel T, Wessjohann L. Tetrahedron Lett. 1997;38:4387–4388. [Google Scholar]; (g) Kurosu M, Lorca M. J Org Chem. 2001;66:1205–1209. doi: 10.1021/jo001293h. [DOI] [PubMed] [Google Scholar]; (h) Evans DA, Tedrow JS, Shaw JT, Downey CW. J Am Chem Soc. 2002;124:392–393. doi: 10.1021/ja0119548. [DOI] [PubMed] [Google Scholar]; (i) Crimmins MT, McDougall PJ. Org Lett. 2003;5:591–594. doi: 10.1021/ol034001i. [DOI] [PubMed] [Google Scholar]; (j) Ghosh AK, Kim JH. Org Lett. 2003;5:1063–1066. doi: 10.1021/ol034086n. [DOI] [PubMed] [Google Scholar]; (k) Hajra S, Giri AK, Karmakar A, Khatua S. Chem Commun. 2007:2408–2410. doi: 10.1039/b618699h. [DOI] [PubMed] [Google Scholar]; (l) Fanjul S, Hulme AN. J Org Chem. 2008;73:9788–9791. doi: 10.1021/jo801720v. [DOI] [PubMed] [Google Scholar]; (m) Dai WM, Feng G, Wu J, Sun L. Synlett. 2008;7:1013–1016. [Google Scholar]; (n) Shinisha CB, Sunoj RB. Org Lett. 2010;12:2868–2871. doi: 10.1021/ol100969f. [DOI] [PubMed] [Google Scholar]; (o) May A, Connell N, Dahlmann H, Hoye T. Synlett. 2010;13:1984–1986. doi: 10.1055/s-0030-1258480. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Grimster NP, Wilton DAA, Chan LKM, Godfrey CRA, Green C, Owen DR, Gaunt MJ. Tetrahedron. 2010;66:6429–6436. [Google Scholar]; (q) Li YJ, Hung HY, Liu YW, Lin PJ, Huang HJ. Tetrahedron. 2011;67:927–935. [Google Scholar]; (r) Chouhan M, Sharma R, Nair VA. Org Lett. 2012;14:5672–5675. doi: 10.1021/ol302653g. [DOI] [PubMed] [Google Scholar]
- 4.(a) Meyers AI, Yamamoto Y. J Am Chem Soc. 1981;103:4278–4279. [Google Scholar]; (b) Masamune S, Sato T, Kim B, Wollmann TA. J Am Chem Soc. 1986;108:8279–8281. [Google Scholar]; (c) Paterson I, Goodman JM, Isaka M. Tetrahedron Lett. 1989;30:7121–7124. [Google Scholar]; (d) Corey EJ, Kim SS. J Am Chem Soc. 1990;112:4976–4977. [Google Scholar]; (e) Parmee ER, Hong Y, Tempkin O, Masamune S. Tetrahedron Lett. 1992;33:1729–1732. [Google Scholar]; (h) Denmark SE, Wynn T, Beutner GL. J Am Chem Soc. 2002;124:13405–13407. doi: 10.1021/ja0282947. [DOI] [PubMed] [Google Scholar]; (i) Yamashita Y, Ishitani H, Shimizu H, Kobayashi S. J Am Chem Soc. 2002;124:3292–3302. doi: 10.1021/ja016293t. [DOI] [PubMed] [Google Scholar]; (j) Jung ME, Zhang T. Org Lett. 2008;10:137–140. doi: 10.1021/ol702729u. [DOI] [PubMed] [Google Scholar]; (k) Díaz-Oltra S, Ruiz P, Falomir E, Murga J, Carda M, Marco JA. Org Biomol Chem. 2012;10:6937–6944. doi: 10.1039/c2ob25803j. [DOI] [PubMed] [Google Scholar]; (l) Sureshkumar D, Kawato Y, Iwata M, Kumagai N, Shibasaki M. Org Lett. 2012;14:3108–3111. doi: 10.1021/ol301200q. [DOI] [PubMed] [Google Scholar]
- 5.For leading references to enantioselective organocatalytic anti-aldol reactions: Northrup AB, MacMillan DWC. J Am Chem Soc. 2002;124:6798–6799. doi: 10.1021/ja0262378.Thayumanavan R, Tanaka F, Barbas CF., III Org Lett. 2004;6:3541–3544. doi: 10.1021/ol0485417.Yang H, Mahapatra S, Cheong PHY, Carter RG. J Org Chem. 2010;75:7279–7290. doi: 10.1021/jo1015008.Li S, Wu C, Fu X, Miao Q. Ind Eng Chem Res. 2011;50:13711–13716.
- 6.(a) Nishiyama H, Shiomi T, Tsuchiya Y, Matsuda I. J Am Chem Soc. 2005;127:6972–6973. doi: 10.1021/ja050698m. [DOI] [PubMed] [Google Scholar]; (b) Ito J, Shiomi T, Nishiyama H. Adv Synth Catal. 2006;348:1235–1240. [Google Scholar]; (c) Shiomi T, Ito J, Yamamoto Y, Nishiyama H. Eur J Org Chem. 2006:5594–5600. [Google Scholar]; (d) Hashimoto T, Ito J, Nishiyama H. Tetrahedron. 2008;64:9408–9412. [Google Scholar]
- 7.Shiomi T, Adachi T, Ito J, Nishiyama H. Org Lett. 2009;11:1011–1014. doi: 10.1021/ol802939u. [DOI] [PubMed] [Google Scholar]
- 8.(a) Evans DA, Fu GC. J Org Chem. 1990;55:5678–5680. [Google Scholar]; (b) Boldrini GP, Mancini F, Tagliavini E, Trombini C, Umani-Ronchi A. J Chem Soc, Chem Commun. 1990:1680–1681. [Google Scholar]; (c) Boldrini GP, Bortolotti M, Mancini F, Tagliavini E, Trombini C, Umani-Ronchi A. J Org Chem. 1991;56:5820–5826. [Google Scholar]; (d) Matsumoto Y, Hayashi T. Synlett. 1991:349–350. [Google Scholar]; (e) Huddleston RR, Cauble DF, Krische MJ. J Org Chem. 2003;68:11–14. doi: 10.1021/jo020629f. [DOI] [PubMed] [Google Scholar]; (f) Ghosh AK, Kass J, Anderson DD, Xu X, Marian C. Org Lett. 2008;10:4811–4814. doi: 10.1021/ol801971t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nuhant P, Allais C, Roush WR. Angew Chem Int Ed. 2013;52 doi: 10.1002/anie.201302535. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corey EJ, Lee DH. Tetrahedron Lett. 1993;34:1737–1740. [Google Scholar]
- 11.Brown HC, Singaram B. J Org Chem. 1984;49:945–947. [Google Scholar]
- 12.(a) Dale JA, Mosher HS. J Am Chem Soc. 1973;95:512–519. [Google Scholar]; (b) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- 13.Ramachandran and Pratihar have previously reported the synthesis of anti-aldols with ≥98:2 dr and 50–66% ee from the Ipc2BOTf mediated aldol reactions of 4a (Ramachandran PV, Pratihar D. Org Lett. 2009;11:1467–1470. doi: 10.1021/ol802850w.). We repeated Ramachandran’s procedure with (lIpc)2BH and cinnamaldehyde as the substrates and obtained 9b with 15:1 dr and 58% ee. However, we also determined that the absolute stereochemistry of the anti-aldols described by Ramachandran have been misassigned, as Mosher ester analysis12 clearly indicated that 9b so obtained was identical to 9b obtained by the reductive aldol reaction presented in Scheme 2.
- 14.This conclusion derives from the fact that the absolute configuration of the hydroxyl groups of the syn-aldols deriving from 1 (see ref. 9) and the anti-aldol reactions deriving from 4, both using (lIpc)2BH as the reducing agent, are opposite.
- 15.Goodman JM, Paton RS. Chem Commun. 2007:2124–2126. doi: 10.1039/b704786j. [DOI] [PubMed] [Google Scholar]
- 16.(a) Paterson I, Goodman JM. Tetrahedron Lett. 1989;30:997–1000. [Google Scholar]; (b) Paterson I, Goodman JM, Anne Lister M, Schumann RC, McClure CK, Norcross RD. Tetrahedron. 1990;46:4663–4684. [Google Scholar]
- 17.Use of the substantially bulkier 2,6-di-tert-butyl-4-methylphenyl acrylate ester led to diminished reaction diastereoselectivity (see Supporting Information).
- 18.Rychnovsky SD, Skalitzky DJ. Tetrahedron Lett. 1990;31:945–948. [Google Scholar]
- 19.Masamune S, Choy W, Petersen JS, Sita LR. Angew Chem Int Ed. 1985;24:1–30. [Google Scholar]
- 20.Nuhant P, Kister J, Lira R, Sorg A, Roush WR. Tetrahedron. 2011;67:6497–6512. doi: 10.1016/j.tet.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lira R, Roush WR. Org Lett. 2007;9:4315–4318. doi: 10.1021/ol7018746. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.