

Significance
With the example of the coarse-grained United Residue model of polypeptide chains, this paper demonstrates that the physics-based approach for protein-structure prediction can lead to exceptionally good results when correct domain packing is an issue, even for a highly homologous target. The reason for this is probably that emphasis is placed on energetically favorable residue–residue interactions, including those with residues in relatively flexible linker regions; these regions are usually very different in the target compared with those of proteins in the databases used for template-based modeling. The results suggest that a combination of bioinformatics and a physics-based approach could result in a major increase in the prediction capacity of existing approaches.
Keywords: protein folding, structure symmetry, multi-domain packing
Abstract
The performance of the physics-based protocol, whose main component is the United Residue (UNRES) physics-based coarse-grained force field, developed in our laboratory for the prediction of protein structure from amino acid sequence, is illustrated. Candidate models are selected, based on probabilities of the conformational families determined by multiplexed replica-exchange simulations, from the 10th Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction (CASP10). For target T0663, classified as a new fold, which consists of two 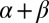 domains homologous to those of known proteins, UNRES predicted the correct symmetry of packing, in which the domains are rotated with respect to each other by 180° in the experimental structure. By contrast, models obtained by knowledge-based methods, in which each domain is modeled very accurately but not rotated, resulted in incorrect packing. Two UNRES models of this target were featured by the assessors. Correct domain packing was also predicted by UNRES for the homologous target T0644, which has a similar structure to that of T0663, except that the two domains are not rotated. Predictions for two other targets, T0668 and T0684_D2, are among the best ones by global distance test score. These results suggest that our physics-based method has substantial predictive power. In particular, it has the ability to predict domain–domain orientations, which is a significant advance in the state of the art.
domains homologous to those of known proteins, UNRES predicted the correct symmetry of packing, in which the domains are rotated with respect to each other by 180° in the experimental structure. By contrast, models obtained by knowledge-based methods, in which each domain is modeled very accurately but not rotated, resulted in incorrect packing. Two UNRES models of this target were featured by the assessors. Correct domain packing was also predicted by UNRES for the homologous target T0644, which has a similar structure to that of T0663, except that the two domains are not rotated. Predictions for two other targets, T0668 and T0684_D2, are among the best ones by global distance test score. These results suggest that our physics-based method has substantial predictive power. In particular, it has the ability to predict domain–domain orientations, which is a significant advance in the state of the art.
Prediction of protein structures from amino acid sequence still remains an unsolved problem of computational biology. Although, since the famous experiments by Anfinsen (1), it is known that a protein adopts the structure which is the (kinetically reachable) global minimum of the free energy of a system, it is not straightforward to implement this physical principle in practice because of the inaccuracy of existing force fields and because of the enormous difficulty to search the conformational space of the system. Therefore, the most effective methods for protein-structure prediction nowadays are knowledge-based approaches, in which database information is incorporated explicitly into the procedure (2). These methods can be divided into three categories, namely, comparative (homology) modeling (3–5), in which the target sequence is compared with the sequences for which experimental structures are known and those structures are usually selected as candidate models for which the greatest similarity is observed; threading (6–8), in which the target sequence is superposed on structures from a database, and those which give the highest score (lowest pseudoenergy) are selected as candidate predictions; and, finally, the fragment-assembly or minithreading method developed by David Baker and colleagues (9, 10), in which the predicted structure is assembled from nine-residue fragments extracted from a protein-structure database, and knowledge- and physics-based filters are applied at each assembly stage. The last method has been used with outstanding success in predicting new protein folds. In many prediction protocols, such as, e.g., MONSSTERR (MOdeling of New Structures from Secondary and TErtiary Restrains) (11), ROSETTA (10, 12), TOUCHSTONE (13), TASSER (Threading ASSEmbly Refinement) (14), and I-TASSER (Iterative Threading ASSEmbly Refinement) (5), some or all of the methods are combined. Another variant of this approach, known as Fragfold, was also developed by Jones (15).
Because the sequences of newly discovered natural proteins are usually similar to those with known structures, comparative modeling covers over 90% of the situations in which a structure is needed and has not been determined (16). If two natural sequences have >20% sequence similarity, they almost certainly have similar structures (5). It should be noted, however, that sequence similarity of natural proteins also indicates that they are closely related evolutionarily and, consequently, their ancestors performed similar functions, which implies a similar structure; organisms, in which a mutation resulted in a major structural change of a vital protein, did not survive. In fact, among artificially mutated proteins, even high sequence similarity is weakly related to structural similarity. A good example are bacterial Ig-binding domains; even a single mutation in the loop regions of these proteins changes the structure from a three-helix bundle into a four-stranded β-sheet with an α-helix packed to it (17). Because sequence-structure correspondence is also, albeit less directly, used by the other two types of knowledge-based method, the above caveat also remains true for the threading and fragment-based approaches.
Even when natural proteins are considered and the structure of a new protein does occur in the database used by knowledge-based methods, situations are met in which these approaches point to a wrong fold. Examples are, e.g., targets T0063 and T0215 of the 3rd Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction (CASP3) and CASP6 experiments, respectively, whose cores are simple three α-helix bundle folds, for which threading found mirror-image folds. Conversely, correct folds were predicted by our physics-based approach based on the physics-based coarse-grained force field (United Residue, UNRES) (18, 19).
Physics-based methods for protein structure prediction, which are based on the Anfinsen thermodynamic hypothesis (1), have also had significant success. Enormous progress has been made in all-atom calculations, due to extensive use of world-distributed computing (20), implementation of all-atom molecular dynamics software on graphical processor units (21), and, most notably, the construction of dedicated machines (22). However, coarse-grained approaches (23–25), in which several atoms are merged into single interaction sites, are used very extensively, because they enable us to treat protein systems at time and dimensional scales, which are orders of magnitude larger (26) than those possible in all-atom computations. During the past 20 y, we have been developing (27–30) a simplified model for proteins termed UNRES, in which two interaction sites per residue are defined, namely, a united side chain and a united peptide group. The effective energy function has been defined as the potential of mean force of polypeptide chains in water. More details of the model are given in the references cited, and a succinct description is given in Materials and Methods.
It should be noted that protein-structure prediction is only one aspect of the application of physics-based methods; however, in our opinion, this exercise, is a necessary step to test physics-based approaches. As stated earlier in this section, these approaches will be needed practically in a fraction of protein structure prediction problems. Most likely, these methods will be needed to predict the structures of the proteins with no sequence similarity to any protein from structural databases or if an artificial protein is considered. Lastly, when there is confidence that the structure is well predicted, and will be used for further procedures, concurrent methods, including physics-based methods, are required. The main scope of implementation of physics-based methods is protein dynamics and thermodynamics, especially with regard to biological applications such as studying functionally important motions and predicting ligand-binding modes.
Physics-based prediction of protein structure was, until recently, understood as finding the global minimum of a potential-energy function, which does not fully result from Anfinsen’s thermodynamic hypothesis because it ignores conformational entropy. The native structure of a protein is not a single rigid structure such as that of, e.g., a polycyclic hydrocarbon molecule but constitutes a statistical ensemble of a very large number of similar but nonidentical conformations. Therefore, the native structure should be sought as the most probable ensemble or the basin in the potential-energy surface that has the lowest free energy. Consequently, we recently (28) developed a procedure based on extensive conformational search of the protein considered in the UNRES representation with the use of the replica-exchange molecular dynamics (REMD) method (20, 31) implemented in the UNRES force field (32, 33), followed by determination of families of structures by the weighted-histogram analysis method (WHAM) (34) and minimum-variance clustering (35). This approach is outlined in Materials and Methods. Recently (36), we parallelized the energy and force calculations in UNRES, which enables us, given access to massively parallel resources, to run MREMD simulations of proteins with sizes up to 1,200 residues in real time.
Our physics-based procedure resulted in some good predictions in the CASP7–CASP9 exercises (37). In particular, in CASP9, UNRES predictions of three targets were outstanding by global distance test total score (GDT_TS), albeit at only >5 Å root-mean-square deviation (rmsd) over α-carbon atoms. In this paper, we present the performance of our physics-based protein structure prediction method in the CASP10 exercise under the Cornell-Gdańsk group name. We demonstrate that use of a physics-based methodology can make a difference in predicting the correct global fold. It should be noted that, in this CASP exercise, UNRES was also used with success in connection with knowledge-based approaches, as in the WEFOLD experiment.
Results and Discussion
As the Cornell-Gdańsk group, we submitted predictions for 21 of 53 targets available to human predictor groups. As in previous CASP exercises (18, 19, 37–39), to use supercomputer resources available to us effectively, we considered primarily the targets that had below 20% sequence similarity with proteins from the Protein Data Bank (PDB), as assessed by the PSIBLAST server (40). The best predictions made with UNRES are described below.
Target T0663.
This protein (PDB ID code 4EXR) consists of two domains, each of which contains a four-stranded antiparallel β-sheet with an N-terminal α-helix packed across the strands. A distorted α-helical segment links the two domains (Fig. 1A). The N-terminal domain is rotated about the axis perpendicular to the β-sheet plane by 180° so that its N-terminal strand is loosely packed against the N-terminal strand of the C-terminal domain. It can be seen that our models 1 and 4 (Fig. 1 B and C) have the same packing topology, although the helices are packed along and not across the β-strands, as opposed to the experimental structure, and the β-sheets are not as curved and are more tightly packed to each other (Fig. 1 B and C). These two predictions were two of only three models submitted to CASP10 with correct domain-arrangement topology. This situation is quite unusual, because the sequence of this protein has high similarity to those of proteins with known structures and clearly falls into the category of comparative-modeling methods with high confidence; still, the structure predicted by comparative-modeling methods is grossly wrong. On the other hand, judging by GDT_TS, comparative-modeling methods seem to have given better results compared with UNRES (Fig. 1D). The reason for this is that the individual domains of this protein are predicted with excellent accuracy by comparative modeling, which accounts for most of the distances in the protein. Only a small fraction of distances correspond to domain packing and, therefore, GDT_TS does not select any model of T0663 predicted by UNRES as outstanding, even though the domains are correctly packed in the UNRES models. Only a combination of GDT_TS with the chirality score (defined as the fraction of tetrahedra of vertices in the Cα atoms of the structure under consideration, which have the same chirality as the corresponding tetrahedra of the reference structure) (41) enabled the assessors to distinguish the UNRES models. The chirality scores for our models 1 and 4 are 0.71 and 0.63, compared with 0.58 (model 5 of group 27), 0.52 (model 1 of group 190), and 0.52 (model 1 of group 388) of the three groups that scored best by means of the GDT_TS measure (see www.predictioncenter.org/casp10/results.cgi for the structure of the models and group information).
Fig. 1.

(A) The experimental 4EXR structure of target T0663. (B) Our model 1. (C) Our model 4. (D) GDT_TS plots of all models of T0663 from all groups with plots corresponding to the Cornell-Gdańsk group models shown as black lines, and models from the other groups shown as orange lines. The N termini in A–C are marked with “N”. The values of GDT_TS are 23.19, 31.98, and 42.80 for model 1 of the whole protein and its domains D1 and D2, respectively, and 22.04, 31.98, and 40.15 for model 4 of the whole protein and its domains D1 and D2, respectively. The respective GDT_TS values of the models with the highest GDT_TS submitted to CASP are 42.93 (model 1 from group 27), 68.61 (model 3 from group 27), and 98.20 (model 4 from group 27). The GDT_TS plots have been reproduced with permission from the CASP10 web site (www.predictioncenter.org/casp10/results.cgi). The drawings of the structures were produced with PYMOL (www.pymol.org).
The correctness of the domain-packing topology also persists in UNRES models 2, 3, and 5 (Fig. S1). Even though the β-sheet is distorted in models 2, 3, and 5, the β-sheet fragments of the N-terminal domain still tend to pack against the N-terminal fragment of the C-terminal domain. However, the chirality scores of these models are low because the α-helices are arranged on the wrong sides of the β-sheet.
Another target, T0644, which is highly homologous to T0663 with a very similar structure, except that the C-terminal part of the N-terminal domain is packed against the N-terminal part of the C-terminal domain, has also been released in CASP10. Our predictions for this target were not very good according to the GDT_TS measure (Fig. 2). It should be noted, however, that the domains are packed as in the experimental structure, i.e., the C terminus of the C-terminal domain is packed to the N terminus of the C-terminal domain (Fig. 2B). The overall handedness score is also remarkable (0.55 for model 2), even though the prediction quality is worse compared with that for T0663 because of swapped strands in the domains (Fig. 2B). For our other model of that target, the domain packing is also correct. It can therefore be concluded that predicting the correct packing topology of T0644 and T0663 is a feature of our UNRES-based approach. Our method did not pick out correctly and incorrectly packed structures with equal probability. It produced structures only with correct domain packing.
Fig. 2.

(A) The experimental 4FR9 structure of target T0644. (B) Our model 3 of this target. The GDT_TS value of this model is 17.38, compared with 85.28 for the best model submitted to CASP (model 3 from group 130). The drawings of the structures were produced with PYMOL (www.pymol.org).
To find out if our approach can rectify the incorrect packing topology, we took three top models according to the GDT_TS measure (model 1 of group 27, model 1 of group 190, and model 1 of group 388) and subjected them to restrained multiplexed REMD (MREMD) simulations with UNRES. In these simulations, restraints were imposed on the Cα … Cα distances within each domain, with reference distance values from the original models, whereas the interdomain distances and linker geometry were unrestricted. The restraints were harmonic, each with a force constant of 0.05 kcal/(mol × Å2). These simulations resulted in repacking the β-sheets to correct symmetry for all starting models. For model 1 of group 27, the chirality score increased from 0.56 to 0.64. For the models from groups 190 and 388, the chirality score did not improve because the helices were packed on the wrong side of the β-sheets. As an illustration, the initial and final structures of model 1 of group 27 are shown in Fig. 3.
Fig. 3.

(A) Model 1 of target T0663 from group 27. (B) Results of UNRES/MREMD simulations starting from this model, with restraints imposed on intradomain Cα…Cα distances. The drawings of the structures were produced with PYMOL (www.pymol.org).
Because the packing of the two domains (the C terminus of the N-terminal domain to the N terminus of the C-terminal domain or the N terminus of the N-terminal domain to the N terminus of the C-terminal domain) is the only feature that distinguishes the structures of T0644 and T0663, and bioinformatics approaches have unambiguously selected the models in which the C terminus of the N-terminal domain is packed to the N terminus of the C-terminal domain for both targets (42) (incorrectly for T0663), as opposed to our physics-based approach, we used the example of T0663 to determine what makes the difference between the two approaches. An analysis of the interactions in the experimental 4EXR structure indicates that the linker interacts with the bottom of the N-terminal β-sheet, which makes the β-sheet of the C-terminal domain pack to the N-terminal part of the β-sheet of the N-terminal domain. This pattern of interactions is preserved in the UNRES model 4; in model 1, which also has correct chirality, the N-terminal β-strand is unwound to join the linker and the extended linker is packed across the two domains (Fig. 1B). In the best models of T0663 according to the GDT_TS measure (Fig. 3A), the linker is not packed to the N-terminal domain, which enables the two domains to assume the more natural C-to-N terminus packing, which results in a lower contact order and, therefore, smaller loss of entropy. This is probably because the linker in the templates is shorter than that in T0663, which does not enable the N-terminal domain to turn around. Because of high sequence homology of T0663 to proteins similar in structure to T0644, the domain packing characteristic of T0644 was selected instead of the correct packing.
Other Predictions.
Our approach has also been featured for the free-modeling target T0740 (an α-helical protein). However, for this target, we obtained the best predictions by making use of extra information, as part of the wfCPUNK group within the WEFOLD initiative directed by Silvia Crivelli (National Institutes of Health).
As the Cornell-Gdańsk group, we also obtained very good results, in terms of the GDT_TS measure, for T0668 and T0684_D2 (the second domain of target T0684). The GDT_TS plots of these targets are shown in Fig. 4 A and B, whereas the experimental structure and the best model of T0684_D2 are shown in Fig. 4 C and D, respectively. The experimental structure of target T0668 has not yet been published in the PDB and, therefore, only the GDT_TS plots are shown for this target (Fig. 4A). T0668 was classified as a hard template-based modeling target, whereas T0684_D2 has been classified as a free-modeling target.
Fig. 4.

(A) The GDT_TS plots of T0668; the blue line on the right corresponds to our model 2 (GDT_TS = 35.90 compared with the highest value of 44.23 obtained by group 190). (B) The GDT_TS plots of T0684_D2; the rightmost blue line corresponds to our model 5 (GDT_TS = 21.58 compared with the highest value of 24.85 obtained by group 45). (C) The experimental structure of T0684_D2 (PDB code: 4FMT). (D) Our model 5 of this protein. The GDT_TS plots have been reproduced with permission from the CASP10 web site (www.predictioncenter.org/casp10/results.cgi). The drawings of the structures were produced with PYMOL (www.pymol.org).
It can be seen from the GDT_TS plots (Fig. 4 A and B) that the UNRES predictions attain high GDT_TS values only when the rmsd cutoff is greater than 5 Å. This suggests that our coarse-grained physics-based approach has only medium resolution. As a result of this medium resolution, our model 5 of T0684_D2 is ranked only 28th, even though it corresponds to the rightmost line in the GDT_TS plot (Fig. 4B), whereas our model 2 of T0668 is ranked only 83rd, even though the corresponding lines are quite shifted to the right (Fig. 4A).
Conclusions
Using our physics-based protocol with the coarse-grained UNRES force field as the main component, we obtained featured predictions of target T0663 in the CASP10 exercise. This success resulted from the fact that UNRES predicted correct domain packing which, in turn, resulted from packing of the interdomain linker to the N-terminal domain (Fig. 1C). It is remarkable that T0663 is largely homologous to known proteins. However, wrong local structure of the linker between the two large domains adopted from largely homologous proteins (in which the linker is shorter than in T0663) resulted in wrong domain packing. UNRES does not have this bias and, therefore, it predicted the correct overall topology, even though knowledge-based methods outperformed it as far as the accuracy of the prediction of each individual domain was concerned. That the result obtained for T0663 was not a fortuitous incident is demonstrated by other good results obtained during the CASP10 exercise (Fig. 4) and in previous CASP exercises (18, 19, 37). It can, therefore, be concluded that our physics-based coarse-grained approach has substantial power to predict protein structures. In particular, it has the ability to predict domain–domain orientations, which is a significant advance in the state of the art. Its main advantage is that it is free of the bias from structural databases.
On the other hand, at present, UNRES provides a resolution of 5 Å on average for a 60- to 80-residue protein or protein fragments with such a size, provided that its topology is predicted correctly (Fig. 4). Therefore, the predictions made with UNRES rise to the top mostly when other methods fail to predict correct topology, as for T0663 in CASP10. This feature of UNRES is also clearly seen in the GDT_TS plots in Fig. 4; the UNRES predictions are distinguished in the GDT_TS plots only at higher rmsd values. One way to correct this deficiency is to improve the force field; the main targets are local interactions, which are recently being enhanced by introducing the terms responsible for the coupling between the backbone-local and side-chain–local conformational states. The other improvement is the replacement of the side-chain–interaction potentials which, at present, are Gay-Berne potentials with spheroidal symmetry. Such a potential function ignores, e.g., the specificity of interactions involving the charged and polar side chains with physics-based potentials that are also being introduced (43, 44).
Finally, the results of the exercise in which the models from knowledge-based predictions were used as starting points for UNRES simulations open another avenue for using UNRES in protein-structure prediction: Segments of a protein with high homology could be predicted in such a manner and then restrained to the resulting conformation, whereas UNRES could work on correct packing of these segments. This route is also being explored in our laboratory.
Materials and Methods
The UNRES force field has been described in detail in refs. 27–30. Briefly, a polypeptide chain is represented by a sequence of α-carbon atoms with united side chains attached to them and peptide groups positioned halfway between two consecutive α-carbons. The effective energy function is defined as the free energy of the chain corresponding to a given coarse-grained conformation plus the surrounding solvent (this free energy is termed a restricted free energy or a potential of mean force) (27, 29). The molecular dynamics equations have been derived by means of the Lagrange formalism (45), with the virtual Cα…Cα and Cα…SC vectors as generalized variables.
The prediction protocol used in this work consisted of the following four steps. In the first step, MREMD (20, 31) simulations were carried out with the use of our coarse-grained UNRES force field (27–30), with which a molecular dynamics method (26, 45) and its multiplexed replica exchange extension (33) were implemented earlier. In the second step, the conformational ensembles obtained by coarse-grained MREMD simulations were analyzed by means of WHAM (34) to determine the heat-capacity profiles and conformational ensembles at any desired temperature, following the procedure described in our earlier work (28). For each system, the heat-capacity profile was analyzed and a temperature of  was selected to analyze conformational ensembles, where Tm is the temperature of the main heat-capacity peak (the “melting temperature”). In the third step, cluster analysis was carried out at the selected temperatures, by means of Ward’s minimum variance method (35). The Cα rmsd was used as a measure of the distance between conformations. For each protein, the rmsd cutoff was selected as a compromise between a small number of families (ideally five, which is the number of models that could be submitted for each target) and grouping similar conformations in a given family. The clusters are ranked according to decreasing probabilities, which are computed from the probabilities of their component conformations calculated by WHAM (28). For each cluster, the conformation closest to the average over the cluster is considered a representative of the whole cluster. The representatives of the five top clusters were selected as prediction candidates and ranked according to decreasing probabilities of the clusters. In step 4, these conformations were converted to all-atom structures by our physics-based method (46, 47). After conversion to the CASP format, the models were submitted.
was selected to analyze conformational ensembles, where Tm is the temperature of the main heat-capacity peak (the “melting temperature”). In the third step, cluster analysis was carried out at the selected temperatures, by means of Ward’s minimum variance method (35). The Cα rmsd was used as a measure of the distance between conformations. For each protein, the rmsd cutoff was selected as a compromise between a small number of families (ideally five, which is the number of models that could be submitted for each target) and grouping similar conformations in a given family. The clusters are ranked according to decreasing probabilities, which are computed from the probabilities of their component conformations calculated by WHAM (28). For each cluster, the conformation closest to the average over the cluster is considered a representative of the whole cluster. The representatives of the five top clusters were selected as prediction candidates and ranked according to decreasing probabilities of the clusters. In step 4, these conformations were converted to all-atom structures by our physics-based method (46, 47). After conversion to the CASP format, the models were submitted.
For each target, 64 trajectories were run for each MREMD simulation, with two trajectories per temperature. The integration time step was 4.89 fs and 20 million to 40 million steps per trajectory were run for each system. This corresponded to about 0.1–0.2 μs simulation time; however, because the fast degrees of freedom are averaged out in the coarse-grained treatment, this corresponds to 0.1–0.2 ms of real time (26). The Berendsen thermostat was used (48) with the coupling constant  . The adaptive multiple time step algorithm (49) was used to integrate the equations of motion. The simulations were run with the parallelized UNRES code (36) available at www.unres.pl. To speed up the search, restraints from secondary structure prediction by PSIPRED (50) were imposed on the virtual-bond geometry.
. The adaptive multiple time step algorithm (49) was used to integrate the equations of motion. The simulations were run with the parallelized UNRES code (36) available at www.unres.pl. To speed up the search, restraints from secondary structure prediction by PSIPRED (50) were imposed on the virtual-bond geometry.
Supplementary Material
Acknowledgments
We thank Dr. B. K. Lee and Dr. T. Taylor, Center of Cancer Research, National Institutes of Health, for supplying the program to compute the chirality score and helpful discussions. This work was supported by grants from the National Institutes of Health (GM-14312) and the National Science Foundation (MCB10-19767), by Grant MPD/2010/5 from the Foundation for Polish Science, and by Grants DEC-2011/01/N/ST4/01772 and DEC-2012/06/A/ST4/00376 from the National Science Center of Poland. This research was supported by an allocation of advanced computing resources provided by the National Science Foundation (www.nics.tennessee.edu/) and the National Science Foundation through TeraGrid resources provided by the Pittsburgh Supercomputing Center. Computational resources were also provided by the supercomputer resources at the Informatics Center of the Metropolitan Academic Network in Gdańsk, the 792-processor Beowulf cluster at the Baker Laboratory of Chemistry, Cornell University, and our 184-processor Beowulf cluster at the Faculty of Chemistry, University of Gdańsk.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1313316110/-/DCSupplemental.
References
- 1.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181(4096):223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Tramontano A. The Ten Most Wanted Solutions in Protein Bioinformatics. London: CRC; 2005. pp. 69–88. [Google Scholar]
- 3.Warme PK, Momany FA, Rumball SV, Tuttle RW, Scheraga HA. Computation of structures of homologous proteins. α-lactalbumin from lysozyme. Biochemistry. 1974;13(4):768–782. doi: 10.1021/bi00701a020. [DOI] [PubMed] [Google Scholar]
- 4.Jones TA, Thirup S. Using known substructures in protein model building and crystallography. EMBO J. 1986;5(4):819–822. doi: 10.1002/j.1460-2075.1986.tb04287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y. Progress and challenges in protein structure prediction. Curr Opin Struct Biol. 2008;18(3):342–348. doi: 10.1016/j.sbi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones D, Thornton J. Protein fold recognition. J Comput Aided Mol Des. 1993;7(4):439–456. doi: 10.1007/BF02337560. [DOI] [PubMed] [Google Scholar]
- 7.Miller RT, Jones DT, Thornton JM. Protein fold recognition by sequence threading: Tools and assessment techniques. FASEB J. 1996;10(1):171–178. doi: 10.1096/fasebj.10.1.8566539. [DOI] [PubMed] [Google Scholar]
- 8.Mirny LA, Shakhnovich EI. Protein structure prediction by threading. Why it works and why it does not. J Mol Biol. 1998;283(2):507–526. doi: 10.1006/jmbi.1998.2092. [DOI] [PubMed] [Google Scholar]
- 9.Bonneau R, et al. Rosetta in CASP4: Progress in ab initio protein structure prediction. Proteins. 2001;(Suppl 5):119–126. doi: 10.1002/prot.1170. [DOI] [PubMed] [Google Scholar]
- 10.Rohl CA, Strauss CEM, Misura KMS, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 11.Skolnick J, Kolinski A, Ortiz AR. MONSSTER: A method for folding globular proteins with a small number of distance restraints. J Mol Biol. 1997;265(2):217–241. doi: 10.1006/jmbi.1996.0720. [DOI] [PubMed] [Google Scholar]
- 12.Misura KM, Chivian D, Rohl CA, Kim DE, Baker D. Physically realistic homology models built with ROSETTA can be more accurate than their templates. Proc Natl Acad Sci USA. 2006;103(14):5361–5366. doi: 10.1073/pnas.0509355103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Kolinski A, Skolnick J. TOUCHSTONE II: A new approach to ab initio protein structure prediction. Biophys J. 2003;85(2):1145–1164. doi: 10.1016/S0006-3495(03)74551-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Skolnick J. The protein structure prediction problem could be solved using the current PDB library. Proc Natl Acad Sci USA. 2005;102(4):1029–1034. doi: 10.1073/pnas.0407152101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones DT. Predicting novel protein folds by using FRAGFOLD. Proteins. 2001;(Suppl 5):127–132. doi: 10.1002/prot.1171. [DOI] [PubMed] [Google Scholar]
- 16. Grishin NV (2012) Predictive landscape of casp. 10th Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction. Available at www.predictioncenter.org/casp10/docs.cgi?view=presentations. Accessed August 6, 2013.
- 17.He YA, Chen YH, Alexander PA, Bryan PN, Orban J. Mutational tipping points for switching protein folds and functions. Structure. 2012;20(2):283–291. doi: 10.1016/j.str.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liwo A, Lee J, Ripoll DR, Pillardy J, Scheraga HA. Protein structure prediction by global optimization of a potential energy function. Proc Natl Acad Sci USA. 1999;96(10):5482–5485. doi: 10.1073/pnas.96.10.5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ołdziej S, et al. Physics-based protein-structure prediction using a hierarchical protocol based on the UNRES force field: Assessment in two blind tests. Proc Natl Acad Sci USA. 2005;102(21):7547–7552. doi: 10.1073/pnas.0502655102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pande VS, et al. Atomistic protein folding simulations on the submillisecond time scale using worldwide distributed computing. Biopolymers. 2003;68(1):91–109. doi: 10.1002/bip.10219. [DOI] [PubMed] [Google Scholar]
- 21.Friedrichs MS, et al. Accelerating molecular dynamic simulation on graphics processing units. J Comput Chem. 2009;30(6):864–872. doi: 10.1002/jcc.21209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw DE, et al. Anton, a special-purpose machine for molecular dynamics simulation. Commun ACM. 2008;51:91–97. [Google Scholar]
- 23.Kolinski A, Gront D, Pokarowski P, Skolnick J. A simple lattice model that exhibits a protein-like cooperative all-or-none folding transition. Biopolymers. 2003;69(3):399–405. doi: 10.1002/bip.10385. [DOI] [PubMed] [Google Scholar]
- 24.Tozzini V. Coarse-grained models for proteins. Curr Opin Struct Biol. 2005;15(2):144–150. doi: 10.1016/j.sbi.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 25.Czaplewski C, Liwo A, Makowski M, Ołdziej S, Scheraga HA. In: Multiscale Approaches to Protein Modeling. Koliński A, editor. Berlin: Springer; 2010. pp. 35–83. [Google Scholar]
- 26.Khalili M, Liwo A, Jagielska A, Scheraga HA. Molecular dynamics with the united-residue model of polypeptide chains. II. Langevin and Berendsen-bath dynamics and tests on model α-helical systems. J Phys Chem B. 2005;109(28):13798–13810. doi: 10.1021/jp058007w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liwo A, Czaplewski C, Pillardy J, Scheraga HA. Cumulant-based expressions for the multibody terms for the correlation between local and electrostatic interactions in the united-residue force field. J Chem Phys. 2001;115:2323–2347. [Google Scholar]
- 28.Liwo A, et al. Modification and optimization of the united-residue (UNRES) potential energy function for canonical simulations. I. Temperature dependence of the effective energy function and tests of the optimization method with single training proteins. J Phys Chem B. 2007;111(1):260–285. doi: 10.1021/jp065380a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liwo A, et al. (2008) Coarse-Graining of Condensed Phase and Biomolecular Systems, ed Voth GA (CRC, Boca Raton, FL) pp 107–122.
- 30.Kozłowska U, Maisuradze GG, Liwo A, Scheraga HA. Determination of side-chain-rotamer and side-chain and backbone virtual-bond-stretching potentials of mean force from AM1 energy surfaces of terminally-blocked amino-acid residues, for coarse-grained simulations of protein structure and folding. II. Results, comparison with statistical potentials, and implementation in the UNRES force field. J Comput Chem. 2010;31(6):1154–1167. doi: 10.1002/jcc.21402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hansmann UHE. Parallel tempering algorithm for conformational studies of biological molecules. Chem Phys Lett. 1997;281:140–150. [Google Scholar]
- 32.Nanias M, Czaplewski C, Scheraga HA. Replica exchange and multicanonical algorithms with the coarse-grained UNRES force field. J Chem Theory Comput. 2006;2(3):513–528. doi: 10.1021/ct050253o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Czaplewski C, Kalinowski S, Liwo A, Scheraga HA (2009) Application of multiplexing replica exchange molecular dynamics method to the UNRES force field: Tests with alpha and alpha+beta proteins. J Chem Theory Comput 5:627–640. [DOI] [PMC free article] [PubMed]
- 34.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comput Chem. 1992;13:1011–1021. [Google Scholar]
- 35.Späth H. Cluster Analysis Algorithms. New York: Halsted Press; 1980. [Google Scholar]
- 36.Liwo A, et al. Implementation of molecular dynamics and its extensions with the coarse-grained UNRES force field on massively parallel systems; towards millisecond-scale simulations of protein structure, dynamics, and thermodynamics. J Chem Theory Comput. 2010;6(3):890–909. doi: 10.1021/ct9004068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liwo A, He Y, Scheraga HA. Coarse-grained force field: General folding theory. Phys Chem Chem Phys. 2011;13(38):16890–16901. doi: 10.1039/c1cp20752k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee J, et al. Hierarchical energy-based approach to protein-structure prediction; blind-test evaluation with casp3 targets. Int J Quantum Chem. 2000;77:90–117. [Google Scholar]
- 39.Pillardy J, et al. Recent improvements in prediction of protein structure by global optimization of a potential energy function. Proc Natl Acad Sci USA. 2001;98(5):2329–2333. doi: 10.1073/pnas.041609598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Altschul SF, et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor TJ, Bai H, Tai CH, Lee BK. Score functions to evaluate casp10 predictions for free-modeling targets. Prot Struct Funct Bioinf. 2013;81(Suppl 11) [Google Scholar]
- 42. Lee BK (2012) Template free modeling assessment in casp10. 10th Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction. Available at www.predictioncenter.org/casp10/docs.cgi?view=presentations. Accessed August 6, 2013.
- 43.Makowski M, et al. Simple physics-based analytical formulas for the potentials of mean force for the interaction of amino acid side chains in water. IV. Pairs of different hydrophobic side chains. J Phys Chem B. 2008;112(36):11385–11395. doi: 10.1021/jp803896b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Makowski M, Liwo A, Scheraga HA. Simple physics-based analytical formulas for the potentials of mean force of the interaction of amino-acid side chains in water. VI. Oppositely charged side chains. J Phys Chem B. 2011;115(19):6130–6137. doi: 10.1021/jp111259e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khalili M, Liwo A, Rakowski F, Grochowski P, Scheraga HA. Molecular dynamics with the united-residue model of polypeptide chains. I. Lagrange equations of motion and tests of numerical stability in the microcanonical mode. J Phys Chem B. 2005;109(28):13785–13797. doi: 10.1021/jp058008o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaźmierkiewicz R, Liwo A, Scheraga HA. Energy-based reconstruction of a protein backbone from its α-carbon trace by a Monte-Carlo method. J Comput Chem. 2002;23(7):715–723. doi: 10.1002/jcc.10068. [DOI] [PubMed] [Google Scholar]
- 47.Kaźmierkiewicz R, Liwo A, Scheraga HA. Addition of side chains to a known backbone with defined side-chain centroids. Biophys Chem 100(1–3):261–280, and erratum. (2003) 2003;106(1):91. doi: 10.1016/s0301-4622(02)00285-5. [DOI] [PubMed] [Google Scholar]
- 48.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 49.Rakowski F, Grochowski P, Lesyng B, Liwo A, Scheraga HA. Implementation of a symplectic multiple-time-step molecular dynamics algorithm, based on the united-residue mesoscopic potential energy function. J Chem Phys. 2006;125(20):204107. doi: 10.1063/1.2399526. [DOI] [PubMed] [Google Scholar]
- 50.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16(4):404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.