

Significance
A considerable fraction of proteins, although functional, does not display a well-ordered native state, posing the structure–function dogma into question. Although different models have been described, the role of protein disorder is still shrouded in mystery. Here, we investigate the mechanism of recognition between an intrinsically unstructured protein and its partner. Unexpectedly, we found the binding to occur with a very high degree of geometrical precision, suggesting that a potential value of disorder is not, in this case, to be searched in the speeding up of the reaction thanks to an increased capture radius. Given that in the cell disordered polypeptides may be more vulnerable than folded domains, the advantage in being natively unfolded remains a conundrum.
Keywords: kinetics, mutagenesis, protein folding
Abstract
A classical dogma of molecular biology dictates that the 3D structure of a protein is necessary for its function. However, a considerable fraction of the human proteome, although functional, does not adopt a defined folded state under physiological conditions. These intrinsically disordered proteins tend to fold upon binding to their partners with a molecular mechanism that is elusive to experimental characterization. Indeed, although many hypotheses have been put forward, the functional role (if any) of disorder in these intrinsically denatured systems is still shrouded in mystery. Here, we characterize the structure of the transition state of the binding-induced folding in the reaction between the KIX domain of the CREB-binding protein and the transactivation domain of c-Myb. The analysis, based on the characterization of a series of conservative site-directed mutants, reveals a very high content of native-like structure in the transition state and indicates that the recognition between KIX and c-Myb is geometrically precise. The implications of our results in the light of previous work on intrinsically unstructured systems are discussed.
In the fourth century B.C., Aristotle claimed: “A thing is more properly said to be when it exists in actuality than when it exists potentially.” For example, a log of wood is potentially many objects but becomes a table only once given its proper shape by an experienced carpenter. Because the form is what sculpts a thing to be what it is, the nature of things lies in their form. Consequently, one of the most intuitive concepts that we develop from childhood is that shape determines function. In the context of the intricate environment of the cell, the specificity that characterizes the interactions between different proteins was generally explained by invoking a precise geometrical complimentarity between the interacting partners. Thus, the recent proposal that up to 40% of the human proteome appears to be essentially disordered highlights a conundrum (1). Is there a potential value for a protein to be disordered? Given that a disordered protein is more vulnerable, is there a functional advantage in being disordered?
A problem of interest is how to reconcile the accepted structure–function dogma with the existence of disorder (2, 3). Because intrinsically disordered proteins (IDPs) exist but are very elusive to characterization, relevant experimental work is still relatively scarce and such hypotheses remain mostly untested. From a thermodynamic perspective, it has been suggested that destabilization of the native protein structure, leading to its unfolding, lowers the affinity of a protein for its ligand, without necessarily abolishing specificity (4–6). Furthermore, the highly dynamic nature of IDPs was suggested to promote alternative binding of the same disordered segments to different partners, the so-called “moonlighting effect” (7, 8), increasing the repertoire of activities. An interesting mechanistic model, proposed by Wolynes and coworkers (9), suggests that the IDPs display an increased capture radius to recruit and bind partners. According to this hypothesis, commonly referred to as the fly-casting mechanism, a disordered protein should form with its physiological partner a high-energy complex that would be locked in place by the coupled folding reaction. An important corollary of the fly-casting mechanism is that a potential advantage for a protein to be disordered is to increase the probability (and speed) to bind the interacting partner, due to its extended conformation. Importantly, the fly-casting was shown to become more effective when the unfolding free-energy barriers of proteins are relatively small (10), which implies an anticorrelation between the binding rate constant and protein stability.
The CREB-binding protein (CBP) is a coactivator that modulates the interaction between DNA-bound activator proteins and the components of the basal transcription complex. A globular domain of CBP, namely the KIX domain, is the principal mediator of such interactions (11). Despite its small size, 87 amino acids, and a relatively simple fold, the KIX domain binds different IDP systems via two distinct, but energetically connected, binding sites, called “c-Myb” and “MLL” sites (named after two characteristic ligands of each site, i.e., the transactivation domain of the protein c-Myb and the mixed lineage leukemia, MLL, protein) (12). We have recently characterized the recognition mechanism by which the KIX domain binds to the transativation domain of c-Myb (13), an IDP that acquires a helical structure when bound to its partner (14). A complete analysis of temperature jump and stopped-flow kinetics in the presence and in the absence of 1,1,1-trifluoro-ethanol, which stabilizes the folded state of c-Myb, revealed that this IDP system recognizes KIX by following a folding-after-binding scenario, whereby it binds in a relatively unstructured conformation, with the locking of the hydrogen bonds of the helical structure occurring only downhill the primary rate-limiting step.
In this work, we present a detailed description of the structural features of the transition state for the binding-induced folding reaction of c-Myb to KIX. The transition state structure was unveiled using the Φ-value analysis (15, 16). By this technique, residue-specific structural information is inferred by comparing the kinetics of the reaction (folding and/or binding) of the wild-type protein with a series of conservative single mutants, yielding the so-called Φ-value that represents an index of native-like structure of the mutated residue in the transition state. Our results reveal that the transition state of c-Myb is well ordered, with an average Φ-value of 0.89. This is consistently higher than what is generally observed for the folding of single-domain proteins (typically displaying an average Φ-value of 0.36), and indicates that the interaction between KIX and c-Myb is characterized by a considerable geometrical fit.
Results and Discussion
Experimental Design and Strategy of Mutagenesis.
Because of their intrinsic nature, transition states never accumulate; thus, their structure must be inferred indirectly. By systematically mutating side chains while probing the effect of the perturbation on the activation- and ground states’ free energies, it is possible to map interaction pattern(s) in metastable states and to unveil their structure (15, 16). In practice, the analysis is performed by measuring the so-called Φ-value, which is formalized as
 |
Thus, the Φ-value is an index that directly reports on the extent of native-like structure in the transition state, approaching 1 and 0 for native- and denatured-like structures, respectively. As detailed below, we applied this methodology to the IDP system c-Myb in its interaction with KIX.
The aim of the mutational analysis is therefore to obtain structural information on c-Myb in the transition state. With the exception of position 21, which is a glycine residue, all positions of c-Myb were mutated. The substitution of an alanine for a glycine typically stabilizes a helix by 0.4–2 kcal mol−1 (17) by means of a complex mechanism, possibly implying multiple factors including differences in backbone conformational entropy in the denatured state, burial of hydrophobic surfaces on folding, and disruption of hydrogen bonding between the protein and the solvent (18). Thus, Ala–Gly scanning at the solvent exposed sites within the helices is an accepted method to monitor the formation of secondary structure in protein folding as exemplified in ref. 19. Therefore, positions 3, 7, 10, 14, 17, 18, and 22 were mutated to both Ala and Gly. In total, 31 mutants were produced, expressed, and characterized (listed in Table 1). Two of the mutants (L8A and L12A) abolished the binding to KIX and were thus excluded from the kinetic analysis. The remaining mutants were subjected to binding-induced folding experiments and, as described below, 20 reliable Φ-values could be calculated.
Table 1.
Binding parameters to KIX for c-Myb* and its site-directed mutants
| Protein | kon,a μM−1 s−1 | koff,b s−1 | ΔΔG#, kcal mol−1 | ΔΔGeq, kcal mol−1 | Φ |
| Wt | 3.11 ± 0.23 | 24 ± 1 | |||
| K1A | 1.03 ± 0.11 | 195 ± 19 | 0.65 ± 0.07 | 1.88 ± 0.06 | 0.35 ± 0.04 |
| E2A | 2.73 ± 0.89 | 32 ± 1 | 0.08 ± 0.19 | 0.24 ± 0.03 | ––– c |
| K3A | 1.06 ± 0.39 | 37 ± 4 | 0.64 ± 0.21 | 0.89 ± 0.06 | 0.71 ± 0.14 |
| R4A | 0.93 ± 0.10 | 45 ± 6 | 0.71 ± 0.07 | 1.08 ± 0.08 | 0.66 ± 0.08 |
| I5A | 3.01 ± 0.36 | 42 ± 2 | 0.02 ± 0.08 | 0.34 ± 0.04 | ––c |
| K6A | 1.32 ± 0.16 | 50 ± 4 | 0.50 ± 0.08 | 0.93 ± 0.05 | 0.54 ± 0.09 |
| E7A | 1.45 ± 0.11 | 91 ± 12 | 0.45 ± 0.06 | 1.23 ± 0.08 | 0.37 ± 0.05 |
| L8Ad | |||||
| E9A | 2.24 ± 0.10 | 42 ± 3 | 0.19 ± 0.05 | 0.51 ± 0.05 | 0.37 ± 0.10 |
| L10A | 6.28 ± 0.88 | 72 ± 6 | −0.42 ± 0.09 | 0.23 ± 0.05 | ––c |
| L11A | 3.34 ± 0.19 | 59 ± 2 | −0.04 ± 0.05 | 0.48 ± 0.03 | −0.09 ± 0.12 |
| L12Ad | |||||
| M13A | 2.05 ± 0.18 | 72 ± 4 | 0.25 ± 0.06 | 0.89 ± 0.04 | 0.28 ± 0.07 |
| S14A | 0.47 ± 0.06 | 30 ± 1 | 1.11 ± 0.08 | 1.24 ± 0.03 | 0.89 ± 0.07 |
| T15S | 4.98 ± 0.62 | 39 ± 4 | −0.28 ± 0.08 | 0.00 ± 0.06 | ––c |
| E16A | 4.81 ± 1.06 | 74 ± 6 | −0.26 ± 0.13 | 0.40 ± 0.05 | ––c |
| N17A | 0.79 ± 0.05 | 39 ± 1 | 0.81 ± 0.05 | 1.10 ± 0.03 | 0.74 ± 0.05 |
| E18A | 3.26 ± 0.16 | 94 ± 7 | −0.03 ± 0.05 | 0.77 ± 0.05 | −0.04 ± 0.07 |
| L19A | 3.49 ± 0.5 | 94 ± 6 | −0.07 ± 0.09 | 0.73 ± 0.04 | −0.09 ± 0.12 |
| K20A | 0.85 ± 0.12 | 37 ± 3 | 0.76 ± 0.09 | 1.00 ± 0.05 | 0.76 ± 0.10 |
| Q22A | 1.72 ± 0.21 | 41 ± 2 | 0.35 ± 0.08 | 0.65 ± 0.04 | 0.53 ± 0.12 |
| Q23A | 3.73 ± 0.06 | 45 ± 4 | −0.11 ± 0.04 | 0.26 ± 0.03 | ––c |
| A24G | 5.86 ± 0.04 | 36 ± 2 | −0.37 ± 0.04 | −0.14 ± 0.04 | ––c |
| L25A | 0.98 ± 0.14 | 68 ± 5 | 0.68 ± 0.09 | 1.29 ± 0.05 | 0.53 ± 0.07 |
| K3Ge | 1.74 ± 0.18 | 117 ± 7 | −0.30 ± 0.07 | 0.38 ± 0.04 | ––c |
| E7Ge | 0.48 ± 0.06 | 227 ± 10 | 0.65 ± 0.08 | 1.19 ± 0.03 | 0.55 ± 0.07 |
| L10Ge | 0.28 ± 0.02 | 99 ± 5 | 1.83 ± 0.06 | 2.02 ± 0.04 | 0.91 ± 0.03 |
| S14Ge | 3.51 ± 0.4 | 67 ± 7 | −1.18 ± 0.08 | −0.71 ± 0.06 | 1.66 ± 0.18 |
| N17Ge | 6.24 ± 0.79 | 69 ± 4 | −1.22 ± 0.08 | −0.89 ± 0.04 | 1.37 ± 0.11 |
| E18Ge | 1.04 ± 0.16 | 132 ± 15 | 0.67 ± 0.10 | 0.88 ± 0.07 | 0.77 ± 0.12 |
| Q22Ge | 2.98 ± 0.37 | 51 ± 2 | −0.32 ± 0.08 | −0.19 ± 0.03 | ––c |
Calculated from the slope of pseudo–first-order binding experiments.
Measured from displacement experiments, as described in the text.
These mutants display binding affinities to pwtKIX similar to c-Myb* (ΔΔGeq <0.4 kcal mol−1), which prevents accurate calculation of Φ-values.
These mutations abolished binding with pwtKIX.
Ala-Gly scanning mutants. Mutants reporting on secondary structure of c-Myb.
Binding Kinetics of Wild-Type c-Myb and Its Site-Directed Mutants.
The binding of c-Myb to KIX is, in theory, a complex reaction involving at least two steps, the folding of c-Myb and a recognition event (14). In a previous study we showed that, despite such a complexity, the observed kinetics appears to follow a two-state mechanism without the accumulation of intermediates (13). An analysis of the experiments obtained in the presence and in the absence of 1,1,1-trifluoro-ethanol, which stabilizes the folded state of c-Myb, revealed complex formation to take place via a folding-after-binding scenario. Accordingly, c-Myb recognizes KIX in a relatively unstructured conformation and the locking of the hydrogen bonds of the main chain occurs only downhill the rate-limiting step. In an effort to provide a structural interpretation of the reaction, we resorted to characterizing the transition state by Φ-value analysis. In particular, wild-type c-Myb and its site directed mutants were analyzed by independent experiments aimed at the measurement of the association and dissociation rate constants.
We have previously shown that an improved signal-to-noise ratio of the fluorescence change associated with binding can be achieved by using an engineered construct containing a Trp in position 72 of the KIX domain, leading to the variant pwtKIX, and a fusion tag protein at the N-terminal region of c-Myb, namely the construct c-Myb*. Independent control experiments indicated that both the presence of Trp in KIX and of the tag in c-Myb had little effect on the observed binding mechanism and dissociation constant; thus these constructs have been used for the Φ-value analysis. We performed pseudo–first-order binding experiments using c-Myb* and the different site-directed mutants by mixing a constant concentration of pwtKIX (5–10 μM) with increasing concentrations of c-Myb*, typically ranging from 20 to 100 μM. Under all conditions, observed kinetics was consistent with a single-exponential behavior, suggesting the reaction follows a two-state mechanism, without the accumulation of intermediates. The observed rate constant for complex formation between KIX and c-Myb* and its mutants as a function of concentrations (Fig. 1) follows bimolecular kinetics. Given the pseudo–first-order approximation, the association rate constants were obtained from the slope of the dependence of the observed rate constant on [c-Myb]. The calculated values are reported in Table 1.
Fig. 1.

Pseudo–first-order kinetics of the binding between pwtKIX and c-Myb* at pH 7.2 and 10 °C. Data were recorded at a constant concentration of pwtKIX, typically between 5 and 10 μM, mixed with variable concentrations of c-Myb* and its site-directed mutants. The linear concentration dependence for wild-type c-Myb* is shown for comparison throughout, depicted in gray dots and broken lines. Observed time courses were consistent with single-exponential behavior in all cases.
Analysis of the data in Fig. 1 allows one, in theory, to calculate the dissociation rate constant by extrapolating the observed rate constants to zero concentration. Very often, however, the experimental error arising from this extrapolation is too high; therefore, accurate determination of dissociation rate constants may demand a different approach. In analogy to classical experiments on myoglobin (20), a powerful method to measure the dissociation rate is to carry out displacement kinetic experiments where a preincubated complex between two partners is challenged with an excess of a competing reactant. In this work, we performed displacement kinetic experiments where a complex between pwtKIX and c-Myb* was rapidly mixed with an excess of wild-type KIX (i.e., the wild-type domain with a Tyr at position 72). The dissociation time course was then measured at different relative concentrations of wild-type KIX, ranging from 2- to 10-fold, and the observed rate constants were found to be insensitive to the concentration for all mutants. The measured dissociation rate constants for wild-type c-Myb and its site-directed mutants are also listed in Table 1.
Φ-Value Analysis and the Structure of the Folding Transition State.
Following Fersht et al. (15), Φ-values were calculated by dividing the effect of mutation on the activation free energy by the change in free energy of the ground state (as formalized in Eq. 1). For each mutant, the changes in free energies were calculated by applying the following equations:
and
Because of experimental uncertainties (21), Φ-values arising from mutations whose 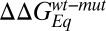 was lower than 0.4 kcal mol−1 were excluded from the analysis. Note that, because of the observed two-state nature of the reaction, it is very difficult to unambiguously assign the rate-limiting step, probed by the Φ-values, to either folding or binding; thus, in theory, the calculated values of Φ may reflect the two different steps involved in the overall process. However, we previously observed that formation of the complex between KIX and c-Myb proceeds via a folding-after-binding mechanism, with transient formation of a high-energy intermediate (13). Under such conditions, folding may either be very rapid and occur downhill the rate-determining barrier, or account for the higher free-energy barrier limiting the overall rate. Whereas in the former situation, with binding being rate limiting, the protein would have to appear rather unstructured in the transition state (as, by definition, folding would only occur after the rate-limiting step), in the latter case we expect to observe rather high values of Φ. Because, as detailed below, the majority of measured Φ values were high, we conclude that folding represents the rate-determining step.
was lower than 0.4 kcal mol−1 were excluded from the analysis. Note that, because of the observed two-state nature of the reaction, it is very difficult to unambiguously assign the rate-limiting step, probed by the Φ-values, to either folding or binding; thus, in theory, the calculated values of Φ may reflect the two different steps involved in the overall process. However, we previously observed that formation of the complex between KIX and c-Myb proceeds via a folding-after-binding mechanism, with transient formation of a high-energy intermediate (13). Under such conditions, folding may either be very rapid and occur downhill the rate-determining barrier, or account for the higher free-energy barrier limiting the overall rate. Whereas in the former situation, with binding being rate limiting, the protein would have to appear rather unstructured in the transition state (as, by definition, folding would only occur after the rate-limiting step), in the latter case we expect to observe rather high values of Φ. Because, as detailed below, the majority of measured Φ values were high, we conclude that folding represents the rate-determining step.
A color-coded representation of secondary and tertiary Φ-values mapped on the 3D structure of the complex between c-Myb and KIX is reported in Fig. 2. In analogy to previous work on folding of single-domain proteins (21), we grouped the Φ-values in three groups (weak, 0 < Φ < 0.3, represented in red; medium, 0.3 < Φ < 0.7, represented in magenta; high, 0.7 < Φ < 1, represented in blue). Inspection of Fig. 2 suggests the transition state to be highly ordered with two native-like folding nuclei displaying medium or high Φ-values located at the N- and C-terminal ends of the helix. Indeed, the region with lower values of Φ was found at the center of the helix, as mirrored by the mutants L11A, M13A, E18A, and L19A. Importantly, some of the residues displaying high Φ-phi values (i.e., K3, S14, and K20) do not make direct contact with KIX in the complex, providing additional support to our proposal that the Φ-values in this case are dominated by the intra- rather than intermolecular contacts and confirming the reaction to be rate limited by the folding of c-Myb rather than by recognition of KIX. The mutants S14A and N17A, located in the same region, display high Φ-values. Interestingly, however, the two secondary mutations, Ala to Gly substitutions in the same positions, returned a Φ > 1, which is classically interpreted as a signature of misfolding in the transition state (22, 23). These observations suggest that, although the side chains of S14 and N17 form some native-like contacts with pwtKIX in the transition state, their main chains are in a partly misfolded conformation and the helix is somewhat distorted in its central part, with native-like structure consolidated at the N- and C termini.
Fig. 2.
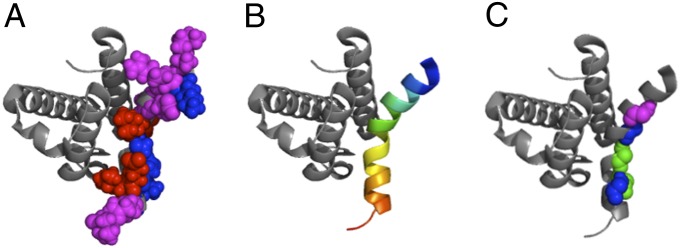
Φ-value analysis on the binding between pwtKIX and c-Myb*. Measured Φ-values are mapped on the 3D structure of the complex, pdb 1SB0 (14). Following Fersht and Sato (21), weak, 0 < Φ < 0.3 are represented in red; medium, 0.3 < Φ < 0.7 are represented in magenta; high, 0.7 < Φ < 1 are represented in blue. Φ-values > 1, reporting misfolding events (22, 23), are indicated in green. A and C refer to tertiary and secondary Φ-values, respectively (as defined in Table 1), whereas in B the structure of bound c-Myb is highlighted in rainbow coloring.
An Ordered Transition State as a Signature of Geometrical Precision.
Linear free-energy relationship (LFER) relates the activation free energy for a reaction with its equilibrium free energy. LFERs were originally introduced to assess the position of the transition state along the reaction coordinate during the formation of a covalent bond (24); in particular, by altering the structure and thus the reactivity of a substrate, the dependence of activation free energy on ground-state free energy generally yields a linear profile. The slope of the observed correlation, classically denoted as α, reflects the position of the transition state along the reaction coordinate. Surprisingly, LFER plots not only apply to simple organic reactions but also to complex reactions stabilized by many noncovalent interactions, and have been then used in enzymology (25), binding reactions involving allosteric control (26, 27), and protein folding (28).
In the case of protein folding, it has been suggested that the linearity of the LFER plot is distinctive of the so-called nucleation–condensation mechanism, whereby the transition state resembles a distorted version of the native state and the whole protein collapses around a weakly formed nucleus (29). Comparative analysis of nearly all Φ-values reported to date revealed that the α-value in protein folding is robust and, for nearly all proteins investigated, α is about 0.36 (30). The mutational work reported in Table 1 is an opportunity to apply the LFER to the recognition reaction between an IDP and its physiological partner (Fig. 3). In agreement with the behavior recalled above for protein folding, also the folding-after-binding reaction of c-Myb* to KIX displays a linear LFER. Unexpectedly, however, the calculated α-value is 0.89, which suggests the transition state to display a very high degree of native-like structure. Therefore, the rate-limiting transition state does not display an increased capture radius of c-Myb*, and it appears to be more ordered than what is typically observed for the folding reaction of globular proteins. These observations allow us to conclude that, despite being an IDP, c-Myb is characterized in its interaction with pwtKIX by a high degree of geometrical precision, and the protein appears by-and-large folded in the transition state. This is in stark contrast with what was recently observed on another IDP system by Jemth and coworkers, who observed an α of about 0.24 for the binding-induced folding of the two proteins NCBD and ACTR (31).
Fig. 3.

Leffler plot for the binding between pwtKIX and c-Myb*. Each individual point refers to a site directed mutant, as reported in Table1. The line is the best fit to a linear function, returning an α-value of 0.89.
Conclusions
We provided a complete structural characterization of the transition state for the folding-after-binding reaction of c-Myb* to KIX. In analogy to classical folding of globular proteins, the reaction occurs in a highly cooperative manner, without accumulation of partly folded intermediates and with a transition state that resembles a slightly distorted version of the native state (29). We observe folding to be triggered by the formation of two nuclei located at the N- and C termini of c-Myb*, with some minor misfolding defects at the center of the helix.
Previous experimental studies have succeeded in describing molecular interactions by means of the fly-casting scenario (32). In particular, in the case of protein-DNA recognition, it has been proposed that the high conformational flexibility of the transition state speeds up a fast direct readout of the DNA sequence that occurs via a monodimensional scan after an initial relatively aspecific complex formation (33). Alternatively, an anticorrelation between binding rate constants and protein stability might be observed. Conversely, we observed that the recognition between the KIX and c-Myb is characterized by a high geometrical precision, as mirrored by the very high content of native-like structure in the transition state, which exceeds what is generally observed in the folding reactions of globular proteins. Furthermore, the binding rate constants are not correlated with the folding stability of c-Myb (Fig. S1). On the basis of these findings, and by considering our earlier observation that the association rate constant is insensitive to the stability of c-Myb (13), we suggest that in this case disorder by itself does not accelerate the recognition events between the IDP and its partner. Given that in the cell disordered polypeptides are generally more vulnerable than folded domains, the selective advantage in being natively unfolded remains an interesting conundrum for the future.
Materials and Methods
Site-Directed Mutagenesis and Protein Expression and Purification.
The site-directed mutant Y72W named “pwtKIX” was obtained by using the QuikChange mutagenesis kit (Stratagene) according to the manufacturer’s instructions. The mutation was confirmed by DNA sequencing. wtKIX and pwtKIX were purified by using a nickel(II)-charged chelating Sepharose FF (Amersham Biosciences) column equilibrated with 40 mM Tris-HCl and 500 mM NaCl, pH 8.5. The His-tagged wtKIX and pwtKIX were eluted with 250 mM imidazole. The samples were then diluted fourfold in 40 mM Tris, pH 8.5 and all minor impurities were removed by the purification step on a Q-Sepharose column equilibrated with 40 mM Tris, pH 8.5. The proteins passed through the Q column; the flow-through containing the protein was collected and concentrated. The purity of the protein was confirmed by SDS/PAGE.
The engineered construct indicated as c-Myb* where c-Myb was fused to the prodomain of subtilisin was cloned into pPAL7 vector (BioRad). c-Myb gene was used as templates to perform site-directed mutagenesis. All mutants were obtained by using the QuikChange mutagenesis kit (Stratagene) according to the manufacturer’s instructions. The mutations were confirmed by DNA sequencing. Proteins were purified by using a cation-exchange chromatography (S-Sepharose column equilibrated with 40 mM Tris-HCl pH 8.5). c-Myb* was eluted with 850 mM NaCl. The sample was then diluted fourfold in 40 mM Tris, pH 8.5 and all minor impurities were removed by the purification step on a nickel(II)-charged chelating Sepharose FF (Amersham Biosciences) column equilibrated with 40 mM Tris-HCl equilibrated pH 8.5. The protein passed through the nickel(II) column; the flow-through containing the protein was collected and concentrated. The purity of the protein was confirmed by SDS/PAGE.
The folding stability of the c-Myb mutants was estimated by titration with 1,2,3-trifluoro-ethanol, in analogy to that reported for the wild-type protein (13). In analogy to what was performed previously, for these experiments we used untagged c-Myb variants.
Stopped-Flow Measurements.
The kinetics of binding and displacement experiments was carried out on a single-mixing SX-18 stopped-flow instrument (Applied Photophysics); the excitation wavelength was 280 nm and the fluorescence emission was collected using a 320-nm–cutoff glass filter.
Binding kinetics.
Pseudo–first-order binding experiments were performed mixing a constant concentration of pwtKIX of 5–10 μM with increasing concentrations of c-Myb* and the different site-directed mutants, typically ranging from 20 to 100 μM. The experiments were carried out at 10 °C and in the following buffer: 50 mM sodium phosphate, 150 mM KCl, 1 mM DTT, pH 7.2. All reagents were of analytical grade.
Displacement kinetics.
The dissociation rate constant was measured by displacement kinetic experiments. An equimolar mixture of pwtKIX and c-Myb* (20 μM pwtKIX and 20 μM c-Myb* or its site-directed mutants) was rapidly mixed with an excess of wild-type KIX (i.e., with a Tyr at position 72); the dissociation process was measured as a function of the concentration of wild-type KIX, ranging from 40 to 400 μM. In all cases, observed kinetics was found to be single exponential and insensitive to the concentration of wtKIX, as expected for a canonical displacement reaction. The experiments were performed at 10 °C and pH 7.2 in 50 mM sodium phosphate buffer in the presence of 150 mM KCl, 1 mM DTT. All reagents were of analytical grade.
Supplementary Material
Acknowledgments
This work was partly supported by grants from the Italian Ministero dell’Istruzione dell’Università e della Ricerca (RBRN07BMCT_007 to M.B. and Progetto di Interesse “Invecchiamento” to S.G.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307337110/-/DCSupplemental.
References
- 1.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804(6):1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dyson HJ. Expanding the proteome: Disordered and alternatively folded proteins. Q Rev Biophys. 2011;44(4):467–518. doi: 10.1017/S0033583511000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tompa P. Unstructural biology coming of age. Curr Opin Struct Biol. 2011;21(3):419–425. doi: 10.1016/j.sbi.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 4.Spolar RS, Record MTJ., Jr Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263(5148):777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- 5.Tompa P, Fuxreiter M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci. 2008;33(1):2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Wright PE, Dyson HJ. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293(2):321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 7.Tompa P, Szász C, Buday L. Structural disorder throws new light on moonlighting. Trends Biochem Sci. 2005;30(9):484–489. doi: 10.1016/j.tibs.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 8.Wells M, et al. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc Natl Acad Sci USA. 2008;105(15):5762–5767. doi: 10.1073/pnas.0801353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc Natl Acad Sci USA. 2000;97(16):8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trizac E, Levy Y, Wolynes PG. Capillarity theory for the fly-casting mechanism. Proc Natl Acad Sci USA. 2010;107(7):2746–2750. doi: 10.1073/pnas.0914727107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radhakrishnan I, et al. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator:coactivator interactions. Cell. 1997;91(6):741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 12.Goto NK, Zor T, Martinez-Yamout M, Dyson HJ, Wright PE. Cooperativity in transcription factor binding to the coactivator CREB-binding protein (CBP). The mixed lineage leukemia protein (MLL) activation domain binds to an allosteric site on the KIX domain. J Biol Chem. 2002;277(45):43168–43174. doi: 10.1074/jbc.M207660200. [DOI] [PubMed] [Google Scholar]
- 13.Gianni S, Morrone A, Giri R, Brunori M. A folding-after-binding mechanism describes the recognition between the transactivation domain of c-Myb and the KIX domain of the CREB-binding protein. Biochem Biophys Res Commun. 2012;428(2):205–209. doi: 10.1016/j.bbrc.2012.09.112. [DOI] [PubMed] [Google Scholar]
- 14.Zor T, De Guzman RN, Dyson HJ, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of c-Myb. J Mol Biol. 2004;337(3):521–534. doi: 10.1016/j.jmb.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 15.Fersht AR, Matouschek A, Serrano L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J Mol Biol. 1992;224(3):771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 16.Matouschek A, Kellis JT, Jr, Serrano L, Fersht AR. Mapping the transition state and pathway of protein folding by protein engineering. Nature. 1989;340(6229):122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 17.Serrano L, Neira JL, Sancho J, Fersht AR. Effect of alanine versus glycine in alpha-helices on protein stability. Nature. 1992;356(6368):453–455. doi: 10.1038/356453a0. [DOI] [PubMed] [Google Scholar]
- 18.Richardson JM, Lopez MM, Makhatadze GI. Enthalpy of helix-coil transition: missing link in rationalizing the thermodynamics of helix-forming propensities of the amino acid residues. Proc Natl Acad Sci USA. 2005;102(5):1413–1418. doi: 10.1073/pnas.0408004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matthews JM, Fersht AR. Exploring the energy surface of protein folding by structure-reactivity relationships and engineered proteins: Observation of Hammond behavior for the gross structure of the transition state and anti-Hammond behavior for structural elements for unfolding/folding of barnase. Biochemistry. 1995;34(20):6805–6814. doi: 10.1021/bi00020a027. [DOI] [PubMed] [Google Scholar]
- 20.Antonini E, Brunori M. Hemoglobin and Myoglobin in Their Reactions with Ligands. Amsterdam: North-Holland; 1971. [Google Scholar]
- 21.Fersht AR, Sato S. Phi-value analysis and the nature of protein-folding transition states. Proc Natl Acad Sci USA. 2004;101(21):7976–7981. doi: 10.1073/pnas.0402684101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ozkan SB, Bahar I, Dill KA. Transition states and the meaning of Phi-values in protein folding kinetics. Nat Struct Biol. 2001;8(9):765–769. doi: 10.1038/nsb0901-765. [DOI] [PubMed] [Google Scholar]
- 23.Gianni S, et al. Structural characterization of a misfolded intermediate populated during the folding process of a PDZ domain. Nat Struct Mol Biol. 2010;17(12):1431–1437. doi: 10.1038/nsmb.1956. [DOI] [PubMed] [Google Scholar]
- 24.Leffler JE. Parameters for the description of transition states. Science. 1953;117(3039):340–341. doi: 10.1126/science.117.3039.340. [DOI] [PubMed] [Google Scholar]
- 25.Toney MD, Kirsch JF. Direct Brønsted analysis of the restoration of activity to a mutant enzyme by exogenous amines. Science. 1989;243(4897):1485–1488. doi: 10.1126/science.2538921. [DOI] [PubMed] [Google Scholar]
- 26.Eaton WA, Henry ER, Hofrichter J. Application of linear free energy relations to protein conformational changes: The quaternary structural change of hemoglobin. Proc Natl Acad Sci USA. 1991;88(10):4472–4475. doi: 10.1073/pnas.88.10.4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edelstein SJ, Changeux JP. Relationships between structural dynamics and functional kinetics in oligomeric membrane receptors. Biophys J. 2010;98(10):2045–2052. doi: 10.1016/j.bpj.2010.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fersht AR. Relationship of Leffler (Bronsted) alpha values and protein folding Phi values to position of transition-state structures on reaction coordinates. Proc Natl Acad Sci USA. 2004;101(40):14338–14342. doi: 10.1073/pnas.0406091101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Itzhaki LS, Otzen DE, Fersht AR. The structure of the transition state for folding of chymotrypsin inhibitor 2 analysed by protein engineering methods: Evidence for a nucleation-condensation mechanism for protein folding. J Mol Biol. 1995;254(2):260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 30.Naganathan AN, Muñoz V. Insights into protein folding mechanisms from large scale analysis of mutational effects. Proc Natl Acad Sci USA. 2010;107(19):8611–8616. doi: 10.1073/pnas.1000988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dogan J, Mu X, Engström A, Jemth P. The transition state structure for coupled binding and folding of disordered protein domains. Sci Rep. 2013;3:2076. doi: 10.1038/srep02076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferreiro DU, Sánchez IE, de Prat Gay G. Transition state for protein-DNA recognition. Proc Natl Acad Sci USA. 2008;105(31):10797–10802. doi: 10.1073/pnas.0802383105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorman J, et al. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol Cell. 2007;28(3):359–370. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.