

Abstract
Transposable elements (TEs) have a major impact on genome evolution, but they are potentially deleterious, and most of them are silenced by epigenetic mechanisms, such as DNA methylation. Here, we report the characterization of a TE encoding an activity to counteract epigenetic silencing by the host. In Arabidopsis thaliana, we identified a mobile copy of the Mutator-like element (MULE) with degenerated terminal inverted repeats (TIRs). This TE, named Hiun (Hi), is silent in wild-type plants, but it transposes when DNA methylation is abolished. When a Hi transgene was introduced into the wild-type background, it induced excision of the endogenous Hi copy, suggesting that Hi is the autonomously mobile copy. In addition, the transgene induced loss of DNA methylation and transcriptional activation of the endogenous Hi. Most importantly, the trans-activation of Hi depends on a Hi-encoded protein different from the conserved transposase. Proteins related to this anti-silencing factor, which we named VANC, are widespread in the non-TIR MULEs and may have contributed to the recent success of these TEs in natural Arabidopsis populations.
Keywords: DNA methylation, epigenetics, evolution, transposon
Introduction
Control of transposable elements (TEs) has been extensively studied in plants. A pioneering early observation in maize is that TE activity often changes between active and inactive states in heritable but reversible manners (McClintock, 1951, 1958). The changes in the TE activity are generally correlated with the DNA methylation status; inactive TEs tend to be more methylated than active TEs (Chandler and Walbot, 1986; Brettell and Dennis, 1991; Fedoroff, 1996; Martienssen, 1996). The importance of DNA methylation in TE control has also been demonstrated using mutants of Arabidopsis; in Arabidopsis mutants with reduced genomic DNA methylation, a variety of silent TEs are de-repressed and mobilized (Miura et al, 2001; Singer et al, 2001; Kato et al, 2003; Lippman et al, 2004; Mirouze et al, 2009; Tsukahara et al, 2009).
Intriguingly, some TEs have mechanisms to counteract DNA methylation and silencing by the host. For example, McClintock’s Suppressor-mutator (Spm) element in maize encodes a protein TnpA, which induces loss of DNA methylation in regions controlling transcript formation in Spm (Schläppi et al, 1994, 1996; Cui and Fedoroff, 2002). An active Spm transiently activates silent Spm copies in trans, a process likely to be mediated by TnpA (Cui and Fedoroff, 2002).
Robertson’s Mutator, another well-characterized TE in maize, spontaneously changes its activity and DNA methylation in a coordinated manner (Chandler and Walbot, 1986; Martienssen and Baron, 1994; Martienssen, 1996). Like Spm, a silent Mutator element loses DNA methylation when an active Mutator is present in the same genome (Brown and Sundaresan, 1992; Lisch et al, 1995, 1999). MuDR, an autonomously mobile copy of maize Mutator family, contains two genes, mudrA and mudrB. The mudrA encodes the MURA protein, which is structurally similar to known transposases of other TEs (Eisen et al, 1994; Lisch, 2002). In addition, mudrA is sufficient for excision of Mutator, further suggesting that MURA functions as a transposase (Lisch et al, 1999). TEs similar to the maize Mutator are widespread in eukaryotes and they are referred to as Mutator-like elements (MULEs) (Jiang et al, 2004). ORFs related to mudrA are generally found in autonomous MULEs. Some of the autonomous MULEs also have additional ORF(s), such as mudrB in MuDR, but the structures of the proteins encoded in these ORFs are diverse and their functions remain largely unknown. In addition, some of the MULEs carry fragments of cellular genes, but their impacts on the TE dynamics and host fitness are still elusive (Talbert and Chandler, 1988; Yu et al, 2000; Jiang et al, 2004; Hoen et al, 2006).
Most of the class II (DNA-type) TEs have long terminal inverted repeat (TIR), but there are a few exceptions (Wicker, 2007). Although the maize Mutator elements have relatively long TIRs of almost identical sequences, subgroups of MULEs with extensively degenerated TIR have been found in the Arabidopsis genome and they are classified as non-TIR MULEs (Le et al, 2000; Yu et al, 2000). Although the theoretical sequence analyses of Arabidopsis genome suggest movement of these non-TIR MULEs in the past (Yu et al, 2000), direct evidence for de novo movements is limited (Hoen et al, 2006; Tsukahara et al, 2009).
We have previously reported that a group of non-TIR MULEs, called VANDAL21, seem to transpose in a background of reduced genomic DNA methylation (Tsukahara et al, 2009). Here, we identified an autonomously mobile copy of VANDAL21, which we renamed Hiun (Hi). Despite the degeneration of TIRs, Hi is competent to excise and transpose in precise manners. Interestingly, a Hi transgene induced loss of DNA methylation, transcriptional activation, and excision of the endogenous Hi copy. Most importantly, these trans-acting effects of Hi do not depend on the protein related to MURA-type transposase but instead depend on another protein encoded by Hi. The function of this novel anti-silencing protein, which we named VANC, will be discussed in the context of TE evolution.
Results
Identification of mobile VANDAL21 copies
The genome sequence of wild-type Col ( http://www.arabidopsis.org/) suggests seven copies of VANDAL21 elements with relatively similar sequences. Other copies of VANDAL21 are distant in the sequences. Consistent with that, Southern analysis revealed seven bands for that group of VANDAL21 (Tsukahara et al, 2009). We have previously shown that additional bands emerge in Arabidopsis plants derived from several rounds of self-pollinations in ddm1 (decrease in DNA methylation 1) mutant backgrounds, suggesting mobility of one or more copies of the VANDAL21 members (Tsukahara et al, 2009). Arabidopsis ddm1 mutation generally induces loss of DNA methylation in TEs, which causes mobilization of diverse TEs (Miura et al, 2001; Singer et al, 2001; Lippman et al, 2004; Mirouze et al, 2009; Tsukahara et al, 2009). In order to know which of VANDAL21 copies are mobile, we used two methods: suppression PCR and whole-genome re-sequencing (see Materials and methods for details).
In total, we identified 72 de novo insertions of VANDAL21s (61 by genome re-sequencing and 14 by suppression PCR with 3 overlaps) in the self-pollinated ddm1 lines (Supplementary Table S1). Of these 72 insertions, 69 correspond to one copy (Figure 1A; AT2TE42810) of VANDAL21 element. The remaining three insertions correspond to another copy (AT4TE15615). For the other five copies of related VANDAL21, no new insertion has been identified. In the following parts, we concentrate on the most active copy, AT2TE42810, which we renamed Hiun (Hi, Japanese for ‘a flying cloud’).
Figure 1.
Mobilization of Hi in ddm1 mutant. (A) Schematic diagram for structure of Hi. Terminal regions, exons, introns, and intergenic regions are shown by white bars, grey bars, grey lines, and black lines, respectively. Regions examined by bisulphite sequencing are shown by L1 and R1 with thick black lines. Region examined by McrBC-qPCR is shown by L2. Region examined for copy number quantification is shown by C1. In most of the transgene constructs, silent mutation is introduced for each ORF, so that the transcripts from the transgene and endogenous copy could be distinguished between. The sites of the silent mutations are shown by vertical bars, with surrounding arrowheads showing regions amplified by RT–PCR. Regions for ORFs deleted in each of the deletion constructs are shown by horizontal bars with two arrowheads. (B) De novo integration sites of Hi in relation to flanking transcription units. The position of integration is normalized by length of the flanking transcription unit. Rightward and leftward arrows indicate insertions with 5′ to 3′ and 3′ to 5′ orientations of Hi, respectively. Insertions flanking pseudogenes and transposon genes are shown by grey arrows, and those flanking canonical genes by black arrows. Sequences of the integration sites are shown in Supplementary Table S1. Four out of the sixty-nine insertions are not included in this figure, because they are further away from transcription units. Genomic locations of all 69 transpositions are shown in Supplementary Figure S2. (C) Excision of Hi in ddm1 plants detected by PCR. Genomic DNA of 11 ddm1 plants (lane numbers from 2 to 12) and 5 wild-type sibling plants (lane numbers from 13 to 17) was used to analyse excision of endogenous Hi by nested PCR. These lines are derived from segregating population in self-pollinated progeny of a DDM1/ddm1-1 heterozygote. Sequences of primers used are shown in Supplementary Table S2.
Source data for this figure is available on the online supplementary information page.

Structure of Hi
Hi is 8177-bp long and includes three ORFs: At2g23500, At2g23490, and At2g23480 (Figure 1A). One ORF (At2g23500; called vanA) encodes a protein with high sequence similarities with MURA-type transposases, which are generally found in MULEs. Proteins encoded by two other ORFs (vanB and vanC) do not have sequence similarity to any characterized proteins. An unorthodox feature is that, unlike other typical mobile DNA-type TEs, the TIRs of this TE are extensively degenerated (Supplementary Figure S1A), showing the characteristics of non-TIR MULEs (Yu et al, 2000).
Integration and excision of Hi
Hi is transposed throughout the genome, although insertions may be more concentrated near the original locus than in unlinked regions (Supplementary Figure S2). Mutator elements in maize preferentially transpose into 5′ region of genes (Hardeman and Chandler, 1989; Dietrich et al, 2002; Liu et al, 2009). That was also the case for Hi; most of the integration sites are localized around transcription start sites of genes (Figure 1B). Interestingly, integration of Hi there had bias in the orientation (Figure 1B). Such a bias in the orientation has not been reported for the maize Mutator (Brown et al, 1989). The bias in the orientation of Hi integration might be related to the asymmetry in its terminal sequences. We could detect 9 bp of target site duplication (TSD) for most of the insertions examined (Supplementary Figure S1B), as is the case for integrations of other MULEs (Yu et al, 2000).
Many DNA-type TEs often transpose with loss of the original copy. Because Hi does not have typical structure of DNA-type TE, an interesting question would be whether Hi excision occurs at defined termini or not. In order to detect somatic excision events, we used PCR with primers for both of the flanking regions of the original Hi locus (details in Materials and methods). Using this assay, we could detect Hi excision in all independent ddm1 lines examined (Figure 1C). We examined the mode of the excisions by sequencing the PCR products (Supplementary Figure S3A). Interestingly, despite the degeneration of TIRs, many of the excision products showed excision around the terminal sites predicted from the integrated copies. Even TSDs are lost in a significant part of the excision product.
In summary, these observations suggest that long perfect TIRs are dispensable not only for integration, but also for reasonably precise excision of this element in the defined termini. The transposition of Hi occurred in a manner comparable to that of typical MULEs with long TIR.
Mobilization of endogenous Hi by transgene
As the ddm1 mutation results in transcriptional de-repression of many repeat sequences (Lippmann et al, 2004; Tsukahara et al, 2009), it is formally possible that mobilization of Hi in ddm1 is triggered by de-repression of other sequence(s). In order to test if expression of proteins encoded by Hi is sufficient for its mobilization, we introduced a cloned Hi copy into wild-type plants by Agrobacterium-mediated transformation. In these transgenic Hi lines, we could detect transcripts corresponding to their three ORFs (Supplementary Figure S4). In these lines, the Hi transgene induced excision of the original copy (Figure 2A). Control transformants with empty vectors did not show excision of the Hi, confirming that Hi transgene triggered mobilization of the endogenous copy. We also examined Hi activity in self-pollinated progeny of one of the Hi transgenic lines. We could detect Hi excision in most of 15 progeny plants that inherited the transgene (Supplementary Figure S6). On the other hand, we could not detect the excision in any of six progeny plants that lost the transgene by segregation, further confirming that the Hi transgene is responsible for the mobilization.
Figure 2.
Introduction of Hi transgene induces loss of DNA methylation and excision of endogenous Hi copy. (A) Excision of endogenous Hi induced by transgene for Hi (Hi TG: lanes 5–18). Lanes 1 and 2–4 are non-transgenic plant (wt) and transformant lines with empty vector (V) used as negative controls, respectively. Excision of Hi copy in the transgene was also detected in some of the transgenic lines (Supplementary Figure S5). (B) DNA methylation status of Hi termini in the transgenic line and progeny. T1 transformant with Hi transgene showed reduction in DNA methylation in both termini, compared to non-transgenic plant (NT) and transformants with empty vector (V). T2/TG+ and T2/TG− are self-pollinated progeny of the T1 with and without transgene, respectively. In both classes, averages and standard deviations of three segregants are shown. We also obtained essentially the same results for a segregating T3 family (Supplementary Figure S7). Regions L1 (upstream of vanA) and R1 (upstream of vanC) were examined (shown in Figure 1). At least 11 clones were examined for each plant.
Source data for this figure is available on the online supplementary information page.

DNA demethylation induced by Hi in trans
The observations described above suggest that expression of Hi transgene induces mobilization of the endogenous copy. Interestingly, in the presence of the transgene, DNA methylation levels were reduced in both termini of the endogenous Hi (Figure 2B). The loss of methylation is more extensive in non-CpG sites than in CpG sites. When the transgene was segregated away in the self-pollinated progeny of the transgenic line (T2/TG− in Figure 2B), Hi was remethylated in the terminal regions, which was associated with its loss of excision activity.
We also tested the trans-acting demethylation effect for transposed Hi copies, using epi-Recombinant Inbred Lines (epi-RILs) from ddm1 mutant (Johannes et al, 2009). The epi-RILs are originated from ddm1 mutant backcrossed to parental wild-type Col to generate DDM1/DDM1 homozygotes, and subsequent self-pollination to fix the heritable epigenetic defects induced by ddm1. The methylation status of Hi at the original locus was examined by PCR after digestion by methylation-sensitive restriction enzyme. As expected from the crossing scheme used to generate the epi-RILs (Johannes et al, 2009), approximately three quarters of the epi-RILs tested had inherited the original Hi copy from wild-type DDM1 parent. Some of these lines have additional transposed Hi copies in trans (Figure 3A). In those lines, Hi at the original locus showed loss of methylation (Figure 3B), despite its wild-type origin. On the other hand, in epi-RILs that did not carry the additional Hi, Hi at the original locus remained methylated to the level comparable to parental wild type. Together, these results suggest that the stable demethylation of Hi at the original locus is due to trans-acting effect of the transposed Hi copies. In addition to its demethylation, the Hi copies present at the original locus showed excision (Figure 3C), and that happened only when the additional trans-acting copies exist. These trans-acting effects of the transposed Hi are consistent with the trans-acting demethylation and mobilization by the Hi transgene.
Figure 3.
Transposed Hi induces loss of DNA methylation and excision of Hi in the original locus. (A) Copy number of Hi in each of the epi-RILs. The copy number was estimated by quantitative PCR using region C1 in Figure 1. Average and standard deviation of two technical replicates are shown in this and next panel. Parental origin of the original Hi locus (wt or ddm1) was determined by methylation status of the linked region (Colomé-Tatché et al, 2012). (B) DNA methylation status in the 5′ region (L2 in Figure 1) of the original Hi locus was estimated by McrBC digestion and subsequent qPCR. Details are described in Materials and methods. Hi in the original locus showed loss of DNA methylation when extra copies of Hi exist. (C) Excision analysis of original Hi copy by nested PCR. (D) Two of the epi-RILs showed germinal transmission of the excised Hi allele. Origin of the Hi locus is wild-type DDM1 for line 166 and ddm1 mutant for line 458. Presence of Hi in the original locus was examined by PCR in the 5′ border of Hi (the primer sequences are shown in Supplementary Table S2). Lack of the signal suggests fixation of the empty allele.
Source data for this figure is available on the online supplementary information page.

Expression of vanC is sufficient for the trans-activation and mobilization of endogenous Hi
The results shown above demonstrate that Hi transgenes induced mobilization of endogenous copy in trans, which is associated with loss of DNA methylation in the terminal regions. In order to further dissect the role for each of the ORFs in Hi, we generated transgenes with deletion in each ORF. Transgene with deletion of the central small ORF (vanB) still caused loss of methylation in both termini of Hi (Figure 4). By contrast, deletion of 3′ ORF (vanC) abolished the demethylation effect for both termini, suggesting that this ORF is essential for the demethylation. Transgene with deletion of 5′ ORF (vanA), which is structurally similar to transposase, still caused loss of methylation in 3′ (vanC side) terminal region of Hi, although the demethylation effect in the 5′ (vanA side) region was less complete than that in the full-length Hi transgenic lines. These results suggest that vanC is important for the demethylation of both of the terminal regions.
Figure 4.

DNA methylation status of endogenous Hi after introduction of Hi transgene and its deletion derivatives. For each of the deletion derivatives, averages and standard deviations of four independent transgenic plant lines are shown. WT and V are from Figure 2.
We then examined the effect of these deletion constructs on excision of endogenous copies (Figure 5A). Very importantly, transgenic lines with deletion in vanA (ΔA-TG) still induced excision of endogenous Hi (Figure 5A). This was surprising because vanA encodes the putative transposase. We then examined expression of endogenous vanA gene in the presence of ΔA-TG. In most of the ΔA-TG lines, endogenous vanA was de-repressed and transcribed, although the expression level was generally lower and less robust than that of the Hi transgene keeping vanA (Figure 5B; Supplementary Figure S8).
Figure 5.
Trans-activation by Hi transgene without putative transposase. (A) Excision of endogenous Hi induced by ΔA transgene. Lanes 1 and 2–6 are non-transgenic plant (wt) and transformant lines with empty vector (V) used as negative controls, respectively. Excision of endogenous Hi induced by ΔB and ΔC transgene is also shown below. The results using additional ΔC lines are shown in Supplementary Figure S9. (B) Transcriptional activation of vanA induced by ΔA transgene. Materials for the same lane number in (A) and (B) are from the same plant, although the DNA and RNA are prepared from different leaves.
Source data for this figure is available on the online supplementary information page.

The results above suggest that expression of vanB and/or vanC can cause de-repression of vanA. We could also detect expression of vanB in ΔB-TG lines, but vanC transcript was undetectable in ΔC-TG lines (Supplementary Figure S8), suggesting possible role of vanC in the trans-activation. In addition, deletion of vanB from the transgene did not affect excision of endogenous Hi by the transgene, while the excision tended to be less robust in ΔC-TG (Figure 5A; Supplementary Figure S9). In order to know if vanB is dispensable for the trans-activation of Hi, we examined the effect of transgene with deletion of both vanA and vanB (ΔAB-TG). Only the vanC ORF remains in the ΔAB-TG construct. The ΔAB-TG also induced demethylation of 3′ (vanC side) terminal regions to the level comparable to the ΔA-TG (Figure 4). In most of the ΔAB-TG lines, we could detect excision of Hi and transcription of vanA and vanB (Figure 6A and B). In T2 generation that originated from self-pollination of a T1 ΔAB-TG plant, all T2 plants with the transgene showed excision, but none of T2 plants without transgene showed excision, confirming that the ΔAB transgene induces the excisions of endogenous Hi (Figure 6C). Taken together, these results demonstrate the key role of vanC for the trans-acting anti-silencing of Hi.
Figure 6.
Expression of vanC is sufficient for the trans-activation. (A) Excision of endogenous Hi induced by ΔAB transgene. (B) Transcriptional activation of vanA and vanB genes induced by ΔAB transgene. Materials for the same lane number in (A) and (B) are from the same plant, although the DNA and RNA are prepared from different leaves. (C) Excision of endogenous Hi induced by ΔAB transgene in the T2 generation. T2 plants from self-pollinated progeny of a T1 (the plant shown in lane 9 of A) were examined after determining the presence/absence of the transgene.
Source data for this figure is available on the online supplementary information page.

Proteins related to the anti-silencing factor are widespread in non-TIR MULEs but not found in TIR MULEs
Non-TIR MULEs in A. thaliana genome are consisted of multiple VANDAL and ARNOLD families (Yu et al, 2000; Figure 7A). Interestingly, the non-TIR MULEs seem to be very successful in the recent proliferation. The phylogenetic analyses revealed recent proliferations in multiple subfamilies of non-TIR MULEs; each of the subfamilies shows terminal proliferations after separation of A. thaliana and A. lyrata lineages. The proliferation rates of the non-TIR MULE clusters are significantly higher than those of TIR-MULE in both A. thaliana and A. lyrata lineages (Figure 7A; Table I).
Figure 7.

Evolution and proliferation of TIR and non-TIR MULE families. (A) Phylogenetic relationship among MULE families in genomes of A. thaliana and A. lyrata. A. lyrata-specific lineages are shown by red lines. An NJ tree made by p-distance is shown. Scale bar is shown in the centre of the tree. The families containing DUF287 or Ulp1 protease domain are indicated in the parentheses. Names of VANDAL members are shown in phylogenetic tree in Supplementary Figure S10. (B) Dot-plot (Harr-plot) analyses among VANDAL families. Regions with nucleotide identities of 15 out of 20 or more are shown by dots. Copies with typical structure were chosen from VANDAL21, 17, 8, 7 and 6 families. From VANDAL21 family, one sequence each from two clusters was used. Coding regions are indicated by pointed thick lines.
Table 1. Comparison of copy numbers per cluster between non-TIR and TIR MULEs.
| #Seqa | #Clusterb | Seq/cluster | t | |
|---|---|---|---|---|
| A. thaliana | ||||
| TIR MULEs | 27 | 24 (21) | 1.125 | 4.42 |
| Non-TIR MULEs | 143 | 34 (13) | 4.206 | P<0.001 |
| A. lyrata | ||||
| TIR MULEs | 85 | 39 (21) | 2.179 | 2.38 |
| Non-TIR MULEs | 219 | 49 (20) | 4.347 | P<0.02 |
Analyses were performed using transposase genes. Therefore, copies without the conserved transposase gene are not included in the analyses. Details for the analyses are described in Materials and methods.
aNumber of sequences analysed.
bNumber of cluster with <0.1 p-distances. In the parentheses, number of clusters with only one sequence is shown.
Notably, all major branches of these proliferating non-TIR MULEs contain TEs with ORFs encoding proteins similar to the anti-silencing factor VANC (protein product of vanC). All the VANC-related proteins contain the domain DUF1985 (domain of unknown function; Supplementary Figure S11). Some of them, including VANC, also contain the domain DUF287 (Figure 7; Supplementary Figure S11). Other VANDAL members, as well as ARNOLD members, have ORFs encoding Ulp1 proteases (Figure 7A; Supplementary Figure S11). Both the Ulp1 and DUF287 types of proteins contain DUF1985, generating a wide distribution of VANC-related proteins. In contrast, such VANC-related proteins were not found in any of the TIR-MULE members. Therefore, association between the VANC-related proteins and the absence of long TIRs is very tight. The non-TIR MULEs seem to keep VANC-related proteins during evolution.
While transposases of the MURA class are generally found in autonomous MULEs, other ORFs in MULEs tend to be diverse. Consistent with that, nucleotide divergence is higher in vanC-related genes than in vanA-related genes, especially between distant groups (Figures 7B and 8). However, that does not seem to be due to weaker constraint on the amino-acid sequence of the VANC-related proteins, because non-synonymous mutation rates are comparable between both families of genes and the overall high divergence seems to reflect the divergence in synonymous sites (Figure 8). The conserved amino-acid sequences in the VANC-related proteins suggest advantage in their functions, at least in short term.
Figure 8.

Sliding window plot analyses of VANDAL21 and the most related family VANDAL17. (A) Divergence between two clusters (seven copies and three copies) of VANDAL21 family in A. thaliana. (B) Divergence between VANDAL21 copies in A. thaliana (three copies, which are orthologous to the A. lyrata copies) and A. lyrata (six copies). (C) Divergence between VANDAL17 family in A. thaliana (four copies) and A. lyrata (three copies). Level of divergence for synonymous and non-synonymous sites in a 200-bp window was plotted in 1 bp intervals. Only coding sequences of complete structures were used. Thin lines and thick lines indicate synonymous and non-synonymous divergences, respectively.
Genome-wide effects of Hi transgene on DNA methylation
Hi transgene affects endogenous Hi sequence. We then examined the effect of Hi transgene genome-wide. DNA methylation was examined, because that could be accessed relatively precisely using whole-genome bisulphite sequencing. In the whole-genome bisulphite sequencing, deep sequencing reads are mapped to the reference genome sequence. In order to reduce possible noise due to repetitive nature of TEs, we used only uniquely mapped reads; reads mapped to multiple loci were not used for the analyses. The analyses revealed that many of the VANDAL21 members showed reduced DNA methylation in the transgenic line (Figure 9A and B; Supplementary Figures S12–S14; Supplementary Table S3). Interestingly, not only terminal regions but also internal regions were affected for both Hi and other VANDAL21 copies (Figure 9B; Supplementary Figure S14). As is the case for the Hi sequence, non-CpG sites tend to be more affected than CpG sites. On the other hand, the demethylating effect was much less in the other TEs and genes (Figure 9A; Supplementary Figure S12), suggesting that the trans-acting effects of Hi are highly specific.
Figure 9.

Demethylation of VANDAL21 members induced by Hi transgene. (A) Effects of Hi transgene on DNA methylation in TEs. Changes in methylation at CpHpG sites and CpHpH sites are plotted. For each of them, significance of decrease in methylation was accessed by the value 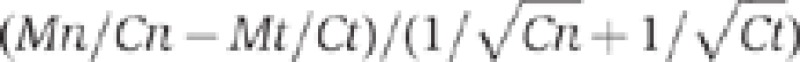 , where Mn, Cn, Mt and Ct are methylated cytosine (M) and total cytosine (C) counts mapped for each TE in the non-transgenic (n) and transgenic (t) plants, respectively. Each of these values is shown in Supplementary Table S3, with the corresponding values for CpG sites. This figure contains 24 282 TEs plotted, while 6910 TEs are not plotted due to lack of mapped cytosine in one or more of the three contexts in non-transgenic or transgenic plant. The lack is mainly because we did not use reads mapped multiple loci. Some of the VANDAL21 family members (surrounded by broken ellipse) showed most significant reduction for both CpHpG and CpHpH sites. The dot for Hi is indicated by an arrow. Similar analyses done on genes are shown in Supplementary Figure S12. (B) Effect of Hi transgene on DNA methylation across each of the VANDAL21 members. Left and right terminals are shown by broken lines for each element. Each point represents proportion of methylated cytosine for a sliding window with seven fractions after separating each TE for 100 fractions. Right and left flanking regions are also analysed by the same conditions. Scale bars for CpG, CpHpG, and CpHpH sites in each panel indicate 1, 0.8, and 0.4, respectively. For some of the VANDAL21 copies, reduced methylation in the terminal region is also confirmed by conventional bisulphite sequencing using primers within and flanking the TE (Supplementary Figure S13). Results of DNA methylation across six additional VANDAL21 members are shown in Supplementary Figure S14. CACTA2 is shown as a negative control.
, where Mn, Cn, Mt and Ct are methylated cytosine (M) and total cytosine (C) counts mapped for each TE in the non-transgenic (n) and transgenic (t) plants, respectively. Each of these values is shown in Supplementary Table S3, with the corresponding values for CpG sites. This figure contains 24 282 TEs plotted, while 6910 TEs are not plotted due to lack of mapped cytosine in one or more of the three contexts in non-transgenic or transgenic plant. The lack is mainly because we did not use reads mapped multiple loci. Some of the VANDAL21 family members (surrounded by broken ellipse) showed most significant reduction for both CpHpG and CpHpH sites. The dot for Hi is indicated by an arrow. Similar analyses done on genes are shown in Supplementary Figure S12. (B) Effect of Hi transgene on DNA methylation across each of the VANDAL21 members. Left and right terminals are shown by broken lines for each element. Each point represents proportion of methylated cytosine for a sliding window with seven fractions after separating each TE for 100 fractions. Right and left flanking regions are also analysed by the same conditions. Scale bars for CpG, CpHpG, and CpHpH sites in each panel indicate 1, 0.8, and 0.4, respectively. For some of the VANDAL21 copies, reduced methylation in the terminal region is also confirmed by conventional bisulphite sequencing using primers within and flanking the TE (Supplementary Figure S13). Results of DNA methylation across six additional VANDAL21 members are shown in Supplementary Figure S14. CACTA2 is shown as a negative control.
Discussion
Trans-activation as a counteraction against genome defense by the host
Here, we report identification of an autonomously mobile copy of MULE that lacks long TIRs. Among three ORFs Hi contains, vanA encodes for a putative transposase conserved among other TEs. Most importantly, vanC, another ORF of Hi, plays a key role in the transcriptional activation of the other ORFs in Hi, and in mobilization of Hi. These trans-acting effects represent efficient means for counteracting DNA methylation and silencing by the host.
Importantly, the anti-silencing mechanism of Hi seems to function specifically, rather than globally (Figure 9). In contrast, some viruses have evolved mechanisms to globally inhibit RNA interference (RNAi)/post-transcriptional gene silencing (PTGS). RNAi/PTGS is an important mechanism for defense against viruses, and the global anti-silencing would facilitate proliferation of the viruses (Zamore, 2004; Bivalkar-Mehla et al, 2011). In contrast to the global anti-silencing mechanisms of these viruses, Hi seems to target related TEs specifically. The specific (rather than global) anti-silencing can be reasonable for a TE. If a TE has global anti-silencing mechanisms, such as genome-wide demethylation, then that would activate diverse TEs, reducing fitness of the host (Lippman et al, 2004; Tsukahara et al, 2012). Reduction of host fitness would be deleterious for survival of the TE itself, given that the TE is an obligatory intra-genomic parasite/symbiont. The strategy of a TE should be different from that of viruses, which can propagate horizontally away from damaged hosts.
Mechanisms for the trans-acting effects
VANC induces both transcriptional activation and loss of DNA methylation in trans. Genome-wide analyses of transgenic plants revealed that the trans-acting demethylation occurs specifically in Hi and other VANDAL21 members. How is the specific loss of methylation induced? In the case of Tam3 transposon of snapdragon, loss of methylation is associated with binding of the transposase (Hashida et al, 2006). It would be informative to know localization of VANC protein in the genome.
Another possible pathway could be demethylation through transcription. Transcription can induce loss of methylation in lysine 9 of histone H3 (H3K9) through the function of histone demethylase IBM1 (Inagaki et al, 2010). The loss of H3K9 methylation induces loss of non-CpG methylation. That would account for the effect of Hi on non-CpG methylation in the internal regions of TEs (Figure 9; Supplementary Figure S14). Generally, DNA methylation in both CpG and non-CpG sites could be involved in silencing of TEs (Johnson et al, 2003; Kato et al, 2003). Considering that active/inactive states of TEs can be stabilized by positive feedback loops, possible interactions between transcription and DNA methylation at CpG and non-CpG sites would be important for understanding TE dynamics (Saze and Kakutani, 2011; Inagaki and Kakutani, 2013; further discussion in Supplementary Figure S15).
Role of VANC and loss of TIR
Non-TIR MULEs found within A. thaliana genome seem to be derived from ancestral TIR MULEs (Yu et al, 2000). Interestingly, evolution of non-TIR MULEs is tightly associated with the VANC-related proteins (Supplementary Figure S11). How has the transition from TIR type to non-TIR type of MULEs occurred? That could be due to loss of restraint to keep long TIRs, possibly reflecting evolution of transposition machinery. Despite degeneration of TIRs, most of the Hi excisions occurred in a manner to keep the original sequence before integration. That was found for both Hi mobilized in ddm1 mutation (Supplementary Figure S3A) and by the Hi transgene (Supplementary Figure S3B). We could detect excision of endogenous Hi in ΔC-TG, but the efficiency tends to be low compared with the full-length TG (Figure 5A; Supplementary Figures S3C and S9). VANC might also have a role in efficient transposition with degenerated TIR. It is also possible that degeneration of the TIR had some advantage for the TE. One possible advantage of losing the TIR would be escape from siRNA-based silencing mechanisms. Long TIRs of MULEs are often associated with siRNA, and generation of siRNA might be less efficient in non-TIR MULEs. Irrespective of the range of functions, the VANC-related proteins are tightly associated with the non-TIR MULE lineages, and the efficient recent proliferations of these TEs (Figure 7A; Table I) may depend on these proteins.
Materials and methods
Plant materials
The ddm1-1 mutant allele was used throughout, except for epi-RILs, which are originated from ddm1-2 mutant. Details for self-pollination of ddm1-1/ddm1-1 mutants and wild-type DDM1/DDM1 siblings were described previously (Kakutani et al, 1996). Generation of epi-RILs is described in Johannes et al (2009). Mutant alleles used for experiments in Supplementary Figure S15 are those described in Sasaki et al (2012).
Identification of mobile VANDAL21 copies
Conditions for suppression PCR were described previously (Miura et al, 2001). To detect de novo insertions from whole-genome sequence data of self-pollinated ddm1 plants (Tsukahara et al, 2012), reads containing terminal sequences of VANDAL21 copies were selected first, and the flanking sequences were subsequently extracted from those reads. In both approaches, each of the VANDAL21 copies can be distinguished by polymorphisms in the terminal regions. Raw sequence data for the self-pollinated ddm1 plants were deposited in the DDBJ (DNA Data Bank of Japan) Sequence Read Archive (DRA; accession nos. DRA000420–000424).
DNA preparation and copy number quantification
DNA was extracted using DNeasy kit (QIAGEN). Copy number of Hi in the genome was quantified at region C1 (Figure 1) by quantitative PCR using Light Cycler 480 machine (Roche) and SYBR green I MasterMix (Roche) and normalized with signals for two single-copy loci, AT5G13440 and AT5G36220. For this and all other experiments, sequences of primers used are listed in Supplementary Table S2.
Detecting excision of endogenous Hi
Excision of Hi was detected using nested PCR with the following conditions. First PCR: 94°C for 2 min, 25 cycles of (94°C for 30 s; 55°C for 30 s; 72°C for 30 s), and 72°C for 2 min. The products diluted to 20 times by H2O were used for second PCR: 94°C for 2 min, 30 cycles of (94°C for 30 s; 55°C for 30 s; 72°C for 30 s), and 72°C for 2 min. The sequence after excision of endogenous Hi was determined after PCR and subsequent cloning by TA-cloning kit (Invitrogen).
DNA methylation analyses
Conventional bisulphite sequence analysis was performed as described previously (Tsukahara et al, 2009). For genome-wide bisulphite sequencing analyses, sequencing libraries (insert size: 300–400 bp) were prepared using TruSeq DNA LT Sample Prep Kit (Illumina) and subjected to bisulphite conversion using MethylCode Bisulfite Conversion Kit (Life Technologies). Bisulphite-treated DNA molecules were PCR amplified with 10 cycles using KAPA HiFi HotStart Uracil+ ReadyMix (2 × ) (Kapa Biosystems) and purified with Agencourt AMPure XP (Beckman Coulter). Conditions of the sequencing are as described previously (Tsukahara et al, 2012). Raw sequence data were deposited in the DDBJ (DNA Data Bank of Japan) Sequence Read Archive (DRA; accession nos. DRA001060 and DRA001061). Reads were mapped to the reference genome (Release 10 of the Arabidopsis Information Resources) using the Bowtie alignment algorithm (Langmead et al, 2009) with conditions described by Chen et al (2010). Only uniquely mapped reads were used; reads mapped more than once were not used. Annotation of TEs is obtained from TAIR (http://www.arabidopsis.org/), which is based on Buisine et al (2008).
Measurement of DNA methylation was also performed by McrBC digestion and subsequent quantitative PCR as described previously (Teixeria et al, 2009). McrBC (New England Biolabs) digestion was performed on 200 ng of genomic DNA. Digested and undigested DNA samples were quantified as described above. The methylation status was estimated by the loss of long DNA after McrBC digestion.
Transcription analysis
Total RNA was isolated by Promega SV Total RNA Isolation System (cat. #Z3100). Reverse transcription reaction was performed using Takara RNA PCR Kit (RR019A) following the manufacturer’s instructions. Oligo dT-Adaptor primer was used to reverse transcribe predicted products of three coding genes of Hi. GAPC was used as a control. qRT–PCR was performed using SYBR Premix Ex Taq II (Takara) on Thermal Cycler Dice_Real Time System TP800 (Takara), with the following conditions; 95°C for 30′, (95°C for 5′, 60°C for 30′ and 72°C for 30′) for 40 times. The UBC gene (At5g25760) was used as an internal control.
Transgene construction
Full-length Hi was recovered by PCR and cloned into vector PZP2H-lac (Fuse et al, 2001). In most constructs, silent mutations were introduced for each of the three ORFs, so that transcripts from the transgene and endogenous copies could be distinguished between (Figure 1). Transgenes with deletion in each of the three ORFs were generated by PCR using the full-length Hi as the template. Primer sequences for generating these constructs are shown in Supplementary Table S2.
Molecular evolutionary analyses
BLAST searches of the A. lyrata genome were conducted with A. thaliana MULE family sequences as the query against assembled genomes by Phytozome ver 8.0 (Goodstein et al, 2012). A conserved transposase region, analysed by Yu et al (2000), was used for estimation of entire MULE family phylogeny. The neighbour-joining trees were constructed by the p-distance in MEGA 5 (Tamura et al, 2011). To analyse the proliferation rate, copy numbers in recently proliferated clusters were estimated. The p-distance of 0.1 was used as a threshold value to classify each copy to clusters and then numbers of copies in each cluster were counted. Average numbers of copies per cluster were compared between non-TIR MULE families and TIR MULE families by t-test. The sliding window analyses of synonymous and non-synonymous distances between subcluster and between species for VANDAL21 and VANDAL17 families were conducted by DnaSP ver. 5 (Librado and Rozas, 2009). Conserved domain was searched by NCBI conserved domain search site (Marchler-Bauer et al, 2011; http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi).
Supplementary Material
Acknowledgments
We thank Akiko Terui and Kazuya Takashima for technical assistance. Special thanks to Damon Lisch for valuable advice about Mutator and TIR, Yasuhiko Sekine for transposition mechanisms, and Yasushi Hiromi for comments on the manuscript. Supported by grants from Mitsubishi Foundation (to TK), Takeda Science Foundation (to TK), Japanese Ministry of Education, Culture, Sports, Science and Technology (19207002 and 19060014, to TK), Systems Functional Genetics Project of the Transdisciplinary Research Integration Center, ROIS, Japan (to AT, AF, YT, and TK), the Agence Nationale de la Recherche (Project EPIMOBILE, NT09_501422 to VC), and the European Union Network of Excellence Epigenesys (to VC). ME was supported by a PhD studentship from the French Ministry of Research.
Author contributions: AK designed and performed sequence analyses (Figures 7 and 8; Supplementary Figures S10 and S11). ME and VC designed and performed experiments using epi-RILs (Figure 3). YF, TI, AT, AF, YT, and TK designed, performed, and analysed whole-genome bisulphite sequencing and genome re-sequencing experiments. YF, YT, and TK designed and performed all other experiments. YF and TK wrote the paper with incorporating comments from other authors.
Footnotes
The authors declare that they have no conflict of interest.
References
- Bivalkar-Mehla S, Vakharia J, Mehla R, Abreha M, Kanwar JR, Tikoo A, Cahuhan A (2011) Viral RNA silencing suppressors (RSS): novel strategy of viruses to ablate the host RNA interference (RNAi) defense system. Virus Res 155: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettell RI, Dennis ES (1991) Reactivation of a silent Ac following tissue culture is associated with heritable alterations in its methylation. Mol Gen Genet 229: 365–372 [DOI] [PubMed] [Google Scholar]
- Brown WE, Robertson DS, Bennetzen JL (1989) Molecular analysis of multiple Mutator-derived alleles of the Bronze locus of maize. Genetics 122: 439–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J, Sundaresan V (1992) Genetic study of the loss and restoration of Mutator transposon activity in maize: evidence against dominant-negative regulator associated with loss of activity. Genetics 130: 889–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisine N, Quesneville H, Colot V (2008) Improved detection and annotation of transposable elements in sequenced genomes using multiple reference sequence sets. Genomics 91: 467–475 [DOI] [PubMed] [Google Scholar]
- Chandler VL, Walbot V (1986) DNA modification of a maize transposable element correlates with loss of activity. Proc Natl Acad Sci USA 83: 1767–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PY, Cokus SJ, Pellegrini M (2010) BS Seeker: precise mapping for bisulfite sequencing. BMC Bioinformatics 11: 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colomé-Tatché M, Cortijo S, Wardenaar R, Morgado L, Lahouze B, Sarazin A, Etcheverry M, Martin A, Feng S, Duvernois-Berthet E, Labadie K, Wincker P, Jacobsen SE, Jansen RC, Colot V, Johannes F (2012) Features of the Arabidopsis recombination landscape resulting from the combined loss of sequence variation and DNA methylation. Proc Natl Acad Sci USA 109: 16240–16245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Fedoroff NV (2002) Inducible DNA demethylation mediated by the maize Suppressor-mutator transposon-encoded TnpA protein. Plant Cell 14: 2883–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich CR, Cui F, Packila ML, Li J, Ashlock DA, Nikolau BJ, Schnable PS (2002) Maize Mu transposons are targeted to the 5′ untranslated region of the gl8 gene and sequences flanking Mu target-site duplications exhibit nonrandom nucleotide composition throughout the genome. Genetics 160: 697–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA, Benito MI, Walbot V (1994) Sequence similarity of putative transposases links the maize Mutator autonomous element and a group of bacterial insertion sequences. Nucleic Acids Res 22: 2634–2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoroff N (1996) Epigenetic regulation of the maize Spm transposable element. InEpigenetic Mechanisms of Gene Regulation Riggs A, Martienssen R, Russo V (eds), pp575–592 Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press [Google Scholar]
- Fuse T, Sasaki T, Yano M (2001) Ti-plasmid vectors useful for functional analysis of rice genes. Plant Biotechnol 18: 219–222 [Google Scholar]
- Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, Rokhsar DS (2012) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40: D1178–D1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashida S, Uchiyama T, Martin C, Kishima Y, Sano Y, Mikami T (2006) The temperature dependent change in Methylation of the Antirrhinum transposon Tam3 is controlled by the activity of its transposase. Plant Cell 18: 104–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardeman KJ, Chandler VL (1989) Characterization of bz1 mutants isolated from mutator stocks with high and low numbers of Mu1 elements. Dev Genet 10: 460–472 [DOI] [PubMed] [Google Scholar]
- Hoen DR, Park KC, Elrouby N, Yu Z, Mohabir N, Cowan RK, Bureau TE (2006) Transposon-mediated expansion and diversification of a family of ULP-like genes. Mol Biol Evol 23: 1254–1268 [DOI] [PubMed] [Google Scholar]
- Inagaki S, Miura A, Nakamura K, Lu F, Cui X, Cao X, Kimura H, Saze H, Kakutani T (2010) Autocatalytic differentiation of epigenetic modifications within the Arabidopsis genome. EMBO J 29: 3496–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki S, Kakutani T (2013) What triggers differential DNA methylation of genes and TEs: contribution of body methylation? Cold Spring Harb Symp Quant Biol 77: 155–160 [DOI] [PubMed] [Google Scholar]
- Jiang N, Bao Z, Zhang X, Eddy SE, Wessler SR (2004) Pack-MULE transposable elements mediate gene evolution in plants. Nature 431: 569–573 [DOI] [PubMed] [Google Scholar]
- Johnson L, Cao X, Jacobsen S (2003) Interplay between two epigenetic marks. DNA methylation and histone H3 lysine 9 methylation. Curr Biol 12: 1360–1367 [DOI] [PubMed] [Google Scholar]
- Johannes F, Porcher E, Teixeira FK, Saliba-Colombani V, Simon M, Agier N, Bulski A, Albuisson J, Heredia F, Audigier P, Bouchez D, Dillmann C, Guerche P, Hospital F, Colot V (2009) Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet 5: e1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakutani T, Jeddeloh JA, Flowers SK, Munakata K, Richards EJ (1996) Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proc Natl Acad Sci USA 93: 12406–12411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Miura A, Bender J, Jacobsen SE, Kakutani T (2003) Role of CG and non-CG methylation in immobilization of transposons in Arabidopsis. Curr Biol 13: 421–426 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le QH, Wright S, Yu Z, Bureau T (2000) Transposon diversity in Arabidopsis thaliana. Proc Natl Acad Sci USA 97: 7376–7381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452 [DOI] [PubMed] [Google Scholar]
- Lippman Z, Gendrel AV, Black M, Vaughn MW, Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau KD, Carrington JC, Doerge RW, Colot V, Martienssen R (2004) Role of transposable elements in heterochromatin and epigenetic control. Nature 430: 810–813 [DOI] [PubMed] [Google Scholar]
- Liu S, Yeh CT, Ji T, Ying K, Wu H, Tang HM, Fu Y, Nettleton D, Schnable PS (2009) Mu transposon insertion sites and meiotic recombination events co-localize with epigenetic marks for open chromatin across the maize genome. PLoS Genet 5: e1000733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch D (2002) Mutator transposons. Trends Plant Sci 7: 498–504 [DOI] [PubMed] [Google Scholar]
- Lisch D, Chomet P, Freeling M (1995) Genetic characterization of the Mutator system in maize: behavior and regulation of Mu transposons in a minimal line. Genetics 139: 1777–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch D, Girard L, Donlin M, Freeling M (1999) Functional analysis of deletion derivatives of the maize transposon MuDR delineates roles for the MURA and MURB proteins. Genetics 151: 331–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL et al. (2011) CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res 39: (Database issue)D225–D229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martienssen R (1996) Epigenetic silencing of Mu transposable elements in maize. InEpigenetic Mechanisms of Gene Regulation Riggs A, Martienssen R, Russo V (eds ), pp593–608 . Cold Spring Harbor, New York:Cold Spring Harbor Laboratory Press [Google Scholar]
- Martienssen R, Baron A (1994) Coordinate suppression of mutations caused by Robertson's mutator transposons in maize. Genetics 136: 1157–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B (1951) Chromosome organization and genic expression. Cold Spring Harbor Symp Quant Biol 16: 13–47 [DOI] [PubMed] [Google Scholar]
- McClintock B (1958) The suppressor-mutator system of control of gene action in maize. Carnegie Institution of Washington Year Book Vol. 57: 415–429 [Google Scholar]
- Mirouze M, Reinders J, Bucher E, Nishimura T, Schneeberger K, Ossowski S, Cao J, Weigel D, Paszkowski J, Mathieu O (2009) Selective epigenetic control of retrotransposition in Arabidopsis. Nature 461: 427–430 [DOI] [PubMed] [Google Scholar]
- Miura A, Yonebayashi S, Watanabe K, Toyama T, Shimada H, Kakutani T (2001) Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 411: 212–214 [DOI] [PubMed] [Google Scholar]
- Saze H, Kakutani T (2011) Differentiation of epigenetic modifications between transposons and genes. Curr Opin Plant Biol 65: 589–599 [DOI] [PubMed] [Google Scholar]
- Sasaki T, Kobayashi A, Saze H, Kakutani T (2012) RNAi-independent de novo DNA methylation revealed in Arabidopsis mutants of chromatin remodeling gene DDM1. Plant J 70: 750–758 [DOI] [PubMed] [Google Scholar]
- Schläppi M, Raina R, Fedoroff N (1994) Epigenetic regulation of the maize Spm transposable element: novel activation of a methylated promoter by TnpA. Cell 77: 427–437 [DOI] [PubMed] [Google Scholar]
- Schläppi M, Raina R, Fedoroff N (1996) A highly sensitive plant hybrid protein assay system based on the Spm promoter and TnpA protein for detection and analysis of transcription activation domains. Plant Mol Biol 32: 717–725 [DOI] [PubMed] [Google Scholar]
- Singer T, Yordan C, Martienssen RA (2001) Robertson's Mutator transposons in A. thaliana are regulated by the chromatin-remodeling gene Decrease in DNA Methylation (DDM1). Genes Dev 15: 591–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert LE, Chandler VL (1988) Characterization of a highly conserved sequence related to mutator transposable elements in maize. Mol Biol Evol 5: 519–529 [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeria FK, Heredia F, Sarazin A, Roudier F, Boccara M, Ciaudo C, Cruaud C, Poulain J, Berdasco M, Fraga MF, Voinnet O, Wincker P, Esteller M, Colot V (2009) A role for RNAi in the selective correction of DNA methylation defects. Science 323: 1600–1604 [DOI] [PubMed] [Google Scholar]
- Tsukahara S, Kobayashi A, Kawabe A, Mathieu O, Miura A, Kakutani T (2009) Bursts of retrotransposition reproduced in Arabidopsis. Nature 461: 423–426 [DOI] [PubMed] [Google Scholar]
- Tsukahara S, Kawabe A, Kobayashi A, Ito T, Aizu T, Shin-i T, Toyoda A, Fujiyama A, Tarutani Y, Kakutani T (2012) Centromere-targeted de novo integrations of an LTR retrotransposon of Arabidopsis lyrata. Genes Dev 26: 705–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8: 973–982 [DOI] [PubMed] [Google Scholar]
- Yu Z, Wright SI, Bureau TE (2000) Mutator-like elements in Arabidopsis thaliana. Structure, diversity and evolution. Genetics 156: 2019–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore PD (2004) Plant RNAi: How a viral silencing suppressor inactivate siRNA. Curr Biol 14: R198–R200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.