

Abstract
The matrix metalloproteinases (MMPs) play a pivotal role in adverse left ventricular (LV) myocardial remodeling. The transmembrane protein extracellular MMP inducer (EMMPRIN) causes increased MMP expression in vitro, and elevated levels occur in patients with LV failure. However, the direct consequences of a prolonged increase in the myocardial expression of EMMPRIN in vivo remained unexplored. Cardiac-restricted EMMPRIN expression (EMMPRINexp) was constructed in mice using the full-length human EMMPRIN gene ligated to the myosin heavy chain promoter, which yielded approximately a twofold increase in EMMPRIN compared with that of the age/strain-matched wild-type (WT) mice; EMMPRINexp (n = 27) and WT (n = 33) mice were examined at 3.2 ± 0.1 or at 13.3 ± 0.5 mo of age (n = 43 and 26, respectively). LV end-diastolic volume (EDV) was similar in young EMMPRINexp and WT mice (54 ± 2 vs. 57 ± 3 μl), but LV ejection fraction (EF) was reduced (51 ± 1 vs. 57 ± 1%; P < 0.05). In old EMMPRINexp mice, LV EDV was increased compared with WT mice values (76 ± 3 vs. 58 ± 3 μl; P < 0.05) and LV EF was significantly reduced (45 ± 1 vs. 57 ± 2%; P < 0.05). In EMMPRINexp old mice, myocardial MMP-2 and membrane type-1 MMP levels were increased by >50% from WT values (P < 0.05) and were accompanied by a twofold higher collagen content (P < 0.05). Persistent myocardial EMMPRINexp in aging mice caused increased levels of both soluble and membrane type MMPs, fibrosis, and was associated with adverse LV remodeling. These findings suggest that EMMPRIN is an upstream signaling pathway that can play a mechanistic role in adverse remodeling within the myocardium.
Keywords: ventricular function
left ventricular (LV) remodeling, defined as changes in the structure and geometry of the LV myocardial wall and chamber, invariably occurs with cardiovascular disease states such as myocardial infarction, hypertension, and cardiomyopathy, as well as a function of age (1, 5, 8, 13, 33). An increased expression of a family of matrix proteases, termed the matrix metalloproteinases (MMPs), occurs in patients and animals with the development and progression LV remodeling (6, 11, 12, 14, 16, 18, 22, 26, 39, 40, 42, 47). Moreover, with the use of transgenic and pharmacological approaches, a cause-effect relationship has been demonstrated between the induction of MMPs and adverse LV remodeling (6, 12, 16, 19, 22, 26, 40). There are a large number of MMP types that are expressed within the myocardium and include the soluble MMP types, such as MMP-2 and MMP-9, as well as the membrane-bound types such as membrane type-1 (MT1)-MMP (28, 38, 48). Ubiquitous signaling molecules such as bioactive peptides, cytokines, and reactive oxygen species have all been clearly demonstrated to induce this portfolio of MMPs in vitro (7, 34, 36, 38). Although the unique functionality for each of these MMP types with respect to the LV remodeling process is beginning to be realized, upstream signaling pathways, which may selectively induce MMPs, remain poorly understood. A transmembrane protein, termed the extracellular MMP inducer (EMMPRIN), was initially described to cause increased MMP transcription in vitro (15, 46). An association between EMMPRIN and MMP induction in tissue remodeling processes such as tumor metastasis has been reported (18, 21, 46). Moreover, increased levels of EMMPRIN have been identified within the myocardium of patients with severe LV remodeling (39). However, whether the increased myocardial induction of EMMPRIN, in and of itself, can cause MMP induction and adverse LV remodeling remains to be established. Accordingly, the present study tested the central hypothesis that persistently elevated EMMPRIN levels within the myocardial compartment of mice, to levels that have been observed previously within the failing human myocardium (39), would adversely affect LV geometry and structure. The present study measured LV function and geometry, relative myocardial MMP content and activity, and collagen content in young and old mice following the selective and persistent myocardial overexpression of EMMPRIN.
METHODS
EMMPRIN overexpression.
A myocardial restricted overexpression construct of EMMPRIN [EMMPRINexp; α-myosin heavy chain promoter (MHC) linked to full-length human EMMPRIN] was established in mice from the C57 background strain. The EMMPRIN full-length human gene sequence (Genebank accession no. NM 001728; 1.6 kb) (15, 42) was cloned into the α-MHC construct (Clone 26, Genebank u71441; courtesy of Jeff Robbins, University of Cincinnati) (42). The incorporation of the MHC-EMMPRIN construct was confirmed by using a PCR protocol from tail-clip DNA. Briefly, digested DNA (2 μg) was subjected to PCR (37 cycles; Bio-Rad iCycler; 170-8720XTU; Hercules, CA) using a forward primer (5′-GGCCAGAAAACGGAGTTCAA-3′; Sigma-Genosys) and a reverse primer (5′-GCGCTTCTCGTAGATGAAGA-3′; Sigma-Genosys) specific to the EMMPRIN transgene. Southern blot analysis was performed to confirm the presence of the transgene. Three independent lines of EMMPRINexp mice were developed, and following backcrossing and stable breeding patterns, ∼50% of each litter was EMMPRINexp positive. The EMMPRINexp-negative mice were used as reference, wild-type (WT) sibling controls. The EMMPRINexp mice displayed no obvious phenotypic abnormalities with respect to growth and developmental characteristics. All animals were treated and cared for in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85-23, Revised 1996). This study was approved by the Medical University of South Carolina Institutional Animal Care and Use Committee (AR1590).
Aging studies.
EMMPRINexp and WT mice were maintained until reaching ∼3 (young) or 13 (old) mo of age, and then LV echocardiography was performed as described in Echocardiography. The latter age category was chosen since past studies have identified that by ∼12 mo old, mice begin to display age-dependent changes in wound healing, immune response, and myocardial remodeling/metabolism (9, 17, 20, 52).
Echocardiography.
For all of the mice included in these studies, transthoracic echocardiography was performed to measure LV geometry and function (19, 26). For these studies, the mice were anesthetized with isoflurane (2% in oxygen) and maintained at ambient body temperature with a heating blanket. Heart rate was determined from a surface electrocardiogram. Ambient heart rate was maintained between 400 and 500 beats/min, and the mice remained normothermic throughout the procedure. Two-dimensional M-mode echocardiographic recordings were obtained using a 40-MHz scanning head with a spatial resolution of 30 μm (Vevo 770; VisualSonics). With the use of long-axis views, LV end-diastolic volume, ejection fraction, and mass were computed (20). Following these studies, the isoflurane was increased to 5%, and the LV was then harvested, quickly weighed, flash frozen, and stored at −70°C for subsequent biochemistry and immunohistochemistry.
EMMPRIN/MMP quantitation.
The relative content of myocardial EMMPRIN was determined by immunoblotting utilizing two distinct approaches. First, an antisera that recognizes both human and mouse EMMPRIN was utilized in quantitative immunoblotting as detailed below. Second, an antisera that recognizes only human EMMPRIN was utilized in parallel immunoblotting studies. In this manner, the specificity and stability of expressing human EMMPRIN within the myocardial compartment could be confirmed, as well as determining the overall increase in total myocardial EMMPRIN achieved in this mouse construct. For immunoblotting, myocardial extracts (10 μg) were loaded onto 4–12% Bis-Tris gels and subjected to electrophoretic separation. The separated proteins were then transferred to a nitrocellulose membrane. Following a blocking and washing step, the membranes were incubated for 30 min at 37°C in mouse antisera directed against amino acids 1–190 of EMMPRIN (1:5,000 dilution; B-5; Santa Cruz Biotechnology) or in rabbit antisera directed specifically against the NH2 terminus of human EMMPRIN (1:5,000 dilution; H-200; Santa Cruz). Initial immunoblotting studies demonstrated that this dilution of the mouse antisera recognized both the human and mouse form of EMMPRIN, whereas there was no cross-reactivity of the rabbit antisera against WT mouse LV extracts. In addition, the relative abundance of the gelatinases (MMP-2 and MMP-9) were performed on LV myocardial extracts using quantitative zymography as described previously (16, 19, 22, 26, 39, 40). Finally, immunoblotting was performed for MMP-13 (1:2,500; No. 3533; BioVision), the predominant rodent interstitial collagenase, and MT1-MMP (1:2,500; No. AB815; Chemicon). Recombinant standards (MT1, Calbiochem No. 475937; EMMPRIN, Santa Cruz-2215; and MMP-13, Biomol No. SE-246) were included on the immunoblots as positive controls.
Myocardial MMP activity.
LV myocardial extracts (10 μg) were incubated with a global MMP fluorogenic substrate (OmniMMP Fluorogenic Substrate, P-126 Biomol) that is cleaved by all active MMPs with equivalent kinetics (10, 29). This MMP fluorogenic peptide will only yield a detectable UV emission when proteolytically processed at a specific amino acid sequence, and the specificity of this approach has been demonstrated previously (10). The LV myocardial extracts were incubated (37°C; 2 h) in the presence and absence of the MMP substrate, and excitation/emission was recorded (328/393; FluoStar Galaxy, BMG Labtech). To convert the fluorescent readings from this in situ assay to relative MMP activity, a recombinant active MMP construct (MMP-2/-9 SE-244/237; Biomol) was utilized. Increasing concentrations of the known MMP-2/-9 catalytic domain (0.15–2.5 ng/ml) were incubated with the MMP substrate to obtain a linear relation between MMP activity and fluorescence. The linear portion of the fluorescence emission-concentration curve (y = 26461x; r2 = 0.99; P < 0.0001) was used to calibrate subsequent fluorescent emission values obtained from the LV myocardial extracts and determine a final MMP activity value (nanograms of MMP catalytic activity per time).
Collagen biochemistry.
Total myocardial collagen was determined by quantifying hydroxyproline content in acid-hydrolyzed samples (40). This biochemical assay was optimized for murine myocardial samples. Briefly, 25 mg of LV myocardial free wall was hydrolyzed overnight in hydrochloric acid. The hydrolyzed samples were reacted with Ehrlich's reagent, measured spectrophotometrically (550 nm), converted to hydroxyproline values using a linearized set of hydroxyproline standards, and expressed as nanograms hydroxyproline per milligrams of myocardial wet weight.
EMMPRIN immunohistochemistry.
LV frozen sections were cut at 9 μm thickness and placed on glass slides. The sections were then immersed in ice-cold acetone for 5 min and washed vigorously with phosphate-buffered saline (PBS). The LV sections were then incubated in 10% normal goat serum for 90 min at room temperature, washed in PBS, and then subsequently incubated in the rabbit antisera directed specifically against the NH2 terminus of human EMMPRIN (H-200; 1:50 dilution) overnight at 4°C. The LV sections were then washed five times with PBS and incubated in 0.3% H2O2/PBS for 30 min. The LV sections were washed three times with PBS and incubated in goat anti-rabbit sera (No. 6101; 1:250 dilution; Vector) for 30 min at room temperature. The LV sections were then subjected to the avidin-biotin reaction (ABC Kit; Vectastain Elite; Vector), and specifically bound EMMPRIN antisera was visualized. The LV sections were then imaged on a Zeiss Axioskop 2 microscope (Plan-Neofluar 63x/1, 25), and images were digitized (Zeiss AxioCam MRc). Negative controls included the substitution of the primary antisera with nonimmune rabbit serum.
Data analysis.
Indexes of LV function and geometry were subjected to a two-way ANOVA in which the treatment effects were EMMPRIN overexpression and age. Pairwise comparisons were performed using a Bonferroni adjusted t-test. Due to myocardial sampling limitations, subsets of mice from the four groups were utilized in the biochemistry and immunohistochemistry studies. Mice were randomized to the different assay protocols following the echocardiographic study. Final sample sizes for each assay are indicated. For the EMMPRIN, MMP, and collagen biochemistry studies, all measurements were performed in duplicate. The zymographic/immunoreactive signals were analyzed using densitometric methods (Gel Pro Analyzer; Media Cybernetics) to obtain two-dimensional integrated optical density (IOD) values. The hydroxyproline values and the EMMPRIN IOD values were subjected to ANOVA and Bonferroni separation. The IOD values were also computed, where appropriate, as a percentage of WT referent control values, where the control values were set to 100%, and comparisons were performed by a separate t-test. Between-group differences in these values were compared using a two-sample t-test. Results are presented as means ± SE. Values of P < 0.05 were considered statistically significant. All statistical procedures were performed using the STATA statistical software package (Statacorp, College Station, TX).
RESULTS
LV function and geometry.
LV function and geometry were assessed by echocardiography in 3 mo (young; 3.2 ± 0.1 mo) WT (n = 33) and EMMPRINexp (n = 27) mice and compared with WT (old; 13.3 ± 0.5 mo; n = 26) and EMMRPINexp (n = 43) mice. Representative long-axis, two-dimensional LV echocardiograms are shown in Fig. 1. Summary results for all of the echocardiographic measurements of LV volumes, function, and mass are shown in Fig. 2. LV end-diastolic volume and mass were similar between the WT and EMMPRINexp young groups, but LV ejection fraction was reduced in the EMMPRINexp group. In the old WT mice, LV end-diastolic volume and ejection fraction were similar to that of young WT mice, but LV mass was increased. In the EMMPRINexp old group, LV end-diastolic volume and mass were significantly increased over respective WT old and young values. Moreover, LV ejection fraction was significantly reduced in the old EMMPRINexp mice when compared with respective old and young values. Thus persistent myocardial EMMPRINexp caused LV dilation, pump dysfunction, and hypertrophy in aging mice.
Fig. 1.
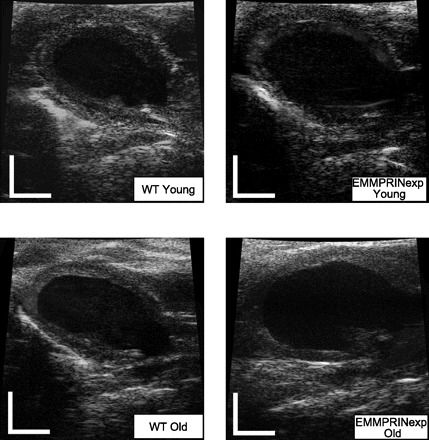
Representative left ventricular (LV) echocardiograms taken from the long-axis, parasternal approach in a young wild-type (WT) mouse, a young extracellular matrix metalloproteinase (MMP) inducer expression (EMMPRINexp) mouse, an old WT mouse, and an old EMMPRINexp mouse. Significant LV dilation was evident in the old EMMPRINexp group, and the summary findings are presented in Fig. 2. Scale bars = 2 mm.
Fig. 2.
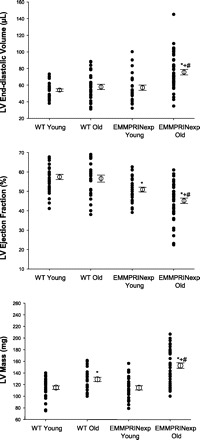
Summary results from the LV echocardiograms for LV end-diastolic volume, ejection fraction, and mass for young (n = 33) and old (n = 26) WT and young (n = 27) and old (n = 43) EMMPRINexp mice. LV end-diastolic volume was significantly increased in the old EMMPRINexp group when compared with respective young or old WT values. LV ejection fraction was lower in the young EMMPRINexp group and fell significantly in the old EMMPRINexp group. LV mass increased in the old WT group and was increased further in the old EMMPRINexp group. P < 0.05 *vs. young WT; #vs. old WT; +vs. young EMMPRINexp.
EMMPRIN and MMP levels.
The results from the EMMPRIN immunoblotting studies are summarized in Fig. 3. Immunoreactive signals for EMMPRIN were identified in LV extracts taken from both WT and EMMPRINexp mice. However, the total EMMPRIN signal was increased by approximately twofold in both the young and old EMMPRINexp mice. Moreover, the increased levels of myocardial EMMPRIN in the EMMPRINexp mice were due to a specific induction of human EMMPRIN. Representative zymograms for MMP-2 and quantitative data for young and old WT and EMMPRINexp mice are shown in Fig. 4. Total MMP-2 levels were reduced in the old WT mice but were substantially increased in the old EMMPRINexp group. The size fractionation of the zymographic signal into the proMMP-2 (72 kDa) and active MMP-2 (64 kDa) form demonstrated increased levels of both in the old EMMPRINexp group. In contrast with MMP-2 levels, MMP-9 was reduced in the old WT mice and was significantly reduced in both the young and old EMMPRINexp groups (Fig. 4). MMP-13 levels were slightly reduced in the EMMPRINexp groups, but this did not reach statistical significance (Fig. 5). However, MT1-MMP myocardial levels were increased in both the young and old EMMPRINexp old groups (Fig. 5).
Fig. 3.
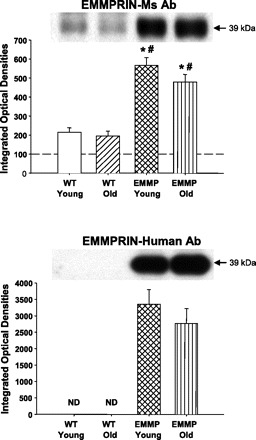
EMMPRIN immunoblotting was performed in young (n = 8) and old (n = 6) WT and young (n = 6) and old (n = 10) EMMPRINexp LV myocardial extracts using 2 different antisera: a mouse EMMPRIN antibody (Ab; EMMPRIN Ms/Human) that recognized both mouse and human EMMPRIN (top) and a rabbit antibody that recognized only human EMMPRIN (EMMPRIN Human; bottom). A significant increase in total LV myocardial EMMPRIN was observed in the EMMPRINexp LV myocardial extracts, which was primarily due to the specific overexpression of human EMMPRIN. P < 0.05 *vs. young WT; #vs. old WT. ND, not determined.
Fig. 4.
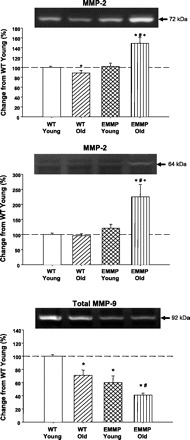
Gelatin zymography was performed on LV myocardial extracts taken from young (n = 14) and old (n = 12) WT and young (n = 12) and old (n = 17) EMMPRINexp mice to identify the abundance and size fractionation for MMP-2 and MMP-9. A significant decrease in the proform of MMP-2 (72 kDa) was observed in old WT mice but was increased in the old EMMPRINexp mice (top). Moreover, an overall increase in the active form of MMP-2 (64 kDa) was observed in the old EMMPRINexp mice (middle). An overall reduction in MMP-9 abundance was observed in the old WT and young EMMPRINexp mice and was further reduced in the old EMMPRINexp mice (bottom). P < 0.05 *vs. young WT; #vs. old WT; +vs. young EMMPRINexp.
Fig. 5.
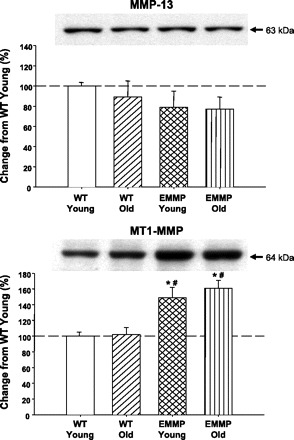
MMP-13 and membrane type (MT)-1-MMP levels were determined by immunoblotting on LV myocardial extracts taken from young WT (n = 14) and old (n = 12) WT and young (n = 12) and old (n = 17) EMMPRINexp mice. There were no changes in relative MMP-13 levels between groups. However, MT1-MMP myocardial levels were significantly increased in the EMMPRINexp mice. P < 0.05 *vs. young WT; #vs. old WT.
Myocardial MMP activity.
Total myocardial MMP activity was determined in crude LV extracts from all groups utilizing a caged fluorogenic peptide where the fluorescence emission was transformed to units of MMP catalytic activity (Fig. 6). Fluorescence emission values obtained in all myocardial extracts fell within the linear portion of the emission-activity curve generated using recombinant active MMP standards. In the old EMMPRINexp group, LV myocardial MMP activity was increased by approximately twofold compared with WT values.
Fig. 6.
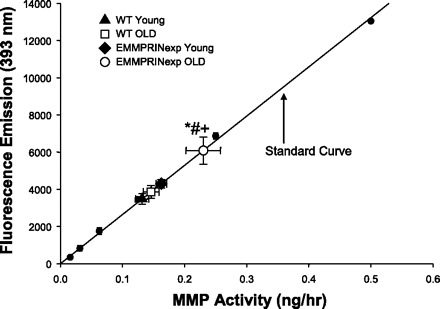
LV myocardial MMP activity was computed by incubating crude myocardial extracts with a global MMP fluorogenic substrate and measuring fluorescent emission. The mean values for LV myocardial MMP activity in the young WT (n = 14) and old (n = 12) WT and young (n = 12) and old (n = 17) EMMPRINexp mice have been plotted with respect to the standard curve. MMP activity was significantly increased in the old EMMPRINexp myocardial samples compared with WT and young EMMPRINexp values. P < 0.05 *vs. young WT; #vs. old WT; +vs. young EMMPRINexp.
Myocardial collagen content.
LV hydroxyproline was similar between the young WT and EMMPRINexp groups (11.9 ± 0.7 vs. 14.3 ± 1.2 ng/mg; n = 8 and 6, respectively) and also similar in the old WT group (13.0 ± 0.7 ng/mg; n = 5). However, LV myocardial hydroxyproline increased significantly in the old EMMPRINexp group compared with either young WT, old WT, or young EMMPRINexp values (26.0 ± 1.6 ng/mg; n = 10; P < 0.05).
EMMPRIN immunohistochemistry.
As with the immunoblotting experiments, the antisera directed against the NH2 terminus of human EMMPRIN failed to produce a positive signal by immunohistochemistry in WT sections. A clear positive signal for EMMPRIN could be detected in the EMMPRINexp mice, which demonstrated a distribution primarily along the sarcolemmal surface of myocytes. Representative images taken from WT and EMMPRINexp old LV sections are shown in Fig. 7. At a higher magnification, a distinct EMMPRIN staining pattern emerged around the sarcolemmal-interstitial interface. Negative controls yielded an absence of a positive immunoreactive signal. Thus, in this EMMPRINexp construct, human EMMPRIN could be identified along the membrane surface of myocytes, consistent with this transmembrane glycoprotein.
Fig. 7.
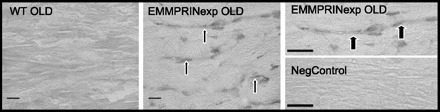
Representative LV sections taken from WT (n = 3) and EMMPRINexp old (n = 6) mice, which were stained using a selective human EMMPRIN antisera. In WT and negative control sections (Neg Control), a complete absence of positive EMMPRIN staining was observed. However, in EMMPRINexp sections, a specific immunoreactive signal was detected along the sarcolemmal surface of myocytes (arrows). At high power (top right), EMMPRIN immunostaining could be appreciated to localize to the sarcolemmal-interstitial interface (arrows). Scale bar = 20 μm.
DISCUSSION
Changes in the expression and activity of the large family of MMPs have been well documented in animal models and in clinical studies of LV remodeling (1, 3–5, 8, 13, 16, 33, 38, 40, 42, 47). Moreover, transgenic models and pharmacological MMP inhibition have demonstrated a cause-effect relation between MMP activity and adverse LV remodeling (7, 13, 36, 47). However, the expression patterns and functionality of the different classes and subtypes of MMPs are quite diverse and are likely to be regulated by specific upstream signaling pathways (7, 14, 34, 36, 38, 47, 49). One such upstream signaling pathway, which appears to be unique for MMPs, is the transmembrane protein, termed EMMPRIN (15, 18, 21, 46, 53). Although past studies have reported associative changes between EMMPRIN levels and adverse tissue remodeling such as tumor metastasis (21, 23, 45, 46, 50, 51, 53), the functional and structural consequences of the cardiac-restricted EMMPRINexp had not been explored previously. In the present study, cardiac-restricted EMMPRINexp was induced in mice and the effects on LV structure and function were examined in mice at ∼3 and 13 mo of age. The relative increase in total myocardial EMMPRIN achieved by EMMPRINexp was similar to that previously measured in patients with severe LV failure (39). Unique findings from this set of investigations were threefold. First, in old EMMPRINexp mice, significant LV remodeling and systolic dysfunction occurred compared with those of age-matched WT controls. Second, EMMPRINexp caused a specific induction of MMP-2 and MT1-MMP within the LV myocardium, which was associated with heightened myocardial MMP activity. Finally, LV myocardial collagen content was significantly increased in old EMMPRINexp mice. Taken together, the results from this study suggest that a persistent myocardial induction of EMMPRIN, to levels that have been observed in patients with severe LV failure and following myocardial infarction (31, 39), can directly contribute to adverse LV remodeling.
EMMPRIN, which has also been known as basigen, CD147, M6, and tumor cell-derived collagenase stimulatory factor, is a glycoprotein that belongs to the immunoglobin superfamily (15, 18, 46). EMMPRIN is a transmembrane protein that has been shown to be heavily enriched on the plasma membrane of cancer cells from a number of different cancer types (21, 23, 45, 50, 51, 53). For example, a robust increase in EMMPRIN has been observed in samples taken from lung, bladder, and renal carcinoma (46). Despite the nomenclature for EMMPRIN, this transmembrane protein has been associated with a number of diverse molecular events, which include tumor cell growth, changes in cell viability such as anoikis, and tumor angiogenesis (21, 23, 45, 46, 50, 51, 53). For example, in a study by Yang et al. (51), it was demonstrated that in breast cancer cells expressing high levels of EMMPRIN, cell size was increased and anoikis was decreased compared with those of cells expressing low levels of EMMPRIN. Thus there is in vitro evidence to suggest that increased EMMPRIN will affect biological processes independent of MMP induction such as cell growth and viability. In the present study, EMMPRINexp within the myocardial compartment for short periods of time (weeks) did not appear to affect myocardial growth, since there was no change in LV mass from WT values. However, with chronic myocardial EMMPRINexp (months), LV mass significantly increased from age-matched WT values. This robust LV hypertrophic response is likely due to several mechanisms. First, the persistent myocardial EMMPRINexp and subsequent MMP induction caused LV dilation and, by extension, increased LV wall stress. The increased LV wall stress, in turn, would serve as a mechanical stimulus for myocardial hypertrophy. Finally, persistent myocardial EMMPRINexp may have had a direct effect of EMMPRIN on myocardial cell growth.
EMMPRIN and MMP induction.
Although the effects of EMMPRIN on tissue remodeling and cell physiological process are likely to be diverse, one predominant feature of this protein is the induction of MMPs (15, 18, 21, 23, 45, 46, 49–51, 53). The MMPs constitute a large family of proteolytic enzymes with subclasses that include the collagenases (i.e., MMP-1, MMP-13), the gelatinases (MMP-2, MMP-9), the stromelysins (MMP-3), and the membrane-type MMPs, such as MT1-MMP (16, 38, 39, 40, 47). It is becoming apparent that there is a diverse enzymatic portfolio for these MMP types, as well as different regulatory mechanisms. One critical control point for MMP synthesis and release is transcriptional regulation, and the promoter region is unique for each MMP type (28, 49). Past in vitro studies have shown that increased EMMPRIN exposure to either cancer or stromal fibroblasts results in a specific rather than a global induction of MMPs (15, 16, 31, 45, 50). For example, increased levels of EMMPRIN from epithelial sarcoma cells caused an induction of MMP-2, but not MMP-9, in peritumoral fibroblasts (23). In an in vivo study, breast cancer clones transfected with EMMPRIN caused an induction of MMP-2 in harvested tissue extracts (53). Moreover, in glioma cell or monocyte cultures, EMMPRIN induced the expression of MT1-MMP (30, 31). In the present study, myocardial EMMPRINexp caused an increase in the levels of specific MMP types, notably MMP-2 and MT1-MMP. The association between the upregulation of these specific MMP types by EMMPRIN is of potential importance for several reasons. First, through the use of a transgenic construct, it has been demonstrated that MMP-2 likely contributes to adverse LV remodeling (26) and is significantly upregulated within the myocardium of patients and animals with LV failure (6, 12, 16, 19, 22, 38–40). Second, MT1-MMP is proteolytically diverse and can likely process a number of matrix proteins and signaling molecules involved in LV remodeling (14, 37, 38, 44). The increased levels of MT1-MMP observed in the present study caused by EMMPRINexp are similar to those found in patients with end-stage LV failure (39). Finally, an important pathway for the conversion of proMMP-2 to active MMP-2 is mediated by MT1-MMP (14, 37, 38, 41). In the present study, long-term myocardial EMMPRINexp was associated with increased MT1-MMP levels as well as higher levels of active MMP-2, which in turn caused higher MMP activity within the myocardium. These past observations along with the present results suggest that EMMPRIN induces a specific portfolio of MMPs within the myocardium, which in turn may contribute to the progression of LV remodeling and failure.
EMMPRIN signaling and activation.
Although it is recognized that EMMPRIN is a transmembrane protein, and increased levels have been associated with MMP induction, the precise cell-signaling pathway remains to be established. However, through the use of recombinant constructs of specific EMMPRIN domains and coculture experiments, it is apparent that the signaling pathway for this glycoprotein is complex (15, 21, 23, 45, 46, 50, 51, 53). One uniform finding is that the soluble extracellular domain of EMMPRIN is capable of interacting with membrane-bound EMMPRIN on nearby stromal cells, which in turn can cause the induction of MMPs (23, 45, 50, 51). For example, Tang et al. (45) reported that the exposure of a soluble EMMPRIN construct induced MMP-2 expression in tumor-associated fibroblasts. Moreover, past studies have suggested that membrane-bound EMMPRIN can undergo proteolytic processing by MMPs to yield a soluble form, which acts in a pericellular fashion (23, 45). These findings suggest that EMMPRIN may act in a paracrine fashion, through interaction with membrane-bound EMMPRIN on nearby cells, such as endothelial cells and fibroblasts. In the present study, human EMMPRIN overexpression was driven by the MHC, which would in effect limit the expression to the cardiac myocyte. However, abundant native mouse EMMPRIN was detected within the myocardium of both WT and transgenic mice. Thus it is likely that enhanced EMMPRIN signaling occurred in all myocardial cell types. However, this issue remains speculative and warrants further study. The intracellular signaling pathways, which are activated following EMMPRIN engagement, are an area of active investigation (2, 31, 32, 51). However, it does appear that the activation of mitogen-activated protein kinases (MAPKs) is an essential intracellular event for mediating the effects of EMMPRIN (2, 32, 51). Another research avenue is to identify upstream stimuli that induce EMMPRIN, which in turn cause MMP upregulation. In a study by Siwik et al. (35), it was demonstrated that the stimulation of the β-adrenergic receptor on isolated rat ventricular myocytes caused an upregulation of EMMPRIN, which in turn increased MMP production. Past studies have reported previously that biologically active molecules such as cytokines, as well as oxidative stress, can induce MMPs in cardiac fibroblasts and myocytes (7, 34, 36). Thus, although it remains speculative, it is likely that increased EMMPRIN levels can be induced by biological signaling cascades that are operative in the context of LV remodeling and failure. Indeed, increased EMMPRIN levels have been reported in patients with cardiomyopathic disease or with myocardial infarction, conditions in which several of these signaling cascades would be operative (31, 39). The findings from the present study provided new mechanistic evidence that increased myocardial levels of EMMPRIN, in and of itself, can cause LV remodeling and failure. Accordingly, identifying the upstream signaling pathways causative for EMMPRIN induction with cardiovascular disease as well as the intracellular signaling pathways by which EMMPRIN induces MMPs would be important avenues of further research.
EMMPRIN and myocardial remodeling.
In the present study, persistent EMMPRINexp caused LV dilation and subsequent dysfunction in aging mice. The LV dilation was likely due to EMMPRIN-mediated MMP induction and subsequently increased MMP activity. Indeed, the present study directly demonstrated that prolonged EMMPRINexp was associated with increased myocardial MMP activity. This increased MMP activity would in turn cause a loss of myocardial matrix stability and myocyte adhesion, which would favor myocyte remodeling and ultimately changes in LV chamber geometry. One of the findings from the present study was that EMMPRINexp was associated with increased total collagen content. Increased myocardial collagen content occurred in the old EMMPRINexp mice, when the determinants of collagen degradation were also increased, suggesting that collagen synthesis rates were higher than overall degradation rates. There are several likely contributory factors for this shift in steady-state collagen content with EMMPRINexp. First, the induction of MMP-2 and MT1-MMP by EMMPRIN would heighten pericellular matrix proteolysis, change local cell-matrix interactions, and thereby affect steady-state synthesis rates. For example, with chronic volume overload, it has been reported that the initial increase in MMP activation and collagen degradation was followed and then followed by collagen accumulation and the progression to LV failure (3, 4). Second, the increased MT1-MMP levels induced by EMMPRIN may cause the proteolysis and the activation of several profibrotic signaling molecules (28, 38, 44). Specifically, MT1-MMP can proteolytically process cytokines and chemokines, which in turn would stimulate collagen synthetic pathways (44). Finally, enhanced EMMPRIN signaling likely caused the heightened activation of intracellular pathways, such as MAPKs, which in turn would cause the formation of transcription factors favoring collagen synthesis (2, 32). Thus, although it remains speculative, it is likely that the increased myocardial collagen content that occurred with prolonged myocardial EMMPRINexp was due to a summation of these proposed mechanisms. The increased myocardial content that occurred in the EMMPRINexp mice would likely yield functional consequences. For example, increased myocardial accumulation within the aging myocardium would increase myocardial stiffness and, in turn, impede diastolic performance. Based upon the findings from this study, future studies that directly examine the physiological consequences of myocardial matrix accumulation in the EMMPRINexp mouse would be warranted.
Study limitations.
The present study utilized a cardiac overexpression model of EMMPRIN driven by a MHC, which achieved an induction of EMMPRIN within the myocardium similar to what has been reported previously in severe LV failure in patients (39). However, it must be recognized that this approach has several limitations. First, this EMMPRIN overexpression approach can be considered artificial and somewhat simplistic in that it is being induced in the absence of an underlying cardiac pathological condition. Nevertheless, this approach provides the means to examine whether the myocardial-restricted overexpression of EMMPRIN would, in and of itself, cause changes in LV myocardial structure and function. Finally, this model system overexpressed the human form of EMMPRIN, and the degree of interaction between human EMMPRIN with the native murine EMMPRIN remains unclear. However, recombinant forms of EMMPRIN have been shown to interact with native, full-length EMMPRIN in several cell systems (23, 31, 45, 51). The present study examined the effects of increased myocardial levels of EMMPRIN, but the selective reduction of this transmembrane molecule was not examined. EMMPRIN gene deletion causes infertility and failure of implantation (24), and therefore utilizing a global deletion model would be problematic. Thus, based upon past studies identifying increased EMMPRIN levels in the failing human myocardium and the results from the present study, more targeted and selective strategies to selectively interrupt EMMPRIN myocardial expression and/or signaling would be warranted.
GRANT
This study was supported by a merit award from the Veterans Affairs Health Administration (to F. G. Spinale).
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Anand IS, Florea VG. Alternations in ventricular structure: role of left ventricular remodeling. In: Heart Failure—A Companion to Braunwald's Heart Disease, edited by Mann BL. Philadelphia, PA: Saunders, 2004, p. 229–245.
- 2.Boulos S, Meloni BP, Arthur PG, Majda B, Bojarski C, Knuckey NW. Evidence that intracellular cyclophilin A and cyclophilin A/CD147 receptor-mediated ERK1/2 signalling can protect neurons against in vitro oxidative and ischemic injury. Neurobiol Dis 25: 54–64, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Brower GL, Chancey AL, Thanigaraj S, Matsubara BB, Janicki JS. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am J Physiol Heart Circ Physiol 283: H518–H525, 2002 [DOI] [PubMed] [Google Scholar]
- 4.Brower GL, Janicki JS. Contribution of ventricular remodeling to pathogenesis of heart failure in rats. Am J Physiol Heart Circ Physiol 280: H674–H683, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Burlew BS. Diastolic dysfunction in the elderly—the interstitial issue. Am J Geriatr Cardiol 13: 29–38, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Chancey AL, Brower GL, Peterson JT, Janicki JS. Effects of matrix metalloproteinase inhibition on ventricular remodeling due to volume overload. Circulation 105: 1983–1988, 2002 [DOI] [PubMed] [Google Scholar]
- 7.Coker ML, Jolly JR, Joffs C, Etoh T, Holder JR, Bond BR, Spinale FG. Matrix metalloproteinase expression and activity in isolated myocytes after neurohormonal stimulation. Am J Physiol Heart Circ Physiol 281: H543–H551, 2001 [DOI] [PubMed] [Google Scholar]
- 8.Colucci WS, Braunwald E. Pathophysiology of heart failure. In: Braunwald's Heart Disease (7th ed.), edited by Zipes DP, Libby P, Bonow RO, and Braunwald E. Philadelphia, PA: Saunders, 2005, p. 509–538.
- 9.Danon D, Kowatch MA, Roth GS. Promotion of wound repair in old mice by local injection of macrophages. Proc Natl Acad Sci USA 86: 2018–2020, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deschamps AM, Apple KA, Leonardi AH, McLean JE, Yarbrough WM, Stroud RE, Clark LL, Sample JA, Spinale FG. Myocardial interstitial matrix metalloproteinase activity is altered by mechanical changes in LV load: interaction with the angiotensin type 1 receptor. Circ Res 96: 1110–1118, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Deschamps AM, Yarbrough WM, Squires CE, Allen RA, McClister DM, Dowdy KB, McLean JE, Mingoia JT, Sample JA, Mukherjee R, Spinale FG. Trafficking of the membrane type-1 matrix metalloproteinase in ischemia and reperfusion: relation to interstitial membrane type-1 matrix metalloproteinase activity. Circulation 111: 1166–1174, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest 106: 55–62, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukuta H, Little WC. Contribution of systolic and diastolic abnormalities to heart failure with a normal and a reduced ejection fraction. Prog Cardiovasc Dis 49: 229–240, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Guo C, Piacentini L. Type I collagen-induced MMP-2 activation coincides with up-regulation of membrane type 1-matrix metalloproteinase and TIMP-2 in cardiac fibroblasts. J Biol Chem 278: 46699–46708, 2003 [DOI] [PubMed] [Google Scholar]
- 15.Guo H, Zucker S, Gordon MK, Toole BP, Biswas C. Stimulation of matrix metalloproteinase production by recombinant extracellular matrix metalloproteinase inducer from transfected Chinese hamster ovary cells. J Biol Chem 272: 24–27, 1997 [PubMed] [Google Scholar]
- 16.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nübe O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med 5: 1135–1142, 1999 [DOI] [PubMed] [Google Scholar]
- 17.Huggins CE, Domenighetti AA, Ritchie ME, Khalil N, Favaloro JM, Proietto J, Smyth GK, Pepe S, Delbridge LM. Functional and metabolic remodelling in GLUT4-deficient hearts confers hyper-responsiveness to substrate intervention. J Mol Cell Cardiol 44: 270–280, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Iacono KT, Brown AL, Greene MI, Saouaf SJ. CD147 immunoglobulin superfamily receptor function and role in pathology. Exp Mol Pathol 83: 283–295, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ikonomidis JS, Hendrick JW, Parkhurst AM, Herron AR, Escobar PG, Dowdy KB, Stroud RE, Hapke E, Zile MR, Spinale FG. Accelerated LV remodeling after myocardial infarction in TIMP-1-deficient mice: effects of exogenous MMP inhibition. Am J Physiol Heart Circ Physiol 288: H149–H158, 2005 [DOI] [PubMed] [Google Scholar]
- 20.Inui T, Nakagawa R, Ohkura S, Habu Y, Koike Y, Motoki K, Kuranaga N, Fukasawa M, Shinomiya N, Seki S. Age-associated augmentation of the synthetic ligand-mediated function of mouse NK1.1 ag+ T cells: their cytokine production and hepatotoxicity in vivo and in vitro. J Immunol 169: 6127–6132, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Jodele S, Blavier L, Yoon JM, DeClerck YA. Modifying the soil to affect the seed: role of stromal-derived matrix metalloproteinases in cancer progression. Cancer Metastasis Rev 25: 35–43, 2006 [DOI] [PubMed] [Google Scholar]
- 22.King MK, Coker ML, Goldberg A, McElmurray JH 3rd, Gunasinghe HR, Mukherjee R, Zile MR, O′Neill TP, Spinale FG. Selective matrix metalloproteinase inhibition with developing heart failure: effects on left ventricular function and structure. Circ Res 92: 177–185, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Koga K, Nabeshima K, Aoki M, Kawakami T, Hamasaki M, Toole BP, Nakayama J, Iwasaki H. Emmprin in epithelioid sarcoma: expression in tumor cell membrane and stimulation of MMP-2 production in tumor-associated fibroblasts. Int J Cancer 120: 761–768, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Kuno N, Kadomatsu K, Fan QW, Hagihara M, Senda T, Mizutani S, Muramatsu T. Female sterility in mice lacking the basigin gene, which encodes a transmembrane glycoprotein belonging to the immunoglobulin superfamily. FEBS Lett 425: 191–194, 1998 [DOI] [PubMed] [Google Scholar]
- 25.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ, Chamber Quantification Writing Group, American Society of Echocardiography's Guidelines, and Standards Committee, European Association of Echocardiography. Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 18: 1440–1463, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K, Kinugawa S, Tsutsui H. Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension 47: 711–717, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Nadon NL. Exploiting the rodent model for studies on the pharmacology of lifespan extension. Aging Cell 5: 9–15, 2006 [DOI] [PubMed] [Google Scholar]
- 28.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res 69: 562–573, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Neumann U, Kubota H, Frei K, Ganu V, Leppert D. Characterization of Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2, a fluorogenic substrate with increased specificity constants for collagenases and tumor necrosis factor converting enzyme. Anal Biochem 328: 166–173, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Sameshima T, Nabeshima K, Toole BP, Yokogami K, Okada Y, Goya T, Koono M, Wakisaka S. Glioma cell extracellular matrix metalloproteinase inducer (EMMPRIN) (CD147) stimulates production of membrane-type matrix metalloproteinases and activated gelatinase A in co-cultures with brain-derived fibroblasts. Cancer Lett 157: 177–184, 2000 [DOI] [PubMed] [Google Scholar]
- 31.Schmidt R, Bültmann A, Ungerer M, Joghetaei N, Bülbül O, Thieme S, Chavakis T, Toole BP, Gawaz M, Schömig A, May AE. Extracellular matrix metalloproteinase inducer regulates matrix metalloproteinase activity in cardiovascular cells: implications in acute myocardial infarction. Circulation 113: 834–841, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Seko Y, Fujimura T, Taka H, Mineki R, Murayama K, Nagai R. Hypoxia followed by reoxygenation induces secretion of cyclophilin A from cultured rat cardiac myocytes. Biochem Biophys Res Commun 317: 162–168, 2004 [DOI] [PubMed] [Google Scholar]
- 33.Sharpe N. Cardiac remodeling in congestive heart failure. In: Congestive Heart Failure (2nd ed.), edited by Hosenpud JD and Greenburg BH. Philadelphia, PA: Saunders, 2000, p. 101–115.
- 34.Siwik DA, Chang DL, Colucci WS. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res 86: 1259–1265, 2000 [DOI] [PubMed] [Google Scholar]
- 35.Siwik DA, Kuster GM, Brahmbhatt JV, Zaidi Z, Malik J, Ooi H, Ghorayeb G. EMMPRIN mediates beta-adrenergic receptor-stimulated matrix metalloproteinase activity in cardiac myocytes. J Mol Cell Cardiol 44: 210–217, 2008 [DOI] [PubMed] [Google Scholar]
- 36.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol 280: C53–C60, 2001 [DOI] [PubMed] [Google Scholar]
- 37.Sodek KL, Ringuette MJ, Brown TJ. MT1-MMP is the critical determinant of matrix degradation and invasion by ovarian cancer cells. Br J Cancer 97: 358–367, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev 87: 1285–1342, 2007 [DOI] [PubMed] [Google Scholar]
- 39.Spinale FG, Coker ML, Heung LJ, Bond BR, Gunasinghe HR, Etoh T, Goldberg AT, Zellner JL, Crumbley AJ. A matrix metalloproteinase induction/activation system exists in the human left ventricular myocardium and is upregulated in heart failure. Circulation 102: 1944–1949, 2000 [DOI] [PubMed] [Google Scholar]
- 40.Spinale FG, Coker ML, Thomas CV, Walker JD, Mukherjee R, Hebbar L. Time-dependent changes in matrix metalloproteinase activity and expression during the progression of congestive heart failure: relation to ventricular and myocyte function. Circ Res 82: 482–495, 1998 [DOI] [PubMed] [Google Scholar]
- 41.Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem 270: 5331–5338, 1995 [DOI] [PubMed] [Google Scholar]
- 42.Subramaniam A, Gulick J, Neumann J, Knotts S, Robbins J. Transgenic analysis of the thyroid-responsive elements in the alpha-cardiac myosin heavy chain gene promoter. J Biol Chem 268: 4331–4336, 1993 [PubMed] [Google Scholar]
- 43.Sundström J, Evans JC, Benjamin EJ, Levy D, Larson MG, Sawyer DB, Siwik DA, Colucci WS, Sutherland P, Wilson PW, Vasan RS. Relations of plasma matrix metalloproteinase-9 to clinical cardiovascular risk factors and echocardiographic left ventricular measures: the Framingham Heart Study. Circulation 109: 2850–2856, 2004 [DOI] [PubMed] [Google Scholar]
- 44.Tam EM, Morrison CJ, Wu YI, Stack MS, Overall CM. Membrane protease proteomics: isotope-coded affinity tag MS identification of undescribed MT1-matrix metalloproteinase substrates. Proc Natl Acad Sci USA 101: 6917–6922, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang Y, Kesavan P, Nakada MT, Yan L. Tumor-stroma interaction: positive feedback regulation of extracellular matrix metalloproteinase inducer (EMMPRIN) expression and matrix metalloproteinase-dependent generation of soluble EMMPRIN. Mol Cancer Res 2: 73–80, 2004 [PubMed] [Google Scholar]
- 46.Toole BP. Emmprin (CD147), a cell surface regulator of matrix metalloproteinase production and function. Curr Top Dev Biol 54: 371–389, 2003 [DOI] [PubMed] [Google Scholar]
- 47.Vanhoutte D, Schellings M, Pinto Y, Heymans S. Relevance of matrix metalloproteinases and their inhibitors after myocardial infarction: a temporal and spatial window. Cardiovasc Res 69: 604–613, 2006 [DOI] [PubMed] [Google Scholar]
- 48.Woessner JF Jr, Nagase H. MMP sequences. In: Matrix Metalloproteinases and TIMPs. New York: Oxford Univ., 2000, p. 11–41.
- 49.Yan C, Boyd DD. Regulation of matrix metalloproteinase gene expression. J Cell Physiol 211: 19–26, 2007 [DOI] [PubMed] [Google Scholar]
- 50.Yan L, Zucker S, Toole BP. Roles of the multifunctional glycoprotein, emmprin (basigin; CD147), in tumour progression. Thromb Haemost 93: 199–204, 2005 [DOI] [PubMed] [Google Scholar]
- 51.Yang JM, O′Neill P, Jin W, Foty R, Medina DJ, Xu Z, Lomas M, Arndt GM, Tang Y, Nakada M, Yan L, Hait WN. Extracellular matrix metalloproteinase inducer (CD147) confers resistance of breast cancer cells to Anoikis through inhibition of Bim. J Biol Chem 281: 9719–9727, 2006 [DOI] [PubMed] [Google Scholar]
- 52.Yang Y, Ma Y, Han W, Li J, Xiang Y, Liu F, Ma X, Zhang J, Fu Z, Su YD, Du XJ, Gao XM. Age-related differences in postinfarct left ventricular rupture and remodeling. Am J Physiol Heart Circ Physiol 294: H1815–H1822, 2008 [DOI] [PubMed] [Google Scholar]
- 53.Zucker S, Hymowitz M, Rollo EE, Mann R, Conner CE, Cao J, Foda HD, Tompkins DC, Toole BP. Tumorigenic potential of extracellular matrix metalloproteinase inducer. Am J Pathol 158: 1921–1928, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]