

Abstract
Altering the splice variant composition of large-conductance Ca2+-activated potassium (BK) channels can alter their activity and apparent sensitivity to Ca2+ and other regulators of activity. We hypothesized that differences in the responsiveness to arachidonic acid of GH3 and GH4 cells was due to a difference in two splice variants, one present in GH3 cells and the other in GH4 cells. The sequences of the two splice variants differ from one another in several ways, but the largest difference is the presence or absence of 27 amino acids in the COOH terminus of the BK α-subunit. Open probability of the variant containing the 27 amino acids is significantly increased by arachidonic acid, while the variant lacking the 27 amino acids is insensitive to arachidonic acid. In addition, sensitivity of BK channels to arachidonic acid depends on cytosolic phospholipase A2 (cPLA2). Here we used the Mammalian Matchmaker two-hybrid assay and two BK α-subunit constructs with [rSlo(27)] and without [rSlo(0)] the 27-amino acid motif to determine whether cPLA2 associates with one construct [rSlo(27)] and not the other. We hypothesized that differential association of cPLA2 might explain the differing responsiveness of the two constructs and GH3 and GH4 cells to arachidonic acid. We found that cPLA2 is strongly associated with the COOH terminus of rSlo(27) and only very weakly associated with rSlo(0). We also found that arachidonic acid has a lower affinity for rSlo(0) than for rSlo(27). We conclude that the lack of response of BK channels in GH4 cells to arachidonic acid can be explained, in part, by the poor binding of cPLA2 to the COOH terminus of the rSlo(0) α-subunit, which is very similar to the splice variant found in the arachidonic acid-insensitive GH4 cells.
Keywords: large-conductance calcium-activated potassium channels, phospholipase A2, protein-protein association
one important type of central nervous system (CNS) ion channel is the large-conductance Ca2+-activated potassium (BK) channel. Like many other potassium channels, BK channels in different cells are often composed of different splice variants, and these splice variants produce at least six distinct functional types of BK channels within the CNS (23). The presence of BK channels formed from different splice variants is important because altering the splice variant composition of BK channels can alter their activity and apparent sensitivity to Ca2+ and other regulators of activity (24, 26). Which splice variants comprise the channel is important in determining whether BK channels are inhibited or activated by reductions in oxygen levels and by acidic intracellular pH as seen in stroke and reperfusion injury (2, 9, 14). In addition, as we show in this article, different splice variants respond to signaling molecules such as arachidonic acid that result from cerebral ischemia.
Over and above any generalized relevance to the CNS as a whole, splice variants are likely to be very important to growth hormone-secreting neurons in the pituitary itself (from which both GH3 and GH4 cells are derived). Neurosecretory neurons control the amount of secretion by striking a balance between ligand-mediated depolarization and the subsequent calcium entry that leads to hormonal secretion, and the hyperpolarization resulting from calcium activation of BK channels modulates hormonal release. Ischemia, trauma, or high levels of reactive oxygen species will strongly depolarize pituitary cells and lead to inappropriate hormonal release but will also lead to production of arachidonic acid. If a cell contains an arachidonic acid-activated BK splice variant, then inappropriate hormonal release will be reduced; otherwise, large amounts of pituitary hormones will be released that will have significant systemic consequences.
Therefore, understanding which splice variants are present in different cells may be important for understanding and pharmacologically manipulating BK channels in the CNS. A specific case is one BK splice variant in GH3 cells (a neurosecretory cell line) that forms channels sensitive to arachidonic acid, while channels formed from an alternative BK splice variant in a subclone of GH3 cells, GH4C1, are not (4, 5, 7, 8). It has been suggested that at least part of this difference in splice variant-dependent sensitivity to channel activators is due to differential association of accessory proteins with different BK channel splice variants that results in some splice variant channels forming specialized signaling complexes while channels formed from other splice variants do not (13). One specific case of such an accessory protein is the so-called BK β-subunit, which associates with the channel-forming α-subunit to alter its voltage and calcium dependence (15). Some β-subunits (β2 and β3) have also been suggested to alter the sensitivity to fatty acids like arachidonic acid.
Besides the β-subunits, there appear to be other accessory proteins. We have shown that cytosolic phospholipase A2 (cPLA2) is required for the optimal response of BK channels to intracellular Ca2+ and arachidonic acid in GH3 cells (7) and that cPLA2 is associated with BK channels since cPLA2 and BK channels can be coimmunoprecipitated and confocal microscopy shows that the two proteins colocalize at the plasma membrane (3). Most wild-type BK channels actually exist as heterotetramers comprised of four α- and four β-subunits. In a sense, the β-subunits can be viewed as accessory protein since four distinct β-subunits have been identified and characterized. Each β-subunit imparts different properties to the overall channel function depending on the anatomic location of the BK channel(s) (1).
As mentioned above, BK channels in GH3 cells are sensitive to arachidonic acid, while GH4 cells are not. GH3 and GH4 cells contain two different BK splice variants whose major difference is the presence in GH3 cells or absence in GH4 cells of a 27-amino acid domain near the COOH terminus of the BK α-subunit. Only GH3 cells containing the splice variant with the 27-amino acid domain are sensitive to arachidonic acid. The simplest explanation for this difference would be that the 27-amino acid region is, itself, the arachidonic acid binding site. However, our previous demonstration that arachidonic acid sensitivity appeared to require close association between cPLA2 and BK channels caused us to hypothesize that the 27-amino acid domain might actually be a site for cPLA2 interaction with the α-subunit of BK channels and that the differential cPLA2 interaction produced the differential sensitivity to arachidonic acid.
MATERIALS AND METHODS
Cell culture.
GH3 and Chinese hamster ovary (CHO) cell lines were obtained from American Type Culture Collection (ATCC, Rockville, MD) and were grown at 37°C in a 5% CO2 atmosphere. GH3 cells were grown in Dulbecco‘s modified Eagle’s medium (DMEM) supplemented with 15% heat-activated horse serum, 2.5% fetal bovine serum, 10 U of penicillin-streptomycin (Pen-Strep), and 2 mM glutamine. GH4C1 cells were originally obtained as a gift from Dr. Gerry Oxford (Univ. of North Carolina, Chapel Hill, NC) and were cultured identically to GH3 cells. Cells from passages 25–45 were used in the experiments described in this article. HEK-293 cells stably expressing the rSlo(27) or rSlo(0) subunit of BK channels (described below) were grown in DMEM supplemented with 10% fetal bovine serum, 10 U of Pen-Strep, and 2 mM glutamine. CHO cells were grown in F12K medium supplemented with 10% fetal bovine serum, 10 U of Pen-Strep, and 2 mM glutamine. CHO cells from passages 1–10 after receipt from ATCC and transfected HEK cells from passages 20–30 were used in the experiments described in this article. Cells for immunoprecipitation experiments were grown to confluence in T-75 culture flasks.
Antibodies and special reagents.
Primary antibodies to BK-α channels (host: rabbit) and cPLA2 (host: mouse) were obtained from Chemicon International (Temecula, CA). A BK channel antibody designed specifically for immunoprecipitation was a generous gift from Dr. Irwin Levitan (University of Pennsylvania, Philadelphia, PA). Primary antibodies to BK channel β1- and β2-subunits (host: rabbit) were obtained from Millipore (formerly Chemicon International). Horseradish peroxidase-conjugated secondary antibodies were obtained from Amersham Biosciences (Piscataway, NJ). A/G agarose beads were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). 5,8,11,14-Eicosatetraynoic acid (ETYA) was obtained from Biomol (Plymouth Meeting, PA).
Transient and stable transfection procedure.
For most transfections, a T-25 flask grown to 80–90% confluence was used. Cells were washed three times with PBS, and serum-free medium was introduced. Transfection was accomplished with a Lipofectamine Plus kit (Invitrogen, Carlsbad, CA). Three micrograms of the cDNA of interest was used according to the manufacturer's directions. For stable transfections, medium containing gentamycin (G-418; 1,000μg/ml) was introduced 96 h after transfection. Selection was allowed to continue for 4 wk, by which time cell numbers had recovered to ∼30% confluence. Gentamycin concentrations were then reduced to 750 μg/ml, where they were maintained. Expression of the desired protein was verified by Western blot. For the Matchmaker experiments, cells were plated in 35-mm culture dishes and allowed to grow to ∼90% confluence before transfection.
Electrophysiological recordings.
All experiments in this study used the excised patch configuration of the patch-clamp technique. Electrodes were fabricated from Corning 7052 glass (Garner Glass, Fullerton, CA) in two steps on a Narishige PP-83 electrode puller (Narishige, Tokyo, Japan). Electrodes were fire polished to a final tip resistance between 3 and 5 MΩ. Recordings were performed at room temperature with a Dagan model 3900 patch-clamp amplifier (Dagan, Minneapolis, MN). All experiments were conducted with the patch depolarized to +20 mV. Single-channel data were digitized with Axoscope-10 software (Molecular Devices, Sunnyvale, CA) at a sampling rate of 5 kHz and filtered at 2 kHz with a 4-pole low-pass Bessel filter.
Arachidonic acid exposure paradigm.
For all experiments, arachidonic acid exposure was accomplished with a gravity perfusion/suction removal technique with a perfusion rate of 2.0 ml/min and a dead volume of <1.0 ml. Previous experiments showed that exchange was 90 ± 7% complete after 0.5 min. After a high-resistance (>25 GΩ) seal was obtained, patches were excised. Control recordings were obtained in K2EGTA-buffered solutions containing the desired ionized Ca2+ (Cai2+) concentration as described below.
Solutions.
The solutions used in all patch-clamp experiments were (mM) 150 KCl, 2 MgCl2, 10 N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), pH 7.30, or 140 KCl, 5 HEPES, 5 K2EGTA, 4.55 CaCl2 (10 μM free Cai2+), pH 7.30 for the pipette and 140 KCl, 15 HEPES, 5 K2EGTA, and 0.1, 1, or 100 μM Cai2+, pH 7.4 for the bath.
Data analysis.
The digitized single-channel data were analyzed in 1-min segments to generate channel activity (NPo, where N is no. of channels and Po is open probability) vs. time plots with pCLAMP 10 software programs (Molecular Devices).
Whole cell lysate immunoprecipitation procedure.
Immunoprecipitation experiments were conducted based on the methods previously described for the precipitation of BK channel α- and β-subunits (12). One flask containing ∼107 cells was lysed with RIPA buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 1.0 mM EDTA, 0.1% SDS, 1.0% Triton X-100, and 1.0% sodium deoxycholate). The protease inhibitors described above were also added. After cell lysis (1 h at 4°C), the suspension was centrifuged at 4,000 g for 10 min to precipitate unlysed cells. For immunoprecipitations, 1 ml of cell lysate and 10 μl of cPLA2 antibody (concn 1.6 mg/ml) were incubated overnight at 4°C. Thirty microliters of protein A/G PLUS-agarose was added and mixed at 4°C for 2 h with gentle agitation. A/G-agarose was pelleted by centrifugation at 4,000 g for 5 min at 4°C. The supernatant was carefully removed and saved for analysis by Western blot. The pellet was washed three times with 30 μl of Laemmli sample buffer (Tris, pH 6.8, 0.1% SDS, 10% glycerol, and 0.025% bromphenol blue). The resuspended sample, which also contained 20 mM DTT, was heated at 95°C for 5 min, and the pellet fraction was centrifuged at 4,000 g for 5 min before gel loading and Western blot analysis.
Western blot analysis.
Western blot analysis was completed as previously described (7). Thirty microliters of the eluent and original supernatant was loaded in each lane on a 7.5% polyacrylamide gel. After electrophoresis (∼1 h at 150 V), the proteins were transferred to nitrocellulose. The nitrocellulose blots were then probed with primary antibodies for BK-α channels (1:1,000). Blots were then washed and incubated with a secondary antibody (1:160,000). The secondary antibody used was goat anti-rabbit IgG, coupled to alkaline phosphatase (Kirkegaard and Perry Laboratories), and the antigen antibody complex was detected by the chemiluminescent detection system CDP-star (Tropix) and the Kodak 2000M camera system (Eastman Kodak, Rochester, NY).
Isolation of BK channel splice variants from GH3 and GH4 cells.
Total RNA was prepared from GH3 and GH4 cells with an RNeasy midi-kit (Qiagen) according to the manufacturer's instructions. Five micrograms of total RNA and 50 μM oligo(dT)20 were used to make cDNA with the ThermoScript RT-PCR system (GIBCO BRL-Invitrogen). PCR primers specific for rat BK channel are as follows: TGTCATGATGACGTCACAGATC, AATTGCTGTCCCACAAGCAAAG for the α-subunit and CCATGGGGAAGAAGCTGGTG, CAGTCAGCAGGAAGGTGGGC for the β-subunit. The PCR products were run on a 0.8% gel and purified with a QIAquick Gel Extraction Kit (Qiagen). The products were cloned into PGEMTeasy (Promega), and the sequence of the insert was verified by sequencing the plasmid.
Quantitative PCR.
Total RNA was prepared from GH3 and GH4 cells with an RNeasy midi-kit (Qiagen) according to the manufacturer's instructions. Five micrograms of total RNA and 50 μM oligo(dT)20 were used to make cDNA with the ThermoScript RT-PCR system (GIBCO BRL-Invitrogen). The following set of sense-antisense PCR primers was used: AGGCGGCTTGAAGATGAGCA-ACTCAGGACAGTCATGGCAGC; CTGATCTATTCCAAGATGTCCA-ACTCAGGACAGTCATGGCAGC TGATCTATTTTGAAGATGAGCAAC-ACTCAGGACAGTCATGGCAGC; and AGGCGGCCCAAGATGTCCAT-ACTCAGGACAGTCATGGCAGC.
The PCR products were run on a gel to confirm that there was a single product obtained for each primer set. The products were cloned into PGEMTeasy (Promega), and the sequence of the insert was verified by sequencing the plasmid. The plasmid was then used to generate a standard curve for real-time PCR using different known concentrations of the plasmid DNA dilutions (100 pg–0.01 pg). Real-time PCR was carried out with the iCycler PCR system (Bio-Rad) in 25-μl reactions on a 96-well plate. Reactions consisted of the following: double-distilled H2O, SYBR Green PCR buffer, MgCl2, dNTPs, AmplitaqGold DNA polymerase (all from Applied Biosystems), PCR primers, and unknown cDNA template or plasmids. The reaction was cycled 40 times, and data were analyzed with the iCycler software and Microsoft Excel.
A melting curve program was run on each primer pair, and only one melting temperature was observed, suggesting one product for each PCR reaction.
Identification of BK channel β-subunits from GH3 and GH4 cells.
Total RNA was prepared from GH3 and GH4 cells with an RNeasy midi-kit (Qiagen) as described above. Five micrograms of total RNA and 50 μM oligo(dT)20 were used to make cDNA with the ThermoScript RT-PCR system (GIBCO BRL-Invitrogen). PCR primers specific for rat β1- and β2-subunits of BK channel are based on published sequences (accession nos. NM019273 for β1 and NM176861 for β2). The primers used were CTGTGCTGCCCCTCTACCA, CAGGAAGGTGGGCCAGAAGA for the β1-subunit and TCAGGGACCATGACCTCCT, CTTCTGGGTCAGAATAGCAGG for the β2-subunit.
The PCR products were run on a 1.5% gel and purified with a QIAquick Gel Extraction Kit (Qiagen). The products were cloned into PGEMTeasy (Promega), and the sequence of the insert was verified by sequencing the plasmid. HEK cell cDNA and mouse brain cDNA were used as positive controls for β1 and β2, respectively. These results were independently confirmed by Western blot.
BK-α splice variant constructs.
A full-length clone of rSlo encoding rSlo(27) was used as the template for constructing cDNA of a specific sequence variant, rSlo(0). rSlo(0) cDNA was constructed by replacing a 1,040-bp DNA fragment of rSlo NheI/XmaI digest (2,783 ± 3,822) with the corresponding 959-bp DNA fragment of mSlo NheI/XmaI digest (2,317 ± 3,275). These regions of rSlo and mSlo cDNA encode identical amino acid sequences, except that rSlo(0) lacks 81 bp, resulting in a deletion of a 27-amino acid sequence. The deletion of the 81-bp section in the rSlo(0) construct was confirmed by subsequent DNA sequencing. The individual rSlo variants were subcloned into pcDNA 3.1(+) (Invitrogen) for expression in HEK-293 cells as described above.
cPLA2 activity assay.
Cells from two confluent T-75 flasks were washed twice with cold PBS (4°C), and the PBS was removed and replaced with 1.5 ml of 250 mM sucrose buffer. Cells were removed from the flasks by scraping. Cells were then lysed by sonication at 4°C. After sonication, the suspensions were centrifuged at 160,000 g for 1 h. The cytosolic supernatant was removed, and the membrane fraction was resuspended in 0.5 ml of sucrose buffer. Protein determinations were made on the cytosol and membrane fractions as described above. PLA2 activity was assessed by the hydrolysis of dipalmitoyl-[2-palmitoyl-1-14C]-l-α-phosphatidylcholine to [14C]palmitic acid, as described previously (7, 21, 22). Each reaction mixture contained 20 μg of protein for cytosolic measurements and 10 μg of protein for membrane measurements.
Mammalian two-hybrid screen.
For two-hybrid experiments, cells were plated in 35-mm culture dishes and allowed to grow to ∼90% confluence before transfection. A full-length clone for cPLA2 was inserted into the Matchmaker BD plasmid, and three different α-subunit clones were inserted into the Matchmaker AD plasmid. The first of these constructs contained amino acids 1-594 from the NH2 terminus, which are identical for both rSlo(27) and rSlo(0). The second construct contained 456 amino acids from the COOH terminus of rSlo(0) (lacking the 27-amino acid domain), and the third contained 491 amino acids from the COOH terminus of rSlo(27) (containing the 27-amino acid domain) In addition, to determine the possible influence of the BK channel β-subunit on any cPLA2-rSlo interaction, a full-length construct for the BK channel β-subunit (β1) was tested for association with the three α-subunit constructs in the presence and absence of cPLA2. The Matchmaker Mammalian Two-Hybrid kit was obtained from Clontech Laboratories (Palo Alto, CA), and the plasmids provided were used for further cloning. The pM plasmid was used to produce one NH2-terminal DNA binding domain fusion protein-encoding plasmid [the same for both rSlo(0) and rSlo(27)] and two COOH-terminal DNA binding domain fusion protein-encoding plasmids [1 for rSlo(0) and 1 for rSlo(27)]. The pVP16 plasmid was used to produce a full-length cPLA2-VP16 or a full-length BK-fusion protein-encoding plasmid. PCR products for rSlo(0), the NH2 terminus of rSlo(0), and rSlo(27) domains were ligated into pM with BaHI at the 5′ end and XbaI at the 3′ end. cPLA2 was ligated into pVP16 with EcoRI at the 5′ end and XbaI at the 3′ end. BK-β was ligated into pVP16 with EcoRI and HindIII. Two-hybrid assays were performed by transfecting and harvesting CHO cells according to the manufacturer's instructions. Typical transfection mixtures contained a total of 22 μg of DNA with 2 μg of PG5CAT plasmid, 10 μg of pM or pM-rSlo, and 10 μg of pVP16 or pVP16-cPLA2. Cells were harvested 48 h after transfection, and six replicate experiments were analyzed for the presence of chloramphenicol acetyltransferase (CAT) with a commercially available UV assay (FAST CAT, Molecular Probes). Blots were visualized and intensities measured with a Kodak 2000M camera system (Eastman Kodak). Measurement areas for each CAT-containing product were defined with intensity minima set at 25% above background.
Arachidonic acid binding assay.
We took advantage of the fact that our anti-BK-α antibody immunoprecipitated rSlo(0) and rSlo(27) constructs equally well. We bound anti-BK-α to A/G beads. We then lysed ∼107 cells from two flasks, one transfected with rSlo(0) and the other with rSlo(27), in a gentle lysis buffer (mM: 150 NaCl, 50 HEPES, 1 EGTA, 1 EDTA, 100 sodium fluoride, and 10 tetrasodium pyrophosphate, with 100 μM sodium orthovanadate, 10% glycerol, 1% TX-100, pH 7.4, 300 nM p-nitrophenylphosphate, 1 mM PMSF, 1 mM Pefablock, 10 mg/ml aprotinin, and 10 mg/ml leupeptin) for 30 min at 4°C. After cell lysis (1 h at 4°C), the suspension was centrifuged at 4,000 g for 10 min to precipitate unlysed cells. Thirty microliters of protein A/G PLUS-agarose beads was added to the supernatants and mixed at 4°C for 2 h with gentle agitation to remove any nonspecifically binding proteins and any nonsolubilized membrane fragments. A/G-agarose was pelleted by centrifugation at 4,000 g for 5 min at 4°C. The supernatants were carefully removed and added to the anti-BK-α A/G beads. The equivalence of the immunoprecipitation of rSlo(0) and rSlo(27) was confirmed by performing Western blots on a small amount of the two sets of beads. Since under these conditions there might be significant nonspecific binding of arachidonic acid to the A/G beads and antibody plus protein complexes, we also prepared beads with anti-GAPDH antibody and GAPDH protein. All of the A/G beads containing antibodies and protein were centrifuged, and the supernatant was removed. We then prepared arachidonic acid by evaporating 100 μl of EtOH containing 10 μCi of [3H]arachidonic acid to dryness under dry N2. We dissolved the arachidonic acid in 1 ml of PBS containing 1% BSA and determined the specific activity. Two hundred fifty microliters of the arachidonic acid in PBS was added to each set of beads [rSlo(27), rSlo(0), and GAPDH (control)] and shaken at room temperature for 15 min, after which the beads were centrifuged and four 50-μl samples of supernatant were removed from each set and counted. The remainder of the supernatant was discarded and replaced with 1 ml of fresh cold PBS with 1% BSA. This procedure was repeated at 15 min, 30 min, 1 h, 2 h, and 3 h. After the supernatant was removed from the last wash, the beads were resuspended in 100 μl of fresh PBS with 1% BSA and four 25-μl samples were counted to determine the arachidonic acid remaining on the beads. The rates and half-lives of unbinding were determined for each condition by calculating the number of counts remaining on the beads at each of the sample times and on the beads at the end of the 3-h period.
RESULTS
Arachidonic acid differentially affects BK channels from GH3 and GH4 cells.
We have shown that BK channels in GH3 cells are directly activated by arachidonic acid, and yet other investigators, using a subclone of GH3 cells (GH4C1) have shown that the BK channels in these cells are not sensitive to arachidonic acid (although they are sensitive to arachidonic acid metabolites) (5, 7, 8). Figure 1 shows that in GH3 cells arachidonic acid produced a significant increase in Po from 0.2 ± 0.05 to 0.57 ± 0.13 (P < 0.001) but in GH4 cells arachidonic acid had no effect on Po (untreated Po was 0.16 ± 0.09, and after treatment with arachidonic acid Po was 0.16 ± 0.1). We hypothesized that the difference between the two cell types was that they contained two different splice variants, one sensitive to arachidonic acid and the other not.
Fig. 1.
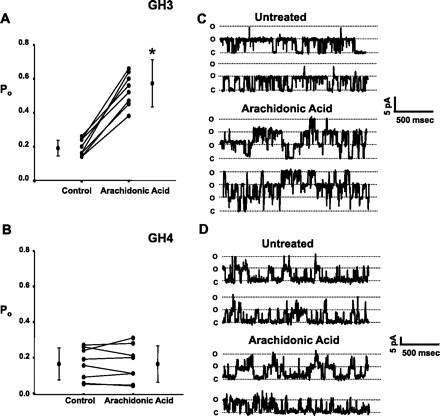
Arachidonic acid differentially affects splice variants in the α-subunit of large-conductance Ca2+-activated potassium (BK) channels from GH3 and GH4 cells. Experiments were conducted with inside-out patches depolarized to +20 mV and exposed to 0.1 μM Ca2+ Open probability (Po) values for individual patches are plotted along with means ± SD, and significant changes in Po are indicated by an asterisk. One micromolar arachidonic acid caused Po to increase from 0.2 ± 0.05 to 0.57 ± 0.13 (P < 0.0001) in GH3 cells (n = 8) (A), while Po in GH4 cells was unaffected by arachidonic acid (0.16 ± 0.09 vs. 0.16 ± 0.10) (n = 7) (B). C and D: representative single-channel records from BK channels in GH3 (C) and GH4 (D) cells. The “o” and “c” denote the open and closed state of the channel(s), respectively.
GH3 and GH4 cells contain different BK channel α-subunit splice variants.
Using PCR with primers described in materials and methods, we isolated β-subunits and the COOH-terminal cytosolic domains of α-subunits.
Examination of the amplified fragments of the α-subunits revealed two different predominant splice variants from the two cell types. Quantitative PCR confirmed the difference in the levels of mRNA for the two splice variants in the two different cell types (Fig. 2). For example, in GH3 cells there is 86 ± 12 times as much of an α-subunit splice variant previously identified in rat brain and rat adrenal chromaffin cells, rnBKα1 (accession nos.: nucleotide AF135265, protein AAD34786), as that of an α-subunit splice variant that has not been previously described in rat but is homologous to a mouse variant, muBKα1 (accession nos.: nucleotide L16912, protein AAA39746). In contrast, in GH4 cells there is 549 ± 80 times as much muBKα1 as rnBKα1. The GH3 splice variant has one small (4 amino acids) and one large (27 amino acids) insertion and one (3 amino acids) deletion not present in the GH4 variant, and the two sequences vary at their COOH-terminal end (Fig. 3).
Fig. 2.
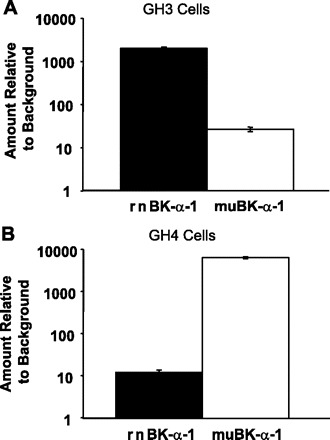
Quantitative PCR (qPCR) reveals a large difference in the relative amounts of 2 different splice variants in GH3 and GH4 cells. Using primers that were based on the clones we had isolated to select different splice variants, we amplified 100- to 200-base regions by qPCR and examined the amount of the 2 splice variants that differed between GH3 and GH4 cells. Relative amounts of the 2 splice variants in GH3 (A) and GH4 (B) cells are shown (note logarithmic scale). In GH3 cells there is 86 ± 12 times (mean ± SD; n = 3) as much of a rat splice variant, rnBKα1, as that of a splice variant previously only observed in mouse, muBKα1. In contrast, in GH4 cells there is 549 ± 80 times (mean ± SD; n = 3) as much muBKα1 as rnBKα1. The larger difference in the GH4 cells may reflect the fact that they are a clonal line derived from GH3 cells.
Fig. 3.

Alignment of cytosolic domains of α-subunit splice variants from GH3 cells and GH4 cells. BK channels in GH3 cells are sensitive to cytosolic arachidonic acid, while BK channels from GH4 cells are not. We hypothesized that the difference could arise because the 2 clonal lines express different splice variants of the α- or β-subunits. Using PCR with degenerate primers as described in materials and methods, we isolated β-subunits and the cytosolic domains of α-subunits. BK β-subunits were identical in both types of cells. Examination of the α-subunits revealed 2 different splice variants from the 2 cell types. The α-subunit of GH3 cells is identical to a splice variant previously identified in rat brain and rat adrenal chromaffin cells (accession nos.: nucleotide AF135265, protein AAD34786). The GH4 α splice variant has not been previously described in rat but is homologous to a mouse variant (muBKα; accession nos.: nucleotide L16912, protein AAA39746).
Identification of BK channel β-subunits from GH3 and GH4 cells.
BK β-subunits were identical in both types of cells and were found to be identical to the sequence for the β1-subunit (accession no. AF020712). To determine whether GH3 or GH4 cells also contained the β2-subunit, total RNA was used to prepare cDNA from GH3 and GH4 cells. PCR primers specific for rat β1 and β2-subunits of BK channel were based on published sequences (accession nos. NM019273 for β1 and NM176861 for β2). Amplification with these primers produced bands of the appropriate size for the β1-subunit from GH3 and GH4 cells but no detectable product for the β2-subunit (although the primers were able to amplify the β2-subunit from rat brain cDNA). Observable PCR products were isolated and cloned, after which the sequence of the PCR products was verified. We conclude that both GH3 and GH4 cells contain the β1- but not the β2-subunit (Fig. 4A). These results were independently confirmed by Western blot for β-subunit protein, which revealed the presence of the β1- but not the β2-subunit in either GH3 or GH4 cells. (Fig. 4, B and C).
Fig. 4.
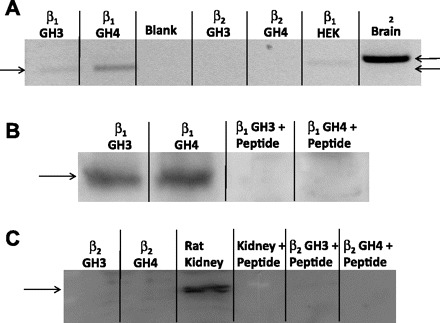
GH3 and GH4 cells contain the β1- but not the β2-subunit of BK channels. A: 1.5% agarose gel containing the PCR products obtained from total RNA from GH3 and GH4 cells and PCR primers specific for rat β1- and β2-subunits of BK channels as described in materials and methods. Gel shows the presence of the β1-subunit in both GH3 and GH4 cells. It is also apparent that the β2-subunit is not present in either GH3 or GH4 cells. HEK cells were used as a positive control for the β1-subunit, and mouse brain was used as a positive control for the β2-subunit. B: GH3 and GH4 cell lysates were probed with a polyclonal antibody to the β1-subunit of BK channels. First 2 lanes show the presence of the β1-subunit in both cell lines. Second 2 lanes are identical to first 2 lanes except that the lysates were probed with the polyclonal antibody to the β1-subunit in the presence of the antigenic peptide to show the successful competition of the β1-subunit band. C: GH3 and GH4 cell lysates (1st 2 lanes) and rat kidney lysate (3rd lane) were probed with a polyclonal antibody to the β2-subunit of BK channels. First and second lanes show the absence of the β2-subunit in both cell lines. On the other hand, 3rd lane shows that the antibody is functional since the β2-subunit was readily visualized in rat kidney. Fourth lane is identical to 3rd lane and 5th and 6th lanes are identical to 1st and 2nd lanes except that the lysates were probed with the polyclonal antibody to the β2-subunit in the presence of the corresponding antigenic peptide, showing the successful competition of the β2-subunit band.
Constructs to examine function of GH3 and GH4 splice variants.
Fortuitously, α-subunit splice variants similar to those in GH3 and GH4 cells have been isolated from rat brain to examine BK channel Ca2+ sensitivity (10). In this work, Park and colleagues examined the role of the large 27-amino acid difference between the GH3 and GH4 splice variants by modifying the mouse Slo sequence to produce two constructs that differed only in the presence or absence of the 27-amino acid region near the COOH terminus that we hypothesized was the arachidonic acid or PLA2 binding site. Figure 5 is a schematic illustrating the differences between the two splice variants. The two variants were designated rSlo(0) (no putative binding site) and rSlo(27) (full 27-amino acid putative binding site). Using excised patches from HEK cells transiently transfected with rSlo(27), we found that arachidonic acid treatment resulted in a significant increase in Po from 0.07 ± 0.03 to 0.19 ± 0.06 (P < 0.005). On the other hand, arachidonic acid treatment of excised patches from HEK cells transiently transfected with rSlo(0) had no significant effect on Po (0.05 ± 0.03 vs. 0.05 ± 0.03). These data are summarized in Fig. 6, and support our hypothesis that the 27-amino acid region in the COOH terminus of the α-subunit is necessary for arachidonic acid sensitivity.
Fig. 5.
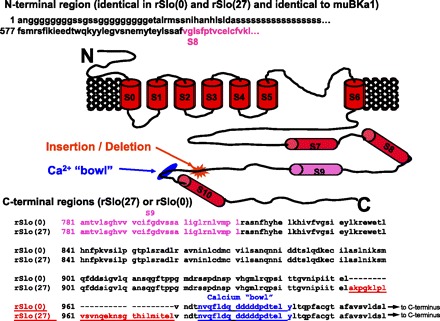
Primary structure and proposed membrane topology of BK channel splice variants. BK channel α-subunits contain 7 putative transmembrane domains, S0–S6, a conserved K+-selective pore region between S5 and S6, and a long COOH-terminal cytosolic tail with 4 additional hydrophobic segments, S7–S10. The splice site that is different in GH3 and GH4 cells near the Ca2+ bowl (blue) is highlighted with a red asterisk. Primary sequence of the COOH terminus is shown at bottom. Sequence of the subunit prior to S9 (pink) is invariant in both constructs [rSlo(27) and rSlo(0)]. The 27 amino acids that are present in GH3 cells and rSlo(27) but missing in GH4 cells and rSlo(0) are shown in red, and the Ca2+ bowl is shown in blue.
Fig. 6.
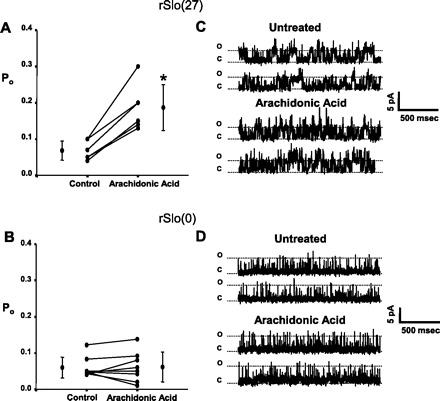
Arachidonic acid differentially affects splice variants rSlo(27) and rSlo(0) isolated from rat brain. Po values for individual patches are plotted along with means ± SD, and significant changes in Po are indicated by an asterisk. When the α-subunit [rSlo(27)], similar to the splice variant from GH3 cells, was transfected into HEK-293 cells, arachidonic acid treatment significantly increased Po from 0.07 ± 0.03 to 0.19 ± 0.06 (P < 0.005) (n = 6) (A). On the other hand, when the α-subunit [rSlo(0)], similar to the splice variant from GH4 cells, was transfected into HEK-293 cells, arachidonic acid treatment had no significant effect on Po (0.05 ± 0.03 to 0.05 ± 0.03) (n = 8) (B). C and D: representative single-channel records from BK channels in HEK-293 cells expressing rSlo(27) (C) and rSlo(0) (D). The “o” and “c” denote the open and closed state of the channel(s), respectively. These data suggest that the effect of arachidonic acid requires the presence of the 27-amino acid sequence contained in rSlo(27), since when it is absent, either in rSlo(0) or in GH4 cells, arachidonic acid has no effect on channel Po.
As is true for endogenous BK-α subunits, coexpressing β-subunits with either of the rSlo constructs does increase NPo. The addition of the β1- or the β2-subunit increased the control Po (0.05 ± 0.03) of the rSlo(0) channels to a level near those from GH4 cells [0.09 ± 0.05 (n = 4) for β1 and 0.13 ± 0.03 (n = 4) for β2]. However, the addition of arachidonic acid to cells expressing α-subunits and either the β1- or the β2-subunit did not result in a significant increase in activity [0.18 ± 0.05 (n = 4; P = 0.125) for β2 and 0.11 ± 0.05 (n = 4; P = 0.092) for β1], implying that the β-subunits were not responsible for producing arachidonic acid sensitivity.
Addition of accessory proteins is required to activate BK channel α-subunits.
While some of the differences in the splice variants found in GH3 and GH4 cells were suggestive of fatty acid binding sites, the 27-amino acid deletion was not obviously a fatty acid binding site. We have previously demonstrated (4, 7) that maximum activation of endogenous BK channels in GH3 cells requires functional cPLA2. We further confirmed that cPLA2 could activate α-subunits as strongly as BK β-subunits. For these experiments, we used excised patches depolarized to +20 mV with four different cytosolic Ca2+ concentrations: 0.1, 1, 10, and 100 μM. BK channels in GH3 cells were compared with HEK cells stably expressing an α-subunit almost identical to the major splice variant in GH3 cells (muBKα) alone or with HEK cells expressing the muBKα subunit and transiently expressing either cPLA2 or the β1-subunit of BK channels or both (Fig. 7) (clones of both subunits were generously provided by Owen McManus at Merck Research Labs (Rahway, NJ), and the cPLA2 clone was a generous gift from John Turk at Washington University, St. Louis, MO). The coexpression of either cPLA2 or the β1-subunit increased the ability of Ca2+ to activate the channel above that of the α-subunit alone. The magnitudes of the increase for the coexpression of the α- and β1-subunits are virtually identical to those reported by McManus et al. (15).
Fig. 7.
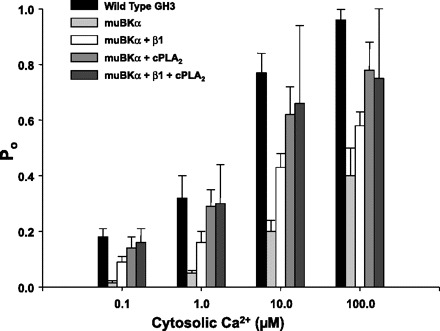
Coexpression of either cytosolic phospholipase A2 (cPLA2) or the β1-subunit with muBKα results in an increase in Ca2+ sensitivity. Groups of 6–8 cells from each treatment were studied at each Ca2+ concentration. For these experiments, inside-out patches depolarized to +20 mV were used. Compared with wild-type GH3 cells, the Ca2+ sensitivity of BK channels derived from stable HEK cell line expressing only the muBKα was greatly diminished. Coexpression with the β1-subunit, cPLA2, or both the β1-subunit and cPLA2 resulted in a sequential marked increase in Ca2+ responsiveness of the channels.
cPLA2 and BK channel α-subunit coimmunoprecipitate from GH3 and GH4 cells.
As we have previously reported (7), any manipulation that prevents activation or expression of cPLA2 shifts the Ca2+ and voltage activation relationships for BK channels to the right (in the absence of cPLA2 full activation requires higher voltages or higher intracellular Ca2+). This implies that cPLA2 and BK channel proteins must be either in extremely close proximity or actually physically associated. We have subsequently shown that cPLA2 and BK channels colocalize at the surface membrane of both GH3 and GH4 cells (3). However, the difference in arachidonic acid sensitivity of GH3 and GH4 suggests some difference in the cPLA2 activation of specific BK channel α-subunit splice variants. There are several possible explanations for the difference: there could be a difference in the cellular expression of cPLA2, a difference in the activity of cPLA2, or a difference in the association of cPLA2 and BK channels. Western blotting (Fig. 8B) shows that cPLA2 protein is equally expressed in both GH3 and GH4 cells, and Fig. 8A shows that there is no significant difference in cPLA2 enzymatic activity (cPLA2 activities for GH3 and GH4 cells were 67.3 ± 7.4 and 62.4 ± 6.9 pmol palmitic acid hydrolyzed·min−1·mg protein−1, respectively). However, when we immunoprecipitated BK channel α-subunits with an antibody (generously supplied by Dr. Irwin Levitan) and then probed the immunoprecipitate for cPLA2, cPLA2 coimmunoprecipitated with the α-subunit of BK channels from both GH3 and GH4 cells, but the amount that immunoprecipitated was much greater in the GH3 cells than in the GH4 cells (Fig. 8C). These data suggest that the BK channel α-subunits in both GH3 and GH4 cells can associate with cPLA2 but that the association is not as strong in GH4 cells.
Fig. 8.
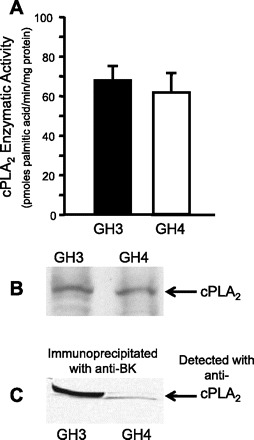
cPLA2 is equally expressed in both GH3 and GH4 cells but associates more strongly with the α-subunit of BK channels from GH3 cells. A: cPLA2 activities for GH3 and GH4 cells were not different [67.3 ± 7.4 and 62.4 ± 6.9 pmol palmitic acid·min−1·mg protein−1, respectively, by assay of PLA2 function in whole cell lysates from GH3 and GH4 cells as described previously (21, 22)]. B: typical Western blot in which whole cell lysates from both GH3 and GH4 cells were probed with a commercially available antibody (Millipore) to cPLA2. This blot shows that cPLA2 is expressed to the same extent in both cell lines (as expected from A). C: we immunoprecipitated BK channel α-subunits, resolved the immunoprecipitate on SDS gels, and probed with a commercially available antibody (Millipore) to cPLA2. cPLA2 was associated with BK channels in both GH3 and GH4 cells but was much more strongly associated in GH3 cells. These data suggest that BK channel α-subunits in both GH3 and GH4 cells are associated with cPLA2, albeit to different extents.
Differential binding of cPLA2 to BK channel α-subunits from GH3 and GH4 cells in mammalian two-hybrid assay.
Coimmunoprecipitation implies an interaction between cPLA2 and some site on the BK channel protein. However, it was unclear how the differences in association were related to the observation that BK channels in the two cell types respond differently to arachidonic acid. Therefore, we decided to examine more specific interactions between cPLA2 and the two different splice variants in the arachidonic acid-sensitive cells (GH3) and the arachidonic acid-insensitive cells (GH4). As mentioned above, we were aided in this investigation since these two splice variants had been previously isolated by us and others (10). The two constructs were identical to the complete mouse Slo gene except that the two constructs differed only in the presence [rSlo(27)] or absence [rSlo(0)] of the 27-amino acid domain near the COOH terminus.
We used these two constructs to test the hypotheses that cPLA2 and BK channels are physically associated and that the association depends on the presence of the 27-amino acid domain in rSlo(27) near the COOH terminus. We also thought that the differences in the association of cPLA2 with the two α-subunit splice variants could explain the differential effects of arachidonic acid. rSlo(27) exhibits an extremely high homology to the major α-subunit of BK channels found in GH3 cells, while rSlo(0) exhibits an extremely high homology to the major α-subunit found in GH4 cells. To test for specific interaction and to identify the region of BK channels that interacts with cPLA2 we used the Mammalian Matchmaker (Clontech) two-hybrid assay.
A full-length clone for cPLA2 was inserted into the Matchmaker BD plasmid, and three different α-subunit clones were inserted into the Matchmaker AD plasmid. The first of these constructs contained amino acids 1-594 from the NH2 terminus, which are identical for rSlo(27) and rSlo(0). The second construct contained 456 amino acids from the COOH terminus of rSlo(0) (lacking the 27-amino acid domain), and the third contained 491 amino acids from the COOH terminus of rSlo(27) (containing the 27-amino acid domain). These constructs and the structure of the BK α-subunit are shown schematically in Fig. 5. In addition, to determine the possible influence of the BK channel β-subunit on any cPLA2-rSlo-interaction, a full-length construct for the BK channel β-subunit (β1) was tested for association with the three α-subunit constructs in the presence and absence of cPLA2. cPLA2 most strongly associated with the COOH terminus of rSlo(27) (Fig. 9A). cPLA2 is also associated with the NH2 terminus of the α-subunit [the region that both rSlo(0) and rSlo(27) have in common], but the binding to the COOH terminus of rSlo(27) is 3.6 ± 1.1 fold greater. cPLA2 is only weakly associated with the COOH terminus of rSlo(0), to the extent that this association was not even detectable in some experiments. In the experiments in which association was weak but detectable, the binding of rSlo(27) was 11 ± 3.3-fold greater than that of rSlo(0). In our hands, the negative controls were below the level of detection for four of the six experiments, clearly demonstrating that for a detectable CAT product to be formed, a physical association between the proteins contained in both the binding and activation domains is required. Statistical comparisons, therefore, were limited to the fold differences between binding to the NH2 terminus and binding to the two COOH termini (Fig. 9B) and to differences in the raw intensity data for binding to the NH2 terminus and to the two COOH termini. Since the data were normally distributed, individual fold changes between the NH2 terminus and the COOH termini of rSlo(0) and rSlo(27) were compared with a Student's t-test. This analysis showed that the association between cPLA2 and rSlo(27) was significantly greater (P < 0.02) than between cPLA2 and rSlo(0). In all experiments, a paired t-test showed that the association was significantly greater for the COOH terminus of rSlo(27) compared with the NH2 terminus (P < 0.03) and significantly weaker for the COOH terminus of rSlo(0) compared with the NH2 terminus (P < 0.01). Somewhat surprisingly, experiments with the β-subunit showed a physical association with all of the rSlo constructs (Fig. 9C).
Fig. 9.
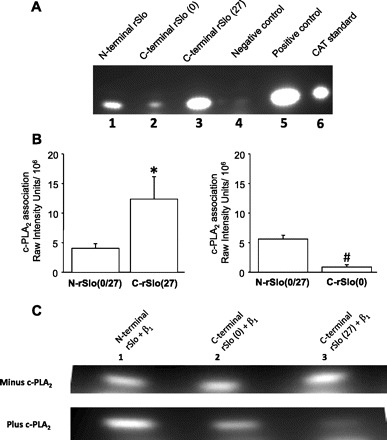
Protein-protein interactions between cPLA2 and splice variants of α-BK channels. A: lane 1 shows result of coexpressing Matchmaker binding domain + full-length cPLA2 and Matchmaker activation domain + NH2-terminal region that rSlo(27) and rSlo(0) have in common. Lane 2 shows result of expressing Matchmaker-cPLA2 and Matchmaker activation domain + COOH-terminal region of rSlo(0). Lane 3 shows result of expressing Matchmaker-cPLA2 and Matchmaker activation domain + COOH-terminal region of rSlo(27). Lane 4 is a negative control resulting from expression of unaltered Matchmaker binding domain and unaltered Matchmaker activation domain, lane 5 is a positive control supplied in the Matchmaker kit, and lane 6 is the unreacted chloramphenicol acetyltransferase (CAT) standard. This blot shows that cPLA2 binds weakly to the NH2 terminus of the α-subunit [which rSlo(27) and rSlo(0) have in common] and binds strongly to the COOH terminus of rSlo(27) but not to the COOH terminus of rSlo(0). B: intensities of expressed CAT resulting from physical association between cPLA2 and the COOH termini of rSlo(27) and rSlo(0) were compared with intensities arising from physical association between cPLA2 and NH2 terminus of rSlo(27) [NH2 terminus is identical for rSlo(27) and rSlo(0)]. All data are presented as means ± SD. In all experiments, the association was significantly greater for the COOH terminus of rSlo(27) compared with the NH2 terminus (*P < 0.04) and significantly weaker for the COOH terminus of rSlo(0) compared with the NH2 terminus (#P < 0.02). C, top: in the absence of cPLA2, BK-β interacts equally well with the NH2 terminus of rSlo, the COOH terminus of rSlo(0), or the COOH terminus of rSlo(27). Lane 1 contains results of expressing Matchmaker binding domain plasmid + BK-β and Matchmaker activation domain + NH2-terminal region that rSlo(27) and rSlo(0) have in common, lane 2 results of expressing Matchmaker BK-β and Matchmaker rSlo(0), and lane 3 results of expressing Matchmaker BK-β and Matchmaker rSlo(27). Since all lanes show equivalent amounts of CAT, activity binding interaction is the same for all constructs. Bottom: all lanes are identical to those at top except that cells were also transfected with cPLA2 in PCDNA3. Top row shows that β-subunit can physically associate with both the NH2 and either of the splice variant COOH termini of the α-subunit. However, when cPLA2 is present, we know that cPLA2 preferentially binds to the COOH terminus of rSlo(27), and this figure shows that when cPLA2 binds to rSlo(27) it also prevents the β-subunit from strongly associating with the COOH terminus of the BK α-subunit to which cPLA2 binds [rSlo(27)].
c-PLA2 binding prevents BK β-subunit from binding to rSlo(27).
We anticipated that the β-subunit would preferentially associate with the NH2 terminus since the NH2 terminus contained the membrane spanning domains that conventional wisdom suggests are the regions with which the β-subunits interact (17). However, the association of cPLA2 with rSlo(27) is so strong that this association prevents association of the β-subunit with rSlo(27). On the other hand, the presence of cPLA2 had no apparent effect on the association between the β-subunit and either the NH2 terminus of rSlo or the COOH terminus of rSlo(0) (Fig. 9C).
rSlo(27) has a higher affinity for arachidonic acid than rSlo(0).
If rSlo(27) is sensitive to arachidonic acid and rSlo(0) is not, then we would anticipate that rSlo(27) should interact more strongly with arachidonic acid than rSlo(0). Since the presumptive arachidonic acid binding site is intracellular (or in the cell membrane) and arachidonic acid is moderately hydrophobic, most whole cell methods and single-channel methods were unsuitable for examining arachidonic acid affinity to BK channels. However, we took advantage of the fact that our BK-α antibody immunoprecipitated rSlo(0) and rSlo(27) constructs equally well. We bound anti-BK-α to A/G beads and then immunoprecipitated rSlo(0) or rSlo(27) from transfected CHO cells. We used the immobilized proteins as targets for [3H]arachidonic acid binding. Since under these conditions there might be significant nonspecific binding of arachidonic acid to the A/G beads and antibody plus protein complexes, we also prepared beads with anti-GAPDH antibody and GAPDH protein. The beads were repeatedly washed, and the rate of release of arachidonic acid from the three different sets of beads was determined. The half-lives of arachidonic acid binding for binding of arachidonic acid to rSlo(27), rSlo(0), and GAPDH (control) were 0.89 ± 0.15, 0.55 ± 0.09, and 0.52 ± 0.11 h, respectively (means ± SE, n = 5) (Fig. 10). The unbinding half-life for rSlo(27) was significantly longer (P < 0.05) than for either rSlo(0) or GAPDH, while the unbinding half-life for rSlo(0) was not significantly different from that for the GAPDH control (ANOVA followed by a Holm-Sidak multiple comparison test). The implication is that rSlo(27), similar to the BK channel variant in GH3 cells, binds arachidonic acid more strongly than rSlo(0), similar to the variant in GH4 cells. Since the only difference between rSlo(27) and rSlo(0) is the 27-amino acid insertion in rSlo(27), the arachidonic acid binding appears to require this region.
Fig. 10.
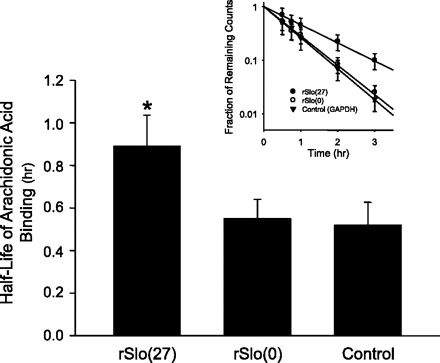
rSlo(27) has a higher affinity for arachidonic acid than rSlo(0). We bound anti-BK-α to A/G beads and then immunoprecipitated rSlo(0) or rSlo(27) from transfected Chinese hamster ovary cells. We used the immobilized proteins as targets for [3H]arachidonic acid binding. Since under these conditions there might be significant nonspecific binding of arachidonic acid to the A/G beads and antibody + protein complexes, we also prepared beads with anti-GAPDH antibody and GAPDH protein. The rate of release of arachidonic acid from the 3 different sets of beads was determined. The half-lives of arachidonic acid binding for binding of arachidonic acid to rSlo(27), rSlo(0), and GAPDH (control) were 0.89 ± 0.15, 0.55 ± 0.09, and 0.52 ± 0.11 h, respectively (means ± SE, n = 5) (see inset for example of typical rate of arachidonic acid loss). It should be noted that the rates of arachidonic acid loss were biexponential. All of the initial rates of loss (1st exponential) were identical and presumably represent nonspecific binding of arachidonic acid to the beads. Only the slower (2nd exponential) rates of loss were significantly different. The unbinding half-life for rSlo(27) was significantly longer (P < 0.05) than either rSlo(0) or GAPDH, while the unbinding half-life for rSlo(0) was not significantly different from the GAPDH control.
DISCUSSION
Arachidonic acid activates BK channels in GH3 cells but does not activate BK channels in GH4 cells.
Arachidonic acid is a critical precursor in several cellular signaling pathways (19) and regulates several types of ion channels within the CNS (11, 16). Arachidonic acid production is primarily controlled by regulating the activity of the membrane-associated phospholipase A2 (cPLA2), an enzyme that hydrolyzes membrane lipids to produce arachidonic acid (18, 20). Once produced, arachidonic acid can itself act on ion channels or can be further metabolized to produce additional signaling molecules. BK channels can be very sensitive to cPLA2 expression and arachidonic acid (6, 8, 29). Arachidonic acid applied to the cytosolic surface of patches formed from GH3 cells strongly activates BK channels, but not all BK channels are so sensitive to arachidonic acid. Arachidonic acid does not activate BK channels in GH4 cells (Fig. 1). We suspected that the difference was because the two different cell types express different splice variants, one of which is sensitive to arachidonic acid and the other insensitive. In fact, when we examined the major splice variants in the two cell types we found two alternative splice variants. We simultaneously confirmed that both cell types expressed the same β-subunit, β1. The variants differ at sites that have been previously described for mammalian BK channels (27). The GH3 splice variant differs from the GH4 variant in having one large 27-amino acid insertion, one smaller 4-amino acid insertion, and one 3-amino acid deletion (Fig. 3), and the two sequences vary at their COOH-terminal end. The large 27-amino acid insertion is adjacent to the Ca2+ “bowl” and has been previously described as altering the Ca2+ sensitivity of the channel (10). The insertion has some positive charge coupled with hydrophobic residues that could be a binding site for anionic lipids like arachidonic acid or a hydrophobic protein (like PLA2). Thus it is possible that the 27-amino acid region could contain both PLA2 and arachidonic acid binding sites. However, one of the insertions (SRKR) or the variation at the COOH terminus (RK) and the adjacent amino acids have a concentration of positive charge coupled with hydrophobic residues that could also contribute to arachidonic acid binding.
Arachidonic acid sensitivity of BK channels could arise either because arachidonic acid failed to interact with the channel or because cPLA2 failed to interact and, therefore, was incapable of delivering high concentrations of arachidonic acid in close proximity to the channel. We have shown that any manipulation that prevents activation or expression of PLA2 in GH3 cells [e.g., treatment with antisense oligonucleotides (4, 7)] makes BK channels less active, but this is not true for GH4 cells. These observations imply several things. First, in GH3 cells, cPLA2 and BK channel proteins at the cell surface must be either in extremely close proximity or actually physically associated. Second, there must be something different about cPLA2 activation of BK channels in GH4 cells. In fact, we have previously shown (3) by immunohistochemistry and confocal microscopy that cPLA2 and BK channel proteins at the plasma membrane are strongly colocalized, implying that, in GH3 cells, cPLA2 is physically associated with BK channels at the cell surface.
On the other hand, BK channels in GH4 cells are not sensitive to arachidonic acid. Originally we thought that it was possible that GH4 cells do not have cPLA2 or that cPLA2 and BK channels do not associate as they do in GH3 cells. This explanation was quickly eliminated because Western blotting showed that both GH3 and GH4 cells had equal amounts of cPLA2 and that the enzyme was equally active (Fig. 8). However, unlike the result in GH3 cells, the α-subunit of BK channels in GH4 cells was only poorly coimmunoprecipitated with cPLA2. These results suggest that cPLA2 does not associate with the BK channel splice variant in GH4 cells as well as it does with the BK channel splice variant in GH3 cells.
We took advantage of the fact that the same splice variants as those in GH3 and GH4 cells had been isolated from rat brain to examine BK channel Ca2+ sensitivity (10). For these studies, parts of the COOH termini of the two splice variants were used to replace the COOH terminus of mSlo, resulting in two constructs whose only difference was the presence [in rSlo(27)] or absence [in rSlo(0)] of the 27-amino acid region near the COOH terminus that we hypothesized was the arachidonic acid or PLA2 binding site. Using excised patches from HEK-293 cells transiently transfected with rSlo(27) or rSlo(0), we found that arachidonic acid treatment resulted in a significant increase in Po in the former but not the latter, implicating the 27-amino acid region in arachidonic acid sensitivity.
rSlo(27) binds both cPLA2 and arachidonic acid much more strongly than rSlo(0).
Because of the differential sensitivity to arachidonic acid and the difference in coimmunoprecipitation, we used a direct binding assay to examine cPLA2 interaction with rSlo(27) and rSlo(0). The mammalian two-hybrid assay that we used can provide quantitative measures of the binding and rules out the possibility of indirect interaction that might occur in coimmunoprecipitation experiments. Using this assay, we found a strong association of cPLA2 with the COOH terminal end of rSlo(27) but not with rSlo(0).
In addition, in separate experiments in which the rSlo constructs were bound to agarose beads, we showed that arachidonic acid bound to rSlo(27) much more strongly than rSlo(0) (Fig. 10). In these experiments, if the nonspecific binding is subtracted from the binding for the two BK constructs, rSlo(27) binds arachidonic acid at least 12-fold more strongly than rSlo(0). This value is interesting since the COOH terminus of rSlo(27) binds cPLA2 11.3-fold better than rSlo(0) and implies that the binding of cPLA2 may be necessary for the binding of arachidonic acid.
The simple interpretation of these results would be that both cPLA2 and arachidonic acid bind to the 27-amino acid insertion in rSlo(27). However, there is an alternative possibility. Since, in the arachidonic acid binding experiments, both of the rSlo constructs were immunoprecipitated from cells transfected with one or the other of the constructs, it is possible that accessory proteins might be coimmunoprecipitated with the rSlo proteins. One candidate for such coimmunoprecipitation, at least for rSlo(27), is cPLA2, since we know it binds strongly to rSlo(27). In this scenario cPLA2 would act as an accessory protein for BK channels in a manner analogous to the better-known β-subunits. In fact, Fig. 7 shows that cPLA2 cotransfected with a mouse α-subunit can increase the activity of the channel as much or more than cotransfection with the β1-subunit. The activity of the wild-type channel obtained from cells that contained rat BKα1, the β1-subunit, and cPLA2 is approximately the same as cells transfected with the α-subunit and cPLA2 alone. This is consistent with our observation that cPLA2 is capable of displacing the β1-subunit from the COOH-terminal end of rSlo(27). In the presence of β1 or cPLA2 alone, either accessory protein binds to the α-subunit and increases its responsiveness to voltage and Ca2+. In the presence of both (as in the native GH3 cell), cPLA2 preferentially binds and the activation is similar to cPLA2 alone (Fig. 7).
In addition to arachidonic acid, cPLA2 and other lipases can produce other cis unsaturated fatty acids in addition to arachidonic acid that increase BK channel activity (5). Physical association of cPLA2 with the BK channel would allow synthesis and efficient delivery of arachidonic acid directly to its binding site on the BK channel protein.
Role of BK channel accessory proteins.
Our results imply that an association between the α-subunit and accessory proteins can result in changes that shift the Ca2+ or voltage response so that lower Ca2+ concentrations and lower voltages activate the channel. While the β-subunit is one example of a molecule that increases Ca2+ sensitivity, cPLA2 is apparently another. We conclude that the lack of response of BK channels in GH4 cells to arachidonic acid is explained, in part, by the poor binding of cPLA2 to the COOH terminus of the α-subunit [rSlo(0)].
Our results contrast with those of Sun et al. (25), who suggested that all arachidonic acid sensitivity of BK channels was conferred by β-subunits and that the α-subunit by itself did not respond to arachidonic acid. We, of course, would agree that some α-subunit isoforms (like the one present in GH4 cells) are not sensitive to arachidonic acid. They do not explicitly mention which human Slo isoform they expressed in Xenopus oocytes, but we presume that it is homologous to rSlo(0). They also used 30 times the amount of arachidonic acid that we use in our work. However, the interesting issue is whether a β-subunit could restore sensitivity to arachidonic acid. We originally examined the effect of the β1-subunit, which does not. They examined the β2-, β3-, and β4-subunits but did not examine β1. Like β1, β4 does not produce arachidonic acid sensitivity. On the other hand, they suggested that the β2-subunit strongly, and the β3-subunit to a lesser extent, confer arachidonic acid sensitivity to channels. If their conclusion that the β2-subunit was solely responsible for the arachidonic acid sensitivity was correct, then we should be able to convert rSlo(0) channels, which are unresponsive to arachidonic acid, to channels that are responsive to arachidonic acid simply by cotransfecting cells expressing rSlo(0) with the β2-subunit. In fact, our results show that, as expected, cotransfecting cells with rSlo(0) and either the β1- or the β2-subunit increased the control Po of the rSlo(0) channels to a level near channels in native GH4 cells (in which α and β are presumably always present). However, the addition of arachidonic acid to these channels did not result in a significant increase in channel activity. Therefore, β-subunits, by themselves, do not seem to induce sensitivity to arachidonic acid. These results are consistent with our hypothesis that the arachidonic acid sensitivity is predominantly dependent on the presence or absence of a 27-amino acid sequence residing on the BK channel α-subunit. One interesting aspect of the study of Sun et al. (25) is their use of ETYA as a lipoxygenase inhibitor. While this agent does produce inhibition, it also acts as a nonmetabolizable arachidonic acid analog, which did not activate channels. This suggests that some of the effect produced by the β-subunits were produced by metabolites. This will be an interesting area for future investigation.
The results presented here reenforce the importance of arachidonic acid as a signaling molecule in the CNS and specific cells. They also show that the expression of different splice variants can have profound effects on the responses of cells to environmental humoral agents. We further suggest that the constructs we have used are an interesting model in which to study the effects of arachidonic acid and possibly its metabolites. Our results also add to the rapidly growing body of evidence that suggests that the pore-forming subunits of many ion channels exist in the membrane as one of the components of a regulatory protein complex along with one or several signaling molecules that contribute to the modulation of the channel.
GRANTS
This work was supported by a National Science Foundation grant (IBN-0091964) to D. D. Denson and D. C. Eaton, an Emory University Research Committee grant to D. D. Denson, and National Institute of Diabetes and Digestive and Kidney Diseases Grant R37-DK-37963 to D. C. Eaton.
DISCLOSURES
The authors are not aware of financial conflict(s) with the subject matter or materials discussed in this manuscript with any of the authors, or any of the authors' academic institutions or employers.
ACKNOWLEDGMENTS
The authors gratefully acknowledge B. J. Duke for skillful technical assistance in the plating and maintenance of the cells used in this investigation.
A portion of this work was presented at the 2007 annual meeting of the Society for Neuroscience in San Diego, CA November 3–7, 2007.
REFERENCES
- 1. Brenner R, Jegla TJ, Wickenden A, Liu Y, Aldrich RW. Cloning and functional characterization of novel large conductance calcium-activated potassium channel beta subunits, hKCNMB3 and hKCNMB4. J Biol Chem 275: 6453–6461, 2000 [DOI] [PubMed] [Google Scholar]
- 2. Church J, Baxter KA, McLarnon JG. pH modulation of Ca2+ responses and a Ca2+-dependent K+ channel in cultured rat hippocampal neurones. J Physiol 511: 119–132, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Denson DD, Li J, Eaton DC. Co-localization of the alpha-subunit of BK-channels and c-PLA2 in GH3 cells. Biochem Biophys Res Commun 350: 39–49, 2006 [DOI] [PubMed] [Google Scholar]
- 4. Denson DD, Wang X, Worrell RT, AlKhalili O, Eaton DC. Cytosolic phospholipase A2 is required for optimal ATP activation of BK channels in GH3 cells. J Biol Chem 276: 7136–7142, 2001 [DOI] [PubMed] [Google Scholar]
- 5. Denson DD, Wang X, Worrell RT, Eaton DC. Effects of fatty acids on BK channels in GH3 cells. Am J Physiol Cell Physiol 279: C1211–C1219, 2000 [DOI] [PubMed] [Google Scholar]
- 6. Denson DD, Worrell RT, Eaton DC. A possible role for phospholipase A2 in the action of general anesthetics. Am J Physiol Cell Physiol 270: C636–C644, 1996 [DOI] [PubMed] [Google Scholar]
- 7. Denson DD, Worrell RT, Middleton P, Eaton DC. Ca2+ sensitivity of BK channels in GH3 cells involves cytosolic phospholipase A2. Am J Physiol Cell Physiol 276: C201–C209, 1999 [DOI] [PubMed] [Google Scholar]
- 8. Duerson K, White RE, Jiang F, Schonbrunn A, Armstrong DL. Somatostatin stimulates BKCa channels in rat pituitary tumor cells through lipoxygenase metabolites of arachidonic acid. Neuropharmacology 35: 949–961, 1996 [DOI] [PubMed] [Google Scholar]
- 9. Gribkoff VK, Starrett JE, JR, Dworetzky SI, Hewawasam P, Boissard CG, Cook DA, Frantz SW, Heman K, Hibbard JR, Huston K, Johnson G, Krishnan BS, Kinney GG, Lombardo LA, Meanwell NA, Molinoff PB, Myers RA, Moon SL, Ortiz A, Pajor L, Pieschl RL, Post-Munson DJ, Signor LJ, Srinivas N, Taber MT, Thalody G, Trojnacki JT, Wiener H, Yeleswaram K, Yeola SW. Targeting acute ischemic stroke with a calcium-sensitive opener of maxi-K potassium channels. Nat Med 7: 471–477, 2001 [DOI] [PubMed] [Google Scholar]
- 10. Ha TS, Jeong SY, Cho SW, Jeon H, Roh GS, Choi WS, Park CS. Functional characteristics of two BKCa channel variants differentially expressed in rat brain tissues. Eur J Biochem 267: 910–918, 2000 [DOI] [PubMed] [Google Scholar]
- 11. Katsuki H, Okuda S. Arachidonic acid as a neurotoxic and neurotrophic substance. Prog Neurobiol 46: 607–636, 1995 [DOI] [PubMed] [Google Scholar]
- 12. Knaus HG, Garcia-Calvo M, Kaczorowski GJ, Garcia ML. Subunit composition of the high conductance calcium-activated potassium channel from smooth muscle, a representative of the mSlo and slowpoke family of potassium channels. J Biol Chem 269: 3921–3924, 1994 [PubMed] [Google Scholar]
- 13. Levitan IB. Signaling protein complexes associated with neuronal ion channels. Nat Neurosci 9: 305–310, 2006 [DOI] [PubMed] [Google Scholar]
- 14. Liu H, Moczydlowski E, Haddad GG. O2 deprivation inhibits Ca2+-activated K+ channels via cytosolic factors in mice neocortical neurons. J Clin Invest 104: 577–588, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McManus OB, Helms LM, Pallanck L, Ganetzky B, Swanson R, Leonard RJ. Functional role of the beta subunit of high conductance calcium-activated potassium channels. Neuron 14: 645–650, 1995 [DOI] [PubMed] [Google Scholar]
- 16. Meves H. Modulation of ion channels by arachidonic acid. Prog Neurobiol 43: 175–186, 1994 [DOI] [PubMed] [Google Scholar]
- 17. Morrow JP, Zakharov SI, Liu G, Yang L, Sok AJ, Marx SO. Defining the BK channel domains required for beta1-subunit modulation. Proc Natl Acad Sci USA 103: 5096–5101, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murakami M, Nakatani Y, Atsumi G, Inoue K, Kudo I. Regulatory functions of phospholipase A2. Crit Rev Immunol 17: 225–283, 1997 [DOI] [PubMed] [Google Scholar]
- 19. Piomelli D. Arachidonic acid in cell signaling. Curr Opin Cell Biol 5: 274–280, 1993 [DOI] [PubMed] [Google Scholar]
- 20. Piomelli D, Greengard P. Bidirectional control of phospholipase A2 activity by Ca2+/calmodulin-dependent protein kinase II, cAMP-dependent protein kinase, and casein kinase II. Proc Natl Acad Sci USA 88: 6770–6774, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ramanadham S, Wolf MJ, Li B, Bohrer A, Turk J. Glucose-responsitivity and expression of an ATP-stimulatable, Ca2+-independent phospholipase A2 enzyme in clonal insulinoma cell lines. Biochim Biophys Acta 1344: 153–164, 1997 [DOI] [PubMed] [Google Scholar]
- 22. Ramanadham S, Wolf MJ, Ma Z, Li B, Wang J, Gross RW, Turk J. Evidence for association of an ATP-stimulatable Ca2+-independent phospholipase A2 from pancreatic islets and HIT insulinoma cells with a phosphofructokinase-like protein. Biochemistry 35: 5464–5471, 1996 [DOI] [PubMed] [Google Scholar]
- 23. Reinhart PH, Chung S, Levitan IB. A family of calcium-dependent potassium channels from rat brain. Neuron 2: 1031–1041, 1989 [DOI] [PubMed] [Google Scholar]
- 24. Shipston MJ. Alternative splicing of potassium channels: a dynamic switch of cellular excitability. Trends Cell Biol 11: 353–358, 2001 [DOI] [PubMed] [Google Scholar]
- 25. Sun X, Zhou D, Zhang P, Moczydlowski EG, Haddad GG. Beta-subunit-dependent modulation of hSlo BK current by arachidonic acid. J Neurophysiol 97: 62–69, 2007 [DOI] [PubMed] [Google Scholar]
- 26. Tian L, Hammond MS, Florance H, Antoni FA, Shipston MJ. Alternative splicing determines sensitivity of murine calcium-activated potassium channels to glucocorticoids. J Physiol 537: 57–68, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tseng-Crank J, Foster CD, Krause JD, Mertz R, Godinot N, Dichiara TJ, Reinhart PH. Cloning, expression, and distribution of functionally distinct Ca2+-activated K+ channel isoforms from human brain. Neuron 13: 1315–1330, 1994 [DOI] [PubMed] [Google Scholar]
- 28. Wallner M, Meera P, Toro L. Determinant for beta-subunit regulation in high-conductance voltage-activated and Ca2+-sensitive K+ channels: an additional transmembrane region at the N terminus. Proc Natl Acad Sci USA 93: 14922–14927, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. White RE, Schonbrunn A, Armstrong DL. Somatostatin stimulates Ca2+-activated K+ channels through protein dephosphorylation. Nature 351: 570–573, 1991. [DOI] [PubMed] [Google Scholar]