

Abstract
There is much interest in developing anti-myostatin agents to reverse or prevent muscle atrophy in adults, so it is important to characterize the effects of reducing myostatin activity after normal muscle development. For assessment of the effect of loss of myostatin signaling on gene expression in muscle, RNA from mice with postdevelopmental myostatin knockout was analyzed with oligonucleotide microarrays. Myostatin was undetectable in muscle within 2 wk after Cre recombinase activation in 4-month-old male mice with floxed myostatin genes. Three months after myostatin depletion, muscle mass had increased 26% (vs. 2% after induction of Cre activity in mice with normal myostatin genes), at which time the expression of several hundred genes differed in knockout and control mice at nominal P < 0.01. In contrast to previously reported effects of constitutive myostatin knockout, postdevelopmental knockout did not downregulate expression of genes encoding slow isoforms of contractile proteins or genes encoding proteins involved in energy metabolism. Several collagen genes were expressed at 20–50% lower levels in the myostatin-deficient muscles, which had ∼25% less collagen than normal muscles as reflected by hydroxyproline content. Most of the other genes affected by myostatin depletion have not been previously linked to myostatin signaling. Gene set enrichment analysis suggested that Smads are not the only transcription factors with reduced activity after myostatin depletion. These data reinforce other evidence that myostatin regulates collagen production in muscle and demonstrate that many of the previously reported effects of constitutive myostatin deficiency do not occur when myostatin is knocked out in mature muscles.
Keywords: collagen, energy metabolism, contractile protein genes, nitric oxide
mice with constitutive knockout of the myostatin gene have a remarkable double-muscled phenotype caused by both muscle fiber hyperplasia and fiber hypertrophy (14). Myostatin also regulates muscle development in other mammals, including humans (6, 15, 18, 22). In addition to causing muscle hypertrophy, lack of myostatin during development has been reported to cause reduced expression of slow isoforms of contractile proteins, diminished force generation per unit of muscle mass, reduced mitochondrial mass and expression of genes involved in mitochondrial energy metabolism, reduced collagen content in muscle, greater susceptibility to stretch-induced muscle injury, and impaired tendon development (1, 10, 16, 17, 24).
There is much interest in developing anti-myostatin agents to counteract muscle atrophy in humans. There already has been a safety trial of an anti-myostatin antibody in human subjects with muscular dystrophies (30). Because anti-myostatin agents would be used mostly in adults, it is important to know more about what happens to mature muscles after loss of myostatin activity. Microarray technology enables a broad overview of the impact of treatments on expression of all known genes, and already this technology has been used to examine the effects of constitutive myostatin deficiency in mice and cattle (4, 20, 24). However, it should not be assumed that loss of myostatin activity after maturity has the same effects as constitutive myostatin deficiency. We therefore used comprehensive oligonucleotide arrays to examine the effect of postdevelopmental myostatin depletion on the gene expression profile of skeletal muscle.
METHODS
Mice with postdevelopmental knockout of floxed myostatin exon 3 (Mstnf/f), via tamoxifen-induced Cre recombinase, have been described previously (32). Both the Mstnf/f (n = 5 males) and control mice (Mstnw/w, n = 5 males) had a Cre recombinase transgene and received a series of tamoxifen injections over a 2 wk period to activate the Cre recombinase when they were 4 mo of age. Based on an estimated myofibrillar protein half life of 3 wk in mature mice (31), we assumed that most of the effect on muscle mass had occurred within 3 mo (4 protein half-lives) after tamoxifen, so the mice were killed for analysis of muscles at that point. The research was approved by the University of Rochester animal research committee.
Mice were killed by CO2 inhalation until cessation of breathing, followed by cervical dislocation. Gastrocnemius muscles were immediately removed and immersed in liquid N2. Quadriceps muscles were then removed and either immersed in liquid nitrogen (for hydroxyproline assays) or frozen in cold isopentane (for histological analysis). Muscles were stored at −70°C.
RNA was extracted from the frozen gastrocnemius samples with TRIzol (Invitrogen) as recommended by the manufacturer. The amount recovered was quantified by UV light (260 nm) absorbance with a Nanodrop spectrophotometer, and was not significantly different between knockout and control mice. RNA preparations were of high quality as reflected by A260/A280 ratios of ∼1.9 and intact 18S and 28S ribosomal RNA according to Bioanalyzer (Agilent) analysis.
Total RNA was converted to biotin-labeled, fragmented cDNA with kits from NuGEN Technologies (Ovation Amplification System V2, FL-Ovation cDNA Biotin Module 2) according to the manufacturer's protocols. The cDNA was hybridized overnight with Mouse Genome 430 2.0 arrays (Affymetrix) as recommended by NuGEN. Arrays were washed, stained with a fluorescent dye that binds to biotin (streptavidin, R-phycoerythrin conjugate), and scanned as recommended by Affymetrix (GeneChip Expression Analysis Technical Manual) using instruments from Affymetrix (Fluidics Station 450, Scanner 3000).
Array normalization and background subtraction were done with the GCRMA procedure (38). The microarray image (cel) files and normalized data for all probe sets are available at the GEO website under accession number GSE15349 (www.ncbi.nlm.nih.gov/geo). Noninformative probe sets, defined as those for which three or more of the samples of the genotype with higher mean expression had no significant difference between perfect-match and mismatch probes at P < 0.10 according to the Affymetrix “detection P” test, were eliminated from the analysis. Nominal P values for differences between Mstnf/f and Mstnw/w mice were determined by t-tests. The significance analysis of microarrays (SAM) method (27) was used to estimate false discovery rate (FDR). Gene set enrichment analysis (GSEA) was used to explore the data for overall trends of upregulation or downregulation of sets of genes defined either by shared biological processes (302 sets after limiting analysis to sets with at least 15 members) or shared promoter motifs (586 sets). With this method (25), each gene set receives an enrichment score that reflects whether the genes within the set tend to be upregulated or downregulated. “Leading-edge” genes are those that have a positive impact on the enrichment score. FDR values are computed from the distribution of enrichment scores generated by permutations of the class labels (i.e., genotypes in this study).
For quantitative RT-PCR, reverse transcription with random hexamer priming was done with the Applied Biosystems high capacity kit. PCR was done in triplicate with a 7900HT thermal cycler/fluorescence detector (Applied Biosystems) using the recommended cycling conditions. Taqman assays for Col1a2 (Assay ID Mm00483937_m1), Nos1(Assay ID Mm00435175_m1), Ddah1 (Assay ID Mm01319453_m1), and Gapdh (catalog number 4308313) cDNAs were purchased from Applied Biosystems. Primers and probe for Myh7 cDNA have been published (identified as myosin heavy chain type 1) (32). Gapdh mRNA was selected as an appropriate reference gene for this study based on the fact that several microarray probes showed differences of <5% between Mstnf/f and Mstnw/w mice.
Hydroxyproline levels in gastrocnemius and quadriceps muscles were measured as an index of collagen content. Muscles were minced and warmed to 60°C in 1 M KCl to dissolve myofibrils. Soluble proteins were removed by aspiration. The insoluble pellets were washed twice with water, dried by vacuum centrifugation, and hydrolyzed in 6 N HCl overnight at 105°C. Concentrations of hydroxyproline in the hydrolyzates were determined by a colorimetric method (37).
Western blots for promyostatin levels in gastrocnemius muscles were performed as described by Anderson et al. (2) with an antibody purchased from R&D Systems (AF1539). The initial blot was done with samples reduced with 5% 2-mercaptoethanol (95°C for 10 min). Subsequently, nonreducing conditions were used (samples heated in Laemmli buffer at 55°C for 10 min with no reducing agent) because the sensitivity was better with nonreduced than with reduced homogenates. Blots were blocked with 5% Blotto. Antibody dilutions were 1,000:1 in 5% BSA for the AF1539 primary antibody and 1,000:1 in 5% Blotto for the anti-sheep IgG-HRP secondary antibody (R&D Systems HAF016). The secondary antibody was detected by chemiluminescence (Pierce SuperSignal West Dura kit).
Histochemical analysis of mitochondrial enzymes (succinate dehydrogenase, SDH; cytochrome oxidase, COX) was done with cryosections of quadriceps muscles (10 μm thickness). Cryosections also were stained for glycogen (periodic acid-Schiff, PAS). Histological analyses were done with established methods (7). Immunofluorescence with an anti-caveolin antibody was used to define sarcolemmal membranes for computation of muscle fiber cross-sectional areas (32).
RESULTS AND DISCUSSION
Efficacy of postdevelopmental myostatin knockout.
As reported previously (32), Mstnf/f mice treated with tamoxifen lost >99% of their myostatin mRNA in gastrocnemius muscle within 5 days after tamoxifen treatment. The Mstn gene encodes promyostatin, from which the active domain of myostatin is cleaved. Promyostatin appears to be the predominant form of myostatin protein in murine skeletal muscle (2). Promyostatin levels were undetectable in Mstnf/f mice within 2 wk after the end of tamoxifen treatment (earliest time point examined) and remained undetectable for >6 mo after treatment (Fig. 1). We could not quantify loss of the active myostatin fragment because only nonspecific bands (i.e., bands present in samples from mice with constitutive myostatin knockout) appeared on immunoblots with several anti-active domain antibodies (JA16, donated by Wyeth Pharmaceuticals; Chemicon AB3239; Santa Cruz SC-6884; Novus NB 100–281; Bethyl A300–401A; R&D Systems AF788). This does not mean that active myostatin is not present in normal muscle tissue, only that levels are below the sensitivity of standard immunoblots with these antibodies.
Fig. 1.
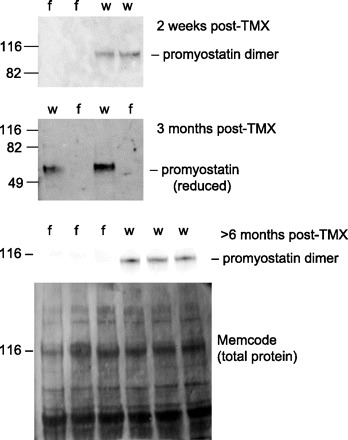
Western blots demonstrate permanent knockout of promyostatin within 2 wk after administration of tamoxifen (TMX) to Mstnf/f mice (f). Age-matched Mstnw/w mice (w) received the same regimen of tamoxifen to induce Cre recombinase activity. Top 2 blots were done with muscle protein samples in which nonglycosylated proteins were removed with concanavalin A (2). Samples in bottom blot were not processed with concanavalin A. Samples used for top and bottom blot were not reduced, and bands are at expected location for propeptide dimer. Samples used for middle blot were reduced, and bands are at expected location for propeptide monomer. Memcode staining was done to confirm even loading and transfer of proteins. Locations of prestained standards (Benchmark, Invitrogen) are denoted by the corresponding molecular weights (kDa).
Muscle mass increased an average of 26% in Mstnf/f mice over the 3 mo period after tamoxifen treatment (24% increase in gastrocnemius mass, 28% increase in quadriceps mass) and did not change significantly after tamoxifen treatment in Mstnw/w mice (1% increase in gastrocnemius mass, 3% increase in quadriceps mass). The mean cross-sectional area of ∼1,000 fibers from quadriceps of five mice with postdevelopmental myostatin knockout was 29% greater than the mean value of ∼1,000 fibers from five control mice.
Number of differentially expressed genes.
At FDR <5% by the SAM procedure, 67 probe sets indicated differential expression in postdevelopmental-knockout versus control mice (the number of affected probe sets is slightly larger than the number of affected genes because of replicate probe sets for some genes). At nominal P < 0.01 by t-tests, 516 probe sets indicated differential expression (these are listed in Supplemental File 11). FDR at nominal P = 0.01 was ∼22%. At nominal P < 0.05, 1,855 probe sets indicated differential expression, but the estimated FDR was very high (∼45%).
Most of the differences were modest in magnitude (Fig. 2). Of the 516 differences at nominal P < 0.01, only 7.2% were >2-fold; 76% of the differences were <1.5-fold, and 31% were <1.2-fold.
Fig. 2.
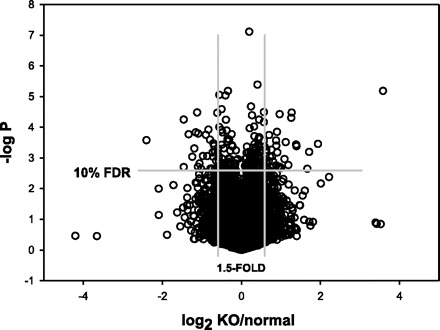
Distribution of gene expression differences between myostatin-knockout (KO) and normal muscles, according to magnitude of mean difference (x-axis is log2 of KO:normal ratio) and nominal P according to t-tests (negative log10 of P value is shown). Each circle represents the mean difference for a single probe set. The total number of probes sets included in the analysis after filtering out noninformative probes (see text) was 32,462. FDR, false discovery rate.
Steelman et al. (24) reported that >2,000 genes were differentially expressed (FDR <5%) in pectoral muscles of young mice with constitutive myostatin knockout versus age-matched control mice (13% >2-fold, 34% >1.5-fold, 87% >1.2-fold). This is far more than the number of affected genes (at the same statistical stringency) in the present study. Because pectoral and gastrocnemius muscles have the same degree of excessive growth in mice with constitutive myostatin knockout (14), and both are primarily fast-twitch muscles, it is unlikely that the vast difference in the number of differently expressed genes in these studies can be explained by examination of different muscles. A more likely explanation is that lack of myostatin during development affects the expression of more genes than does loss of myostatin after maturity. Another factor that contributes to the greater number of effects in the study of Steelman et al. is that they examined replicate arrays after pooling RNA from several mice of each genotype. Hence the within-genotype variance was smaller because mouse-to-mouse variability was not included as a source of variance.
Genes encoding proteins involved in muscle contraction.
Genes encoding slow isoforms of several contractile proteins are downregulated 4- to 10-fold in pectoral muscle of mice with constitutive myostatin deficiency (Table 1). A similar reduction in Myh7 gene expression (this gene encodes the slowest myosin heavy chain isoform, type 1) was observed by real-time PCR in extensor digitorum longus (EDL) muscle of 6-month-old mice with constitutive myostatin knockout, and a twofold reduction was observed in soleus muscle (10). The decline in Myh7 expression in soleus corresponded to a reduced number of type 1 fibers. Reduced MYH7 expression (about fivefold) also was observed in muscles of myostatin-deficient late bovine fetuses (4). Although Myh7 and other genes encoding slow isoforms of contractile proteins had modestly lower mean expression in mice with postdevelopmental myostatin knockout, none of the effects was statistically significant (Table 1). This result is consistent with the observation that administration of an anti-myostatin antibody to mice for 12 wk (starting at 10 wk of age) did not alter the pattern of expression of the genes encoding different isoforms of myosin heavy chain (10). Thus, unlike constitutive lack of myostatin, loss of myostatin activity in mature muscle does not significantly reduce the expression of genes encoding slow isoforms of contractile proteins.
Table 1.
Effect of postdevelopmental myostatin knockout on expression of contractile protein genes that are downregulated more than fourfold in mice with constitutive myostatin deficiency
| Gene | Symbol | Affymetrix probe ID | Constitutive KO/Normal | P | Postdevelopmental KO/Normal | P |
|---|---|---|---|---|---|---|
| Myosin heavy polypeptide 7 (type 1 myosin heavy chain) | Myh7 | 1448554_s_at | 0.09 | <10−6 | 0.74 | 0.47 |
| 1448553_at | 0.14 | <10−6 | 0.79 | 0.37 | ||
| RealtimePCR | 0.68 | 0.27 | ||||
| Myosin light polypeptide 2 | Myl2 | 1448394_at | 0.18 | <10−6 | 0.86 | 0.19 |
| Myosin light polypeptide 3 | Myl3 | 1427768_s_at | 0.21 | <10−6 | 0.98 | 0.84 |
| 1428266_at | 0.25 | <10−6 | 1.00 | 0.99 | ||
| 1427769_x_at | 0.30 | <10−6 | 1.01 | 0.94 | ||
| Troponin C, cardiac/slow skeletal | Tnnc1 | 1418370_at | 0.15 | <10−6 | 0.86 | 0.48 |
| Troponin I, skeletal, slow 1 | Tnni1 | 1450813_a_at | 0.20 | <10−6 | 0.74 | 0.47 |
| Troponin T1, skeletal, slow | Tnnt1 | 1419606_a_at | 0.14 | <10−6 | 0.82 | 0.48 |
| Tropomyosin 3, gamma | Tpm3 | 1449997_at | 0.12 | <10−6 | 0.83 | 0.57 |
| 1449996_a_at | 0.13 | <10−6 | 0.82 | 0.63 |
Data for mice with constitutive knockout (KO) are from Steelman et al. (24), in which the same microarray platform used in the present study was used to examine RNA from pectoral muscles of 5 wk old mice. P refers to significance of difference between knockout and control groups by t-tests without adjustment for multiple comparisons.
When EDL muscles are stimulated directly ex vivo, there is a deficit in the amount of force generated per unit of muscle cross sectional area in mice with constitutive myostatin deficiency (1, 17). This deficit might be caused by an abnormal angle of pennation of the fibers due to the double muscling (17). An abnormal angle of pennation is less likely to be problematic with the more modest muscle growth after postdevelopmental loss of myostatin. After several weeks of anti-myostatin antibody treatment, normal mice have increased grip strength, but the percent increase in strength is less than the percent increase in muscle mass (34). Although this result could mean that the enlarged muscles generated less force per unit of cross-sectional area during grip strength testing, such testing depends on the motivation of the mouse to hold the bar tightly and therefore does not assess maximum force-generating capacity. We did not observe any gene expression changes that would be expected to influence contraction force, with the possible exception of reduced expression of two genes, Nos1 and Ddah1, involved in nitric oxide (NO) synthesis. Nos1 encodes neuronal nitric oxide synthase (also known as nNOS or NOS-I), a sarcolemmal protein that catalyzes synthesis of NO from arginine. Nos1 mRNA expression was 69% of normal (P < 0.01) after myostatin depletion. Ddah1 encodes dimethylarginine dimethylaminohydrolase 1, which metabolizes asymmetric dimethylarginine, an inhibitor of NOS. Ddah1 mRNA expression was 50% of normal (P < 0.001) after myostatin depletion. Reduced expression of these genes was confirmed by real-time PCR (Nos1/Gapdh mRNA ratio 60% of normal, P < 0.0001; Ddah1/Gapdh mRNA ratio 40% of normal, P < 0.0001). Moreover, short-term (4 day) inhibition of myostatin activity with an antibody (33) reduced Nos1 and Ddah1 expression (Nos1 expression 63% of normal; Ddah1 expression 45% of normal, P < 0.005 for both genes). Pharmacologic inhibition of NO synthesis reduces muscle twitch force (8), so if reduced expression of Nos1 and Ddah1 reduces NO production it is possible that this could impair the capacity for force generation. Increased fatigability is another potential consequence of reduced NO production (11, 19). However, genes encoding other isoforms of NOS were not differentially expressed, and we did not determine whether the NO production was reduced after myostatin depletion. Dynll1, another gene downregulated after myostatin depletion (70% of normal expression, FDR <5%), encodes a protein (dynein light chain LC8-type 1) that inhibits nNOS activity (39). Reduced Dynll1 expression could counteract any effect that reduced expression of Nos1 and Ddah1 might have on NO production.
Genes encoding mitochondrial proteins and other proteins involved in energy metabolism.
Constitutive myostatin knockout reduces the number of oxidative fibers (assessed by histochemical staining for SDH activity), the number of mitochondria per unit cross-sectional area, the amount of mitochondrial DNA, and expression of genes involved in mitochondrial electron transport and energy metabolism (1, 10, 24). We therefore analyzed a group of 938 probe sets that target transcripts encoding mitochondrial proteins (based on annotations provided by Affymetrix, which were obtained from Gene Ontology, GenMAPP, and KEGG databases) by the SAM procedure. As illustrated in Fig. 3, there was no evidence for overall downregulation of this group of genes after myostatin depletion. Only two genes showed reduced expression after myostatin depletion at nominal P < 0.01, and both effects were modest in magnitude (7% decrease in expression of Psap, 19% decrease in expression of Htra2). Neither of these genes encodes a protein directly involved in energy metabolism (see Table 2 for comments on gene function). Four genes in this group were modestly (<1.4-fold) upregulated at FDR < 5% (Hibch, Glul, Cyb5b, Acadsb). Only one was upregulated more than twofold (LOC632297), and this is a chromosome 1 gene that might have been affected by its proximity to the Mstn locus rather than a specific effect of myostatin depletion (more on this issue below).
Fig. 3.
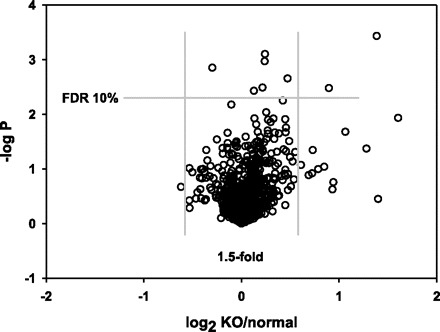
Subset of data from Fig. 2; 938 probes targeting transcripts encoding mitochondrial proteins.
Table 2.
Additional information for gene abbreviations mentioned in text
| Symbol | Full Name | Function of Encoded Protein |
|---|---|---|
| Nos1 | nitric oxide synthase 1 | NO synthesis |
| Ddah1 | dimethylarginine dimethylaminohydrolase 1 | catalyzes metabolism of ADMA, an inhibitor of nitric oxide synthase |
| Dynll1 | dynein light chain LC8-type 1 | inhibits nNOS activity |
| Psap | prosaposin | catabolism of glycosphingolipids with short oligosaccharide groups |
| Htra2 | HtrA serine peptidase 2 | serine protease |
| Hibch | 3-hydroxyisobutyryl-coenzyme A hydrolase | hydrolysis of HIBYL-CoA, a valine catabolite, and of beta-hydroxypropionyl-CoA, an intermediate in a minor pathway of propionate metabolism |
| Glul | glutamate-ammonia ligase (glutamine synthetase) | gltuamine synthesis |
| Cyb5b | cytochrome b5 type B | localized in outer mitochondrial membrane; metabolism of various compounds |
| Acadsb | acyl-coenzyme A dehydrogenase, short/branched chain | dehydrogenation of acyl-CoA derivatives in the metabolism of fatty acids or branch chained amino acids |
| LOC632297 | cimilar to UDP glycosyltransferase family | UDP glycosyltransferases convert small lipophilic molecules to excretable, water-soluble molecules |
| Ppargc1a | peroxisome proliferator-activated receptor gamma, coactivator 1 alpha | transcriptional coactivator that permits the interaction of PPARγ with multiple transcription factors. Provides a link between external physiological stimuli and the regulation of mitochondrial biogenesis, and regulates muscle fiber type determination. |
| Ppp1r3c | protein phosphatase 1, regulatory (inhibitor) subunit 3C | dephosphorylation of proteins |
| Acsl1 | acyl-CoA synthetase long-chain family member 1 | converts free long-chain fatty acids into fatty acyl-CoA esters |
| Eln | elastin | extracellular matrix component |
| Pcolce | procollagen C-endopeptidase enhancer protein | enhances cleavage of type I procollagen |
| Zmynd17 | zinc finger, MYND-type containing 17 | no references available |
| Fam19a5 | family with sequence similarity 19 (chemokine (C-C motif)-like), member A5 | putative chemokine |
| Cercam | cerebral endothelial cell adhesion molecule | cell adhesion |
| Btbd1 | BTB (POZ) domain containing 1 | binds DNA topoisomerase I; essential for myogenic differentiation |
| Rrad | Ras-related associated with diabetes | Ras-related small G protein |
| Myl4 | myosin, light chain 4, alkali | myosin alkali light chain that is found in embryonic muscle |
| Pmepa1 | prostate transmembrane protein, androgen induced 1 | inhibits androgen receptor protein expression in prostate cancer cells |
| Sult5a1 | sulfotransferase family 5A, member 1 | conjugation of compounds with sulfate |
| Krt18 | keratin 18 | component of keratin filaments |
| Dntt | deoxynucleotidyltransferase, terminal | template-independent DNA polymerase |
| Zfp318 | zinc finger protein 318 | modulates androgen receptor activity |
| Ccl25 | chemokine (C-C motif) ligand 25 | chemokine that binds to chemokine receptor CCR9 |
| Reep6 | receptor accessory protein 6 | might be involved in intracellular membrane trafficking |
| Rhpn2 | rhophilin, Rho GTPase binding protein 2 | binds both GTP- and GDP-bound RhoA and GTP-bound RhoB |
| Npnt | nephronectin | extracellular matrix protein |
| Cdh4 | cadherin 4 | calcium-dependent cell-cell adhesion glycoprotein |
Information in this table was extracted from Entrez Gene (http://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene) and references cited at the Entrez Gene web site.
Ppargc1a (Table 2) encodes a protein that is a key regulator of mitochondrial biogenesis. In mice with constitutive myostatin knockout, this gene is downregulated 1.5-fold (24). Mice with postdevelopmental knockout expressed Ppargc1a at a normal level (P = 0.43 vs. control mice).
Several hundred probe sets had annotations relating the target transcripts to glucose, glycogen, or fatty acid metabolism. There was some overlap of this list with the mitochondrial category discussed above. No gene in these categories was downregulated at nominal P < 0.01, and only three were modestly (<1.25-fold) upregulated (Ppp1r3c, Acsl1, Acadsb; see Table 2 for comments on functions of these genes).
The failure of postdevelopmental myostatin depletion to influence genes encoding mitochondrial proteins is consistent with the report that several weeks of administration of an anti-myostatin antibody in adult mice did not affect SDH activity (histochemical method) (10). Histochemistry of quadriceps sections from mice examined in the present study (representative sections shown in Supplemental File 2) confirmed that there is no deficit in SDH activity after myostatin depletion. Moreover, sections stained for COX activity and glycogen (PAS) were consistent with the conclusion that loss of myostatin did not lead to metabolic changes in muscle.
Genes encoding collagen.
Myostatin increases expression of several collagen genes and promotes collagen synthesis in cultured myotubes, C3H10T1/2 cells, and muscle fibroblasts (3, 12, 17). Total collagen abundance, as reflected by hydroxyproline content, is reduced in mature mice with constitutive myostatin knockout (17). In myostatin-deficient bovine fetuses, expression of three collagen genes (COL1A1, COL1A2, COL3A1) was reduced more than twofold (4). However, Steelman et al. (24) did not observe reduced expression of collagen genes in pectoral muscles of young mice with constitutive myostatin knockout. In adult bulls with a myostatin promoter polymorphism that reduced myostatin expression ∼1.5-fold, expression of COL1A2, COL6A1, and COL6A3 was modestly increased (20).
In the present study, myostatin depletion led to reduced expression of 11 collagen genes at nominal P < 0.01 (Table 3). The effect on Col1a2 gene expression was confirmed by quantitative PCR (Col1a2/Gapdh mRNA ratio 56% of normal, P < 0.02). Six other collagen genes were expressed at a lower level after myostatin depletion at nominal P < 0.05 (Col4a2, Col4a5, Col5a3, Col8a1, Col12a1, Col22a1). Two other genes involved in extracellular matrix formation also were expressed at lower levels after myostatin depletion: Eln (−43%, P < 0.01) and Pcolce (−34%, P < 0.01) (Table 2).
Table 3.
Collagen genes downregulated at P < 0.01 after myostatin knockout in mature mice
| Collagen Type | Symbol | Affymetrix Probe ID | KO/Normal | Nominal P | FDR |
|---|---|---|---|---|---|
| I,α1 | Col1a1 | 1423669_at | 0.46 | 0.00003 | 0.00 |
| 1455494_at | 0.61 | 0.0009 | 0.08 | ||
| I,α2 | Col1a2 | 1423110_at | 0.45 | 0.0001 | 0.03 |
| 1450857_a_at | 0.56 | 0.0001 | 0.03 | ||
| III,α1 | Col3a1 | 1427883_a_at | 0.76 | 0.003 | 0.15 |
| IV,α1 | Col4a1 | 1426348_at | 0.75 | 0.004 | 0.17 |
| 1452035_at | 0.82 | 0.002 | 0.12 | ||
| IV,α4 | Col4a4 | 1440250_at | 0.65 | 0.004 | 0.17 |
| V,α1 | Col5a1 | 1416740_at | 0.79 | 0.007 | 0.20 |
| 1434479_at | 0.66 | 0.006 | 0.19 | ||
| V,α2 | Col5a2 | 1422437_at | 0.54 | 0.0002 | 0.03 |
| VI,α1 | Col6a1 | 1488590_at | 0.76 | 0.00001 | 0.00 |
| VI,α2 | Col6a2 | 1426947_x_at | 0.66 | 0.0005 | 0.05 |
| 1452250_a_at | 0.71 | 0.00003 | 0.00 | ||
| VI,α3 | Col6a3 | 1424131_at | 0.68 | 0.00001 | 0.00 |
| XVI,α1 | Col16a1 | 1427986_a_at | 0.71 | 0.008 | 0.20 |
KO/Normal refers to ratio of mean normalized fluorescence intensities for 5 Mstnf/f mice and 5 Mstnw/w mice 3 mo after treatment with tamoxifen to induce Cre recombinase activity. Nominal P refers to significance of difference between KO and control groups by t-tests without adjustment for multiple comparisons. FDR is false discovery rate estimate from the significance analysis of microarrays procedure.
The five most abundant collagen mRNAs according to fluorescence intensities on the microarrays (Col1a1, Col1a2, Col3a1, Col4a1, and Col4a2) all were downregulated in mice with postdevelopmental myostatin knockout. Those most affected by loss of myostatin (Col1a1 and Col1a2) encode the proteins of type I collagen, which is a major component of the endomysium and perimysium of skeletal muscle (13). The decline in expression of mRNAs encoding type III and type IV collagens (Col3a1, Col4a1, Col4a2) was only half as much as the decline in Col1a1 and Col1a2 expression. Like type I collagen, type III is major component of endomysium and perimysium. Type IV collagen is a component of the basement membrane surrounding muscle fibers (13).
Hydroxyproline levels in protein hydrolyzates of gastrocnemius and quadriceps muscles, an index of collagen abundance, were 25–30% less in myostatin-deficient mice (Fig. 4). This effect is much less than the 75% reduction in hydroxyproline content of EDL muscles of mice with constitutive myostatin deficiency, but similar to the 24% reduction in hydroxyproline in soleus muscles of these mice (17).
Fig. 4.
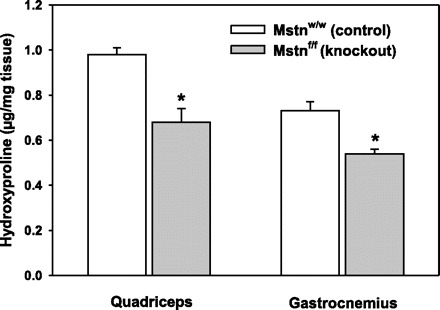
Reduced collagen content in muscles of myostatin-KO (Mstnf/f) vs. control mice (Mstnw/w) as reflected by hydroxyproline content of tissue protein hydrolyzates. *P < 0.02 for comparison between Mstnf/f and Mstnw/w.
In situ hybridization has indicated that the mRNAs encoding type I and type III collagens in muscle tissue are produced mostly in fibroblasts (35, 36). Thus, if the total mRNA concentration within muscles remains constant while they increase in size, and the total number of muscle fibroblasts remains constant or declines (12), then there should be reduced collagen mRNA expression (normalized to total tissue mRNA) simply because fibroblast mRNA is a smaller proportion of the total. It is not possible to quantify based on the available information the extent to which this factor explains the decline in collagen gene expression after myostatin depletion. This proportion may vary for different collagen genes depending on the extent to which the genes are expressed in fibroblasts versus muscle fibers or other types of cells.
The amount of collagen that surrounds a muscle fiber or fascicle is proportional to the circumference of the fiber or fascicle. Circumference is proportional to fiber diameter, whereas overall size (cross-sectional area) is proportional to the square of the diameter. Thus, muscle fiber growth should cause some reduction in the amount of collagen per mg tissue if normal collagen levels in the extracellular matrix and normal interstitial spaces are maintained. However, we would have expected a decline of only ∼11% in the amount of collagen per mg tissue based on fiber hypertrophy alone (see Supplemental File 3 for assumptions and calculations). The observed decrease in hydroxyproline abundance was greater than this, suggesting that muscle fibers and/or fascicles were surrounded by less collagen after myostatin depletion.
Effects of large magnitude (twofold or more).
All of the genes affected more than fourfold (nominal P < 0.01) were chromosome 1 genes near enough to the Mstn locus that the effects probably are related to the gene targeting method rather than loss of myostatin activity. This issue is discussed in a separate section below. Three of the genes downregulated about twofold, Col1a1, Col1a2, and Ddah1, were mentioned above. Only a few other annotated genes not located in chromosome 1 were downregulated twofold or more: Zmynd17, Fam19a5, Cercam, Btbd11, Rrad, Myl4, Pmepa1, Sult5a1, and Krt18 (Table 2). Even fewer annotated genes not located in chromosome 1 were upregulated twofold or more: Dntt, Zfp318, Ccl25, Reep6, Rhpn2, Npnt, and Cdh4 (Table 2). For the most part, the potential impact of altered expression of these genes on muscle phenotype cannot be predicted based on current knowledge of the functions of the encoded proteins. Pmepa1 encodes a protein that inhibits androgen receptor protein expression in prostate cancer cells. If it has a similar action in muscle, it is possible that reduced Pmepa1 gene expression could promote muscle growth by enhancing sensitivity to the anabolic effect of testosterone. Zfp318 encodes a protein that can modulate the effect of the androgen receptor on gene transcription, depending on the splice variant (26). Rrad encodes the GTPase Rad (Ras associated with diabetes). Rad-deficient mice have increased susceptibility to cardiac hypertrophy caused by activation of CaMKII, but baseline cardiac mass is normal (5). Whether Rad has a role in skeletal muscle hypertrophy is not known.
GSEA.
GSEA identified downregulation (FDR <5%) of three curated pathway gene sets: “extracellular matrix-receptor interaction,” “cell communication,” and “intrinsic pathway.” All three of these sets had a high proportion of collagen genes among the leading-edge genes. No other process/pathway gene sets were influenced at FDR <5%.
Nine promoter motif gene sets were downregulated (FDR <5%) after myostatin depletion (Table 4). Except for Smad4, the transcription factors that might be responsible for these effects have not been linked previously to myostatin. TGF-β increases expression of one of these transcription factors, Foxj1, in T cells (23), but the only microarray probe for Foxj1 mRNA indicated that it is not expressed in muscle. The reduced expression of a Smad4 promoter motif gene set is consistent with the fact that Smads are involved in myostatin signaling (21). Smad4 participates in transcriptional regulation after forming a complex with phosphorylated Smad2 or Smad3 (9). A Smad3 promoter motif (TGTCTGTCT) gene set also showed a trend (FDR = 11%) for downregulation after myostatin depletion. Smads promote the transcription of collagen genes (29), so reduced Smad activity after myostatin depletion is the likely explanation for reduced collagen gene expression. The minimal Smad binding element is 5′-GTCT-3′ (or its reverse complement 5′-AGAC-3′), but other DNA sequences can bind Smads (9). Because the GSEA promoter motif database does not include sequences with fewer than six bases (40), there are no gene sets linked to this minimal element. Affinity of Smads for the minimal binding element is low, and generally the Smads affect transcription by forming complexes with other transcription factors that have high affinity DNA binding (9). Thus, even though Smads are not known to bind to most of the motifs listed in Table 4, they might interact with other transcription factors that have a high affinity for these motifs.
Table 4.
Promoter motif gene sets downregulated in mice with postdevelopmental myostatin knockout (GSEA method)
| Motif | Potential Transcription Factor | FDR, % | Gene Set Size | Leading Edge Genes | Leading Edge Genes Differentially Expressed at P < 0.01 |
|---|---|---|---|---|---|
| WWWWNGMCACGTCATYNYW | X-box binding protein 1 (Xbp1) | 0.0 | 47 | 16 | Col1a2, Creb3l1, Sec61a1 |
| GKSRKKCAGMCANCY | Smad4 | 1.0 | 148 | 55 | Col1a2, Notch3, Mylk |
| RYTAAWNNNTGAY | unknown | 1.3 | 36 | 13 | Col16a1 |
| RYAAAKNNNNNNTTGW | unknown | 2.0 | 51 | 28 | none |
| GGCTCYATCAYC | zinc finger and BTB domain containing 6 (Zbtb6) | 2.7 | 136 | 56 | Skil, Sesn3 |
| CCANNAGRKGGC | unknown | 2.9 | 63 | 17 | Eln |
| RTAATNA | POU domain, class 2, transcription factor 1 (Pou2f1) | 3.4 | 142 | 42 | Skil |
| CTGYNNCTYAA | unknown | 3.7 | 49 | 9 | Cercam |
| AAAYWAACM | forkhead box J1 (Foxj1) | 4.7 | 165 | 62 | Itgbl1, Fstl1, Angptl1, Stard13 |
Promoter motifs were defined as 6-base or longer sequences conserved in promoters (within 2 kb of transcription start sites) across different mammalian species (40). Potential transcription factors were derived from the TRANSFAC database, but it is possible that other factors bind to these sequences. Leading edge genes are those that contribute to the enrichment score generated by the gene set enrichment analysis (GSEA) method. Note that most of the leading edge genes are not differentially expressed at nominal P < 0.01.
Some effects could be consequences of the gene targeting method rather than myostatin depletion.
Interpretation of gene targeting experiments can be complicated by strain differences if the embryonic stem cell strain used for gene targeting (129S6/SvEvTac in this study) and the background strain (C57BL/6 in this study) are not the same (28). Even 10 crosses into the background strain may be insufficient to eliminate DNA polymorphisms near the targeted gene. Moreover, cis-acting effects of the Cre-mediated DNA deletion might affect gene expression. Thus we examined whether chromosome 1 genes near Mstn were more likely to be differentially expressed than genes on other chromosomes. The number of differentially expressed genes at nominal P < 0.01 on chromosome 1, relative to the number of probes sets for this chromosome, was more than twofold greater than the number expected from the overall average across all chromosomes (P < 10−8 by χ2 test). A poorly annotated transcript from a locus within 10 Mb of Mstn had the largest difference in gene expression (12-fold) between the control and postdevelopmental-knockout mice. Of the 34 genes with the largest expression differences (more than twofold in magnitude and nominal P < 0.01), 12 were located within ∼50 Mb of the Mstn locus. A small percentage of the DNA on other chromosomes could have had 129S6/SvEvTac polymorphisms when we started inbreeding the Mstnf/f line. It is possible that a few differences in gene expression could be related to this factor rather than the loss of myostatin.
General Discussion
Myostatin knockout induced after development affects the expression of fewer genes than does constitutive myostatin knockout. Most of the genes that are affected by postdevelopmental myostatin depletion have only modest changes in expression (<1.5-fold). These results do not raise concerns that inhibiting myostatin activity would have adverse effects related to abnormal expression of genes encoding proteins involved in energy metabolism or muscle contraction. A possible exception to this conclusion is the reduced expression of two genes involved in NO production. If studies of force generation or fatigability in these mice reveal any deficits associated with myostatin deficiency, NO production could be increased pharmacologically to determine whether deficits are reversed.
Loss of myostatin reduced the expression of several collagen genes and reduced the amount of collagen per mg muscle tissue. In some settings, such as muscular dystrophies or muscle injuries in which there can extensive fibrosis, reduced collagen production could be beneficial. However, if myostatin inhibition were to be used in an attempt to counteract atrophy that is not associated with fibrosis, the reduced collagen production might weaken the endomyium or perimysium and render muscles more susceptible to damage induced by stretch (17).
A potential value of microarray studies is the discovery of novel biomarkers. Molecular markers of reduced myostatin activity could be useful to study new agents that might be developed to inhibit myostatin activity. Use of molecular markers could shorten the time needed to determine appropriate doses in preclinical or early clinical trials, since it might take several weeks for muscle mass to change significantly after myostatin activity is suppressed. Ideally, a biomarker is affected within a short period of time, is affected persistently, and is affected to a large enough extent that detecting a change is not too challenging. For mRNA biomarkers, this means that the effect should be roughly twofold or more. The present study, together with microarray data describing the early effects of inhibiting myostatin with an antibody (33) and microarray data describing gene expression in growing mice with constitutive myostatin knockout (24), points to Ddah1 mRNA as a candidate biomarker. Remarkably, Ddah1 is the only gene affected twofold or more at P < 0.01 in all three of these studies. Moreover, the effect has been prospectively confirmed by PCR in Mstnf/f mice in which a different Cre recombinase transgene was used to knock out myostatin after normal muscle development (unpublished data). Whether or not reduced expression of the Ddah1 gene has any functional significance, Ddah1 appears to be the best “reporter gene” for detecting loss of myostatin signaling.
GRANTS
This work was supported by National Institutes of Health Grants AR-054366 and RR-024160.
Supplementary Material
Acknowledgments
We thank Sangeeta Mehta, Bharati Shah, and Meghann McBennett for technical assistance.
Footnotes
Address for reprint requests and other correspondence: S. Welle, Univ. of Rochester Medical Center, 601 Elmwood Ave., Box 693, Rochester, NY 14642 (e-mail: stephen_welle@urmc.rochester.edu).
The online version of this article contains supplemental material.
REFERENCES
- 1.AmthorHAmthor H, Macharia R, Navarrete R, Schuelke M, Brown SC, Otto A, Voit T, Muntoni F, Vrbova G, Partridge T, Zammit P, Bunger L, Patel K. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc Natl Acad Sci USA 104: 1835–1840, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.AndersonSBAnderson SB, Goldberg AL, Whitman M. Identification of a novel pool of extracellular pro-myostatin in skeletal muscle. J Biol Chem 283: 7027–7035, 2008. [DOI] [PubMed] [Google Scholar]
- 3.ArtazaJNArtaza JN, Singh R, Ferrini MG, Braga M, Tsao J, Gonzalez-Cadavid NF. Myostatin promotes a fibrotic phenotypic switch in multipotent C3H 10T1/2 cells without affecting their differentiation into myofibroblasts. J Endocrinol 196: 235–249, 2008. [DOI] [PubMed] [Google Scholar]
- 4.Cassar-MalekICassar-Malek I, Passelaigue F, Bernard C, Leger J, Hocquette JF. Target genes of myostatin loss-of-function in muscles of late bovine fetuses. BMC Genomics 8: 63, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.ChangLChang L, Zhang J, Tseng YH, Xie CQ, Ilany J, Bruning JC, Sun Z, Zhu X, Cui T, Youker KA, Yang Q, Day SM, Kahn CR, Chen YE. Rad GTPase deficiency leads to cardiac hypertrophy. Circulation 116: 2976–2983, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.ClopAClop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibe B, Bouix J, Caiment F, Elsen JM, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet 38: 813–818, 2006. [DOI] [PubMed] [Google Scholar]
- 7.DubowitzVDubowitz V, Sewry CA. Muscle Biopsy A Practical Approach. Elsevier, 2007.
- 8.EuJPEu JP, Hare JM, Hess DT, Skaf M, Sun J, Cardenas-Navina I, Sun QA, Dewhirst M, Meissner G, Stamler JS. Concerted regulation of skeletal muscle contractility by oxygen tension and endogenous nitric oxide. Proc Natl Acad Sci USA 100: 15229–15234, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.FengXHFeng XH, Derynck R. Specificity and versatility in TGF-beta signaling through Smads. Annu Rev Cell Dev Biol 21: 659–693, 2005. [DOI] [PubMed] [Google Scholar]
- 10.GirgenrathSGirgenrath S, Song K, Whittemore LA. Loss of myostatin expression alters fiber-type distribution and expression of myosin heavy chain isoforms in slow- and fast-type skeletal muscle. Muscle Nerve 31: 34–40, 2005. [DOI] [PubMed] [Google Scholar]
- 11.KobayashiYMKobayashi YM, Rader EP, Crawford RW, Iyengar NK, Thedens DR, Faulkner JA, Parikh SV, Weiss RM, Chamberlain JS, Moore SA, Campbell KP. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature 456: 511–515, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LiZBLi ZB, Kollias HD, Wagner KR. Myostatin directly regulates skeletal muscle fibrosis. J Biol Chem 283: 19371–19378, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LightNLight N, Champion AE. Characterization of muscle epimysium, perimysium and endomysium collagens. Biochem J 219: 1017–1026, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McPherronACMcPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 387: 83–90, 1997. [DOI] [PubMed] [Google Scholar]
- 15.McPherronACMcPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA 94: 12457–12461, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MendiasCLMendias CL, Bakhurin KI, Faulkner JA. Tendons of myostatin-deficient mice are small, brittle, and hypocellular. Proc Natl Acad Sci USA 105: 388–393, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MendiasCLMendias CL, Marcin JE, Calerdon DR, Faulkner JA. Contractile properties of EDL and soleus muscles of myostatin-deficient mice. J Appl Physiol 101: 898–905, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MosherDSMosher DS, Quignon P, Bustamante CD, Sutter NB, Mellersh CS, Parker HG, Ostrander EA. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet 3: e79, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.PercivalJMPercival JM, Anderson KN, Gregorevic P, Chamberlain JS, Froehner SC. Functional deficits in nNOSmu-deficient skeletal muscle: myopathy in nNOS knockout mice. PLoS ONE 3: e3387, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.SadkowskiTSadkowski T, Jank M, Zwierzchowski L, Siadkowska E, Oprzadek J, Motyl T. Gene expression profiling in skeletal muscle of Holstein-Friesian bulls with single-nucleotide polymorphism in the myostatin gene 5′-flanking region. J Appl Genet 49: 237–250, 2008. [DOI] [PubMed] [Google Scholar]
- 21.SartoriRSartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, Sandri M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol 296: C1248–C1257, 2009. [DOI] [PubMed] [Google Scholar]
- 22.SchuelkeMSchuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 350: 2682–2688, 2004. [DOI] [PubMed] [Google Scholar]
- 23.SelaUSela U, Dayan M, Hershkoviz R, Cahalon L, Lider O, Mozes E. The negative regulators Foxj1 and Foxo3a are up-regulated by a peptide that inhibits systemic lupus erythematosus-associated T cell responses. Eur J Immunol 36: 2971–2980, 2006. [DOI] [PubMed] [Google Scholar]
- 24.SteelmanCASteelman CA, Recknor JC, Nettleton D, Reecy JM. Transcriptional profiling of myostatin-knockout mice implicates Wnt signaling in postnatal skeletal muscle growth and hypertrophy. FASEB J 20: 580–582, 2006. [DOI] [PubMed] [Google Scholar]
- 25.SubramanianASubramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.TaoRHTao RH, Kawate H, Ohnaka K, Ishizuka M, Hagiwara H, Takayanagi R. Opposite effects of alternative TZF spliced variants on androgen receptor. Biochem Biophys Res Commun 341: 515–521, 2006. [DOI] [PubMed] [Google Scholar]
- 27.TusherVGTusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.ValorLMValor LM, Grant SG. Clustered gene expression changes flank targeted gene loci in knockout mice. PLoS ONE 2: e1303, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.VerrecchiaFVerrecchia F, Mauviel A. Transforming growth factor-beta and fibrosis. World J Gastroenterol 13: 3056–3062, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.WagnerKRWagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, Flanigan KM, Pestronk A, Tawil R, Wolfe GI, Eagle M, Florence JM, King WM, Pandya S, Straub V, Juneau P, Meyers K, Csimma C, Araujo T, Allen R, Parsons SA, Wozney JM, Lavallie ER, Mendell JR. A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol 63: 561–571, 2008. [DOI] [PubMed] [Google Scholar]
- 31.WelleSWelle S, Bhatt K, Pinkert CA. Myofibrillar protein synthesis in myostatin-deficient mice. Am J Physiol Endocrinol Metab 290: E409–E415, 2006. [DOI] [PubMed] [Google Scholar]
- 32.WelleSWelle S, Bhatt K, Pinkert CA, Tawil R, Thornton CA. Muscle growth after postdevelopmental myostatin gene knockout. Am J Physiol Endocrinol Metab 292: E985–E991, 2007. [DOI] [PubMed] [Google Scholar]
- 33.WelleSWelle S, Burgess K, Mehta S. Stimulation of skeletal muscle myofibrillar protein synthesis, p70 S6 kinase phosphorylation, and ribosomal protein S6 phosphorylation by inhibition of myostatin in mature mice. Am J Physiol Endocrinol Metab 296: E567–E572, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.WhittemoreLAWhittemore LA, Song K, Li X, Aghajanian J, Davies M, Girgenrath S, Hill JJ, Jalenak M, Kelley P, Knight A, Maylor R, O'Hara D, Pearson A, Quazi A, Ryerson S, Tan XY, Tomkinson KN, Veldman GM, Widom A, Wright JF, Wudyka S, Zhao L, Wolfman NM. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem Biophys Res Commun 300: 965–971, 2003. [DOI] [PubMed] [Google Scholar]
- 35.WilsonVJWilson VJ, Rattray M, Thomas CR, Moreland BH, Schulster D. Growth hormone increases IGF-I, collagen I and collagen III gene expression in dwarf rat skeletal muscle. Mol Cell Endocrinol 115: 187–197, 1995. [DOI] [PubMed] [Google Scholar]
- 36.WilsonVJWilson VJ, Rattray M, Thomas CR, Moreland BH, Schulster D. Effects of hypophysectomy and growth hormone administration on the mRNA levels of collagen I, III and insulin-like growth factor-I in rat skeletal muscle. Growth Horm IGF Res 8: 431–438, 1998. [DOI] [PubMed] [Google Scholar]
- 37.WoessnerJFWoessner JF Jr. The determination of hydroxyproline in tissue and protein samples containing small proportions of this amino acid. Arch Biochem Biophys 93: 440–447, 1961. [DOI] [PubMed] [Google Scholar]
- 38.WuZWu Z, Irizarry RA. Preprocessing of oligonucleotide array data. Nat Biotechnol 22: 656–658, 2004. [DOI] [PubMed] [Google Scholar]
- 39.XiaYXia Y, Berlowitz CO, Zweier JL. PIN inhibits nitric oxide and superoxide production from purified neuronal nitric oxide synthase. Biochim Biophys Acta 1760: 1445–1449, 2006. [DOI] [PubMed] [Google Scholar]
- 40.XieXXie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434: 338–345, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.