

Abstract
Cooperative interactions between growth factor signaling pathways are important elements in carcinoma progression. A model system combining transforming growth factor-β1 (TGF-β1) and EGF was developed to investigate mechanisms underlying induced epithelial-to-mesenchymal transition (EMT) in ras-transformed human (HaCaT II-4) keratinocytes. Dual stimulation with TGF-β1+EGF resulted in keratinocyte “plasticity” and pronounced colony dispersal. The most highly expressed transcript, identified by mRNA profiling, encoded plasminogen activator inhibitor-1 (PAI-1; SERPINE1). PAI-1 negatively regulates plasmin-dependent matrix degradation, preserving a stromal scaffold permissive for keratinocyte motility. Mitogen-activated extracellular kinase (MEK)/extracellular signal-regulated kinase (ERK) and p38 signaling were required for maximal PAI-1 upregulation and TGF-β1+EGF-stimulated cell locomotion, as pharmacologic disruption of MEK/p38 activity ablated both responses. Moreover, PAI-1 knockdown alone effectively inhibited TGF-β1+EGF-dependent cell scattering, indicating a functional role for this SERPIN in the dual-growth factor model of induced motility. Moreover, EGFR signaling blockade or EGFR knockdown attenuated TGF-β1-induced PAI-1 expression, implicating EGFR transactivation in TGF-β1-stimulated PAI-1 expression, and reduced colony dispersal in TGF-β1+EGF-treated cultures. Identification of such cooperative signaling networks and their effect on specific invasion-promoting target genes, such as PAI-1, may lead to the development of pathway-specific therapeutics that affect late-stage events in human tumor progression.
INTRODUCTION
The emergence of highly aggressive squamous cell carcinomas (SCCs) in mouse models is causally linked to p53 mutation, Ha-ras activation, and increased expression of transforming growth factor-β1 (TGF-β1) (Portella et al., 1998; Yuspa, 1998; Akhurst and Balmain, 1999; Boukamp, 2005; Smoller, 2006). Although typically antiproliferative for normal and early-transformed epithelial cells, TGF-β1 has protumorigenic effects on late-stage murine malignancies due, at least in part, to ras-dependent antagonism of growth inhibition and apoptosis programs (Cui et al., 1996; Go et al., 1999; Kretzschmar et al., 1999; Derynck et al., 2001; Han et al., 2005; Kim et al., 2005). Recent findings suggest a mechanism for such signaling dichotomy. TGF-β-dependent COOH-terminal Smad3 phosphorylation initiates growth restriction in normal cells, whereas oncogenic Ha-ras activates c-Jun N-terminal kinase-mediated Smad2/3 linker region phosphorylation, resulting in the upregulation of several important tumor progression genes (Sekimoto et al., 2007). This is particularly relevant to TGF-β1-initiated “plasticity” in susceptible epithelial cells (usually referred to as epithelial-to-mesenchymal transition (EMT)), a response linked to ras signaling (Oft et al., 1996, 2002; Tian et al., 2007; Samarakoon et al., 2008; Joo et al., 2008; Uttamsingh et al., 2008). Indeed, acquisition of mesenchymal-like traits with creation of a morphologically restructured, motile phenotype (see, e.g., Heldin et al., 2009; Thiery and Sleeman, 2006) is thought to facilitate development of invasive carcinomas (Cui et al., 1996; Portella et al., 1998; Zavadil et al., 2001; Zavadil and Bottinger, 2005; Peinado et al., 2007).
Data mining of the repertoire of TGF-β1-regulated “plasticity” genes in human malignant keratinocytes has produced some remarkable consistencies. The most prominently induced transcript encodes plasminogen activator inhibitor type-1 (PAI-1; SERPINE1), the major physiologic regulator of the pericellular plasmin-generating cascade (Akiyoshi et al., 2001; Zavadil et al., 2001; Sekimoto et al., 2007). PAI-1 is highly induced in epithelial cells undergoing an EMT-like conversion upon expression of the E-cadherin transcriptional repressors Snail, Slug, or E47 (Moreno-Bueno et al., 2006). In incipient epidermal SCC, PAI-1 often localizes in tumor cells and myofibroblasts at the invasive front (Wilkins-Port et al., 2007), implying involvement as a positive modulator of cellular invasive potential (Christensen et al., 1996; Offersen et al., 2003; Illemann et al., 2004). Elevated PAI-1 tumor levels, in fact, signal a poor prognosis and reduced disease-free survival in patients with breast, lung, ovarian, gastric, and oral malignancies (Pedersen et al., 1994; Andreasen et al., 1997; Herszènyi et al., 1999; Chen et al., 2004; Vairaktaris et al., 2005; Lindberg et al., 2006; Sakakibara et al., 2006; Sternlicht et al., 2006). Moreover, in vivo modeling in knockout mice identified PAI-1 as an essential element in cutaneous cancer invasion and the associated angiogenic response (see, e.g., Bajou et al., 1998; Bajou et al., 2001). PAI-1 likely maintains an angiogenic “scaffold,” stabilizes nascent capillaries, and regulates tumor cell invasion through focalized control of the proteolytic environment (Bacharach et al., 1998; Bajou et al., 2001, 2004, 2008; Davis et al., 2001; Maillard et al., 2005) as well as by competitive interactions with matrix vitronectin (for integrin binding) and cell surface LDL receptor-related protein (Degryse et al., 2004).
Identifying mechanisms underlying TGF-β signaling that affect expression of tumor progression genes is complicated and involves the engagement of both canonical and noncanonical pathways downstream of the activated TGF-β receptors (see, e.g., Zhang, 2009). In several cell types, including human (HaCaT) keratinocytes, for example, EGFR transactivation upon TGF-β1 stimulation contributes to signal bifurcation (Kim and Joo, 2002; Joo et al., 2008; Samarakoon et al., 2008). This has significant pathophysiologic ramifications. During cutaneous SCC development, for example, elevated TGF-β1 levels in the tumor microenvironment are accompanied by EGFR amplification and/or dysregulated signaling (Rho et al., 1994; O-Charoenrat et al., 2000, 2002). Therefore, to more specifically define inductive controls and potential functions for plasticity-associated PAI-1 expression in human cutaneous SCC, a physiologically relevant model system was established. Transformed human keratinocytes (HaCaT II-4 cells), which possess UV signature mutations in both p53 alleles (C→T and CC→TT at codons 179 and 281/282) (Lehman et al., 1993; Elmageed et al., 2009) as well as an activated Ha-rasval–12 gene, were stimulated with TGF-β1+EGF to closely mimic pathway co-activation typical of late-stage malignancies. Simultaneous addition of TGF-β1+EGF (when compared with either growth factor alone) was required to develop the full plastic response involving E-cadherin downregulation, induction of mesenchymal markers (vimentin, α-smooth muscle actin, and N-cadherin), increased cell motility (i.e., colony “scattering”), and maximal PAI-1 expression. Small interfering (siRNA)-directed PAI-1 knockdown effectively inhibited dual-growth factor-stimulated colony dispersal, supporting a role for this SERPIN in tumor cell migration. Moreover, pharmacologic blockade of EGFR signaling (with AG1478) and shRNA-mediated EGFR reduction effectively inhibited both TGF-β1-induced PAI-1 expression and cell scattering. A model is presented in which TGF-β1 activates two distinct but cooperative downstream pathways consistent with the known relationship between ras→mitogen-activated protein (MAP) kinase signaling and TGF-β1-initiated EMT.
RESULTS
TGF-β1+EGF co-stimulation elicits a plastic response in HaCaT II-4 cells
To assess the consequences of TGF-β+EGF signaling, p53 mutant, ras-transformed human (HaCaT II-4) keratinocytes, grown as small colonies, were serum starved for 3 days to initiate a G0 arrest (growth kinetics detailed in Qi et al., 2006, 2008) before stimulation with TGF-β1 (1ngml−1) and EGF (10 ng ml−1) (Figure 1a). Within 48 hours, the discrete colonies of normally sedentary, cuboidal-shaped, and tightly juxta-posed keratinocytes underwent pronounced morphological and motile changes (Figure 1b and c),a phenotypic conversion accompanied by E-cadherin downregulation and the acquisition of prominent but heterogeneously distributed N-cadherin immunoreactivity (Figure 1d). Colony dispersal (Figure 1b and c) was coincident with de novo expression of α-smooth muscle actin and vimentin (with construction of a vimentin-rich intermediate filament network; Figure 1d), both established markers of epithelial cell plasticity (reviewed in Moustakas and Heldin, 2007). The synthesis of vimentin similarly characterizes the invading epithelial cohort at the periphery of HaCaT II-4 transplants in immunocompromized mice (Tomakidi et al., 2003).
Figure 1. TGF-β1+EGF induce a plastic response in HaCaT II-4 cells.

(a) A model system was devised in which small colonies of HaCaT II-4 cells seeded on tissue culture plastic were serum starved, followed by stimulation with a combination of transforming growth factor-β1 (TGF-β1; 1 ngml−1) and EGF (10 ng ml−1). (b, c) Acquisition of a spindle-shaped, highly migratory phenotype resulted in marked colony dispersal within 24–48 hours. Data plotted in c is the mean±SD of ImageJ measurements on 10 arbitrarily selected colonies; ***P≤0.001. The pixel area of unscattered and dispersed colonies was measured using NIH ImageJ software for cultures stimulated with TGF-β1+EGF for 48 hours (e.g., as in b) after a 3-day serum-free incubation or maintained under control (quiescence) conditions for a comparable period. (d) Cell scattering was accompanied by the loss of E-cadherin (green) immunostaining at cell–cell junctions (48 hours), and the gain of several mesenchymal markers, such as N-cadherin (48 hours; although heterogeneously), α-smooth muscle actin (α-SMA, 24 hours), and vimentin (72 hours) (all red). Scale bars are as follows: in b=100 μm; in d (E-cadherin) =10 μm, (N-Cadherin, α-SMA, and Vimentin) =20 μm. Nuclei are visualized with 4,6-diamidino-2-phenylindole (DAPI); the red counterstain in E-Cadherin panels (d) is Texas red–X phalloidin.
TGF-β1 when used alone promoted a more flattened morphology with loss of cell–cell contact and induced N-cadherin expression (Figure 2a and b), whereas EGF clearly initiated a proliferative response (Figure 2a and c). Optimal colony dispersal required both growth factors, although the expression of certain EMT “markers” (such as N-cadherin) was TGF-β1 dependent (Figure 2a and b). Moreover, TGF-β1+EGF-stimulated keratinocyte scattering was proliferation independent; TGF-β1 retained its growth-suppressive effects even in the presence of mitogenic levels of EGF, as determined by quantitative assessments of the cell cycle markers cyclin A and Ki-67 (Figure 2c). This is consistent with recent findings that TGF-β1 suppresses EGF-induced S-phase entry in HaCaT II-4 cells (Wilkins-Port et al., 2009) and indicates, moreover, that TGF-β1+EGF-stimulated plasticity does not require concomitant cell proliferation. Furthermore, deprivation of fetal bovine serum for 3 days did not initiate an apoptotic response or permanent growth arrest, as quiescent HaCaT II-4 cells re-entered the proliferative cycle upon reintroduction of serum (not shown).
Figure 2. TGF-β1 and EGF differentially affect HaCaT II-4 phenotype.
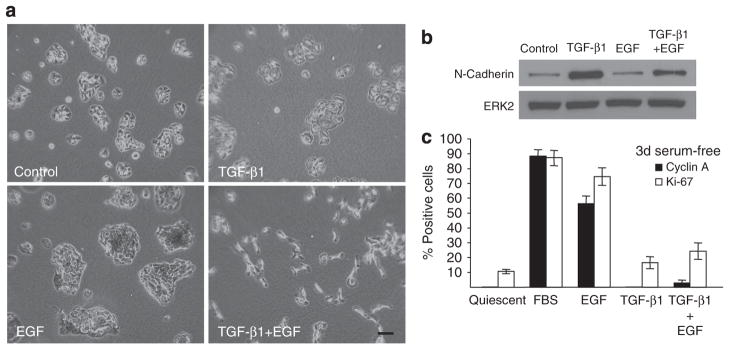
When used individually for 48 hours, transforming growth factor-β1 (TGF-β1) generated flat, less closely juxtaposed cells (a) and high-level N-cadherin expression (b). EGF stimulated cell proliferation (a, c) but not N-cadherin synthesis (b). Colony dispersal in TGF-β1+EGF-stimulated keratinocyte cultures (a) occurs in the absence of induced proliferation (c). Fetal Bovine Serum (FBS; 10% final concentration), EGF (10 ng ml−1), TGF-β1 (1ngml−1), or the combination of TGF-β1+EGF was added to 3-day serum-deprived HaCaT II-4 cells and the fraction of cyclin A- or Ki-67 positive keratinocytes was assessed using immunocytochemistry 24 hours later (c). FBS (used as a positive control) and EGF stimulation significantly increased the number of cyclin A- and Ki67-expressing cells. Simultaneous addition of TGF-β1 in the presence of EGF (EGF+TGF-β1) effectively attenuated the EGF-induced increase in both cell cycle markers. Data plotted (c) are the mean+SD of triplicate assessments. Scale bar in a=100 μm.
Colony dispersal in TGF-β1+EGF-stimulated HaCaT II-4 keratinocytes involves p38, MEK/ERK, and upstream EGFR signaling
As cell scattering is an in vitro correlate of invasive behavior, it was important to define signaling pathways underlying TGF-β1+EGF-stimulated motility. p38 and mitogen-activated extracellular kinase (MEK)/extracellular signal-regulated kinase (ERK) phosphorylation both increased within 15 minutes of TGF-β1+EGF addition, although the duration of response differed (Figure 3a). Although phospho-p38 returned to approximately basal levels by 12 hours, ERK1/2 remained phosphorylated throughout the entire course of growth factor treatment (48 hours), paralleling the complete emergence of plasticity-related markers and full colony dispersal. Preincubation with the MEK inhibitor U0126 significantly attenuated TGF-β1+EGF-initiated Ha-CaT II-4 cell motility and retained E-cadherin staining at cell–cell junctions, as did blockade of p38 kinase activity with either PD169316 or SB220025 (Figure 3b and c), coincident with decreased phosphorylation of the phosphop38 downstream target HSP27 (Figure 3d). It was established previously that EGF and TGF-β1 (when used separately) activated MAP kinase pathways in HaCaT II-4 cells, and this response is sensitive to the EGFR inhibitor AG1478 (Kutz et al., 2006; Wilkins-Port et al., 2009). In conformity with these results, inhibition of EGFR signaling by pretreatment with AG1478 completely prevented TGF-β1+EGF-stimulated colony dispersal and the associated E-cadherin loss at cell–cell contact points (Figure 4a). Collectively, these data suggest an EGFR→MAP kinase axis in the dual-growth factor model of induced keratinocyte migration. To verify participation of the EGFR in the TGF-β1+EGF-stimulated scattering response, HaCaT II-4 cells were infected with EGFR short hairpin RNA lentiviruses to reduce cellular EGFR levels. Consistent with the pharmacologic approach, targeted EGFR knockdown (Figure 4b) effectively attenuated colony scattering after TGF-β1+EGF addition (Figure 4c).
Figure 3. TGF-β1+EGF-induced colony dispersal requires both p38 and MEK/ERK signaling.

(a) Assessments of pERK1/2 and phospho-p38 (pp38) levels indicated that the mitogen-activated extracellular kinase (MEK)/extracellular signal-regulated kinase (ERK) and p38 signaling pathways are activated within 15 minutes of transforming growth factor-β1 (TGF-β1) and EGF addition. ERK1/2 phosphorylation was sustained over 48 hours, whereas pp38 returned to basal levels by 12 hours after stimulation. Protein loading levels were confirmed by immunoblotting for total p38 and ERK2. Preincubation with the MEK inhibitor, U0126, as well as the p38 inhibitors, PD169316 and SB220025, completely blocked TGF-β1+EGF-stimulated keratinocyte motility and colony dispersal in 48-hour TGF-β1+EGF-treated cultures (b), and prevented loss of E-cadherin from cell–cell junctions (c). ImageJ analysis of colony area verified the visual assessments (not shown) in b. Lack of colony dispersal in cultures pretreated with kinase inhibitors was not due to associated toxicity, as removal of drugs followed by addition of DMEM/10% fetal bovine serum (FBS) promoted a rapid re-entry into exponential phase growth. Phosphorylation of the pp38 target protein HSP27 in response to a 1-hour TGF-β1+EGF stimulation was reduced to near-basal levels by pretreatment (for 0.5 or 1 hour) with PD169316 and SB220025. The expected pp38 activation in the presence of PD169316 or SB220025 and decreased HSP27 phosphorylation confirmed that these compounds function in HaCaT II-4 cells to effectively inhibit p38 downstream activity (c). Scale bars in b=100 μm; in c=10 μm.
Figure 4. Participation of the EGFR in TGF-β1+EGF-induced colony scattering.

(a) Inhibition of EGFR kinase activity with AG1478 effectively eliminated transforming growth factor-β1 (TGF-β1) and EGF-stimulated cell scattering (left panels) and retained E-cadherin immunolocalization (right panels) at cell–cell junctions. (b) EGFR knockdown by infection of HaCaT II-4 cells with EGFR short hairpin RNA (shRNA)-bearing lentiviruses markedly reduced TGF-β1+ EGF-induced colony dispersal compared with keratinocytes infected with control, GFP-expression construct, or lentiviruses. Scale bars in a (left panels), c=100 μm.
PAI-1 is a prominent member of the TGF-β1+EGF-upregulated gene set and required for colony dispersal
Quantitative reverse transcriptase-PCR (qRT-PCR) array profiling of HaCaT II-4 keratinocytes stimulated with TGF-β1 and/or EGF provided an initial assessment of potential effectors of TGF-β1+EGF-induced cell scattering. PAI-1 (SERPINE1), a regulator of keratinocyte migration (see, e.g., Providence et al., 2008), was identified as the most prominently upregulated transcript (Figure 5), an increase verified by RT-PCR (data not shown) and western blotting (Figure 6a). Individually, both EGF and TGF-β1 enhanced PAI-1 mRNA expression (10- and 74-fold, respectively). However, combining TGF-β1 and EGF resulted in a 170-fold increase in PAI-1 mRNA and protein levels, suggesting that induction associated with dual-growth factor treatment was more than simply additive (Figures 5 and 6a). Moreover, kinetic analysis indicated that PAI-1 induction was first evident within 1 hour of TGF-β1+EGF addition and sustained for over 24 hours (Figure 6b). Therefore, the initiation of PAI-1 expression preceded and, over the long term, correlated temporally with the development of cellular plasticity and colony dispersal.
Figure 5. Plasminogen activator inhibitor-1 (PAI-1) is a prominent member of the TGF-β1+EGF-upregulated gene set.

Of the several upregulated and downregulated transcripts identified by focused microarray analysis at 6 hours after stimulation with TGF-β1+EGF, the most highly upregulated is PAI-1 (170-fold over control). TGF-β1 and EGF, when used alone, induced a 74-fold and 10-fold increase, respectively. Other genes commonly associated with cancer progression and metastasis were also upregulated, as well as members of the proteolytic cascade (i.e., several MMPs) and plasminogen activation system (i.e., uPA and uPAR). Relative expression levels for each gene was normalized to that of GAPDH using the PCR Array Data Analysis Web Portal to calculate ΔΔCt-based fold-change in transcript abundance. ANGPT1, angiopoietin 1; BRCA1, breast cancer 1, early onset; CDK2, cyclin-dependent kinase 2; CDKN1A, cyclin-dependent kinase inhibitor 1A (p21, Cip1); IFN-α1, interferon-α1; IFN-β1, interferon-β1, fibroblast; ITGα1, integrin α1; ITGα2, integrin α2; ITGβ1, integrin β1; ITGβ3, integrin β3; ITGβ5, integrin β5; MMP-1, matrix metallopeptidase 1; MMP-9, matrix metallopeptidase 9; MTA1, metastasis-associated 1; MTA2, metastasis-associated 1 family, member 2; MTSS1, metastasis suppressor 1; PDGFβ, platelet-derived growth factor β polypeptide; PLAU, urokinase plasminogen activator; PLAUR, urokinase plasminogen activator receptor; SERPINE1, serpin peptidase inhibitor, clade E (plasminogen activator inhibitor-1); TGF-β1, transforming growth factor-β1; TGF-βR1, transforming growth factor-β receptor 1; THBS1, thrombopsondin 1; TNFRSF10B, tumor necrosis factor receptor superfamily, member 10b; TNFRSF25, tumor necrosis factor receptor superfamily, member 25; VEGFA, vascular endothelial growth factor A.
Figure 6. Plasminogen activator inhibitor-1 (PAI-1) induction occurs early after TGF-β1+EGF stimulation and requires MEK/ERK, p38, and EGFR signaling.

(a) Consistent with the array data (Figure 5), the PAI-1 protein-inductive response to a 5-hour transforming growth factor-β1(TGF-β1) and EGF co-stimulation was significantly greater when compared with either growth factor (EGF or TGF-β1) alone. (b) Time course analysis indicates that the first detectable increase in PAI-1 protein occurred 1 hour after TGF-β1+EGF addition and was sustained throughout 24 hours, correlating with the onset of morphological changes associated with colony scattering (e.g., Figure 1). (c) Prior treatment with the p38 inhibitors, PD169316 and SB220025, and the MEK inhibitor, U0126, effectively attenuated PAI-1 expression in response to TGF-β1+EGF; PD169316 or SB220025 in combination with U0126 completely ablated PAI-1 induction in TGF-β1+EGF-stimulated cultures. (d) Preincubation with AG1478 blocked not only EGF-induced PAI-1 expression (as expected) but also significantly attenuated the PAI-1 response to TGF-β1 and TGF-β1+EGF.
Similar to requirements for TGF-β1+EGF-stimulated colony dispersal (Figure 3b), TGF-β1+EGF-induced PAI-1 expression was also attenuated by inhibitors of MAP kinase signaling (Figure 6c) as well as the EGFR inhibitor AG1478 (Figure 6d), suggesting a potential link between PAI-1 expression and cell scattering. Moreover, simultaneous inhibition of both the MEK/ERK and p38 MAP kinase pathways completely ablated PAI-1 expression (Figure 6c). Pretreatment of HaCaT II-4 keratinocytes with AG1478 abolished EGF-dependent PAI-1 induction (as expected), and also markedly attenuated TGF-β1-stimulated PAI-1 expression (Figure 6d). This latter finding reflects the increased EGFR activation (phosphorylation) at the Y845 site and internalization evident in TGF-β1-stimulated HaCaT II-4 cells (Figure 7a and b) and the requirement for EGFR presence for maintenance of the PAI-1 response (Figure 7c). Importantly, the synergistic increase in PAI-1 levels in TGF-β1+EGF-treated cultures was also significantly reduced by EGFR inhibition with AG1478 (Figure 6d) as was colony dispersal (Figure 4a), suggesting an important role for EGFR signaling in both events. HaCaT II-4 cells with reduced EGFR levels also had significantly lower levels of PAI-1 after growth factor treatment, either singularly or in combination (not shown). These data highlight the TGF-β and EGF receptor signaling synergies reported previously in other cell types (Tian et al., 2007; Uttamsingh et al., 2008).
Figure 7. The EGFR is phosphorylated at Y845 in response to TGF-β1 and required for plasminogen activator inhibitor-1 (PAI-1) induction.

(a) Stimulation of quiescent HaCaT II-4 cells with transforming growth factorβ1 (TGF-β1) resulted in phosphorylation of the EGFR on the Y845 src kinase target residue, although the kinetics of phosphorylation was delayed relative to EGF-treated cultures. (b) While the level of phosphorylation was also reduced compared with EGF-stimulated keratinocytes, suggesting perhaps the involvement of a subset of receptors (a), both EGF- and TGF-β1-treated HacaT II-4 cells effectively internalized the activated EGFR (b). To assess the requirement for EGFR receptor presence for TGF-β1-induced PAI-1 expression, EGFR−/− mouse embryonic fibroblasts (MEFs) were infected with adenoviruses bearing either control (GFP) or wild-type human EGFR expression constructs. EGFR−/− MEFs or cells engineered to re-express a wild-type EGFR (EGFR+/+) were maintained in growth factor-free medium (−TGF-β1) or stimulated with TGF-β1 (+TGF-β1; 1 ngml−1) for 5 hours. (c) Western blots of PAI-1 levels in cellular extracts were quantified by densitometry; data are the mean ±SD of triplicate independent experiments normalized to total extracellular signal-regulated kinase 2 (ERK2) levels. Scale bar in b=10 μm.
As PAI-1 stimulates directional migration in response to monolayer scrape injury (Degryse et al., 2004; Providence et al., 2008), it was important to determine whether PAI-1 also contributed to TGF-β1+EGF HaCaT II-4 keratinocyte motility. Pretreatment with a function-blocking (i.e., neutralizing) antibody to PAI-1 significantly reduced the percentage of scattered colonies in dual-growth factor-treated cultures (Figure 8a). siRNA-mediated PAI-1 knockdown (Figure 8b) similarly decreased TGF-β1+EGF-stimulated colony dispersal (Figure 8c), confirming involvement of this SERPIN in at least several models of induced planar motility (cf., Degryse et al., 2004; Providence et al., 2008). Finally, the Oris cell invasion assay confirmed the necessity for PAI-1 in basement membrane extract barrier penetration. TGF-β1+EGF significantly increased invasion of HaCaT II-4 cells as did exogenous addition of PAI-1 alone; inhibition of EGFR or p38/MEK signaling attenuated TGF-β1+EGF-dependent migration (Figure 9a). HaCaT II-4 keratinocytes transfected with a control (CTL) or PAI-1 (PAI) siRNA were subsequently used in a long-term Oris invasion assay. In this system, PAI-1 knockdown (by approximately 80%; a consequence of the protracted time course of this evaluation when compared with the almost complete attenuation of PAI-1 expression in a shorter time frame context used in the scattering assay in Figure 8b) reduced both basal invasion as well as TGF-β1+EGF-stimulated migration by 50% (Figure 9b and c). Collectively, these data establish the requirement for PAI-1 expression for TGF-β1+EGF-stimulated colony dispersal as well as barrier invasion in HaCaT II-4 keratinocytes.
Figure 8. Plasminogen activator inhibitor-1 (PAI-1) is necessary for growth factor-stimulated colony scattering.

(a) The fraction of scattering colonies in cultures incubated in preimmune serum (1% final serum concentration) was approximately 40%; the addition of function-blocking (neutralizing) PAI-1 antibodies significantly reduced the incidence of dispersed colonies to just 5%. Data plotted in a are the mean ±SD of four independent assessments, each consisting of measurements of 25 individual colonies. (b) Western analysis confirmed that transfection of a pool of four PAI-1 targeting small interfering RNAs (siRNAs) significantly reduced transforming growth factor-β1 (TGF-β1) and EGF-stimulated PAI-1 expression in HaCaT II-4 keratinocytes. (c) PAI-1 knockdown effectively attenuated TGF-β1+EGF-induced cell motility/colony dispersal. (d) Quantitative assessment of a limited colony scatter assay for HaCaT II-4 keratinocytes transfected with control (CTL) or PAI-1 siRNA constructs confirmed the visual data (c). Scale bars in c=100 μm.
Figure 9. Plasminogen activator inhibitor-1 (PAI-1) is required for optimal barrier penetration by TGF-β1+EGF-stimulated HaCaT II-4 cells.

(a) Invasive ability was evaluated using the Oris cell invasion assay that provides visual assessment of three-dimensional (3D) penetration of a basement membrane gel. Dashed line (black arrowhead) indicates the initial cellular “front”; white solid line (white arrowheads) depicts the extent of maximal gel migration by transforming growth factor-β1 (TGF-β1) and EGF-stimulated HaCaT II-4 cells. Cell location was determined by 4,6-diamidino-2-phenylindole (DAPI) staining. TGF-β1 had a small positive effect on gel penetration, whereas EGF increased invasive ability to approximately 50% of that of cells exposed to both growth factors; PAI-1 (20 nM) when used alone also stimulated basal barrier penetration (a, top panels). In all cases, simultaneous addition of AG1478, PD169316, or U0126 with TGF-β1+EGF significantly attenuated the migratory response (a, bottom panels). (b) HaCaT II-4 cells were transfected with PAI-1 small interfering RNA (siRNA) or a control (CTL) siRNA construct for use in the Oris assay. At the completion of the extended assay period (96 hours), the cells were extracted for western analysis of PAI-1 levels in response to a TGF-β1+EGF challenge. PAI-1 knockdown was still significant (i.e., approximately 80%) even at this longer time point after initial introduction of siRNA, although the PAI-1 knockdown was not as effective as that attained at 48 hours after transfection (Figure 8). When evaluated in a longer-term invasion assay to assess the effects of PAI-1 siRNA on both basal and TGF-β1+EGF-stimulated barrier penetration, it was obvious that PAI-1 knockdown significantly attenuated both processes (c).
DISCUSSION
Aggressive SCCs often emerge from specific transdifferentiation events that switch normally polarized epithelial cells to a more “plastic,” invasive phenotype. To define mechanisms associated with this pathological response in a cutaneous system, ras-transformed HaCaT II-4 keratinocytes were stimulated with a combination of TGF-β1+EGF to recapitulate the increase in TGF-β1 expression and amplification/dysregulation of EGFR signaling frequently observed in metastatic carcinomas (Rho et al., 1994; Derynck et al., 2001; Shimizu et al., 2001; Weeks et al., 2001).
The transcriptional profile of TGF-β1+EGF-treated HaCaT II-4 keratinocytes was relatively unique compared with cultures exposed to either growth factor alone, suggesting cooperativity in downstream signaling as a consequence of TGF-βR/EGFR activation. Although TGF-β1 initiates EMT-related events in the HaCaT system (Zavadil et al., 2001), the extent of cell scattering was not as profound as that associated with the combination of TGF-β1+EGF. Moreover, this response was not unique to HaCaT II-4 keratinocytes as a similar effect of the combined TGF-β1+EGF treatment was also evident in SCC-25 oral squamous carcinoma and A431 cells (not shown). In this regard, the expression of several plasticity markers, moreover, was most apparent with dual-growth factor stimulation, suggesting that EGF supports, or augments, development of the plastic phenotype. Indeed, both EGFR ligand-dependent (i.e., ADAM mediated) and -independent TGF-βR/EGFR cross-talk has been reported in several cell systems, including HaCaT keratinocytes (Wang et al., 2008; Joo et al., 2008), and may contribute specifically to PAI-1 expression in much the same way as ADAM-17 may enhance PAI-1 expression in phorbol ester-treated cells (Arts et al., 1999; Mussoni et al., 2000; Horiuchi et al., 2007).
Cross-talk between TGF-β1 (or HGF) and EGFR results in a MAP kinase-dependent upregulation of several proteases (Tian et al., 2007; Zhou et al., 2007; Uttamsingh et al., 2008). In this system, synergistic increases in matrix metalloproteinases 1 and 9, as well as the serine proteinase inhibitor PAI-1 in response to TGF-β1+EGF addition, were evident. PAI-1, which modulates downstream MMP activation and stromal remodeling (Wilkins-Port et al., 2009), was the most highly upregulated transcript and causally linked to TGF-β1+EGF-induced cell scattering. This study shows for the first time a fundamental role for PAI-1 in growth factor-induced motility and transdifferentiation in malignant human keratinocytes. The p38 and MEK/ERK pathways were both activated upon TGF-β1+EGF stimulation and necessary for maximal induction of PAI-1, as well as the development of a migratory phenotype. MAP kinase signaling in other systems also affects motility-related molecules such as Snail (Grotegut et al., 2006) and myosin light chain kinase (Klemke et al., 1997). Although TGF-β1 and EGF are known promoters of MAP kinase signaling, this study establishes a direct correlation between TGF-β1+EGF-dependent MAP kinase pathway stimulation, PAI-1-dependent HaCaT II-4 cell scattering, and EMT, consistent with recent studies (Zuo and Chen, 2009). It is apparent that a complicated, cooperative interaction exists between intracellular events orchestrated by TGF-β1-activated pathways and the EGFR, which specifically lead to epithelial tumor plasticity. PAI-1 induction in response to TGF-β1 involves a complex network of signaling intermediates and requires the activities of the MEK, p21ras, and pp60c–src in addition to the EGFR (Kutz et al., 2006), with pp60c–src a critical intermediate in the cascade leading to MEK signaling, PAI-1 transcription, and subsequent phenotypic responses (e.g., Samarakoon et al., 2005; Sato et al., 2005). The src family kinase inhibitors and dominant-negative pp60c–src constructs effectively attenuate TGF-β1-induced PAI-1 expression in HaCaT cells (Kutz et al., 2006), confirming the generality of src kinase involvement in PAI-1 gene regulation. The effective blockade of TGF-β1-stimulated ERK1/2 phosphorylation and PAI-1 transcription by interference with src kinase activity as well as with the EGFR inhibitor AG1478, and the requirement for MEK/ERK signaling for the full inductive effect of TGF-β1, suggests that pp60c–src, perhaps through phosphorylation of the Y845 src-specific EGFR target residue, regulates the EGFR→MEK/ERK-dependent PAI-1 expression transduction cascade. Although the role of individual MAP kinases remains to be determined, ERK activation through TGF-β1/EGFR cross-talk may involve Sprouty2, which prolongs EGF signaling by titering EGFR ubiquitination (Ding et al., 2007). Sprouty2 levels decrease after TGF-β1 stimulation, an effect that could not be rescued with additional EGF (Ding et al., 2007) despite the ability of EGF to upregulate Sprouty2 expression (Ozaki et al., 2001). A similar dominant inhibitory effect was observed in HaCaT II-4 cells, as EGF stimulation was unable to overcome the growth-suppressive effects of TGF-β1. Therefore, the potential contribution of Sprouty2 to TGF-β1/EGF-dependent events in the present system warrants further investigation.
The continued definition of specific molecular mechanisms underlying control of tumor progression genes is an essential element in the ultimate design of targeted, clinically relevant options for treatment of human cutaneous SCCs. Indeed, the emerging appreciation that cooperative EGFR signaling is an essential aspect of TGF-β1-stimulated PAI-1 expression provides, to our knowledge, previously unreported insights as to the effect of TGF-β1 in late-stage human tumor progression, and underscores the potential diversity of new molecular targets that can be exploited for therapeutic benefit. Refining this understanding of PAI-1 gene regulation, as well as its signaling pathways, may lead to the design of transcription-focused “therapeutics” to manage human cutaneous malignancies.
MATERIALS AND METHODS
Cell culture and immunocytochemistry
Subconfluent cultures of human HaCaT II-4 keratinocytes were washed twice with phosphate-buffered saline (PBS) and maintained in serum-free DMEM for 1–3 days before stimulation with TGF-β1 (1 ng ml−1) and/or EGF (10 ng ml). EGFR−/− and EGFR+/+ mouse embryonic fibroblasts were provided by Dr Jennifer R. Grandis (University of Pittsburgh Medical Center). For immunocytochemistry, keratinocytes were fixed in paraformaldehyde (3.2%), permeabilized in 0.5% Triton X-100/PBS for 10 minutes, blocked in 5% goat serum/0.3% Triton X-100/PBS, and incubated with antibodies to E-cadherin, N-cadherin (BD Transduction Laboratories, San Jose, CA; 610182 and 610921, respectively), α-smooth muscle actin (Sigma, St Louis, MO; a5228), vimentin (Abcam, Cambridge, MA; ab45939), cyclin A, Ki-67 (both from Santa Cruz Biotechnology, Santa Cruz, CA), pEGFRY845 (Cell Signaling, Danvers, MA; 2231), or PAI-1 (Wilkins-Port et al., 2009) for 1 hour at room temperature. After three washes with 0.3% Triton X-100/PBS, cells were incubated with appropriate secondary antibodies for 1 hour, followed by a final series of 0.3% Triton X-100/PBS washes and coverslips mounted with ProLong Gold antifade reagent (Invitrogen, Carlsbad, CA) with 4,6-diamidino-2-phenylindole. Texas red–X phalloidin was used to visualize actin microfilament structures. Paraformaldehyde-fixed cells were stained with 0.5% crystal violet/20% methanol/PBS for morphologic assessments. The pharmacologic agents U0126, PD169316, and SB220025 (10 μM final concentrations) were added 30 minutes before stimulation; cells were incubated with AG1478 (2.5 μM final concentration) for 45 minutes before growth factor addition. For inhibition of PAI-1 by functionblocking (neutralizing) antibodies, cells were seeded at a low density to form widely spaced individual colonies, serum starved, and PAI-1 polyclonal antiserum or preimmune serum (1% final concentration) added simultaneously with TGF-β1+EGF; 24–48 hours later, the number of scattered colonies were counted. Colony dispersal was quantified by the NIH ImageJ (http://rsb.info.nih.gov/ij/) analysis of crystal violet-stained cultures at T0 and after TGF-β1+EGF stimulation. Careful seeding of single cells at low population densities resulted in formation of rather uniform-sized colonies of closely juxtaposed cells that approximated an area of 3–6×104 pixels. “Scattered” colonies, in contrast, had several distinguishing characteristics, including loss of cell–cell contact, mesenchymal or migratory phenotype, and area occupancy of 15–20×104 pixels. PAI-1 siRNA constructs were from Dharmacon (Lafayette, Co). The SMARTpool A-019376-13 to 16 SERPINE1 target sequences were as follows: 5′-CCGACAUGUUCAGACAGUU-3′, 5′-CCAAUGU GUUCAAUAGAUU-3′, 5′-GGGGUGUACCUAAAUAUUU-3′, and 5′-GGGUUAUUUUGGAGUGUAG-3′.
Western blotting
For detection of EGFR, pEGFRY845, pERK1/2, p38, phospho-p38, or pHSP27, HaCaT II-4 cells were washed with cold PBS and scrape-harvested on ice in cold lysis buffer (50mM HEPES, pH 7.5, 0.5% deoxycholate, 1% Triton X-100, 1% NP-40, 150mM NaCl, 50mM NaF, 1mM sodium orthovanadate, 2% protease inhibitor cocktail, 1mM p-nitrophenyl phosphate, 20 nM calyculin A, and 50 nM okadaic acid). For the assessment of cellular PAI-1 protein levels, cells were washed with PBS, trypsin-detached and collected by centrifugation before lysis with RIPA buffer (50mM Tris pH 8.0, 150mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1% NP-40, and 1% protease inhibitor cocktail). Mouse embryonic fibroblasts were disrupted in 4% SDS/PBS for 10 minutes, and lysates vortexed briefly, and boiled for 5 minutes. All extracts were clarified by centrifugation and 10–30 μg of protein was separated on denaturing Tris-HCl SDS/10% PAGE gels. Upon nitrocellulose transfer, membranes were blocked in 5% milk/0.05% Triton X-100/PBS for 1 hour, incubated with indicated antibodies, washed three times (in 0.05% Triton X-100/PBS) for 10 minutes each, and incubated with horseradish peroxidase-conjugated secondary antibodies. After three 10-minute rinses, blots were incubated with enhanced chemiluminescent substrate and exposed to X-ray film before stripping for reprobing with ERK2 antibodies to assess overall protein loading.
Transcriptional profiling
qRT-PCR analysis used the RT2 Profiler Cancer Pathway Finder qPCR Array System (SABiosciences, Frederick, MD). RNA was isolated using the RT2 qPCR-Grade RNA Isolation Kit and complementary DNA was prepared from 0.5 mg RNA samples using RT2 PCR Array First Strand Kit. Real-time PCR used the Bio-Rad MyiQ Detection System (Hercules, CA) with the following two-step cycling program: a 10-minute incubation at 95 °C to activate the DNA polymerase, followed by 40 cycles of 15 seconds at 95 °C and 1 minute at 60 °C. SYBR Green fluorescence was recorded for every well during the annealing step of each cycle, and fold changes in gene expression were calculated based on the threshold cycle (Ct) values. Relative quantification for each gene in the array was normalized to that of the housekeeping gene GAPDH (glyceraldehyde-3-phosphate dehydrogenase) using the SABiosciences integrated web-based software to perform all ΔΔCt-based fold-change qPCR data calculations.
EGFR “rescue” and knockdown
EGFR−/− mouse embryonic fibroblasts were infected (multiplicity of infection =50–100) with adenoviruses bearing either control (GFP) or wild-type human EGFR1 expression constructs (gift of Dr Paula McKeown-longo, Albany Medical College) in low-serum-containing medium. Stimulation with TGF-β1 (1ngml−1) was for 4 hours before cell extraction. Adenovirus infectivity was >90% (assessed by GFP fluorescence). HaCaT II-4 cells were similarly infected with EGFR short hairpin RNA-bearing lentiviruses (Sigma) using the TRCN000012120 2-6 clone set.
Oris cell invasion assay
Assessments of the ability of HaCaT II-4 keratinocytes to invade through a three-dimensional extracellular matrix barrier were carried out essentially as described (Platypus Technologies Oris Cell Invasion Assay, Madison, WI) (Soltaninassab et al., 2008; Vasilaki et al., 2010). A 96-well plate was coated with Oris basement membrane extract, allowed to dry, and a polymeric insert was fitted to the inside of the wells to prevent cells from entering the inner analytic zone before seeding 1×105 HaCaT II-4 cells per well. After the cells became confluent (2 days), they were serum-deprived (1 day) and the stopper inserts were removed creating a vacant invasion zone in the center of the well. The cells were overlaid with basement membrane extract to form a three-dimensional matrix barrier. Although cells may continue to proliferate in the peripheral regions of the well under the basement membrane extract overlay, proteolytic activity is required to invade the barrier and enter the analytic area (Soltaninassab et al., 2008). TGF-β1 and/or EGF with or without inhibitors were added to serum-free DMEM after serum starvation and incubated for 72–96 hours before fixation with 3.2% paraformaldehyde and 4,6-diamidino-2-phenylindole staining. Recombinant PAI-1, when used, was added exogenously as described (Providence et al., 2008). An optical mask was applied to the plate bottom to block from view all cells except those that had invaded into the center analytic zone before photography. For siRNA treatment, cells were transfected, trypsinized, and seeded as described above.
Acknowledgments
This work was supported by NIH grant GM57242 to PJH.
Abbreviations
- EMT
epithelial-to-mesenchymal transition
- ERK
extracellular signal-regulated kinase
- MAP
mitogen-activated protein
- MEK
mitogen-activated extracellular kinase
- PAI-1
plasminogen activator inhibitor-1
- PBS
phosphate-buffered saline
- SCC
squamous cell carcinoma
- siRNA
small interfering RNA
- TGF-β1
transforming growth factor-β1
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
References
- Akhurst RJ, Balmain A. Genetic events and the role of TGFβ in epithelial tumour progression. J Pathol. 1999;187:82–90. doi: 10.1002/(SICI)1096-9896(199901)187:1<82::AID-PATH248>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Akiyoshi S, Ishii M, Kawabata M, et al. Targets of transcriptional regulation by transforming growth factor-β: expression profile analysis using oligonucleotide arrays. Jpn J Cancer Res. 2001;92:257–68. doi: 10.1111/j.1349-7006.2001.tb01090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen PA, Kjøller L, Christensen L, et al. The urokinase-type plasminogen activator system in cancer metastasis: a review. Int J Cancer. 1997;72:1–22. doi: 10.1002/(sici)1097-0215(19970703)72:1<1::aid-ijc1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Arts J, Grimbergen J, Toet K, et al. On the role of c-Jun in the induction of PAI-1 gene expression by phorbol ester, serum, and IL-1α in HepG2 cells. Arterioscler Thromb Vasc Biol. 1999;19:39–46. doi: 10.1161/01.atv.19.1.39. [DOI] [PubMed] [Google Scholar]
- Bacharach E, Itin A, Keshet E. Apposition-dependent induction of plasminogen activator inhibitor type 1 expression: a mechanism for balancing pericellular proteolysis during angiogenesis. Blood. 1998;92:939–45. [PubMed] [Google Scholar]
- Bajou K, Noël A, Gerard RD, et al. Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nat Med. 1998;4:923–8. doi: 10.1038/nm0898-923. [DOI] [PubMed] [Google Scholar]
- Bajou K, Masson V, Gerard RD, et al. The plasminogen activator inhibitor PAI-1 controls in vivo tumor vascularization by interaction with proteases, not vitronectin: implications for antiangiogenic strategies. J Cell Biol. 2001;152:777–84. doi: 10.1083/jcb.152.4.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajou K, Maillard C, Jost M, et al. Host-derived plasminogen activator inhibitor-1 (PAI-1) concentration is critical for in vivo tumoral angiogenesis and growth. Oncogene. 2004;23:6986–90. doi: 10.1038/sj.onc.1207859. [DOI] [PubMed] [Google Scholar]
- Bajou K, Peng H, Laug WE, et al. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell. 2008;14:324–34. doi: 10.1016/j.ccr.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukamp P. UV-induced skin cancer: similarities-variations. J Dtsch Dermatol Ges. 2005;3:493–503. doi: 10.1111/j.1610-0387.2005.05037.x. [DOI] [PubMed] [Google Scholar]
- Chen YJ, Lin SC, Kao T, et al. Genome-wide profiling of oral squamous cell carcinoma. J Pathol. 2004;204:326–32. doi: 10.1002/path.1640. [DOI] [PubMed] [Google Scholar]
- Christensen L, Wiborg Simonsen AC, Heegaard CW, et al. Immunohistochemical localization of urokinase-type plasminogen activator, type-1 plasminogen-activator inhibitor, urokinase receptor and α2-macroglobulin receptor in human breast carcinomas. Int J Cancer. 1996;66:441–52. doi: 10.1002/(SICI)1097-0215(19960516)66:4<441::AID-IJC6>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Cui W, Fowlis DJ, Bryson S, et al. TGFβ1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell. 1996;86:531–42. doi: 10.1016/s0092-8674(00)80127-0. [DOI] [PubMed] [Google Scholar]
- Davis GE, Pintar Allen KA, Salazar R, et al. Matrix metalloproteinase-1 and -9 activation by plasmin regulates a novel endothelial cell-mediated mechanism of collagen gel contraction capillary tube regression in three-dimensional collagen matrices. J Cell Sci. 2001;114:917–30. doi: 10.1242/jcs.114.5.917. [DOI] [PubMed] [Google Scholar]
- Degryse B, Neels JG, Czekay RP, et al. The low density lipoprotein receptor-related protein is a motogenic receptor for plasminogen activator inhibitor-1. J Biol Chem. 2004;279:22595–604. doi: 10.1074/jbc.M313004200. [DOI] [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–29. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- Ding W, Shi W, Bellusci S, et al. Sprouty2 downregulation plays a pivotal role in mediating crosstalk between TGF-β1 signaling and EGF as well as FGF receptor tyrosine kinase-ERK pathways in mesenchymal cells. J Cell Physiol. 2007;212:796–806. doi: 10.1002/jcp.21078. [DOI] [PubMed] [Google Scholar]
- Elmageed ZYA, Gaur RL, Williams M, et al. Characterization of coordinated immediate responses by p16INK4A and p53 pathways in UVB-irradiated human skin cells. J Invest Dermatol. 2009;129:175–83. doi: 10.1038/jid.2008.208. [DOI] [PubMed] [Google Scholar]
- Go C, Li P, Wang XJ. Blocking transforming growth factor β signaling in transgenic epidermis accelerates chemical carcinogenesis: a mechanism associated with increased angiogenesis. Cancer Res. 1999;59:2861–8. [PubMed] [Google Scholar]
- Grotegut S, von Schweinitz D, Christofori G, et al. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J. 2006;25:3534–45. doi: 10.1038/sj.emboj.7601213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han G, Lu SL, Li AG, et al. Distinct mechanisms of TGF-β1-mediated epithelial-to-mesenchymal transition and metastasis during skin carcinogenesis. J Clin Invest. 2005;115:1714–23. doi: 10.1172/JCI24399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin CH, Landström M, Moustakas A. Mechanism of TGF-β signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009;21:1–11. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- Herszènyi L, Plebani M, Carraro P, et al. The role of cysteine and serine proteases in colorectal carcinoma. Cancer. 1999;86:1135–42. doi: 10.1002/(sici)1097-0142(19991001)86:7<1135::aid-cncr6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Horiuchi K, Le Gall S, Schulte M, et al. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell. 2007;18:176–88. doi: 10.1091/mbc.E06-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illemann M, Hansen U, Nielsen HJ, et al. Leading-edge myofibroblasts in human colon cancer express plasminogen activator inhibitor-1. Am J Clin Pathol. 2004;122:256–65. doi: 10.1309/F32X-WQ20-T568-H8VP. [DOI] [PubMed] [Google Scholar]
- Joo CK, Kim HS, Park JY, et al. Ligand release-independent transactivation of epidermal growth factor receptor by transforming growth factor-β involves multiple signaling pathways. Oncogene. 2008;27:614–28. doi: 10.1038/sj.onc.1210649. [DOI] [PubMed] [Google Scholar]
- Kim ES, Kim MS, Moon A. Transforming growth factor (TGF)-β in conjunction with H-ras activation promotes malignant progression of MCF10A breast epithelial cells. Cytokine. 2005;29:84–91. doi: 10.1016/j.cyto.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Klemke RL, Cai S, Giannini AL, et al. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137:481–92. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JT, Joo CK. Involvement of cell-cell interactions in rapid stimulation of Cas tyrosine phosphorylation and Src kinase activity by transforming growth factor-β1. J Biol Chem. 2002;277:31938–48. doi: 10.1074/jbc.M201178200. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Timokhina I, et al. A mechanism of repression of TGFβ/Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–16. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutz SM, Higgins CE, Samarakoon R, et al. TGF-β1-induced PAI-1 expression is E-box/USF-dependent and requires EGFR signaling. Exp Cell Res. 2006;312:1093–105. doi: 10.1016/j.yexcr.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Lehman TA, Modali R, Boukamp P, et al. p53 mutations in human immortalized epithelial cell lines. Carcinogenesis. 1993;14:833–9. doi: 10.1093/carcin/14.5.833. [DOI] [PubMed] [Google Scholar]
- Lindberg P, Larsson, Nielsen BS. Expression of plasminogen activator inhibitor-1, urokinase receptor and laminin γ-2 chain is an early coordinated event in incipient oral squamous cell carcinoma. Int J Cancer. 2006;118:2948–56. doi: 10.1002/ijc.21568. [DOI] [PubMed] [Google Scholar]
- Maillard C, Jost M, Rømer MU, et al. Host plasminogen activator inhibitor-1 promotes human skin carcinoma progression in a stagedependent matter. Neoplasia. 2005;7:57–66. doi: 10.1593/neo.04406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Bueno G, Cubillo E, Sarrió D, et al. Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition. Cancer Res. 2006;66:9543–56. doi: 10.1158/0008-5472.CAN-06-0479. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007;98:1512–20. doi: 10.1111/j.1349-7006.2007.00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussoni L, Banfi C, Sironi L, et al. Fluvastatin inhibits basal and stimulated plasminogen activator inhibitor 1, but induces tissue type plasminogen activator in cultured human endothelial cells. Thromb Haemost. 2000;84:59–64. [PubMed] [Google Scholar]
- O-Charoenrat P, Rhys-Evans P, Modjtahedi H, et al. Overexpression of epidermal growth factor receptor in human head and neck squamous carcinoma cell lines correlates with matrix metalloproteinase-9 expression and in vitro invasion. Int J Cancer. 2000;86:307–17. doi: 10.1002/(sici)1097-0215(20000501)86:3<307::aid-ijc2>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- O-Charoenrat P, Rhys-Evans PH, Archer DJ, et al. C-erbB receptors in squamous cell carcinomas of the head and neck: clinical significance and correlation with matrix metalloproteinases and vascular endothelial growth factors. Oral Oncol. 2002;38:73–80. doi: 10.1016/s1368-8375(01)00029-x. [DOI] [PubMed] [Google Scholar]
- Offersen BV, Nielsen BS, Høyer-Hansen G, et al. The myofibroblast is the predominant plasminogen activator inhibitor-1-expressing cell type in human breast carcinomas. Am J Pathol. 2003;163:1887–99. doi: 10.1016/S0002-9440(10)63547-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oft M, Akhurst RJ, Balmain A. Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat Cell Biol. 2002;4:487–94. doi: 10.1038/ncb807. [DOI] [PubMed] [Google Scholar]
- Oft M, Peli J, Rudaz C, et al. TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996;10:2462–77. doi: 10.1101/gad.10.19.2462. [DOI] [PubMed] [Google Scholar]
- Ozaki K, Kadomoto R, Asato K, et al. ERK pathway positively regulates the expression of Sprouty genes. Biochem Biophys Res Commun. 2001;285:1084–8. doi: 10.1006/bbrc.2001.5295. [DOI] [PubMed] [Google Scholar]
- Pedersen H, Brunner N, Francis D, Østerlind K, et al. Prognostic impact of urokinase, urokinase receptor, and type 1 plasminogen activator inhibitor in squamous and large cell lung cancer tissue. Cancer Res. 1994;54:4671–5. [PubMed] [Google Scholar]
- Peinado H, Olmeda D, Cano A. Snail, ZEB, and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- Portella G, Cumming SA, Liddell J, et al. Transforming growth factor β is essential for spindle cell conversion of mouse skin carcinoma in vivo: implications for tumor invasion. Cell Growth Differ. 1998;9:393–404. [PubMed] [Google Scholar]
- Providence KM, Higgins SP, Mullen A, et al. SERPINE1 (PAI-1) is deposited into keratinocyte migration “trails” and required for optimal monolayer wound repair. Arch Dermatol Res. 2008;300:303–10. doi: 10.1007/s00403-008-0845-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Allen RR, Higgins CE, et al. PAI-1 transcriptional regulation during the G0G1 transition in human epidermal keratinocytes. J Cell Biochem. 2006;99:495–507. doi: 10.1002/jcb.20885. [DOI] [PubMed] [Google Scholar]
- Qi L, Higgins SP, Lu Q, et al. SERPINE1 (PAI-1) is a prominent member of the early G0G1 transition “wound repair” transcriptome in p53 mutant human keratinocytes. J Invest Dermatol. 2008;128:749–53. doi: 10.1038/sj.jid.5701068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho O, Beltrán LM, Gimenez-Conti IB, et al. Altered expression of the epidermal growth factor receptor and transforming growth factor-α during multistage skin carcinogenesis in SENCAR mice. Mol Carcinog. 1994;11:19–28. doi: 10.1002/mc.2940110105. [DOI] [PubMed] [Google Scholar]
- Sakakibara T, Hibi K, Koike M, et al. Plasminogen activator inhibitor-1 as a potential marker for the malignancy of gastric cancer. Cancer Sci. 2006;97:395–9. doi: 10.1111/j.1349-7006.2006.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarakoon R, Higgins CE, Higgins SP, et al. Plasminogen activator inhibitor type-1 gene expression and induced migration in TGF-β1-stimulated smooth muscle cells is pp60c–src/MEK-dependent. J Cell Physiol. 2005;204:236–46. doi: 10.1002/jcp.20279. [DOI] [PubMed] [Google Scholar]
- Samarakoon R, Higgins SP, Higgins CE, et al. TGF-β1-induced plasminogen activator-1 expression in vascular smooth muscle cells requires pp60c-src/EGFRY845 and Rho/ROCK signaling. J Mol Cell Cardiol. 2008;44:527–38. doi: 10.1016/j.yjmcc.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Kawai-Kowase K, Sato H, et al. c-Src and hydrogen peroxide mediate transforming growth factor-β1-induced smooth muscle cell-gene expression in 10T1/2 cells. Arterio Thromb Vasc Biol. 2005;25:341–7. doi: 10.1161/01.ATV.0000152608.29351.8f. [DOI] [PubMed] [Google Scholar]
- Sekimoto G, Matsuzaki K, Yoshida K, et al. Reversible Smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res. 2007;67:5090–6. doi: 10.1158/0008-5472.CAN-06-4629. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Izumi H, Oga A, et al. Epidermal growth factor receptor overexpression and genetic aberrations in metastatic squamous-cell carcinoma of the skin. Dermatology. 2001;202:203–6. doi: 10.1159/000051637. [DOI] [PubMed] [Google Scholar]
- Smoller BR. Squamous cell carcinoma: from precursor lesions to high-risk variants. Mod Pathol. 2006;19:S88–92. doi: 10.1038/modpathol.3800509. [DOI] [PubMed] [Google Scholar]
- Soltaninassab SR, Anhalt KM, Bonds MD, et al. A novel 96-well assay for assessing chemokinetic modulators of cell migration and invasion. Proc Am Assoc Cancer Res. 2008;2008:1178. [Google Scholar]
- Sternlicht MD, Dunning AM, Moore DH, et al. Prognostic value of PAI1 in invasive breast cancer: evidence that tumor-specific factors are more important than genetic variation in regulating PAI1 expression. Cancer Epidemiol Biomarkers Prev. 2006;15:2107–14. doi: 10.1158/1055-9965.EPI-06-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–42. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Tian YC, Chen YC, Chang CT, et al. Epidermal and transforming growth factor-β1 enhance HK-2 cell migration through a synergistic increase of matrix metalloproteinase and sustained activation of ERK signaling pathway. Exp Cell Res. 2007;313:2367–77. doi: 10.1016/j.yexcr.2007.03.022. [DOI] [PubMed] [Google Scholar]
- Tomakidi P, Stark H-J, Herold-Mende C, et al. Discriminating expression of differentiation markers evolves in transplants of benign and malignant human skin keratinocytes through stromal interactions. J Pathol. 2003;200:298–307. doi: 10.1002/path.1366. [DOI] [PubMed] [Google Scholar]
- Uttamsingh S, Bao X, Nguyen KT, et al. Synergistic effect between EGF and TGF-β1 in inducing oncogenic properties of intestinal epithelial cells. Oncogene. 2008;27:2626–34. doi: 10.1038/sj.onc.1210915. [DOI] [PubMed] [Google Scholar]
- Vairaktaris E, Yapijakis C, Serefoglou Z, et al. Plasminogen activator inhibitor-1 polymorphism is associated with increased risk for oral cancer. Oral Oncol. 2005;42:888–92. doi: 10.1016/j.oraloncology.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Vasilaki E, Papadimitriou E, Tajadura V, et al. Transcriptional regulation of the small GTPase RhoB gene by TGFβ-induced signaling pathways. FASEB J. 2010;24:891–905. doi: 10.1096/fj.09-134742. [DOI] [PubMed] [Google Scholar]
- Wang SE, Xiang B, Guix M, et al. Transforming growth factor β engages TACE and ErbB3 to activate phosphatidylinositol-3 kinase/Akt in ErbB2-overexpressing breast cancer and desensitizes cells to trastuzumab. Mol Cell Biol. 2008;28:5605–20. doi: 10.1128/MCB.00787-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks BH, He W, Olson KL, et al. Inducible expression of transforming growth factor β1 in papillomas causes rapid metastasis. Cancer Res. 2001;61:7435–43. [PubMed] [Google Scholar]
- Wilkins-Port CE, Higgins CE, Freytag J, et al. PAI-1 is a critical upstream regulator of the TGF-β1/EGF-induced invasive phenotype in mutant p53 human cutaneous squamous cell carcinoma. J Biomed Biotechnol. 2007;2007:85208. doi: 10.1155/2007/85208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins-Port CE, Ye Q, Mazurkiewicz JE, et al. TGF-β1+EGF-initiated invasive potential in transformed human keratinocytes is coupled to a plasmin/MMP-10/MMP-1-dependent collagen remodeling axis: role for PAI-1. Cancer Res. 2009;69:4081–91. doi: 10.1158/0008-5472.CAN-09-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuspa SH. The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis. J Dermatol Sci. 1998;17:1–7. doi: 10.1016/s0923-1811(97)00071-6. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Böttinger EP. TGF-β and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–74. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Bitzer M, Liang D, et al. Genetic programs of epithelial cell plasticity directed by transforming growth factor-β. Proc Natl Acad Sci USA. 2001;98:6686–91. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YE. Non-Smad pathways in TGF-β signaling. Cell Res. 2009;19:128–39. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HY, Pon YL, Wong AS. Synergistic effects of epidermal growth factor and hepatocyte growth factor on human ovarian cancer cell invasion and migration: role of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase. Endocrinology. 2007;148:5195–208. doi: 10.1210/en.2007-0361. [DOI] [PubMed] [Google Scholar]
- Zuo W, Chen YG. Specific activation of mitogen-activated protein kinase by transforming growth factor-β receptors in lipid rafts is required for epithelial cell plasticity. Mol Biol Cell. 2009;20:1020–9. doi: 10.1091/mbc.E08-09-0898. [DOI] [PMC free article] [PubMed] [Google Scholar]