

Tpl2 kinase regulates FcγR-induced MEK/ERK1/2 activation and effector functions in myeloid cells; a potential therapeutic target for immune-complex mediated diseases.
Keywords: macrophages, MAPK, immune complex
Abstract
The MAPK3 Tpl2 controls innate and adaptive immunity by regulating TLR, TNF-α, and GPCR signaling in a variety of cell types. Its ablation gives rise to an anti-inflammatory phenotype characterized by resistance to LPS-induced endotoxin shock, DSS-induced colitis, and TNF-α-induced IBD. Here, we address the role of Tpl2 in autoimmunity. Our data show that the ablation and the pharmacological inhibition of Tpl2 protect mice from antiplatelet antibody-induced thrombocytopenia, a model of ITP. Thrombocytopenia in this model and in ITP is caused by phagocytosis of platelets opsonized with antiplatelet antibodies and depends on FcγR activation in splenic and hepatic myeloid cells. Further studies explained how Tpl2 inhibition protects from antibody-induced thrombocytopenia, by showing that Tpl2 is activated by FcγR signals in macrophages and that its activation by these signals is required for ERK activation, cytoplasmic Ca2+ influx, the induction of cytokine and coreceptor gene expression, and phagocytosis.
Introduction
FcγRs are expressed widely throughout the hematopoietic system and play a central role in the activation of innate immunity by specific high-affinity antibodies produced by the adaptive immune system [1]. In humans, polymorphisms in the genes encoding various FcγRs predispose to autoimmune diseases, including SLE and rheumatoid arthritis [2, 3]. In mice, FcγRs have been linked to the pathogenesis of several models of systemic autoimmunity, including the antiplatelet, antibody-induced thrombocytopenia, the KRN serum transfer arthritis, and the nephritis of the NZBxWF1 model of SLE. Deletion of FcγRs or their signaling targets prevented the development of cytotoxicity and inflammation associated with these models [4–6]. FcγR cross-linking by IgG-opsonized particles or IgG-containing immune complexes activates the NF-κB, MAPKs, PI3K/Akt, and PLCγ/cytoplasmic Ca2+ influx pathways and regulates important biological functions, such as phagocytosis, secretion of soluble mediators, and induction of ROS. In addition, it up-regulates membrane costimulatory molecules [1, 7].
MAPK activation involves three-tiered kinase cascades, in which MAPKs are activated by MEKs, which in turn, are activated by MAP3Ks [8]. Tpl2 is a serine/threonine kinase that belongs to the MAP3K family (MAP3K8) [9]. It has been shown to regulate MAPK activation downstream of TLRs, members of the TNFR family, the IL-1R, and Gαi2-coupled GPCRs, including the proteinase-activated receptor 1 and the receptor for sphingosine-1-phosphate, in a variety of cell types [10–14]. In addition, it has been shown that Tpl2 is required for the induction of cytoplasmic Ca2+ influx in response to GPCR and IL-1R signals [13]. As a result of the signaling abnormalities induced by Tpl2 ablation, Tpl2-deficient macrophages, DCs, and T cells exhibit defects in cytokine and chemokine secretion [10, 15–17]. As a result, Tpl2 KO mice are resistant to LPS-induced endotoxin shock, to DSS-induced experimental colitis, and to TNF-induced IBD [15, 18, 19].
In this study, we addressed the role of Tpl2 in antiplatelet, antibody-induced thrombocytopenia in mice, an animal model of the autoimmunity syndrome ITP. In this model and in ITP, thrombocytopenia results from phagocytosis of antibody-opsonized platelets, by splenic and hepatic myeloid cells, and depends on FcγR activation. Data presented in this report show that the genetic and pharmacological inhibition of Tpl2 protects from antiplatelet, antibody-induced thrombocytopenia. Furthermore, Tpl2 is activated by FcγR signals in macrophages, and its activation is a prerequisite for the transduction of FcγR ERK activation signals, cytoplasmic Ca2+ influx, phagocytosis of IgG-opsonized latex particles, and the induction of cytokine and coreceptor gene expression. Given the importance of FcγR signals in the pathogenesis of antibody-induced thrombocytopenia, these data mechanistically explain the role of Tpl2 in this syndrome and suggest that the Tpl2 kinase may be an excellent therapeutic target for (auto)antibody-mediated pathologies.
MATERIALS AND METHODS
Antibodies, immunodetection reagents, recombinant proteins, and the Tpl2 inhibitor
The antibody used for Tpl2 immunoprecipitation and Western blotting was from Santa Cruz Biotechnology (sc-720; Santa Cruz, CA, USA). The antibody against phosphorylated ERK1/2 was from Sigma (M 8159; St. Louis, MO, USA). Antibodies against ERK1/2, IκBα, SAPK/JNK, phospho(T183/Y185)-SAPK/JNK, MEK1/2, phospho(S217/S221)-MEK1/2, p38, and phospho(T180/Y182)-p38, and phospho(Y705)-STAT3 were from Cell Signaling Technology (Danvers, MA, USA). Antibodies for STAT3 Western blotting were from BD PharMingen (San Diego, CA, USA). Secondary antibodies used in Western blotting were purchased from Jackson ImmunoResearch Laboratories (anti-rabbit HRP; West Grove, PA, USA), as well as from Millipore (anti-mouse HRP; Billerica, MA, USA). The blots were developed using ECL (Thermo Fisher Scientific, Rockford, IL, USA). Anti-CD41 antibody and the IgG1 isotype control antibody used for antiplatelet, antibody-induced thrombocytopenia in mice were purchased from BD PharMingen. GST [GST-MEK (kinase dead)] protein was kindly provided by Aris Eliopoulos (Institute of Molecular Biology and Biotechnology, Foundation for Research and Technology-Hellas, Crete, Greece). The A Sepharose beads used for the Tpl2 in vitro kinase assay were from Upstate Biotechnology (Lake Placid, NY, USA). IgG1, used to cross-link FcγRs, was purchased from Sigma (M7894). Tpl2 kinase inhibitor {4-(3-chloro-4-fluorophenylamino)-6-(pyridine-3-yl-methylamino)-3-cyano- [1,7]-naphthyridine} and the MEK/ERK1/2 inhibitor U0126 [1,4-diamino-2,3-dicyano-1,4-bis (2-amino-phenylthio) butadiene] were purchased from Calbiochem (San Diego, CA, USA).
Mice and primary cells
Animals (C57/Bl6 and Tpl2 KO mice) [15] were housed in the animal facility of the Medical School of the University of Crete (Greece). Experiments in these mice were approved by the Animal Utilization and Care Committee of the University of Crete. BMDMs were generated by culturing BM cells from these mice in DMEM, supplemented with L cell-conditioned medium (20%). The resulting cultures consisted of macrophages (>95% purity), as determined by staining for F4/80 and flow cytometry.
Protein isolation for immunoprecipitation and Western blotting
BMDMs were transferred for the times indicated onto plates coated with 50 μg/ml IgG1. Cells were washed once in PBS prior to lysis in a 1% Nonidet P-40-containing lysis buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0,25% sodium deoxycholate, 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF, plus a mixture of protease inhibitor cocktail; Roche (Indianapolis, IN, USA)]. Tpl2 was immunoprecipitated from these lysates by incubation with protein A-Sepharose-coupled anti-Tpl2 antibody for 16 h at 4°C. Beads were washed once in kinase lysis buffer and three times in reaction buffer [50 mM Tris (pH 7.6), 150 mM NaCl, 3 mM EGTA, 5 mM β-glycerophosphate, 10 mM MgCl2, 1 mM sodium vanadate, 0.03% Brij35]. The washed beads were resuspended in 30 μl reaction buffer, supplemented with 1 mM ATP and 0.5 μg of the Tpl2 kinase substrate [GST-MEK (kinase dead)]. Following 30 min incubation at 30°C, the samples were diluted in protein-loading buffer, boiled for 10 min, and analyzed by SDS-PAGE (10%) and Western blotting. Phosphorylation of the substrate was monitored by probing the Western blots with an antiphospho-MEK antibody.
Flow cytometry
Cells were incubated for 20 min at 4°C with conjugated fluorescent antibodies for cell-surface markers. Stained cells were monitored by flow cytometry (FACSCalibur; BD Biosciences, San Jose, CA, USA). Data were analyzed using BD CellQuest Pro software.
ELISA
FcγR cross-linking in WT and Tpl2 KO C57BL/6 BMDMs was induced by treatment with IgG. Culture supernatants were collected 3 and 24 h later and analyzed for the abundance of mouse IL-6, TNF, and IL-12 p70, using ELISA kits (BD PharMingen), according to the instructions of the manufacturer.
Antiplatelet antibody ITP
WT and Tpl2 KO mice were injected i.p. with 2 μg of an IgG1 rat anti- mouse CD41(GpIIa) antibody, diluted in 200 μl PBS or with control rat IgG1, also diluted in 200 μl PBS. Twenty-four hours later, platelets were counted using a Coulter LH780 analyzer (Beckman Coulter, Brea, CA, USA). In some of these experiments, before the injection of the anti-CD41 antibody or the IgG1 control, WT mice were injected i.p. with the Tpl2 inhibitor {4-(3-chloro-4-fluorophenylamino)-6-(pyridine-3-yl-methylamino)-3-cyano- [1,7]-naphthyridine; 2 mg/kg and 10 mg/kg} or with IVIg (2 g/kg). Platelets were counted again 24 h later.
Measurement of intracellular Ca2+ concentration
Intracellular calcium concentration was measured using the Fluo-4 NW Calcium Assay Kit (Molecular Probes, Invitrogen, Carlsbad, CA, USA). WT and Tpl2 KO BMDMs were incubated with mouse IgG1 (50 μg/ml) for 30 min. Following this treatment, the cells were washed with the assay buffer (20 mM HEPES, 1× HBSS), plated into 96-well plates (30,000 cells/well), and incubated with the dye-loading solution for 30 min at 37°C. Anti-mouse IgG (Fab-specific) was added to the cells robotically (100 μg/ml), and the influx of Ca2+ into the cytosol was measured over a period of 15 min using a FlexStation 3 fluorescence reader (Molecular Devices, Sunnyvale, CA, USA; excitation at 494 nm and emission at 516 nm).
Phagocytosis of opsonized latex beads and phalloidin staining
WT and Tpl2 KO BMDMs were platted on glass coverslips (50,000 cells/well) and incubated overnight in complete medium. Twenty-four hours later, polystyrene latex beads (3.0 μm mean particle size) were diluted in PBS and incubated with mouse IgG1 (1 mg/ml) for 30 min at room temperature. Following incubation, the beads were centrifuged, washed with PBS, diluted in medium, and added to the cells. Cells were incubated at 37°C for 1.5 h, rinsed in PBS, and fixed in 4% formaldehyde at 25°C for 20 min. Coverslips were then stored at 4°C in formaldehyde. Prior to staining, cells were permeabilized for 5 min in 0.2% Triton X-100 and rinsed three times in PBS. To stain the permeabilized cells, coverslips were incubated for 40 min at room temperature with a 50-μg/ml fluorescent phalloidin-FITC (Sigma) solution in PBS. Subsequently, the coverslips were rinsed five times in PBS to remove unbound phalloidin-FITC and mounted on glass slides. Images were obtained using a Nikon Eclipse 80i microscope with a 40× objective and a Spot charge-coupled device camera (Spot Imaging Solutions, Diagnostic Instruments, Sterling Heights, MI, USA). The number of beads internalized by 100 cells was determined. The phagocytosis index represents the number of internalized beads/macrophage.
RESULTS AND DISCUSSION
Tpl2 deficiency and the pharmacological inhibition of Tpl2 protect mice from antiplatelet antibody-induced thrombocytopenia, an experimental model of acute ITP
Antiplatelet, antibody-induced thrombocytopenia depends on FcγR activation by IgG-opsonized platelets and has been used as a model of acute ITP [6]. In this animal model, i.p. injection of an antiglycoprotein IIb (anti-CD41) antibody reproducibly induces severe thrombocytopenia in mice. Platelets opsonized with the IgG antibody are removed by macrophage-dependent phagocytosis in spleen and liver. Development of thrombocytopenia depends on FcγR activation in macrophages and is independent of T and B lymphocytes [20]. Experiments addressing the role of Tpl2 in antiplatelet, antibody-induced thrombocytopenia showed that whereas i.p. injection of the antiplatelet antibody in WT mice induces severe thrombocytopenia, 24 h later, injection of the same antibody in Tpl2-deficient mice has a minimal or no effect on the platelet count (Fig. 1A).
Figure 1. Tpl2 inhibition protects mice from antiplatelet, antibody-induced thrombocytopenia.
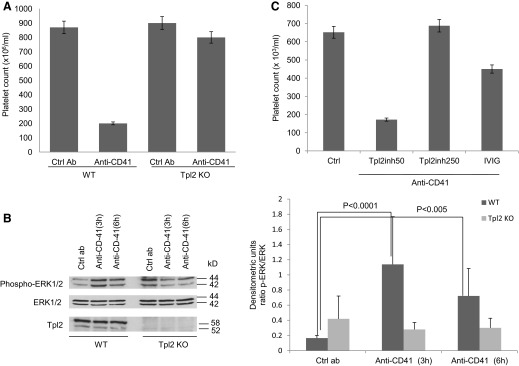
(A) WT and Tpl2 KO mice were injected i.p. with an anti-CD41 IgG1 antibody or the isotype control IgG1 (2 μg/mouse). Platelet counts were evaluated 24 h later. Cumulative results from three independent experiments (total number of mice/group, n=15; bars are sem) are shown. Ctrl Ab, Control antibody. (B) ERK1/2 phosphorylation in spleen protein extracts from mice injected with the anti-CD41 antibody or the isotype control IgG1. Western blots of the cell lysates were probed with antibodies against the phosphorylated or total ERK1, ERK2. To confirm the ablation of Tpl2 in Tpl2 KO mice, the same lysates were also probed with the anti-Tpl2 antibody. (Right) Densitometry analysis of the bands at left [ratio of phosphorylated (p)-ERK1/2:total ERK1/2]. Data are representative of three experiments (error bars, sd of five mice/group). (C) WT mice were injected i.p. with escalating doses of the Tpl2 inhibitor (inh; 2 mg/kg and 10 mg/kg). Each mouse received one injection/day for 2 sequential days. Alternatively, the mice were injected with IVIg (2 g/kg). Twenty-four hours after the pretreatment with the Tpl2 inhibitor or IVIg, the mice were injected i.p. with the anti-CD41 antibody or with the isotype antibody control. Platelet counts were determined 24 h later. The results shown are cumulative results of three independent experiments (total number of mice/group; n=15; bars are sem).
Probing Western blots of whole spleen lysates of anti-CD41 and control antibody-treated WT and Tpl2 KO mice revealed that the anti-CD41 antibody induces ERK1/ERK2 MAPK phosphorylation only in the WT mice (Fig. 1B).
Pretreatment of WT mice with a Tpl2-specific pharmacological inhibitor protected them from thrombocytopenia in a dose-dependent manner and was more effective than pretreatment with IVIg (Fig. 1C), a well-established and effective treatment of acute ITP in mice and humans [21].
FcγR cross-linking in macrophages activates Tpl2, which regulates ERK1 and ERK2 phosphorylation and cytoplasmic Ca2+ influx
As the platelet removal in antiplatelet, antibody-induced thrombocytopenia depends on FcγR activation and FcγR-induced phagocytosis by splenic and hepatic macrophages, we hypothesized that Tpl2 is required for FcγR signaling. Cross-linking of FcγRs in macrophages induces a rapid increase in the concentration of cytoplasmic Ca2+ and activates multiple signaling pathways, including the MAPK, NF-κB, and PI3K/Akt pathways. To evaluate the involvement of Tpl2 in FcγR signaling, we first examined its enzymatic activation in FcγR-stimulated BMDMs from WT C57/Bl6 mice. Tpl2, immunoprecipitated from whole-cell lysates, harvested before and after FcγR cross-linking with plate-bound IgG, was used to carry out in vitro kinase assays, with GST-MEK as the kinase substrate. The results showed that FcγR cross-linking activates Tpl2 in primary macrophages (Fig. 2A).
Figure 2. Tpl2 is activated by FcγR signals and is required for FcγR-induced MEK/ERK1/2 MAPK phosphorylation and cytoplasmic Ca2+ influx.
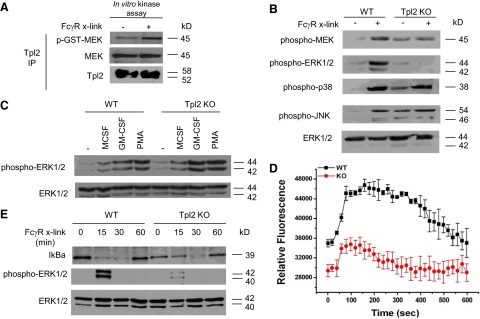
(A) FcγR cross-linking activates Tpl2, which was immunoprecipitated (IP) from WT BMDMs after FcγR cross-linking (x-link), and the immunoprecipitates were used in an in vitro kinase reaction, using a GST-MEK peptide as the substrate. Phosphorylation (p) of the GST-MEK peptide was determined by probing the products of the in vitro kinase reaction with an antiphospho-MEK and an anti-MEK antibody. (B) Phosphorylation of MEK, ERK1 and ERK2, p38MAPK, and JNK upon FcγR cross-linking in WT and Tpl2 KO BMDMs. Phosphorylation was monitored by probing Western blots of cell lysates harvested 30 min after the start of the stimulation with IgG1. (C) Tpl2 is not required for ERK phosphorylation in cells stimulated with MCSF, GM-CSF, or PMA. Phosphorylation was monitored by probing Western blots of cell lysates harvested 30 min after the start of the stimulation with the indicated cytokines. (D) WT and Tpl2 KO BMDMs were loaded with the fluorescent Ca2+ indicator, Fluo-4 NW. Following incubation with mouse IgG for 30 min, FcγRs were cross-linked with a goat anti-mouse IgG (Fab-specific). Cytoplasmic Ca2+ influx was monitored fluorimetrically (excitation at 494 nm and emission at 516 nm). Results are expressed as mean fluorescence ± se of the mean, and they are calculated by combining the results of two independent experiments. Each experiment included triplicate measurements for each time-point (n=6). (E) Degradation of IκBα upon FcγR cross-linking in WT and Tpl2 KO BMDMs. Molecular mass values represent the estimated molecular mass of the proteins. Each panel is representative of three to six independent experiments, all of which gave similar results.
Tpl2 has been established as the specific MAP3K activating the MEK and ERK1 and ERK2 kinases downstream of most TLRs. In parallel experiments, Western blots of cell lysates harvested before and after FcγR cross-linking were probed with antibodies against phosphorylated MEK, ERK, JNK, and p38 MAPK, as well as total ERK, which was used as the loading control. The results showed that the FcγR-activated Tpl2, similar to Tpl2 activated by TLR, TNF-α/CD40, or GPCR signals [10, 11, 13–15, 17], is required for MEK and ERK but not JNK or p38 MAPK activation in macrophages (Fig. 2B). These findings add FcγR to the receptors using Tpl2 to transducer ERK activation signals. It is noteworthy that ERK activation is not impaired in Tpl2 KO macrophages stimulated with M-CSF, GM-CSF, or PMA (Fig. 2C), suggesting that the list of ERK activation receptor signals that depend on Tpl2 is finite.
To determine whether cytoplasmic Ca2+ influx in FcγR-stimulated macrophages [1] also depends on Tpl2, WT and Tpl2 KO BMDMs were incubated with mouse IgG and loaded with the fluorescent Ca2+ indicator Fluo-4 NW, and FcγRs were cross-linked with a goat anti-mouse IgG (Fab-specific). Changes in the cytoplasmic levels of Ca2+ were measured by monitoring the intensity of cytoplasmic fluorescence. This analysis showed that FcγR signals, similarly to signals elicited by GPCR or IL-1β stimulation [13], also depend on Tpl2 for the induction of cytoplasmic Ca2+ influx (Fig. 2D). Based on these results, we conclude that Tpl2 plays a central role in the activation of MEK-ERK1/ERK2 and the mobilization of intracellular Ca2+ by FcγR signals in macrophages.
Other signaling pathways activated after FcγR cross-linking include the NF-κB and PI3K/Akt pathways. To evaluate the role of Tpl2 in the activation of NF-κB, we examined the protein levels of IκBα before and after FcγR cross-linking in WT and Tpl2 KO BMDMs. The phosphorylation of ERK was monitored as a control for the FcγR cross-linking. The results showed that the steady-state levels of IκBα were lower in Tpl2 KO macrophages. Moreover, the degradation of IκBα upon FcγR cross-linking was delayed in Tpl2-deficient macrophages (Fig. 2E). Parallel experiments revealed that Tpl2 is dispensable for Akt phosphorylation induced by FcγR cross-linking in macrophages (data not shown).
Tpl2 regulates FcγR-induced phagocytosis, cytokine production, and the induction of costimulatory molecules in myeloid cells
Ligation of FcγR induces phagocytosis of IgG-opsonized particles and endocytosis of immune complexes, but signaling pathways regulating this process are not well defined [22]. It has been suggested that activation of ERK1/ERK2 MAPKs regulates FcγR-induced phagocytosis in myeloid cells. To evaluate the role of Tpl2 in FcγR-induced phagocytosis, we examined the phagocytosis of IgG-opsonized latex beads by WT and Tpl2 KO macrophages in vitro. The results showed that phagocytosis was impaired significantly in Tpl2 KO macrophages (Fig. 3A). Quantification of the phagocytosis index in four separate experiments showed a robust decrease in phagocytosis in Tpl2 KO macrophages, by comparison with the WT macrophages (Fig. 3B).
Figure 3. Tpl2 regulates FcγR-induced phagocytosis.
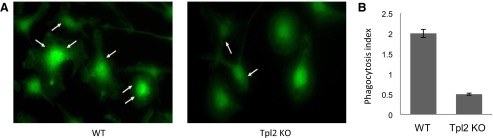
(A) WT and Tpl2 KO macrophages were seeded on glass coverslips, and they were incubated with IgG-opsonized polystyrene latex beads for 1.5 h. Cells were fixed, and actin was visualized with phalloidin-FITC. Representative pictures of two independent experiments. Original magnification scale, ×40. Arrows indicate phagocytosed opsonized latex beads. (B) The phagocytosis index was calculated as the mean number of engulfed IgG-opsonized latex beads/macrophage.
Ligation of FcγR in myeloid cells induces the secretion of cytokines and chemokines, the up-regulation of costimulatory molecules, and the activation of important biological functions that regulate the immune responses and inflammation [23]. To evaluate the effect of Tpl2 on the FcγR-induced cytokine production, we used ELISA assays to measure TNF-α, IL-6, IL-10, and IL-12 in culture supernatants of WT and Tpl2 KO macrophages, harvested before and after FcγR cross-linking. The results showed that FcγR-stimulated Tpl2 KO BMDMs secrete significantly lower amounts of TNF-α, IL-10, and IL-6 and significantly higher amounts of IL-12 (Fig. 4A). To confirm that the effects of Tpl2 deficiency on FcγR-induced cytokine production resulted from abrogation of ERK activation, WT cells were treated with the MEK inhibitor U0126 to block ERK activation, while leaving p38 or JNK activation unaffected. U0126 reduced production of IL-6 and IL-10 by FcγR-stimulated BMDMs (Fig. 4B), similar to the effects of Tpl2 deficiency in this cell type (Fig. 4A). We conclude that the effects of Tpl2-transduced FcγR signals on cytokine secretion in macrophages are similar to the effects of Tpl2-transduced TLR signals [10, 15, 24].
Figure 4. Tpl2 regulates FcγR-induced cytokine production and costimulatory molecule expression.
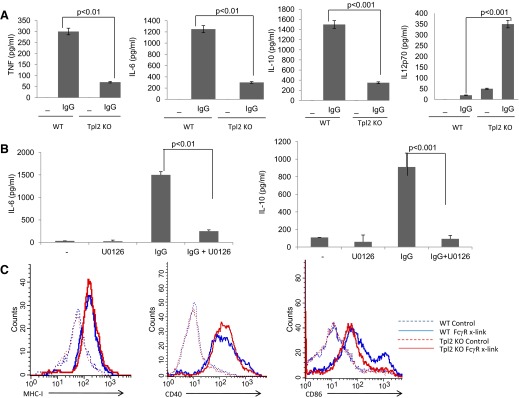
(A) WT and Tpl2 KO BMDMs were stimulated with plate-bound IgG (50 μg/ml). Cytokine levels were determined by ELISA in culture supernatants from cells stimulated for 3 h for TNF and for 24 h for the other cytokines. (B) Before stimulation with plate-bound IgG (50 μg/ml), WT BMDMs were incubated for 30 min with 2.5 μΜ U0126 or vehicle control (DMSO). Cytokine levels were determined by ELISA in culture supernatants form cells stimulated for 24 h. Results are representative of triplicate cultures from at least three independent experiments. (C) BMDMs isolated from WT and Tpl2 KO mice were stimulated with plate-bound IgG (50 μg/ml). Up-regulation of MHC-I, CD40, and CD86 was monitored by flow cytometry.
Ligation of FcγRs by immune complexes causes up-regulation of costimulatory molecules in APCs. Signaling pathways that regulate this process, however, are not well-defined. FcγR-induced activation of the tyrosine kinase Syk and the STAT1 transcription factor have been identified as major regulators of costimulatory molecule expression in myeloid cells [23, 25]. To evaluate the effects of FcγR-induced up-regulation of costimulatory molecules, we used FACS to analyze the expression of MHC class I, CD40, and CD86 in WT and Tpl2 KO BMDCs. The results showed that whereas Tpl2 deficiency does not affect FcγR-induced up-regulation of MHC class I and CD40, it interferes with the up-regulation of CD86, which undergoes only partial induction in these cells (Fig. 4C).
Overall, the data presented in this report demonstrate that FcγR activation induces Ca2+ influx and MAPK activation via Tpl2-transduced signals. FcγR is activated by immune complexes and Ig-opsonized cells, which are observed in the course of autoimmunity and contribute to the phenotype. The importance of FcγR and Tpl2 activation in the autoimmunity phenotype was confirmed here, with experiments showing that the ablation and the pharmacological inhibition of Tpl2 prevent the development of thrombocytopenia in the murine ITP model of antiplatelet, antibody-induced immune thrombocytopenia. These results identify Tpl2 as a potential therapeutic target for immune thrombocytopenia and perhaps other autoimmune syndromes in humans.
ACKNOWLEDGMENTS
This work was supported by grants from the Hellenic Secretariat for Research and Technology (Pythagoras I and II fund; to D.T.B.), the European Union (Autocure, Sixth Framework Programme and BTCure, Innovative Medicines Initiative; to D.T.B. and I.T.), the Hellenic Society of Rheumatology (to I.T.), the “ARISTEIA” Action of the “Operational Programme Education and Lifelong Learning” and is cofunded by the European Social Fund (ESF) and National Resources (to D.T.B. and I.T.), and U.S. National Institutes of Health grant R01 CA124835 (to P.N.T.).
We thank Raymond Dattwyler for critically reviewing the manuscript.
Footnotes
- BMDM
- bone marrow-derived macrophage
- DSS
- dextran sodium sulfate
- IBD
- inflammatory bowel disease
- ITP
- immune thrombocytopenic purpura
- IVIg
- i.v. Ig
- KO
- knockout
- MAP3K
- MEK kinase
- SLE
- systemic lupus erythematosus
- Tpl2
- tumor progression locus-2
AUTHORSHIP
I.K., I.T., D.T.B., and P.N.T. designed the study, performed experiments, and wrote the manuscript. M.I. and M.H. performed experiments and wrote the manuscript.
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1. Nimmerjahn F., Ravetch J. V. (2008) Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34–47 [DOI] [PubMed] [Google Scholar]
- 2. Salmon J. E., Millard S., Schachter L. A., Arnett F. C., Ginzler E. M., Gourley M. F., Ramsey-Goldman R., Peterson M. G., Kimberly R. P. (1996) Fc γ RIIA alleles are heritable risk factors for lupus nephritis in African Americans. J. Clin. Invest. 97, 1348–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Salmon J. E., Pricop L. (2001) Human receptors for immunoglobulin G: key elements in the pathogenesis of rheumatic disease. Arthritis Rheum. 44, 739–750 [DOI] [PubMed] [Google Scholar]
- 4. Clynes R., Dumitru C., Ravetch J. V. (1998) Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science 279, 1052–1054 [DOI] [PubMed] [Google Scholar]
- 5. Ji H., Ohmura K., Mahmood U., Lee D. M., Hofhuis F. M., Boackle S. A., Takahashi K., Holers V. M., Walport M., Gerard C., Ezekowitz A., Carroll M. C., Brenner M., Weissleder R., Verbeek J. S., Duchatelle V., Degott C., Benoist C., Mathis D. (2002) Arthritis critically dependent on innate immune system players. Immunity 16, 7–168 [DOI] [PubMed] [Google Scholar]
- 6. Takai T., Li M., Sylvestre D., Clynes R., Ravetch J. V. (1994) FcR γ chain deletion results in pleiotrophic effector cell defects. Cell 76, 519–529 [DOI] [PubMed] [Google Scholar]
- 7. Nimmerjahn F., Ravetch J. V. (2006) Fcγ receptors: old friends and new family members. Immunity 24, 19–28 [DOI] [PubMed] [Google Scholar]
- 8. Kyriakis J. M., Avruch J. (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81, 807–869 [DOI] [PubMed] [Google Scholar]
- 9. Patriotis C., Makris A., Chernoff J., Tsichlis P. N. (1994) Tpl-2 acts in concert with Ras and Raf-1 to activate mitogen-activated protein kinase. Proc. Natl. Acad. Sci. USA 91, 9755–9759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaiser F., Cook D., Papoutsopoulou S., Rajsbaum R., Wu X., Yang H. T., Grant S., Ricciardi-Castagnoli P., Tsichlis P. N., Ley S. C., O'Garra A. (2009) TPL-2 negatively regulates interferon-β production in macrophages and myeloid dendritic cells. J. Exp. Med. 206, 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eliopoulos A. G., Wang C. C., Dumitru C. D., Tsichlis P. N. (2003) Tpl2 transduces CD40 and TNF signals that activate ERK and regulates IgE induction by CD40. EMBO J. 22, 3855–3864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eliopoulos A. G., Das S., Tsichlis P. N. (2006) The tyrosine kinase Syk regulates TPL2 activation signals. J. Biol. Chem. 281, 1371–1380 [DOI] [PubMed] [Google Scholar]
- 13. Hatziapostolou M., Koukos G., Polytarchou C., Kottakis F., Serebrennikova O., Kuliopulos A., Tsichlis P. N. (2011) Tumor progression locus 2 mediates signal-induced increases in cytoplasmic calcium and cell migration. Sci. Signal. 4, ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hatziapostolou M., Polytarchou C., Panutsopulos D., Covic L., Tsichlis P. N. (2008) Proteinase-activated receptor-1-triggered activation of tumor progression locus-2 promotes actin cytoskeleton reorganization and cell migration. Cancer Res. 68, 1851–1861 [DOI] [PubMed] [Google Scholar]
- 15. Dumitru C. D., Ceci J. D., Tsatsanis C., Kontoyiannis D., Stamatakis K., Lin J. H., Patriotis C., Jenkins N. A., Copeland N. G., Kollias G., Tsichlis P. N. (2000) TNF-α induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell 103, 1071–1083 [DOI] [PubMed] [Google Scholar]
- 16. Watford W. T., Hissong B. D., Durant L. R., Yamane H., Muul L. M., Kanno Y., Tato C. M., Ramos H. L., Berger A. E., Mielke L., Pesu M., Solomon B., Frucht D. M., Paul W. E., Sher A., Jankovic D., Tsichlis P. N., O'Shea J. J. (2008) Tpl2 kinase regulates T cell interferon-γ production and host resistance to Toxoplasma gondii. J. Exp. Med. 205, 2803–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiao N., Eidenschenk C., Krebs P., Brandl K., Blasius A. L., Xia Y., Khovananth K., Smart N. G., Beutler B. (2009) The Tpl2 mutation Sluggish impairs type I IFN production and increases susceptibility to group B streptococcal disease. J. Immunol. 183, 7975–7983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lawrenz M., Visekruna A., Kuhl A., Schmidt N., Kaufmann S. H., Steinhoff U. (2012) Genetic and pharmacological targeting of TPL-2 kinase ameliorates experimental colitis: a potential target for the treatment of Crohn's disease? Mucosal Immunol. 5, 129–139 [DOI] [PubMed] [Google Scholar]
- 19. Kontoyiannis D., Boulougouris G., Manoloukos M., Armaka M., Apostolaki M., Pizarro T., Kotlyarov A., Forster I., Flavell R., Gaestel M., Tsichlis P., Cominelli F., Kollias G. (2002) Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn's-like inflammatory bowel disease. J. Exp. Med. 196, 1563–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Song S., Crow A. R., Freedman J., Lazarus A. H. (2003) Monoclonal IgG can ameliorate immune thrombocytopenia in a murine model of ITP: an alternative to IVIG. Blood 101, 3708–3713 [DOI] [PubMed] [Google Scholar]
- 21. Clynes R. (2007) Protective mechanisms of IVIG. Curr. Opin. Immunol. 19, 646–651 [DOI] [PubMed] [Google Scholar]
- 22. Sanchez-Mejorada G., Rosales C. (1998) Signal transduction by immunoglobulin Fc receptors. J. Leukoc. Biol. 63, 521–533 [DOI] [PubMed] [Google Scholar]
- 23. Dhodapkar K. M., Banerjee D., Connolly J., Kukreja A., Matayeva E., Veri M. C., Ravetch J. V., Steinman R. M., Dhodapkar M. V. (2007) Selective blockade of the inhibitory Fcγ receptor (FcγRIIB) in human dendritic cells and monocytes induces a type I interferon response program. J. Exp. Med. 204, 1359–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sugimoto K., Ohata M., Miyoshi J., Ishizaki H., Tsuboi N., Masuda A., Yoshikai Y., Takamoto M., Sugane K., Matsuo S., Shimada Y., Matsuguchi T. (2004) A serine/threonine kinase, Cot/Tpl2, modulates bacterial DNA-induced IL-12 production and Th cell differentiation. J. Clin. Invest. 114, 857–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sedlik C., Orbach D., Veron P., Schweighoffer E., Colucci F., Gamberale R., Ioan-Facsinay A., Verbeek S., Ricciardi-Castagnoli P., Bonnerot C., Tybulewicz V. L., Di Santo J., Amigorena S. (2003) A critical role for Syk protein tyrosine kinase in Fc receptor-mediated antigen presentation and induction of dendritic cell maturation. J. Immunol. 170, 846–852 [DOI] [PubMed] [Google Scholar]