

Abstract
Objective:
To use principal component analyses (PCA) of Pittsburgh compound B (PiB) PET imaging to determine whether the pattern of in vivo β-amyloid (Aβ) in Parkinson disease (PD) with cognitive impairment is similar to the pattern found in symptomatic Alzheimer disease (AD).
Methods:
PiB PET scans were obtained from participants with PD with cognitive impairment (n = 53), participants with symptomatic AD (n = 35), and age-matched controls (n = 67). All were assessed using the Clinical Dementia Rating and APOE genotype was determined in 137 participants. PCA was used to 1) determine the PiB binding pattern in AD, 2) determine a possible unique PD pattern, and 3) directly compare the PiB binding patterns in PD and AD groups.
Results:
The first 2 principal components (PC1 and PC2) significantly separated the AD and control participants (p < 0.001). Participants with PD with cognitive impairment also were significantly different from participants with symptomatic AD on both components (p < 0.001). However, there was no difference between PD and controls on either component. Even those participants with PD with elevated mean cortical binding potentials were significantly different from participants with AD on both components.
Conclusion:
Using PCA, we demonstrated that participants with PD with cognitive impairment do not exhibit the same PiB binding pattern as participants with AD. These data suggest that Aβ deposition may play a different pathophysiologic role in the cognitive impairment of PD compared to that in AD.
Synucleinopathy with Lewy bodies and neurites, neuronal loss, and gliosis constitute key neuropathologic features of Parkinson disease (PD) and cortical synucleinopathy is associated with cognitive impairment.1–3 Cortical β-amyloid (Aβ) deposition, but rarely florid tauopathy, also occurs in PD-related dementia, thus distinguishing β-amyloidosis in PD from Alzheimer disease (AD).2,4 Notably, cortical Aβ deposition is associated with faster progression to dementia4,5 and shorter survival,2,6 suggesting that Aβ deposition in PD may act synergistically with synucleinopathy. Identifying patients with PD with elevated Aβ may provide a strategy to test therapeutic interventions.
One biomarker for Aβ deposition is the Pittsburgh compound B (PiB) PET radioligand, which binds to fibrillar Aβ.7 Elevated PiB binding occurs in 20%–30% of patients with PD with dementia8–12 and is associated with the APOE ε4 gene, lower CSF Aβ42,10 and cognitive decline.13 Increased PiB binding in the pons and mesencephalon may occur in PD without elevated cortical binding,9 suggesting that PiB binding may differ between PD and AD. However, the typical analysis focuses on cortical PiB binding and would not capture these differences.
The purpose of this study was to compare PiB binding patterns in participants with AD and PD with cognitive impairment using principal component analyses (PCA). PCA identifies independent components that best account for data variance and provides a sensitive method for detecting PiB binding patterns.14 We first established an AD binding pattern, then tested for a unique PD pattern, and finally directly compared PD and AD.
METHODS
Standard protocol approvals, registrations, and patient consents.
The Human Research Protection Office at Washington University in St. Louis, Missouri, approved this study and participants provided written informed consent.
Participants.
Participants with PD with cognitive impairment, defined as Clinical Dementia Rating15 (CDR) ≥0.5 (n = 53), were selected from 70 volunteers with PD (17 were cognitively intact; CDR 0) recruited through the Movement Disorders Center (MDC) at Washington University and the community between January 2006 and December 2011. Age-matched controls (MDC controls; n = 29) were recruited through participants with PD and the community. Participants with PD had a clinical diagnosis of idiopathic PD (n = 43) based on modified United Kingdom Parkinson's Disease Society Brain Bank criteria with clear response to levodopa16 or idiopathic PD with Lewy body dementia17 (DLB; n = 10). Exclusion criteria were 1) head injury with loss of consciousness >5 minutes; 2) neurologic diagnosis other than PD; 3) psychiatric disorders other than depression or anxiety; 4) inability to complete PET and MRI. Everyone consented to brain donation upon death. MDC controls had a normal neurologic examination, CDR = 0, no family history of PD, and a normal PiB scan based upon mean cortical binding potential (MCBP).18 Two controls were excluded due to elevated PiB scans (MCBP ≥ 0.18).18 See table 1 for clinical and demographic information.
Table 1.
Participant demographic and clinical informationa
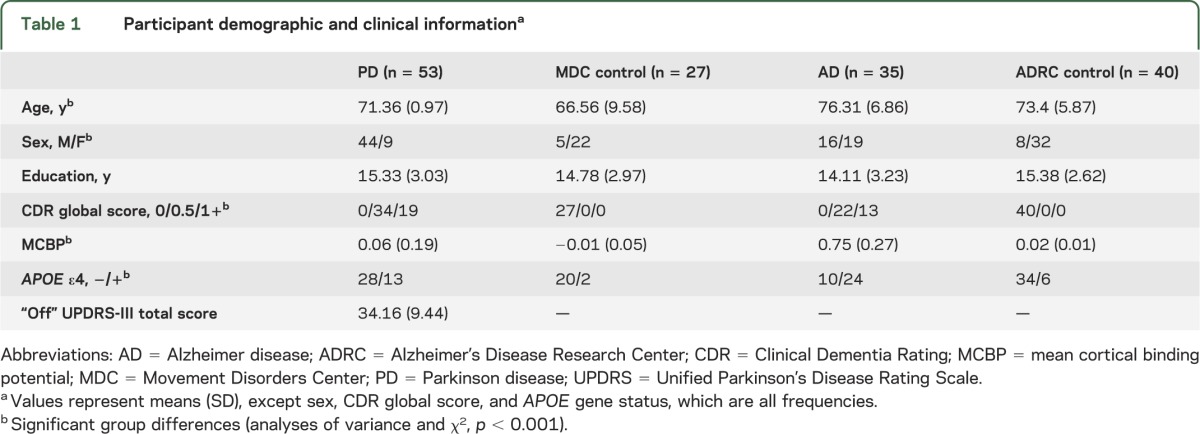
Data for the participants with symptomatic AD (n = 35) and age-matched controls (n = 40) were obtained from the Knight Alzheimer's Disease Research Center (ADRC) at Washington University. Participants with AD had CDR ≥0.5 and elevated PiB binding (MCBP ≥ 0.18); ADRC control participants had CDR = 0 and normal PiB scans (table 1). Any control with a positive PiB (MCBP ≥ 0.18) was excluded to avoid including participants with presymptomatic AD.19,20
PiB PET scans.
PET was done on a Siemens HR or HR+ ECAT PET scanner in 3D mode (CTI, Knoxville, TN). [11C]-PiB was synthesized as described.21 Approximately 12 mCi of radiotracer (range 10.4–14.5; specific activity ≥1,200 Ci/mmol) was injected and a 60-minute, dynamic PET scan was collected in 53 frames (25 5-second frames, 9 20-second frames, 10 60-second frames, 9 5-minute frames). Emission data were corrected for attenuation, scatter, and randoms, with a reconstructed resolution of 6-mm full width half maximum (FWHM). Frame alignment was corrected for head motion and coregistered to each person's T1-weighted magnetization-prepared rapid gradient echo MRI scan (1.5 or 3 T Siemens Trio). Time-activity curves for selected regions of interest were analyzed using Logan graphical analysis, with cerebellar cortex as the reference tissue input function.18,22 Binding potentials (BP) were calculated from the distribution volume ratio (DVR) as BP = DVR − 1. MCBP was computed as the average of BPs from 4 bilateral cortical regions (prefrontal cortex, gyrus rectus, lateral temporal cortex, and precuneus).18
PCA analyses.
We applied PCA to PiB PET scans to differentiate patterns of uptake across groups. PCA is a data reduction method that determines a set of linearly uncorrelated components (images) that account for variability in a dataset. The eigenvalue associated with each component yields the fraction of the variability associated with that component. The components are traditionally ordered by degree of variability explained; the first component accounts for as much of the data variability as possible. The procedure also yields component weights or coefficients (loadings) relating the data for each subject to the principal components (PCs) (see equation 3).
For PCA, composite images were made from the last 30 minutes of the PET scan and then smoothed to 8-mm FWHM. These images were normalized by each participant's respective mean cerebellum value yielding the standard uptake value ratio,14 an approximation of DVR. All negative pixel values were set to zero and a common mask was used that included only those voxels shared by all participants.
PET data were stored in a 2-dimensional matrix, X (M, N), where M is the number of participants and N is the number of pixels (after masking). PCs were computed from the singular-valued decomposition of the matrix after centering the data by subtracting the mean of each image. The singular value decomposition method, denoted in MATLAB by sdv (X, “econ”) (for N > M), decomposes the data into 3 matrices (U, S, V):
where the M × M matrix U is the eigenvector matrix, S is a real M × M diagonal matrix containing the singular values (square root of the eigenvalues), and V is a N × M matrix. The PCs are given by
where each PC (P) is an image. The contribution of the jth PC to the ith participant is given by the ijth element of the U matrix. We refer to these coefficients as component weights.
The PCs associated with any reference dataset can be used to determine whether other groups of participants have similar component weights. The new data are processed to form a new data matrix X′, where each row represents data for a participant. The estimated or predicted component weight (wij) for the ith participant relative to the jth PC of the reference dataset is computed using the PCs and associated eigenvalue of the reference set by
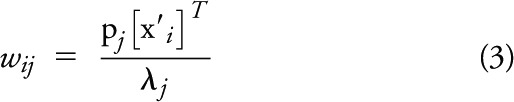 |
where pj is the jth PC, λj is the associated eigenvalue, and x′i is the ith row vector of the data matrix X′. The estimated component weights are computed assuming that PiB uptake conforms to certain patterns (PCs) as determined by the reference PCA.
APOE genotype.
To examine possible differences in PiB binding based on APOE genotype, we obtained APOE genotypes (n = 137).23
Neuropathology.
Seven participants with PD died during this longitudinal study and had neuropathologic assessment as described.2 Lewy body stage was assessed using a PD staging scale24 (range: 0, 1–6) and McKeith et al.17,25 staging. AD pathology was rated using amyloid plaque stage (range: 0, A–C) and neurofibrillary tangle (tauopathy) stage (range: 0, I–VI).3,26
Statistical analyses.
To establish the AD pattern of PiB binding (PCA1), AD and ADRC control participants were randomly split into “training” and “test” sets. PCA was done on the training set and then those components were applied to the test set for validation. AD-defined components then were applied to participants with PD to determine their relative expression of the AD pattern.
To determine a possible PD-specific PiB binding pattern, PCA was used to compare participants with PD and MDC controls (PCA2). To avoid potential bias toward the PD group due to larger sample size, a subset of participants with PD was selected based on age and education to match the smaller sample of MDC controls (table 2).
Table 2.
Group demographic and clinical information by specific PCA analysisa
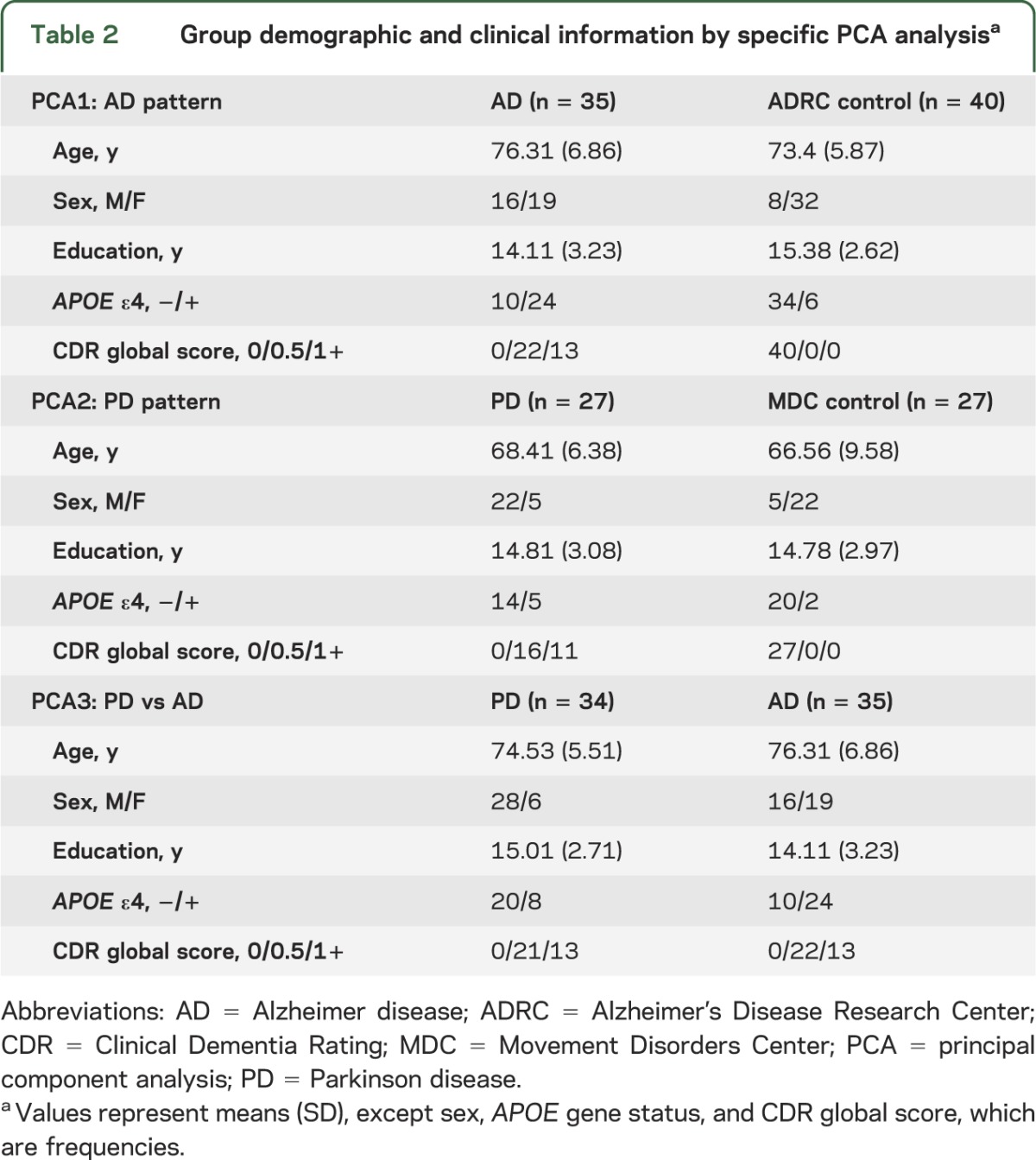
Finally, participants with AD and participants with PD were directly compared with PCA to determine group differences in PiB binding (PCA3). These analyses were conducted on age-, education-, and CDR-matched AD and PD groups (table 2).
Component weights for the PCs that accounted for at least 5% of the variance were compared between groups using t tests or nonparametric Mann-Whitney U and analyses of covariance, including age and sex as covariates. Component weights for the AD-defined components also were compared based on APOE genotype for each group. Data were analyzed with PASW version 18 (IBM, Chicago, IL). All tests were 2-tailed and p < 0.05 was considered statistically significant.
RESULTS
MCBP.
We used the standard approach of MCBP ≥ 0.18 to indicate elevated PiB binding (PiB+).18 An inclusion criterion for controls was normal PiB scans; all participants with AD were PiB+. Only 9 (17%) participants with PD had elevated MCBP, which was lower than the AD group (Mann-Whitney U = 47, z = −3.22, p < 0.001; see figure e-1 on the Neurology® Web site at www.neurology.org for PiB binding images). Although some report greater PiB binding in DLB,8,11 the proportion of PiB+ participants was similar across DLB (20%) and PD (16%) (χ2 = 0.08, df = 1, p = 0.78); therefore, all participants with PD with cognitive impairment were included in the analyses.
PCA1: AD PiB pattern.
We first established the AD pattern with a training set of AD and ADRC control PiB scans and then confirmed this in a test set of AD and ADRC controls. Comparing AD and control training sets, the first 2 components accounted for the majority of the variance (PC1 = 61%; PC2 = 15%; PC3 = 3%; PC4 = 2%; PC5 = 2%; PC6–8 = 1%) and differed between groups (figure 1A and table e-1; PC1: t [27.18] = −8.97, p < 0.001; PC2: t [23.04] = −15.40, p < 0.001). As expected, PC1 and PC2 were driven by PiB binding in cortical regions with PC2 demonstrating greater contribution from prefrontal cortex (PFC) and posterior cingulate cortex (PCC) and much less contribution from thalamus, midbrain, and brainstem (figure 2A).
Figure 1. Distribution of principal component analysis component weights across groups.

Scatterplots represent the component weights of the primary principal components (PC1 and PC2) for (A) establishing and validating the Alzheimer disease (AD) Pittsburgh compound B binding pattern (PCA1), (B) the expression of the AD pattern by participants with Parkinson disease (PD), (C) the expression of the AD pattern by participants with PD with autopsy confirmation, (D) comparison of PD and Movement Disorders Center controls (PCA2), (E) the expression of the PD-defined components by the participants with AD, and (F) the direct comparison of participants with PD and participants with AD (PCA3).
Figure 2. Principal component images.

Images represent the principal components for the (A) comparison of Alzheimer disease (AD) and Alzheimer's Disease Research Center controls (PCA1); (B) comparison of Parkinson disease (PD) and Movement Disorders Center controls (PCA2); (C) comparison of participants with PD and participants with AD (PCA3). The color bar indicates the relative intensities at each voxel for that principal component.
These components then were applied to the test set of AD and control participants. The estimated component weights for the test sets replicated the training set patterns (figure 1A) and separated the groups (PC1: t [19.66] = −8.75, p < 0.001; PC2: t [16.94] = −9.68, p < 0.001). Thus, we combined the component weights of the AD and control training and test sets for comparison with participants with PD.
Comparison with participants with PD.
Next, we examined the relative expression of the AD-defined pattern by participants with PD. The first 2 components were applied to PD and MDC control participants to estimate component weights. Both PC1 and PC2 component weights were greater in AD than PD (table e-1; PC1: F1,87 = 109.66, p < 0.001; PC2: F1,87 = 121.76, p < 0.001) but similar across control groups (table e-1). Comparing PD and ADRC controls revealed a difference on PC2 (F1,92 = 17.61, p < 0.001) but not on PC1 (p = 0.95). The majority of participants with PD had component weights similar to ADRC controls (figure 1B). Those few PD (n = 9) with component weights in the lower range of the AD group had elevated MCBP, consistent with the emphasis on cortical binding expressed by PC2. Comparing participants with PD with elevated MCBP and participants with AD revealed differences on both components (Mann-Whitney; PC1: U = 57, z = −2.92, p = 0.003; PC2: U = 54, z = −3.01, p = 0.003).
APOE genotype.
APOE genotyping was done on 34 participants with AD and 41 participants with PD. A greater proportion of participants with AD (24 of 34; 71.4%) than participants with PD (13 of 41; 31.7%) had at least one APOE ε4 allele (χ2 = 11.24, df = 1, p = 0.001). Gene status did not affect either component for participants with AD (PC1: p = 0.81; PC2: t [32] = −1.78, p = 0.09) or participants with PD (p > 0.12) (figure e-2).
Autopsy confirmation.
All 7 participants with PD who died had dementia and had typical findings of PD with loss of substantia nigra neurons, α-synuclein-immunoreactive Lewy bodies and Lewy neurites, and gliosis in affected areas. Three had predominant cortical synucleinopathy without substantial neocortical β-amyloidosis or tauopathy. The others had cortical synucleinopathy and abnormal neocortical Aβ plaques (Braak amyloid plaque stages B–C) without substantial neocortical tauopathy (Braak neurofibrillary tangle stages ≤4).2 The 4 with abnormal Aβ deposits at autopsy fell within the lower range of PiB PCA distribution of the participants with AD and had elevated MCBP, while the 3 without abnormal Aβ at autopsy had normal MCBP and component weights similar to controls (figure 1C).
PCA2: PD PiB pattern.
To determine a unique PD pattern, PCA was applied to matched groups of PD and MDC controls (table 2). The first 2 components accounted for the majority of the variance (PC1 = 71%; PC2 = 6%; PC3 = 2%; PC4–8 = 1%), but only PC2 differed between PD and MDC controls (table e-1 and figure 1D; PC1: p = 0.89; PC2: t [31.74] = −3.19, p = 0.003). Interestingly, PC1 largely reflects PiB binding in white matter, thalamus, and brainstem, whereas PC2 reflects cortical PiB binding especially in PFC and PCC (figure 2B). However, the difference on PC2 was largely driven by a small number (n = 6) of participants with PD (figure 1D) with elevated MCBP, consistent with the emphasis on cortical binding expressed in PC2. In fact, PC2 did not differ between groups after removing these 6 participants with PD (t [46] = −1.69, p = 0.10).
We then determined the relative expression of these components in AD. Both components differed between the PD and AD groups (figure 1E; PC1: F1,62 = 99.67, p < 0.001; PC2: F1,62 = 52.58, p < 0.001).
PCA3: AD vs PD PiB pattern.
Finally, we directly compared matched groups of participants with AD and participants with PD (table 2) to determine potential differences in PiB binding patterns. The first 2 components accounted for most of the variance (PC1 = 62%; PC2 = 12%; PC3 = 4%; PC4–8 = 1%) and differed between AD and PD (table e-1 and figure 1F; PC1: t [67] = 9.84, p < 0.001; PC2: t [67] = 11.60, p < 0.001), with participants with AD expressing higher component weights. PC1 appears to largely reflect differences in PiB binding between cortex and cerebellum; however, PC2 emphasizes differences in the PFC, PCC, white matter tracts, thalamus, and midbrain (figure 2C). Again, participants with PD with elevated MCBP (n = 9) had component weights in the lower range of participants with AD, but still differed from AD (PC1: Mann-Whitney U = 45, z = −2.97, p = 0.003; PC2: Mann-Whitney U = 40, z = −3.12, p = 0.002).
DISCUSSION
PCA demonstrates a clear difference in PiB binding between participants with PD with cognitive impairment and participants with symptomatic AD. In all 3 PCA analyses, the first 2 components separated participants with AD and participants with PD with PC2 reflecting cortical PiB binding. Specifically, participants with AD had the expected cortical PiB binding pattern reflected by PC2, while the majority of participants with PD exhibited a PiB binding pattern consistent with controls and significantly different from participants with AD. In the present study, 17% of participants with PD had elevated PiB binding, indicated by component weights and MCBP. However, even those participants with PD with elevated MCBP expressed component weights significantly lower than participants with AD, despite matching for cognitive impairment. This indicates that the PiB binding pattern in PD differs from AD. We found no evidence of a unique noncortical pattern of PiB binding in PD. These results further emphasize that cognitive impairment associated with PD can occur independently of Aβ burden and suggest that the role of Aβ in those participants with PD with elevated cortical Aβ may differ from that in AD.
Our data suggest that cortical PiB binding differs between PD and AD, as indicated by the differences in PC2 component weights. Participants with PD consistently had component weights lower than participants with AD (figure 1), even when the components were established by comparison of PD and MDC control groups. This reflects the emphasis of PC2 on cortical binding, which was relatively low in participants with PD. Most importantly, PC1 and PC2 significantly differed between participants with AD and participants with PD with elevated MCBP. These differences were not likely due to the effects of cortical atrophy despite the lack of atrophy correction since the PiB binding pattern, as expressed by the PCs, was not isolated to brain edges. Nevertheless, given the small sample size of PD with elevated cortical PiB binding, it is unclear whether this reflects differences in the amount of PiB binding, the cortical pattern of PiB binding, or both.
These results support previous PiB PET reports of relatively few participants with PD having increased PiB.8–12 Although limited evidence suggests possible increased midbrain and brainstem PiB binding in PD,9 our whole-brain PCA approach did not reveal unique noncortical binding patterns in PD. Overall, the PiB binding pattern in PD was similar to controls and significantly different from AD. Our results highlight that even with a more sensitive, whole-brain PCA approach, the pattern of PiB binding appears to reflect cortical binding.
Recent neuropathologic studies report more frequent Aβ burden in PD.2,5,6,27,28 Although our PCA results agree with our postmortem evaluations (figure 1C), the overall frequency of elevated PiB binding in our PD sample was lower than typically reported in autopsy studies. This difference may be attributed to methodologic differences. PiB binds in vivo to fibrillar Aβ and has less sensitivity to Aβ burden than direct in vitro immunohistochemistry that labels diffuse and fibrillar Aβ. Aβ burden increases with age,29 which may help explain higher rates of Aβ burden at autopsy. Later stages of PD with greater spread of Lewy body pathology may predispose to increased cerebral β-amyloidosis. Also, aggregation of α-synuclein may disrupt protein homeostasis,30 leading to Aβ deposition. Increased aggregation of α-synuclein may reduce clearance of Aβ,31 thus increasing Aβ deposition. The possible interaction between α-synuclein and Aβ warrants further investigation.
Although not implicated as the primary pathology for cognitive impairment associated with PD, Aβ may contribute to shorter survival rates2,32 and more rapid cognitive decline.4,5,13 Identification of patients with PD with elevated Aβ burden is important for evaluation of potential treatment with anti-amyloid therapies should they prove effective in clinical trials for AD.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the Knight Alzheimer's Disease Research Center at Washington University School of Medicine for sharing the AD and ADRC control data and for their time in training and certifying members of the authors’ team in CDR administration.
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADRC
Alzheimer's Disease Research Center
- BP
binding potentials
- CDR
Clinical Dementia Rating
- DLB
Lewy body dementia
- DVR
distribution volume ratio
- FWHM
full width half maximum
- MCBP
mean cortical binding potential
- MDC
Movement Disorders Center
- PC
principal component
- PCA
principal component analysis
- PCC
posterior cingulate cortex
- PD
Parkinson disease
- PFC
prefrontal cortex
- PiB
Pittsburgh compound B
Footnotes
Editorial, page 516
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Study concept and design (M.C.C., J.M., T.O.V., J.S.P.), analysis and interpretation of data (M.C.C., J.M., H.P., J.H., A.G., N.J.C., T.O.V., J.S.P.), drafting/revising the manuscript for content (M.C.C., J.M., A.G., N.J.C., T.O.V., J.S.P.).
STUDY FUNDING
Supported by grants from NINDS (NS075321, NS41509, NS058714, NS48924), NIH NCRR (UL1RR024992), and NIA (P50 AG05681, P01 AG03991); the American Parkinson Disease Association (APDA) Advanced Research Center for Parkinson Disease at Washington University in St. Louis; the Greater St. Louis Chapter of the APDA; and the Barnes Jewish Hospital Foundation (Elliot Stein Family Fund and Parkinson Disease Research Fund). The funders had no role in the design and conduct of the study; collection, management, analysis, or interpretation of the data; or preparation, review, or approval of the manuscript.
DISCLOSURE
M. Campbell, J. Markham, and H. Flores report no disclosures. J. Hartlein receives honoraria from PESI. A. Goate is a consultant for Finnegan, receives royalties from Taconic for a tau mutation patent, received honoraria from Pfizer, Genentech, and Amgen, receives research support from Genentech and Pfizer, and has provided expert testimony for Howrey & Associates, Finnegan HC, and Dickstein Shapiro. N. Cairns, T. Videen, and J. Perlmutter report no disclosures. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Hurtig HI, Trojanowski JQ, Galvin J, et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson's disease. Neurology 2000;54:1916–1921 [DOI] [PubMed] [Google Scholar]
- 2.Kotzbauer PT, Cairns NJ, Campbell MC, et al. Pathologic accumulation of alpha-synuclein and A beta in Parkinson disease patients with dementia. Arch Neurol 2012;69:1326–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braak H, Muller CM, Rub U, et al. Pathology associated with sporadic Parkinson's disease: where does it end? J Neural Transm Suppl 2006;89–97 [DOI] [PubMed] [Google Scholar]
- 4.Ballard C, Ziabreva I, Perry R, et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology 2006;67:1931–1934 [DOI] [PubMed] [Google Scholar]
- 5.Compta Y, Parkkinen L, O'Sullivan SS, et al. Lewy- and Alzheimer-type pathologies in Parkinson's disease dementia: which is more important? Brain 2011;134:1493–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sabbagh MN, Adler CH, Lahti TJ, et al. Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis Assoc Disord 2009;23:295–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain 2008;131:1630–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edison P, Rowe CC, Rinne JO, et al. Amyloid load in Parkinson's disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry 2008;79:1331–1338 [DOI] [PubMed] [Google Scholar]
- 9.Maetzler W, Reimold M, Liepelt I, et al. [11C]PIB binding in Parkinson's disease dementia. Neuroimage 2008;39:1027–1033 [DOI] [PubMed] [Google Scholar]
- 10.Maetzler W, Liepelt I, Reimold M, et al. Cortical PIB binding in Lewy body disease is associated with Alzheimer-like characteristics. Neurobiol Dis 2009;34:107–112 [DOI] [PubMed] [Google Scholar]
- 11.Gomperts SN, Rentz DM, Moran E, et al. Imaging amyloid deposition in Lewy body diseases. Neurology 2008;71:903–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foster ER, Campbell MC, Burack MA, et al. Amyloid imaging of Lewy body-associated disorders. Mov Disord 2010;25:2516–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomperts SN, Locascio JJ, Rentz DM, et al. Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology 2013;80:85–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fripp J, Bourgeat P, Acosta O, et al. Appearance modeling of 11C PiB PET images: characterizing amyloid deposition in Alzheimer's disease, mild cognitive impairment and healthy aging. Neuroimage 2008;43:430–439 [DOI] [PubMed] [Google Scholar]
- 15.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414 [DOI] [PubMed] [Google Scholar]
- 16.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 2005;65:1863–1872 [DOI] [PubMed] [Google Scholar]
- 18.Mintun MA, LaRossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology 2006;67:446–452 [DOI] [PubMed] [Google Scholar]
- 19.Morris JC, Roe CM, Grant EA, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol 2009;66:1469–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Resnick SM, Sojkova J, Zhou Y, et al. Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology 2010;74:807–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathis CA, Wang YM, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of C-11-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem 2003;46:2740–2754 [DOI] [PubMed] [Google Scholar]
- 22.Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab 1996;16:834–840 [DOI] [PubMed] [Google Scholar]
- 23.Morris JC, Roe CM, Xiong C, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 2010;67:122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res 2004;318:121–134 [DOI] [PubMed] [Google Scholar]
- 25.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 1996;47:1113–1124 [DOI] [PubMed] [Google Scholar]
- 26.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259 [DOI] [PubMed] [Google Scholar]
- 27.Lashley T, Holton JL, Gray E, et al. Cortical alpha-synuclein load is associated with amyloid-beta plaque burden in a subset of Parkinson's disease patients. Acta Neuropathol 2008;115:417–425 [DOI] [PubMed] [Google Scholar]
- 28.Irwin DJ, White MT, Toledo JB, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 2012;72:587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price JL, Morris JC. Tangles and plaques in nondemented aging and "preclinical" Alzheimer's disease. Ann Neurol 1999;45:358–368 [DOI] [PubMed] [Google Scholar]
- 30.Kikis EA, Gidalevitz T, Morimoto RI. Protein homeostasis in models of aging and age-related conformational disease. Adv Exp Med Biol 2010;694:138–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deleidi M, Maetzler W. Protein clearance mechanisms of alpha-synuclein and amyloid-beta in Lewy body disorders. Int J Alzheimers Dis 2012;2012:391438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jellinger KA, Wenning GK, Seppi K. Predictors of survival in dementia with Lewy bodies and Parkinson dementia. Neurodegener Dis 2007;4:428–430 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.