

Abstract
Aging markedly affects mitochondrial biogenesis and functions particularly in tissues highly dependent on the organelle’s bioenergetics capability such as the brain’s frontal cortex. Calorie restriction (CR) diet is, so far, the only intervention able to delay or prevent the onset of several age-related alterations in different organisms. We determined the contents of mitochondrial transcription factor A (TFAM), mitochondrial DNA (mtDNA), and the 4.8-kb mtDNA deletion in the frontal cortex from young (6-month-old) and aged (26-month-old), ad libitum-fed (AL) and calorie-restricted (CR), rats. We found a 70 % increase in TFAM amount, a 25 % loss in mtDNA content, and a 35 % increase in the 4.8-kb deletion content in the aged AL animals with respect to the young rats. TFAM-specific binding to six mtDNA regions was analyzed by mtDNA immunoprecipitation and semiquantitative polymerase chain reaction (PCR), showing a marked age-related decrease. Quantitative real-time PCR at two subregions involved in mtDNA replication demonstrated, in aged AL rats, a remarkable decrease (60–70 %) of TFAM-bound mtDNA. The decreased TFAM binding is a novel finding that may explain the mtDNA loss in spite of the compensatory TFAM increased amount. In aged CR rats, TFAM amount increased and mtDNA content decreased with respect to young rats’ values, but the extent of the changes was smaller than in aged AL rats. Attenuation of the age-related effects due to the diet in the CR animals was further evidenced by the unchanged content of the 4.8-kb deletion with respect to that of young animals and by the partial prevention of the age-related decrease in TFAM binding to mtDNA.
Keywords: Aging rat frontal cortex mitochondria, Calorie restriction diet, TFAM amount, mtDNA content, mtDNA 4.8-kb deletion content, mtDNA–TFAM binding
Introduction
Aging is a very complex process that involves the progressive functional decline of tissues, and several theories have been suggested to explain it. The “mitochondrial theory of aging” (Harman 1972; Lee and Wei 2007; Linnane et al. 1989; Miquel and Fleming 1986) implies that the age-dependent dysfunction of the mitochondrial respiratory complexes leads to a reduced amount of synthesized adenosine triphosphate (ATP) and the progressive decline in the bioenergetics capability of the cell. The mitochondrial involvement in aging is tissue-specific (Park and Prolla 2005) and is particularly relevant within tissues such as the brain, heart, and skeletal muscle that have a high dependence on oxidative metabolism (Chabi et al. 2008; Wallace 2005). The other relevant and common feature of aging is an increased presence of reactive oxygen species (ROS), by-products of the mitochondrial respiratory complexes. This results in oxidative damage to mitochondrial DNA (mtDNA), proteins, and lipids and contributes to genome instability and the aging phenotype. Eventually, the increased presence of ROS and the related oxidative damages lead to cell loss through mitochondrial- or death receptor-mediated apoptosis (Circu and Aw 2010; Pollack and Leeuwenburgh 2001). As a consequence of oxidative stress, mtDNA can undergo quantitative and qualitative alterations (bases modifications, abasic sites, single- and double-strand breaks, point mutations, and large-sized deletions) that affect its structure and function. Age-related changes in the mtDNA content have often been found in various mammalian tissues (Barazzoni et al. 2000; Barrientos et al. 1997; Dinardo et al. 2003; Lee et al. 1998; McInerny et al. 2009; Pesce et al. 2001, 2005; Short et al. 2005), as well as in single cells (Blokhin et al. 2008). Large-sized deletions are among the most investigated qualitative alterations of mtDNA. They can remove up to more than one half of the mitochondrial genome and have been causally related to the aging process (Wiesner et al. 2006) and the onset of age-related disorders (Krishnan et al. 2008). These deletions are often delimited by two 12- to 16-nucleotide-long direct repeats (direct repeat 1 and direct repeat 2) and accumulate with aging, although at different levels in postmitotic (e.g., brain and skeletal muscle) (Pesce et al. 2001, 2005; Soong et al. 1992) as well as mitotic tissues (e.g., liver, lung, and kidney) (Gadaleta et al. 1992; Yowe and Ames 1998). Rat mtDNA carries a 4.8-kb-long deletion, delimited by two 16-nucleotide-long DRs, which accumulates in various tissues, including aged brain (Cassano et al. 2004; Kang et al. 1998).
To date, calorie restriction (CR) diet is the only experimental approach that consistently delays or prevents the onset of several age-related alterations in organisms, ranging from yeast to mammals (Civitarese et al. 2007; Guarente 2008; Masoro 2000). Such treatment (Carter et al. 2007; Fontana 2009) induces both changes shared by all tissues as well as marked tissue-specific effects (Anderson and Weindruch 2010; Kim et al. 2008; Opalach et al. 2010; Rangaraju et al. 2009; Seo et al. 2006). The common changes include: (a) coordinated increase in the expression of genes involved in the mitochondrial energy metabolism; (b) improved sensitivity of insulin signaling; (c) increased levels of the circulating adipokine, adiponectin, which counteract some well-recognized features of the aging process. An analysis of various CR possible mechanisms indicates that they are all rooted in the response to the decreased energy availability and lead, through metabolic reprogramming, to an altered metabolic state that facilitates longevity. Such mechanisms include sirtuins activation, increase of PGC-1α expression and activity, activation of the energy-sensitive (AMPK) or nutrient-sensitive (mTOR) regulators (reviewed in Anderson and Weindruch 2010) and might imply very consistently the reduction in the age-related oxidative stress (Barja 2007; Guarente 2008) and its effects (Aspnes et al. 1997; Bua et al. 2004; Cassano et al. 2004, 2006; Kang et al. 1998; Stuart et al. 2004).
A key component involved in mtDNA replication and maintenance is mitochondrial transcription factor A (TFAM) (Kanki et al. 2004). TFAM is a high-mobility group protein that was discovered and characterized initially for its function in mitochondrial transcription (Fisher and Clayton 1988). Further studies demonstrated that TFAM is also implicated in the regulation of mtDNA copy number (Larsson et al. 1998). In fact, in the various tissue-specific TFAM knockout mutant mice, a direct relationship between TFAM amount and mtDNA content was reported (Ekstrand et al. 2004; Silva et al. 2000; Wang et al. 1999). Furthermore, TFAM has been shown to be intricately involved in the constitution of mtDNA nucleoids and the maintenance of the organelle genome (Kaufman et al. 2007). TFAM has also been suggested to be a member of a system responsible for the sensing and repair of oxidative damage to mtDNA (Canugovi et al. 2010; Yoshida et al. 2002). Data about changes of TFAM content in various tissues during aging show a trend toward an increase (Dinardo et al. 2003; Lezza et al. 2001). Pesce et al. (2005) found in the soleus skeletal muscle, a tissue highly dependent on mitochondrial oxidative metabolism, a relevant age-related increase in TFAM, which was, however, associated with a reduction in mtDNA content.
Here, we decided to measure not only TFAM amount and mtDNA content, but also the in vivo binding activity of TFAM in rat frontal cortex, one among the brain areas mostly affected by the age-related mitochondrial dysfunction (Navarro et al. 2008; Navarro and Boveris 2010). In aged AL rats, we found age-related increased amount of TFAM, loss of mtDNA content, increased content of the 4.8-kb deletion, and decreased TFAM binding to mtDNA. CR animals showed an attenuation of the age-related alterations; in particular, they presented a 4.8-kb deletion content similar to that of young animals and a partially preserved TFAM binding to mtDNA. These data support a mechanism in which the lack of correspondence between TFAM and mtDNA contents in aging depends on decreased binding of TFAM to mtDNA with CR able to attenuate the age-related effects.
Materials and methods
Samples
The study was approved by the Institutional Animal Care and Use Committee at the University of Florida. All procedures were performed in accordance with the National Institutes of Health (NIH) guidelines for the care and use of laboratory animals. Frontal cortex samples used in this study were from male Fischer 344 rats obtained from the NIH barrier-raised rodent colony and housed at the Division of Biology of Aging, Department of Aging and Geriatric Research, College of Medicine, University of Florida, Gainesville, FL, USA. The animals consisted of the following groups: 6-month-old ad libitum-fed (YOUNG, n = 6), 26-month-old ad libitum-fed (AL, n = 6), and 26-month-old calorie-restricted (CR, n = 6) rats. Specifically, for the immunoprecipitation experiments, we examined all six animals of each group, but in the process of setting up such technique, we completely finished the samples of two rats from every group. Therefore, the content of TFAM, mtDNA, and the 4.8-kb deletion, as well as the TFAM-bound mtDNA amounts, was determined using the samples from the remaining four rats for each group. CR diet had been initiated at 3.5 months of age (10 % restriction), raised to 25 % restriction at 3.75 months, and kept at 40 % restriction from 4 months until the end of each animal’s life. Such composition of the CR diet, implying decreasing percentages of food intake over time, has been often utilized (Barger et al. 2008) and its rationale (Pugh et al. 1999) mainly lies in the possibility for animals to get gradually acclimated to the final reduction of food in the CR diet. Animals were anesthetized before being sacrificed and samples from the frontal cortex immediately removed, snap-frozen in isopentane cooled by liquid nitrogen, and stored in liquid nitrogen until further use.
Western blotting
Total proteins were extracted from frontal cortex samples obtained from young and aged AL and CR animals. Approximately 100 mg of each frozen sample was ground with a mortar and pestle and suspended in 600 μl of lysis buffer (220 mM mannitol, 70 mM sucrose, 20 mM Tris–HCl pH 7.4, 5 mM MgCl2, 5 mM ethylene glycol tetraacetic acid (EGTA), and 1 mM ethylenediaminetetraacetic acid (EDTA)). Cell lysates were precleared by centrifugation in an Eppendorf microfuge at 12,000 rpm for 10 min and the supernatant fraction containing proteins was recovered. Proteins were quantified with the Bradford method (Bio-Rad Laboratories Inc., Hercules, CA, USA) according to the supplier’s instructions. Total proteins (10 μg) were separated by gel electrophoresis on 4–12 % Bis-Tris Criterion XT precast gels (Bio-Rad Laboratories Inc., Hercules, CA, USA) and electroblotted onto polyvinylidene fluoride membranes (Amersham Pharmacia Biotech Inc., Piscataway, NJ, USA). The membranes were blocked for 1 h in 5 % milk in 1X phosphate-buffered saline (PBS)/Tween 20 (0.15 M NaCl, 0.1 mM KH2PO4, 3 mM Na2HPO4, 0.1 % Tween 20) and probed with TFAM (1:25,000) and β-actin (1:10,000; Sigma-Aldrich Corp., St. Louis, MO, USA). Membranes were then incubated with antirabbit secondary antibody conjugated with horseradish peroxidase at a dilution of 1:10,000 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Membranes were washed in PBS (three times of 15 min each) and proteins were subsequently visualized with an enhanced chemiluminescence kit (ECL-Plus; Amersham Pharmacia Biotech Inc., Piscataway, NJ, USA). Autoradiographs were analyzed by laser densitometry with the Chemi Doc System and Quantity One software (Bio-Rad Laboratories Inc., Hercules, CA, USA). Preliminary titration experiments allowed us to establish the amounts of protein extracts (2.5–25 μg) which gave a signal in the linear range of the relation: densitometric value/blotted proteins. Different exposures of each western blot, in the time range between 1 s and 5 min, were taken to ensure the linearity of the response for all assayed proteins. The densitometric value of the optical density (OD) units of each TFAM band was then related to the OD units number of the respective band of β-actin (in the corresponding lane).
Measurement of mtDNA content and mtDNA 4.8-kb deletion content
mtDNA content was measured using quantitative real-time polymerase chain reaction (qRT-PCR). RT-PCR amplification reactions were performed via SYBR Green chemistry on an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The primers were accurately designed with the Primer Express software (Applied Biosystems, Foster City, CA, USA), respectively, for the rat mitochondrial D-loop region (numbering is according to GenBank™ accession number AY172581) and for the rat nuclear β-actin gene (numbering is according to GenBank™ accession number VO1217.1) and are listed in Table 1. The method has been validated by primer-limiting experiments to determine the proper primer concentrations (200 nM for each primer pair) and by evaluating the equal reaction efficiency of the two amplicons. Amplification specificity was controlled by melting curve analysis and gel electrophoresis. Each sample was analyzed in triplicate in 25 μl of final volume and fluorescence spectra were monitored by the ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The reaction mixture consisted of iTaq SYBR Green Supermix PCR 1X Master Mix (Bio-Rad Laboratories Inc., Hercules, CA, USA), 0.2 μM forward and reverse primers, and DNA template (2.5 μl of diluted 1:10). After 10 min of denaturation at 95 °C, amplification proceeded for 40 cycles, each consisting of denaturation at 95 °C for 15 s, annealing, and extension at 60 °C for 1 min. The quantification of the relative mtDNA content in AL and CR rats, compared to young rats, all normalized to β-actin, was performed according to the Pfaffl mathematical model (Pfaffl 2001).
Table 1.
Oligonucleotide primer sequences
| Primer set | Forward primer | Reverse primer | (nps) | (nps) |
|---|---|---|---|---|
| Primers for PCR of mIP | ||||
| 7.1 For/7.3 Rev | 5′-CACTAATACTAACAACAAAACTAAC-3′ | 5′-GGGATTATGTAGGAGTCAAAGC-3′ | (7,133–7,157) | (7,376–7,355) |
| 8.0 For/8.2 Rev | 5′-CAACCGACTACACTCATTTCAAC-3′ | 5′-CTCATAGGGGGATGGCTATGC-3′ | (8,035–8,057) | (8,246–8,226) |
| 8.8 For/9.1 Rev | 5′-CATTTTATCATTCCAGCCTAGTTC-3′ | 5′-GATGTTTCGAAATACTCTGAGGC-3′ | (8,897–8,920) | (9,152–9,130) |
| 15.0 For/15.2 Rev | 5′-GAGTCGTAGCCCTAATCTTATC-3′ | 5′-GAATGAGGATAATTGAAAAGTAGC-3′ | (15,004–15,025) | (15,228–15,205) |
| 16.0 For/16.2 Rev | 5′-GCTCGAAAGACTATTTTATTCATG-3′ | 5′-GCTAAGATTTAAGTTAAAATTTTGTG-3′ | (16,015–16,038) | (16,226–16,201) |
| 5.0 For/5.3Rev | 5′-GGATTCAAACCTACGAAAATTTAG-3′ | 5′-GTGGTTAGTTGAAAAGAGTCAAC-3′ | (5,092-5,115) | (5,358-5,336) |
| Primers for RT-PCR | ||||
| mtDNA For/mtDNA Rev | 5′-GGTTCTTACTTCAGGGCCATCA-3′ | 5′-TGATTAGACCCGTTACCATCGA-3′ | (15,785–15,806) | (15,868–15,847) |
| Beta-actin For/Beta-actin Rev | 5′-CCCAGCCATGTACGTAGCCA-3′ | 5′-CGTCTCCGGAGTCCATCAC-3′ | (2,181–2,200) | (2,266–2,248) |
| 4.8 Del For/4.8 Del Rev | 5′-AAGGACGAACCTGAGCCCTAATA-3′ | 5′-CGAAGTAGATGATGCGTATACTGTA-3′ | (8,109–8,131) | (13,020–12,996) |
| RT-Dloop For/RT-Dloop Rev | 5′-CACCCCCTACACCTGAAACTT-3′ | 5′-TTTGTGTCGGGAAATTTTACCAAT-3′ | (16,092–16,112) | (16,250–16,227) |
| RT-OriL For/RT-OriL Rev | 5′-CAGCTAAATACCCTACTTACTGG-3′ | 5′-GCCCCCTTTTTACCAAAAAGCC-3′ | (5,120–5,142) | (5,270–5,249) |
The content of the 4.8-kb deletion was measured by qRT-PCR. RT-PCR amplification reactions were performed via SYBR Green chemistry on an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The primers were accurately designed with the Primer Express software (Applied Biosystems, Foster City, CA, USA), respectively, for the rat mtDNA 4.8-kb deleted region and for the rat mtDNA, generally undeleted, D-loop region (numbering is according to GenBank™ accession number AY172581) and are listed in Table 1. The method has been validated by primer-limiting experiments to determine the proper primer concentrations (200 nM for each primer pair) and by evaluating the equal reaction efficiency of the two amplicons. Amplification specificity was controlled by melting curve analysis and gel electrophoresis. Each sample was analyzed in triplicate in 25 μl of final volume and fluorescence spectra were monitored by the ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The reaction mixture consisted of iTaq SYBR Green Supermix PCR 1X Master Mix (Bio-Rad Laboratories Inc., Hercules, CA, USA), 0.2 μM forward and reverse primers, and DNA template (2.5 μl of diluted 1:10). After 10 min of denaturation at 95 °C, amplification proceeded for 40 cycles, each consisting of denaturation at 95 °C for 15 s, annealing, and extension at 60 °C for 1 min. The relative abundance of the 4.8-kb deleted mtDNA in AL and CR animals, compared to young rats, all normalized with respect to total mtDNA was calculated according to the Pfaffl mathematical model (Pfaffl 2001).
Mitochondrial DNA immunoprecipitation
The binding of TFAM to specific regions of mtDNA was analyzed using mitochondrial DNA immunoprecipitation (mIP) following the procedure described by Lu et al. (2007) with some modifications. At the beginning, to 100 mg of frozen tissue, 250 μl of a cold formaldehyde cross-linking solution 1X (1 % formaldehyde, 5 mM HEPES pH 8.0, 10 mM NaCl, 0.1 mM EDTA pH 8.0, 0.05 mM EGTA) were added and incubated for 15 min at room temperature. Cross-linking was terminated by adding 0.125 M glycine and incubating at room temperature for 10 min. Tissues were then briefly homogenized (Heidolph DIAX 900, Heidolph Instruments GmbH & Co. KG, Schwabach, Germany) and centrifuged twice at 3,000 rpm for 5 min. The pellet was washed with 1 ml of PBS (135 mM NaCl, 10 mM Na2HPO4, 2.7 mM KCl, 1.8 mM KH2PO4 pH 7.4), suspended with 1 ml of homogenization buffer (100 mM Tris–HCl pH 8.0, 100 mM NaCl, 30 mM MgCl2, 0.1 % NP-40, 0.1 mM phenylmethylsulfonyl fluoride (PMSF)) and manually homogenized with a pestle. The homogenized sample was centrifuged at 3,000 rpm for 5 min and the pellet was incubated in lysis buffer (0.5 % Triton X-100, 300 mM NaCl, 50 mM Tris–HCl pH 7.4, containing 100 μg/ml leupeptin and 200 μM PMSF) for 10 min at room temperature. Cellular DNA was sheared by sonication using a Branson Sonifier (Hielscher Ultrasonics GmbH, Teltow, Germany) fitted with a microtip (Power 6, duty cycle constant) for 30 s followed by a pause for 30 s and repeated ten times. The size of produced DNA fragments ranged mainly between 500 and 900 bp and was checked by electrophoresis on a 1.2 % agarose gel containing ethidium bromide (0.5 μg/ml) in 1X TAE buffer (40 mM Tris–HCl, 20 mM acetic acid, 1 mM EDTA pH 8.3). Each sample was diluted with three volumes of frozen storage buffer (5 mM EDTA, 20 mM Tris–HCl pH 7.5, 50 mM NaCl). Tissues extracts were precleared with 75 μl of protein A agarose/salmon sperm 50 % DNA (Upstate, distributed by Millipore Corporate Headquarters, Billerica, MA, USA) for 2 ml of sample on a rotator at 4 °C for 30 min. After centrifugation at 1,000 rpm for 1 min, each sample was divided into four aliquots: the first one (100 μl) was not immunoprecipitated and represented the input, at this point stored at −80 °C until the cross-linking reversal; the other three aliquots (100 μl each) were incubated overnight at 4 °C, respectively, with a rabbit anti-TFAM antibody (1:50 dilution), a nonspecific rabbit anti-β-actin antibody (1:100 dilution), and without antibody (−Ab). The following day, 15 μl of protein A agarose were added to each sample for 2 h at 4 °C to isolate protein–DNA complexes. The samples were centrifuged at 1,000 rpm for 1 min and the pellets were washed: three times with 1 ml of radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris–HCl pH 8.0, 1 % Triton X-100, 0.1 % sodium dodecyl sulfate (SDS), 0.1 % deoxycholate, 1 mM EDTA) containing 140 mM NaCl; three times with 1 ml of RIPA buffer containing 500 mM NaCl; three times with 1 ml of LiCl buffer (10 mM Tris–HCl pH 8.0, 0.25 M LiCl, 0.5 % NP-40, 0.5 % deoxycholate, 1 mM EDTA); and twice with 1 ml of TE (10 mM Tris–HCl pH 8.0, 1 mM EDTA). Each wash lasted 5 min with rotation at 4 °C and was followed by centrifugation at 1,000 rpm for 1 min. The last pellets were resuspended with TE containing 0.5 % SDS (200 μl), and together with the input, taken back from the storage at −80 °C, were incubated at 65 °C for 6 h for the thermal reversal of the cross-links. The DNA samples treated with specific antibodies and the −Ab samples were all precipitated by adding 20 μl of 3 M sodium acetate pH 5.2, 1 μl of 20 μg/μl glycogen, and 500 μl of cold absolute ethanol. The input DNAs (100 μl volume) were precipitated by adding 10 μl of 3 M sodium acetate pH 5.2, 1 μl of 20 μg/μl glycogen, and 250 μl of cold absolute ethanol. All samples were kept at −80 °C overnight and the next day centrifuged at 13,000 rpm for 15 min at room temperature. The pellets were resuspended in 300 μl of cold 70 % ethanol, centrifuged at 13,000 rpm for 5 min at room temperature, dried, and resuspended in 200 μl of sterile ultrapure H2O. Ten micrograms of RNase A (50 μg/μl) was added to each sample and incubated for 1 h at 37 °C. Then, 20 μg of proteinase K (100 μg/μl) and SDS (0.25 % final concentration) were added and incubated at 37 °C overnight. The following day, phenol extraction with an equal volume mixture of phenol/chloroform/isoamyl alcohol (25:24:1, respectively) and a second extraction with an equal volume mixture of chloroform/isoamyl alcohol (respectively 24:1) were performed. All DNAs were precipitated overnight at −80 °C by adding 20 μg of glycogen, sodium acetate pH 5.2 (0.3 M final concentration), and two volumes of cold absolute ethanol. The next day, all the samples were centrifuged at 12,000 rpm for 20 min at 4 °C, resuspended in 300 μl of cold 70 % ethanol, centrifuged at 12,000 rpm for 10 min at 4 °C, dried, and resuspended in 60 μl of 10 mM Tris–HCl pH 8.0. Control and mIP mtDNAs were subjected to PCR analysis using six primer pairs. Table 1 shows the primer sequences and the numerical assignment of primer pairs. PCRs were performed with 20 pmol of each primer in a 25-μl mixture (200 μM dNTPs, 1.5 mM MgCl2, 1X Taq Buffer, and 1 U Taq polymerase by Roche, F. Hoffmann-La Roche, Basel, Switzerland) using a Mastercycler PCR (Eppendorf Scientific, Hinz GmbH, Hamburg, Germany). Amplification conditions were as follows: one cycle at 94 °C for 5 min followed by 30 cycles at 94 °C for 1 min, 58 °C for 1 min, 72 °C for 1 min, and then finally one cycle at 72 °C for 5 min. Reactions were analyzed on an ethidium bromide-stained 1.3 % agarose gel in TAE buffer (20 mM Tris acetate, 50 mM EDTA pH 8.3). The intensity of each DNA band visualized on agarose gels was analyzed by densitometry and used for the detection of specific TFAM binding by subtracting the value of the intensity of the aliquot precipitated without primary antibody from that of the TFAM-immunoprecipitated aliquot, both normalized to the value of the respective input aliquot made equal to 1. Such calculated value was compared with the intensity of the nonspecific β-actin immunoprecipitated aliquot that resulted always below 1 % of that with anti-TFAM antibody.
Quantitative RT-PCR of mIP DNA
The measurement of the relative amounts of mtDNA immunoprecipitated by TFAM was carried out by qRT-PCR. In order to analyze some of the mtDNA regions bound by TFAM, we designed two pairs of specific primers for the D-loop and OriL regions. The qRT-PCR amplification reactions were performed via SYBR Green chemistry on an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The primers for each region (Table 1) were accurately designed with the Primer Express software (Applied Biosystems, Foster City, CA, USA). The method has been validated by primer-limiting experiments to determine the proper primer concentrations (200 nM for each primer pair) and by evaluating the equal reaction efficiency of the amplicon in input, immunoprecipitated with anti-TFAM, and without antibodies DNA aliquots. Amplification specificity was controlled by melting curve analysis and gel electrophoresis. Each sample was analyzed in triplicate in 25 μl of final volume and fluorescence spectra were monitored by the ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The reaction mixture consisted of iTaq SYBR Green Supermix PCR 1X Master Mix (Bio-Rad Laboratories Inc., Hercules, CA, USA), 0.2 μM forward and reverse primers, and 2.5 μl of the input or immunoprecipitated with anti-TFAM or without antibodies (diluted 1:10) DNA aliquots. After a 10-min denaturation at 95 °C, samples were amplified for 40 cycles, each consisting of denaturation at 95 °C for 15 s, annealing, and extension at 60 °C for 1 min. The calculation of the amount of TFAM-bound mtDNA was performed according to the formula  , where
, where 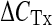 is the difference between the CT values of the input and the immunoprecipitated sample and
is the difference between the CT values of the input and the immunoprecipitated sample and 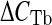 is the CT difference between the CT values of the input and the no-antibody sample (Vercauteren et al. 2006).
is the CT difference between the CT values of the input and the no-antibody sample (Vercauteren et al. 2006).
Statistics
The statistical significance of differences observed between groups of animals was assessed by means of analysis of variance using Tukey’s honestly significant difference post hoc test with the SPSS Base 11.5 software (SPSS Inc., Chicago, IL, USA). Data represented in the box-plot format were subsequently analyzed by Student’s t test. Statistical significance for all tests was set at p < 0.05.
Results
TFAM amount in brain tissue from aged, ad libitum-fed, and CR-treated rats
The experimental system chosen to investigate age-related alterations of mitochondrial biogenesis in rat was the frontal cortex, a tissue that largely depends on mitochondrial energy supply. The amount of TFAM in the frontal cortex of young, aged, AL, and CR animals was measured by western blot experiments. As reported in Fig. 1a, b, we found a statistically significant increase (70 %) in TFAM amount in aged AL animals with respect to young rats. CR rats exhibited a lower increase (42 %) with respect to young animals; this, however, was not statistically different from that of AL rats.
Fig. 1.

Age- and CR-related changes of TFAM amount in the rat frontal cortex. a Representative western blot carried out in the assayed samples. The bands from top to bottom show, respectively, the signals from TFAM and β-actin. b The histogram shows the relative amount of TFAM in AL and CR rats, compared to young rats, all normalized with respect to β-actin. Bars represent the mean (±SD) of the values obtained, respectively, from analysis in triplicate of the protein extract, from each young and aged AL and CR-treated rat. *p < 0.05 versus the value of the young rats (fixed as 1); n number of analyzed animals
mtDNA content and 4.8-kb deletion content in brain tissue from aged, ad libitum-fed, and CR-treated rats
Given the close relationship between TFAM and mtDNA copy number (Larsson et al. 1998), we determined, by RT-PCR, the mtDNA content in the three groups of previously analyzed rats. Figure 2a shows a statistically significant decrease (25 %) in mtDNA content in the aged AL animals compared to the young ones. A slightly lower mtDNA decrease (17 %) was also observed in the CR animals, although the reduction was not statistically different compared to that of the AL rats. This result indicates an age-related effect leading to mtDNA loss. We also measured, by RT-PCR, the content of the 4.8-kb mtDNA deletion in the three groups of animals since it is considered a good marker of age-related mtDNA damage (Kujoth et al. 2007; Pesce et al. 2001, 2005). We found (Fig. 2b) that aged AL rats exhibited, with respect to the young animals, a statistically significant increase (35 %) of the mean deletion content. In contrast, aged CR rats showed a lower and not statistically significant increase. This result indicates the presence of a more severe damage in the AL animals and a preventive effect by CR that is able to maintain a value similar to that of the young animals.
Fig. 2.

Age- and CR-related changes of the relative mtDNA content and of the 4.8-kb deletion content. a The histogram shows the mean value of the ratio mtDNA/nuclear DNA in AL and CR rats, compared to young rats. Bars represent the mean (±SD) obtained, respectively, from analysis in triplicate of total nucleic acids from each young and aged AL and CR-treated rat. *p < 0.05 versus the value of the young rats (fixed as 1); n number of analyzed animals. b The histogram shows the mean value of the ratio deleted mtDNA/total mtDNA in AL and CR rats, compared to young animals. Bars represent the mean (±SD) of the values obtained, respectively, from analysis in triplicate of the total nucleic acids from each young and aged AL and CR-treated rat. *p < 0.05 versus the value of the young rats (fixed as 1); n number of analyzed animals
Binding of TFAM to specific regions of mtDNA in brain from aged, ad libitum-fed, and CR-treated rats
The results about TFAM amount and mtDNA content led us to analyze a possible modulation of TFAM binding to mtDNA with aging, also because recent findings showed that a chemically induced oxidative stress may alter the binding to mtDNA of another mitochondrial protein, namely, the Lon protease (Lu et al. 2007). To determine whether aging and CR may influence the in vivo binding of TFAM to some regions of mtDNA in the frontal cortex, we carried out mIP experiments. With this assay, we analyzed six mtDNA regions that included a part of the D-loop with the OriH origin of replication; the OriL origin together with a portion of the CO I gene; a part of the cytochrome b gene; and three sequences upstream of, downstream of, and containing the direct repeat 1 of the 4.8-kb deletion (Fig. 3a). The D-loop and the OriL-containing regions were selected because of their role in mtDNA replication, involving TFAM binding. The Cyt b gene section represented a control for the binding of TFAM along mtDNA in regions not involved in particular functions. The three regions, respectively, upstream of, including, and downstream of the direct repeat 1, were chosen because of the possible involvement of TFAM in the origin of mtDNA deletions through the most recent proposed models (Fukui and Moraes 2009; Krishnan et al. 2008). All tested sequences were far enough apart from each other (600 bp on average) to consistently rule out the possibility of a false-positive result deriving from the amplification of too close, overlapping sequences physically immunoprecipitated by the same TFAM molecule (Ohgaki et al. 2007). TFAM binding was analyzed by means of a semiquantitative PCR assay in six animals for each group. Figure 3b shows the representative results of a rat, out of every group, whereas Fig. 3c reports a quantitative evaluation of the binding for all tested rats. Young rats showed a similar TFAM-specific binding at all analyzed regions. The only exception was the CO III region where TFAM binding was very low. TFAM binding was altered in the aged AL rats, being reduced at all regions. The age-related decreased binding of TFAM was largely prevented by CR since all tested rats presented a more intense signal for TFAM binding than the AL animals at every region. We believe that the reduced TFAM binding at the CO III region might be due to a poor accessibility of the protein because, although TFAM is known to bind mtDNA in a nonsequence-specific fashion, this does not necessarily imply that all regions show the same binding affinity.
Fig. 3.

Age- and CR-related changes of TFAM binding to rat mtDNA regions by mIP assay. a Rat mtDNA consists of a heavy-strand (outer circle) and a light-strand (inner circle) that code for the 12S and 16S rRNAs and 22 tRNAs genes (single-letter codes indicated under the corresponding tRNA genes) and subunits of the oxidative phosphorylation complexes: NADH dehydrogenase subunits (ND1–6 and ND4L), cytochrome b (Cyt b), cytochrome c oxidase subunits (CO I–III), and ATP synthase subunits (A6, A8). The thick arches extending out of the circles represent the six mIP amplified regions delimited, respectively, by the primer pairs: 16.0 For/16.2 Rev (D-loop), 5.0 For/5.3 Rev (OriL), 7.1 For/7.3 Rev (CO II), 8.0 For/8.2 Rev (DR1), 8.8 For/9.1 Rev (CO III), and 15.0 For/15.2 Rev (Cyt b), listed in Table 1, and corresponding to the indicated nucleotides. Numbering is according to GenBank™ accession number AY172581. The outmost arch represents the 4.8-kb deletion, delimited by direct repeat 1 (DR1) and direct repeat 2 (DR2). b Semiquantitative PCR of mIP assay. Representative results of PCR amplifications of the mIP-derived templates from a young rat, an AL aged rat, and a CR aged rat for the indicated regions. The specific primer pairs and the amplified genetic region are indicated on the left side of the gel. The PCRs contained either input DNA (i) or mIP mtDNA immunoprecipitated with TFAM (T) or mIP mtDNA immunoprecipitated with β-actin (A) or mIP mtDNA immunoprecipitated with no antibody (−Ab). c Quantitative evaluation of TFAM binding results in the three groups of animals after the mIP assay performed at five mtDNA regions (the results for the CO III region were not evaluated due to the very reduced TFAM binding at this region in all groups of animals). The groups of assayed animals included, respectively, six young, six AL, and six CR rats and their values were represented in the box-plot format. The box contains the middle 50 % of the data (with the upper edge and the lower edge representing the 75th and 25th percentiles, respectively); the horizontal line within the box represents the median value. The filled lines indicate the minimum and maximum data values for each rat group, unless data points extend further than 1.5 times the interquartile range, in which case these values are represented as individual points
Quantitative determination of TFAM-bound mtDNA in brain from aged, ad libitum-fed, and CR-treated rats
To deepen the analysis, we determined, by RT-PCR, the relative amount of TFAM-bound mtDNA at two subregions derived from those assayed with the semiquantitative PCR approach. Such regions encompassed: (1) the part of the D-loop including the light strand transcription promoter (LSP) and (2) the tRNA genes stretch enclosing OriL. The analysis was performed in the same rats that had been assayed for the various parameters previously reported in this study. We found (Fig. 4) that the mean amount of TFAM-bound mtDNA in the D-loop region of aged AL rats exhibited a statistically significant decrease (73 %) with respect to that in the young animals. This aging-related effect was partially prevented by CR, as in these animals, TFAM-bound mtDNA decreased by only 48 % with respect to the amount in the young animals. This value was significantly different also from the AL rats counterpart. Similar results were obtained for the OriL region, where a 61 % reduction in the mean amount of TFAM-bound mtDNA was found in the aged AL animals with respect to the value in the young rats. Also, in this case, CR rats exhibited a lower decrease in TFAM-bound mtDNA (48 %), although it was not statistically different from the value of the AL animals.
Fig. 4.

Age- and CR-related changes of the relative mtDNA amount bound by TFAM in two mtDNA regions determined by RT-PCR. The amplified products enclose sections, respectively, of the D-loop region and of the OriL region delimited by the primer pairs listed in the bottom part of Table 1. The calculation of the amount of TFAM-bound mtDNA was performed according to the formula  , where
, where 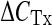 is the difference between the CT values of the input and of the immunoprecipitated sample and
is the difference between the CT values of the input and of the immunoprecipitated sample and 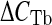 is the CT difference between the CT values of the input and of the no-antibody sample. The obtained results were then normalized with respect to the mean value of TFAM-bound mtDNA in the D-loop region from young animals. Bars represent the mean (±SD) of the relative amounts of TFAM-bound mtDNA derived from each of the assayed animals in the specific group. *p < 0.05 versus the value of the young rats (fixed as 1); #p < 0.05 versus the amount from the same region of aged AL rats; n number of analyzed animals
is the CT difference between the CT values of the input and of the no-antibody sample. The obtained results were then normalized with respect to the mean value of TFAM-bound mtDNA in the D-loop region from young animals. Bars represent the mean (±SD) of the relative amounts of TFAM-bound mtDNA derived from each of the assayed animals in the specific group. *p < 0.05 versus the value of the young rats (fixed as 1); #p < 0.05 versus the amount from the same region of aged AL rats; n number of analyzed animals
Discussion
The present study reports the effects of aging and CR on the content of TFAM, mtDNA, and the 4.8-kb deletion, as well as on TFAM binding to specific regions of mtDNA in the rat frontal cortex. This brain area depends considerably on mitochondrial functionality as documented by the large number of papers describing age-related mitochondrial dysfunctions such as decreased rate of electron transfer in complex I and complex IV, decreased membrane potential, and increased content of macromolecular oxidized products (Bagh et al. 2011; Frenzel et al. 2010; Gilmer et al. 2010; Guevara et al. 2009, 2011; Navarro and Boveris 2010; Navarro et al. 2008). In this brain area, we found that AL rats presented an age-related increase in TFAM amount, which was not associated with an increase in mtDNA content, but with a mtDNA decrease. Moreover, the in vivo binding capacity of TFAM to mtDNA was markedly reduced in AL animals. AL rats also showed an increase in the 4.8-kb deletion content with respect to the young rats’ value. These data were obtained in a limited number of animals (four to six for each group); however, the determined results were homogenous and produced clear, statistically significant data.
The increase in TFAM with aging can be due either to an enhancement of its expression or to a slower degradation. The age-related increased expression and binding activity of NRF-1, a positive regulator of TFAM expression, was previously described in human skeletal muscle (Lezza et al. 2001). On the other hand, reduced TFAM degradation can also occur and it may be linked to the decreased expression and activity with aging of the Lon mitochondrial protease (Bakala et al. 2003; Bota et al. 2002). Support for this possibility comes from recent findings in Drosophila Schneider cells where Lon protease appears to control mtDNA copy number through selective degradation of TFAM (Matsushima et al. 2010).
Regarding the mtDNA decrease with aging, previous findings from some of us did not show any age-related change in the whole-brain mtDNA content (Cassano et al. 2006); however, they are not comparable to the present results for two main reasons. Firstly, those data were obtained in whole cortex samples and not in the frontal cortex as in this paper. It is now well-established that determinations performed in the whole brain tissue, rather than in specific areas, lead to mean tissue values that may overlook specific brain area differences (see Bender et al. 2008; McInerny et al. 2009; Meissner et al. 2008). Secondly, Cassano et al. (2006) used, as counterpart in the comparison with the mtDNA content of old animals, 12-month-old rats; this age, according to recent reports (Kadish et al. 2009), can be placed in the midlife age range. On the contrary, rats here used as the AL counterpart are 6 months of age and can be truly considered as young animals.
In addition, our data reporting mtDNA loss with aging in the rat frontal cortex are in agreement with previous reports, such as that by McInerny et al. (2009), who described mtDNA decrease in rat cerebral cortex, and with several other reports in various rat tissues such as skeletal muscle and liver (Barazzoni et al. 2000) as well as in humans (Short et al. 2005).
It may be argued that, here, the reported changes in TFAM and mtDNA could also depend on age-related alterations in the cell composition of the frontal cortex such as an increase in astrocytes density and/or a neuronal loss. This possibility seems unlikely for several reasons. Reports on variations of astrocytes density during aging are conflicting, with papers showing either increase, decrease, or no change (see Mansour et al. 2008). At most, Cotrina and Nedergaard (2002) reported a 20 % increased density with aging. Considering that astrocytes contain a threefold to fourfold lower amount of mitochondria than neurons (Alano et al. 2007), their eventual slight increase with aging should not particularly affect the content of mtDNA and of mitochondrial proteins such as TFAM. For what concerns neurons, it is now widely accepted that brain aging does not imply a loss of neurons, especially in the rat frontal cortex, but rather structural changes of neurons that alter their function (Grill and Riddle 2002; Peinado et al. 1993; Turlejski and Djavadian 2002). Therefore, we can conclude that the changes in TFAM and mtDNA content are not the result of a variation in cellular population during aging, but are truly due to the effect of age. A novel finding that comes out from this paper concerns the age-related decrease in TFAM binding to mtDNA; this can provide an explanation for the discrepancy between increased TFAM and mtDNA loss, a result that was also previously reported by Pesce et al. (2005) in rat soleus muscle. The lower binding to the D-loop region, where the two HSP and LSP promoters are localized, could determine a reduced rate of mitochondrial transcription. Interestingly, such an event was described in the cerebral hemispheres of aged rats (Gadaleta et al. 1990). Decreased mitochondrial transcription can severely affect the production of the primer for H-strand synthesis and then mtDNA content; this is consistent with the 25 % mtDNA loss that we observed in the aged AL animals. The reduced TFAM binding to mtDNA may also alter its histone-like protective function, leading to increased oxidative damage to mtDNA, indicated by the raised content of the 4.8-kb deletion. The decreased binding might also affect TFAM repair activity, as the protein has been recently identified as a member of the mitochondrial BER system and the regulation of its binding to mtDNA has been suggested to be involved in the modulation of the repair activity and in the maintenance of mtDNA integrity (Canugovi et al. 2010). The mechanism responsible for TFAM reduced binding is not yet known and needs further investigations. In principle, it might be due to the oxidative stress that can induce either mtDNA damage (reviewed in Druzhyna et al. 2008) and/or age-dependent protein modifications (Dinardo et al. 2003; Musicco et al. 2009; Ugarte et al. 2010).
Regarding the effect of CR, we found that it did not significantly affect mtDNA content and TFAM amount compared to AL feeding, although CR rats exhibited a trend in approaching the values of the young. However, the attenuation by CR was significant looking both at the content of the 4.8-kb deletion, which was similar to that of young rats, and at the binding activity of TFAM, which clearly showed an improvement in CR animals with respect to AL ones. Altogether, these results, even interpreted in a cautious manner, underlie a cortex-specific, mild protective effect by CR in old animals. Support for the protective action by CR on mitochondrial functions comes from the data by Sanz et al. (2005), showing that CR in brain mitochondria reduces the age-related increased production of ROS at complex I (20 % decrease) by improving the efficiency of mitochondria in avoiding electron leaks at that complex as well as from other studies (Aspnes et al. 1997; Barja 2007; Bua et al. 2004; Cassano et al. 2004, 2006; Kang et al. 1998; Quintas et al. 2012; Stuart et al. 2004). Additional studies, performed in different areas of the rat brain, including some cortical regions, support the hypothesis about the attenuation of some age-induced alterations by CR (Kaur et al. 2008; Shi et al. 2007). In particular, Shi et al. (2007) identified a brain-specific possible mechanism of CR action through the induction of stability or homeostasis of some crucial neural parameters, usually declining with aging. Such CR-induced homeostasis should involve an enhanced defense also against external insults as the age-related, accumulating oxidative stress. The enhanced neuroprotection might be obtained by CR also through the upregulation of neuronal survival factors and neurotrophic factors, active in modulating the expression of apoptotic regulatory proteins (Hiona and Leeuwenburgh 2004).
In conclusion, this work shows that mtDNA content might be regulated not only by the amount of the factors involved in mtDNA replication and maintenance, such as TFAM, but also by means of their activity. Further work will be needed to clarify the mechanisms responsible for the reduced binding of the protein as well as whether other proteins, such as those involved in mtDNA replication and in mitochondrial dynamics (Seo et al. 2010), change their activity during aging and in response to CR.
Acknowledgments
This research was supported by grants to VP (Contributo d’Ateneo Bari 2009), MNG (Contributo d’Ateneo Bari 2008, fiRSt s.r.l. 2009 and FIRB-MERIT 2008 No. RBNE08HWLZ_012), AMSL (Contributo d’Ateneo Bari 2007), and CL (NIA R01 AG17994), the University of Florida Institute on Aging, and the Claude D. Pepper Older Americans Independence Center (1 P30 AG028740). We thank Dr. C.F. Minervini for the helpful discussions, Dr. F.P. Fallacara for the assistance with the experiments, and Ms. R. Longo for the expert secretarial assistance.
References
- Alano CC, Tran A, Tao R, Ying W, Karliner JS, Swanson RA. Differences among cell types in NAD(+) compartmentalization: a comparison of neurons, astrocytes, and cardiac myocytes. J Neurosci Res. 2007;85:3378–3385. doi: 10.1002/jnr.21479. [DOI] [PubMed] [Google Scholar]
- Anderson RM, Weindruch R. Metabolic reprogramming, caloric restriction and aging. Trends Endocrinol Metab. 2010;21:134–141. doi: 10.1016/j.tem.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspnes LE, Lee CM, Weindruch R, Chung SS, Roecker EB, Aiken JM. Caloric restriction reduces fiber loss and mitochondrial abnormalities in aged rat muscle. FASEB J. 1997;11:573–581. doi: 10.1096/fasebj.11.7.9212081. [DOI] [PubMed] [Google Scholar]
- Bagh MB, Thakurta IG, Biswas M, Behera P, Chakrabarti S. Age-related oxidative decline of mitochondrial functions in rat brain is prevented by long term oral antioxidant supplementation. Biogerontology. 2011;12:119–131. doi: 10.1007/s10522-010-9301-8. [DOI] [PubMed] [Google Scholar]
- Bakala H, Delaval E, Hamelin M, Bismuth J, Borot-Laloi C, Corman B, Friguet B. Changes in rat liver mitochondria with aging. Lon protease-like reactivity and N(epsilon)-carboxymethyllysine accumulation in the matrix. Eur J Biochem. 2003;270:2295–2302. doi: 10.1046/j.1432-1033.2003.03598.x. [DOI] [PubMed] [Google Scholar]
- Barazzoni R, Short KR, Nair KS. Effects of aging on mitochondrial DNA copy number and cytochrome c oxidase gene expression in rat skeletal muscle, liver, and heart. J Biol Chem. 2000;275:3343–3347. doi: 10.1074/jbc.275.5.3343. [DOI] [PubMed] [Google Scholar]
- Barger JL, Kayo T, Vann JM, Arias EB, Wang J, Hacker TA, Wang Y, Raederstorff D, Morrow JD, Leeuwenburgh C, Allison DB, Saupe KW, Cartee GD, Weindruch R, Prolla TA. A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS One. 2008;3(6):e2264. doi: 10.1371/journal.pone.0002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barja G. Mitochondrial oxygen consumption and reactive oxygen species production are independently modulated: implications for aging studies. Rejuvenation Res. 2007;10:215–224. doi: 10.1089/rej.2006.0516. [DOI] [PubMed] [Google Scholar]
- Barrientos A, Casademont J, Cardellach F, Estivill X, Urbano-Marquez A, Nunes V. Reduced steady-state levels of mitochondrial RNA and increased mitochondrial DNA amount in human brain with aging. Brain Res Mol Brain Res. 1997;52:284–289. doi: 10.1016/S0169-328X(97)00278-7. [DOI] [PubMed] [Google Scholar]
- Bender A, Schwarzkopf RM, McMillan A, Krishnan KJ, Rieder G, Neumann M, Elstner M, Turnbull DM, Klopstock T. Dopaminergic midbrain neurons are the prime target for mitochondrial DNA deletions. J Neurol. 2008;255:1231–1235. doi: 10.1007/s00415-008-0892-9. [DOI] [PubMed] [Google Scholar]
- Blokhin A, Vyshkina T, Komoly S, Kalman B. Variations in mitochondrial DNA copy numbers in MS brains. J Mol Neurosci. 2008;35:283–287. doi: 10.1007/s12031-008-9115-1. [DOI] [PubMed] [Google Scholar]
- Bota DA, Van Remmen H, Davies KJA. Modulation of Lon protease activity and aconitase turnover during aging and oxidative stress. FEBS Lett. 2002;532:103–106. doi: 10.1016/S0014-5793(02)03638-4. [DOI] [PubMed] [Google Scholar]
- Bua E, McKiernan SH, Aiken JM. Calorie restriction limits the generation but not the progression of mitochondrial abnormalities in aging skeletal muscle. FASEB J. 2004;18:582–584. doi: 10.1096/fj.03-0668fje. [DOI] [PubMed] [Google Scholar]
- Canugovi C, Maynard S, Bayne AC, Sykora P, Tian J, de Souza-Pinto NC, Croteau DL, Bohr V. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair. 2010;9:1080–1089. doi: 10.1016/j.dnarep.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CS, Hofer T, Seo AY, Leeuwenburgh C. Molecular mechanisms of life- and health-span extension: calorie restriction and exercise intervention. Appl Physiol Nutr Metab. 2007;32:954–966. doi: 10.1139/H07-085. [DOI] [PubMed] [Google Scholar]
- Cassano P, Lezza AM, Leeuwenburgh C, Cantatore P, Gadaleta MN. Measurement of the 4,834-bp mitochondrial DNA deletion level in aging rat liver and brain subjected or not to caloric restriction diet. Ann N Y Acad Sci. 2004;1019:269–273. doi: 10.1196/annals.1297.045. [DOI] [PubMed] [Google Scholar]
- Cassano P, Sciancalepore AG, Lezza AM, Leeuwenburgh C, Cantatore P, Gadaleta MN. Tissue-specific effect of age and caloric restriction diet on mitochondrial DNA content. Rejuvenation Res. 2006;9:211–214. doi: 10.1089/rej.2006.9.211. [DOI] [PubMed] [Google Scholar]
- Chabi B, Ljubicic V, Menzies KJ, Huang JH, Saleem A, Hood DA. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell. 2008;7:2–12. doi: 10.1111/j.1474-9726.2007.00347.x. [DOI] [PubMed] [Google Scholar]
- Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civitarese AE, Carling S, Heilbronn LK, Hulver MH, Ukropcova B, Deutsch A, Smith SR, Ravussin E. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4:e76. doi: 10.1371/journal.pmed.0040076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Nedergaard M. Astrocytes in the aging brain. J Neurosci Res. 2002;67:1–10. doi: 10.1002/jnr.10121. [DOI] [PubMed] [Google Scholar]
- Dinardo MM, Musicco C, Fracasso F, Milella F, Gadaleta MN, Gadaleta G, Cantatore P. Acetylation and level of mitochondrial transcription factor A in several organs of young and old rats. Biochem Biophys Res Commun. 2003;301:187–191. doi: 10.1016/S0006-291X(02)03008-5. [DOI] [PubMed] [Google Scholar]
- Druzhyna NM, Wilson GL, LeDoux SP. Mitochondrial DNA repair in aging and disease. Mech Ageing Dev. 2008;129:383–390. doi: 10.1016/j.mad.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13:935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- Fisher RP, Clayton DA. Purification and characterization of human mitochondrial transcription factor 1. Mol Cell Biol. 1988;8:3496–3509. doi: 10.1128/mcb.8.8.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L. The scientific basis of caloric restriction leading to longer life. Curr Opin Gastroenterol. 2009;25:144–150. doi: 10.1097/MOG.0b013e32831ef1ba. [DOI] [PubMed] [Google Scholar]
- Frenzel M, Rommelspacher H, Sugawa MD, Dencher NA. Ageing alters the supramolecular architecture of OxPhos complexes in rat brain cortex. Exp Gerontol. 2010;45:563–572. doi: 10.1016/j.exger.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Fukui H, Moraes CT. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum Mol Genet. 2009;18:1028–1036. doi: 10.1093/hmg/ddn437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadaleta MN, Petruzzella V, Renis M, Fracasso F, Cantatore P. Reduced transcription of mitochondrial DNA in the senescent rat. Tissue dependence and effect of l-carnitine. Eur J Biochem. 1990;187:501–506. doi: 10.1111/j.1432-1033.1990.tb15331.x. [DOI] [PubMed] [Google Scholar]
- Gadaleta MN, Rainaldi G, Lezza AM, Milella F, Fracasso F, Cantatore P. Mitochondrial DNA copy number and mitochondrial DNA deletion in adult and senescent rats. Mutat Res. 1992;275:181–193. doi: 10.1016/0921-8734(92)90022-H. [DOI] [PubMed] [Google Scholar]
- Gilmer LK, Ansari MA, Roberts KN, Scheff SW. Age-related changes in mitochondrial respiration and oxidative damage in the cerebral cortex of the Fisher 344 rat. Mech Ageing Dev. 2010;131:133–143. doi: 10.1016/j.mad.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill JD, Riddle DR. Age-related and laminar-specific dendritic changes in the medial frontal cortex of the rat. Brain Res. 2002;937:8–21. doi: 10.1016/S0006-8993(02)02457-5. [DOI] [PubMed] [Google Scholar]
- Guarente L. Mitochondria—a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132:171–176. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guevara R, Santandreu FM, Valle A, Gianotti M, Oliver J, Roca P. Sex-dependent differences in aged rat brain mitochondrial function. Free Radic Biol Med. 2009;46:169–175. doi: 10.1016/j.freeradbiomed.2008.09.035. [DOI] [PubMed] [Google Scholar]
- Guevara R, Gianotti M, Roca P, Oliver J. Age and sex-related changes in rat brain mitochondrial function. Cell Physiol Biochem. 2011;27:201–206. doi: 10.1159/000327945. [DOI] [PubMed] [Google Scholar]
- Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- Hiona A, Leeuwenburgh C. Effects of age and caloric restriction on brain neuronal cell death/survival. Ann NY Acad Sci. 2004;1019:96–105. doi: 10.1196/annals.1297.018. [DOI] [PubMed] [Google Scholar]
- Kadish I, Thibault O, Blalock EM, Chen K-C, Gant JC, Porter NM, Landfield PW. Hippocampal and cognitive aging across the lifespan: a bioenergetics shift precedes and increased cholesterol trafficking parallels memory impairment. J Neurosci. 2009;29:1805–1816. doi: 10.1523/JNEUROSCI.4599-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang CM, Kristal BS, Yu BP. Age-related mitochondrial DNA deletions: effect of dietary restriction. Free Radic Biol Med. 1998;24:148–154. doi: 10.1016/S0891-5849(97)00204-9. [DOI] [PubMed] [Google Scholar]
- Kanki T, Ohgaki K, Gaspari M, Gustafsson CM, Fukuoh A, Sasaki N, Hamasaki N, Kang D. Architectural role of mitochondrial transcription factor A in maintenance of human mitochondrial DNA. Mol Cell Biol. 2004;24:9823–9834. doi: 10.1128/MCB.24.22.9823-9834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, Grutter P, Shoubridge EA. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol Biol Cell. 2007;18:3225–3236. doi: 10.1091/mbc.E07-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur M, Sharma S, Kaur G. Age-related impairments in neuronal plasticity markers and astrocytic GFAP and their reversal by late-onset short term dietary restriction. Biogerontology. 2008;9:441–454. doi: 10.1007/s10522-008-9168-0. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kwak HB, Leeuwenburgh C, Lawler JM. Lifelong exercise and mild (8 %) caloric restriction attenuate age-induced alterations in plantaris muscle morphology, oxidative stress and IGF-1 in the Fischer-344 rat. Exp Gerontol. 2008;43:317–329. doi: 10.1016/j.exger.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, Wanrooij S, Spelbrink JN, Lightowlers RN, Turnbull DM. What causes mitochondrial DNA deletions in human cells? Nat Genet. 2008;40:275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Bradshaw PC, Haroon S, Prolla TA. The role of mitochondrial DNA mutations in mammalian aging. PLoS Genet. 2007;3(2):e24. doi: 10.1371/journal.pgen.0030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- Lee HC, Wei YH. Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med. 2007;232:592–606. [PubMed] [Google Scholar]
- Lee HC, Lu CY, Fahn HJ, Wei YH. Aging- and smoking-associated alteration in the relative content of mitochondrial DNA in human lung. FEBS Lett. 1998;441:292–296. doi: 10.1016/S0014-5793(98)01564-6. [DOI] [PubMed] [Google Scholar]
- Lezza AMS, Pesce V, Cormio A, Fracasso F, Vecchiet J, Felzani G, Cantatore P, Gadaleta MN. Increased expression of mitochondrial transcription factor A and nuclear respiratory factor-1 in skeletal muscle from aged human subjects. FEBS Lett. 2001;501:74–78. doi: 10.1016/S0014-5793(01)02628-X. [DOI] [PubMed] [Google Scholar]
- Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1:642–645. doi: 10.1016/S0140-6736(89)92145-4. [DOI] [PubMed] [Google Scholar]
- Lu B, Yadav S, Shah PG, Liu T, Tian B, Pukszta S, Villaluna N, Kutejová E, Newlon CS, Santos JH, Suzuki CK. Roles for the human ATP-dependent Lon protease in mitochondrial DNA maintenance. J Biol Chem. 2007;282:17363–17374. doi: 10.1074/jbc.M611540200. [DOI] [PubMed] [Google Scholar]
- Mansour H, Chamberlain CG, Weible MW, II, Hughes S, Chu Y, Chan-Ling T. Aging-related changes in astrocytes in the rat retina: imbalance between cell proliferation and cell death reduces astrocyte availability. Aging Cell. 2008;7:526–540. doi: 10.1111/j.1474-9726.2008.00402.x. [DOI] [PubMed] [Google Scholar]
- Masoro EJ. Caloric restriction and aging: an update. Exp Gerontol. 2000;35:299–305. doi: 10.1016/S0531-5565(00)00084-X. [DOI] [PubMed] [Google Scholar]
- Matsushima Y, Goto Y, Kaguni LS. Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM) Proc Natl Acad Sci USA. 2010;107:18410–18415. doi: 10.1073/pnas.1008924107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerny SC, Brown AL, Smith DW. Region-specific changes in mitochondrial D-loop in aged rat CNS. Mech Ageing Dev. 2009;130:343–349. doi: 10.1016/j.mad.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Meissner C, Bruse P, Mohamed SA, Schulz A, Warnk H, Storm T, Oehmichen M. The 4977 bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: a useful biomarker or more? Exp Gerontol. 2008;43:645–652. doi: 10.1016/j.exger.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Miquel J, Fleming JE (1986) Theoretical end experimental support for an “oxygen radical mitochondrial injury” hypothesis of cell aging. In: Johnson J, Walford R, Harman D, Miquel J (eds) Free radicals, aging, and degenerative diseases. Alan R. Liss, New York, pp 51–74
- Musicco C, Capelli C, Pesce V, Timperio AM, Calvani M, Mosconi L, Zolla L, Cantatore P, Gadaleta MN. Accumulation of overoxidized Peroxiredoxin III in aged rat liver mitochondria. Biochim Biophys Acta. 2009;1787:890–896. doi: 10.1016/j.bbabio.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Navarro A, Boveris A. Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front Aging Neurosci. 2010;2:1–11. doi: 10.3389/fnagi.2010.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Lopez-Cepero JM, Bandez MJ, Sanchez-Pino MJ, Gomez C, Cadenas C, Boveris A. Hippocampal mitochondrial dysfunction in rat aging. Am J Physiol Regul Integr Comp Physiol. 2008;294:R501–R509. doi: 10.1152/ajpregu.00492.2007. [DOI] [PubMed] [Google Scholar]
- Ohgaki K, Kanki T, Fukuoh A, Kurisaki H, Aoki Y, Ikeuchi M, Kim SH, Hamasaki N, Kang D. The C-terminal tail of mitochondrial transcription factor A markedly strengthens its general binding to DNA. J Biochem. 2007;141:201–211. doi: 10.1093/jb/mvm020. [DOI] [PubMed] [Google Scholar]
- Opalach K, Rangaraju S, Madorsky I, Leeuwenburgh C, Notterpek L. Lifelong calorie restriction alleviates age-related oxidative damage in peripheral nerves. Rejuvenation Res. 2010;13:65–74. doi: 10.1089/rej.2009.0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Prolla TA. Lessons learned from gene expression profile studies of aging and caloric restriction. Ageing Res Rev. 2005;4:55–65. doi: 10.1016/j.arr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Peinado MA, Martinez M, Pedrosa JA, Quesada A, Peinado JM. Quantitative morphological changes in neurons and glia in the frontal lobe of the aging rat. Anat Rec. 1993;237:104–108. doi: 10.1002/ar.1092370110. [DOI] [PubMed] [Google Scholar]
- Pesce V, Cormio A, Fracasso F, Vecchiet J, Felzani G, Lezza AM, Cantatore P, Gadaleta MN. Age-related mitochondrial genotypic and phenotypic alterations in human skeletal muscle. Free Radic Biol Med. 2001;30:1223–1233. doi: 10.1016/S0891-5849(01)00517-2. [DOI] [PubMed] [Google Scholar]
- Pesce V, Cormio A, Fracasso F, Lezza AM, Cantatore P, Gadaleta MN. Age-related changes of mitochondrial DNA content and mitochondrial genotypic and phenotypic alterations in rat hind-limb skeletal muscles. J Gerontol A Biol Sci Med Sci. 2005;60:715–723. doi: 10.1093/gerona/60.6.715. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack M, Leeuwenburgh C. Apoptosis and aging: role of the mitochondria. J Gerontol A Biol Sci Med Sci. 2001;56:B475–B482. doi: 10.1093/gerona/56.11.B475. [DOI] [PubMed] [Google Scholar]
- Pugh TD, Klopp RG, Weindruch R. Controlling caloric consumption: protocols for rodents and rhesus monkeys. Neurobiol Aging. 1999;20:157–165. doi: 10.1016/S0197-4580(99)00043-3. [DOI] [PubMed] [Google Scholar]
- Quintas A, de Solìs AJ, Dìez-Guerra FJ, Carrascosa JM, Begonez E. Age-associated decrease of SIRT1 expression in rat hippocampus. Prevention by late onset caloric restriction. Exp Gerontol. 2012;47:198–201. doi: 10.1016/j.exger.2011.11.010. [DOI] [PubMed] [Google Scholar]
- Rangaraju S, Hankins D, Madorsky I, Madorsky E, Lee WH, Carter CS, Leeuwenburgh C, Notterpek L. Molecular architecture of myelinated peripheral nerves is supported by calorie restriction with aging. Aging Cell. 2009;8:178–191. doi: 10.1111/j.1474-9726.2009.00460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz A, Caro P, Ibanez J, Gomez J, Gredilla R, Barja G. Dietary restriction at old age lowers mitochondrial oxygen radical production and leak at complex I and oxidative DNA damage in rat brain. J Bioenerg Biomembr. 2005;37:83–90. doi: 10.1007/s10863-005-4131-0. [DOI] [PubMed] [Google Scholar]
- Seo AY, Hofer T, Sung B, Judge S, Chung HY, Leeuwenburgh C. Hepatic oxidative stress during aging: effects of 8 % long-term calorie restriction and lifelong exercise. Antioxid Redox Signal. 2006;8:529–538. doi: 10.1089/ars.2006.8.529. [DOI] [PubMed] [Google Scholar]
- Seo AY, Joseph A-M, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci. 2010;123:2533–2542. doi: 10.1242/jcs.070490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Adams MM, Linville MC, Newton IG, Forbes ME, Long AB, Riddle DR, Brunso-Bechtold JK. Caloric restriction eliminates the aging-related decline in NMDA and AMPA receptor subunits in the rat hippocampus and induces homeostasis. Exp Neurol. 2007;206:70–79. doi: 10.1016/j.expneurol.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JP, Köhler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet. 2000;26:336–340. doi: 10.1038/81649. [DOI] [PubMed] [Google Scholar]
- Soong NW, Hinton DR, Cortopassi G, Arnheim N. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat Genet. 1992;2:318–323. doi: 10.1038/ng1292-318. [DOI] [PubMed] [Google Scholar]
- Stuart JA, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA base excision repair are affected differently by caloric restriction. FASEB J. 2004;18:595–597. doi: 10.1096/fj.03-0890fje. [DOI] [PubMed] [Google Scholar]
- Turlejski K, Djavadian R. Life-long stability of neurons: a century of research on neurogenesis, neuronal death and neuron quantification in adult CNS. Prog Brain Res. 2002;136:39–65. doi: 10.1016/S0079-6123(02)36006-0. [DOI] [PubMed] [Google Scholar]
- Ugarte N, Petropoulos I, Friguet B. Oxidized mitochondrial protein degradation and repair in aging and oxidative stress. Antioxid Redox Signal. 2010;13:539–549. doi: 10.1089/ars.2009.2998. [DOI] [PubMed] [Google Scholar]
- Vercauteren K, Pasko RA, Gleyzer N, Marino VM, Scarpulla RC. PGC-1-related coactivator: immediate early expression and characterization of a CREB/NRF-1 binding domain associated with cytochrome c promoter occupancy and respiratory growth. Mol Cell Biol. 2006;26:7409–7419. doi: 10.1128/MCB.00585-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Brüning JC, Kahn CR, Clayton DA, Barsh GS, Thorén P, Larsson NG. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet. 1999;21:133–137. doi: 10.1038/5089. [DOI] [PubMed] [Google Scholar]
- Wiesner RJ, Zsurka G, Kunz WS. Mitochondrial DNA damage and the aging process: facts and imaginations. Free Radic Res. 2006;40:1284–1294. doi: 10.1080/10715760600913168. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Izumi H, Ise T, Uramoto H, Torigoe T, Ishiguchi H, Murakami T, Tanabe M, Nakayama Y, Itoh H, Kasai H, Kohno K. Human mitochondrial transcription factor A binds preferentially to oxidatively damaged DNA. Biochem Biophys Res Commun. 2002;295:945–951. doi: 10.1016/S0006-291X(02)00757-X. [DOI] [PubMed] [Google Scholar]
- Yowe DL, Ames BN. Quantitation of age-related mitochondrial DNA deletions in rat tissues shows that their pattern of accumulation differs from that of humans. Gene. 1998;209:23–30. doi: 10.1016/S0378-1119(97)00628-8. [DOI] [PubMed] [Google Scholar]