

Abstract
We investigated the use of integrase-defective lentiviral vectors (IDLVs) for transient delivery of zinc finger nucleases (ZFNs) and donor templates for site-specific modification of the human adenosine deaminase (hADA) gene. Initially, we constructed IDLVs carrying ZFN monomers (Single-IDLVs) and found them to be able to deliver their gene-editing payload to K562 cells successfully upon cotransduction, with minimal cytotoxicity. To simplify delivery, we designed an IDLV construct to deliver both ZFN monomers from the same vector (Double-IDLV). However, this construct in its original state was prone to rearrangements of the vector genome, resulting in greatly reduced functionality; this was due to recombination between highly similar ZFN monomers arranged in tandem. We modified the Double-IDLV constructs to reduce recombination and restored simultaneous delivery of both ZFNs. We also tested an IDLV construct for delivery of donor templates and demonstrated its efficacy for gene modification. In summary, we highlighted the importance of modifying vector design for co-delivery of highly similar sequences inherent to genome-editing nucleases, and demonstrated significant improvement in the use of IDLVs for delivery of ZFNs and donor templates for genome modification.
Introduction
Zinc finger nucleases (ZFNs) are chimeric endonucleases containing sequence-specific ZF motifs coupled with FokI endonuclease domains. Upon dimerization of ZFN monomers, they can introduce sequence-specific double-strand breaks (DSBs).1 ZFN-induced DSBs can be repaired by one of two major cellular DNA repair pathways: nonhomologous end joining or homology-directed repair. Nonhomologous end joining-mediated repair of DSBs often introduces mutations, and this can be exploited to knockout ZFN-targeted genes. In contrast, homology-directed repair-mediated repair is more accurate because it uses a homologous donor template to repair DSBs. By using a donor template that has homologous sequences flanking the ZFN target site and containing desired changes or additional sequences, it is possible to introduce precise site-specific changes to the genome.2
Because of its tremendous potential, ZFN technology is rapidly becoming popular for site-specific genome modification3 and has been successfully used in the context of proof-of-concept for gene therapy.4 Many of the gene therapy approaches investigated so far have been for disorders of the hematopoietic system, such as X-linked severe combined immunodeficiency,1 X-linked chronic granulomatous disease,5 targeting hematopoietic stem/progenitor cells for ZFN-mediated genome modification. In addition, T-lymphocytes have been used as a target cell type, especially for CCR5 disruption to provide resistance to HIV infection.6,7 Multiple gene therapy approaches have envisioned the use of human embryonic stem cells or human induced pluripotent stem cells, where gene-modified cells can be cloned, characterized, and used to produce mature cell types for regenerative medicine applications.8
One of the major hurdles to using ZFNs effectively is the delivery of ZFNs and donor templates to target cells. ZFNs and donor templates can be delivered using various gene delivery methods. The most commonly used methods for transient gene delivery to cell lines are lipid-based transfection or electroporation. Although these methods work very efficiently for cell lines, they may lack the required efficiency or be prohibitively cytotoxic for primary cells. Among the viral methods of gene delivery, chimeric adenoviral vectors,7 adeno-associated vectors,9 and integrase-defective lentiviral vectors (IDLVs, also called Nonintegrating lentiviral vectors)10,11 have been investigated for delivery of ZFNs and donors to target cells. Adeno-associated vectors and adenoviral vectors have limited cell tropism depending on their capsid proteins but are capable of delivering transgenes at high levels.12,13
LVs are able to deliver transgenes at high efficiency with low cytotoxicity and have the added advantage of broad cell tropism if pseudotyped with the vesicular stomatitis virus-G protein. However, LVs integrate into the genome in a semi-random manner and hence carry risks of insertional oncogenesis. As a solution, if LVs are packaged with a catalytically inactive integrase, these IDLVs can still transduce cells efficiently, while minimizing genomic integration.14 IDLVs exist as episomal linear or circular double-stranded DNA forms. These proviral genomes can be transcribed, albeit at a lower efficiency compared with the corresponding integrated forms. Episomal forms of IDLVs dilute out with cell division and thus they can achieve short-lived expression,15,16 which is required for ZFNs to act in a “hit and run” manner. The ability to transduce a broad range of human cell types at high efficiency makes IDLV a promising gene delivery vehicle for delivering ZFN and donor templates to a wide variety of target cells including human hematopoietic stem/progenitor cells, human embryonic stem cells, and human induced pluripotent stem cells.
Previous studies,10,17 most significantly those by Lombardo et al.,10 have reported the use of IDLVs for genome modification. However, there has not been an in-depth investigation of the issues regarding ZFN or donor delivery by IDLVs. In this study, we investigated the use of IDLVs to deliver ZFNs and donor templates for site-specific gene modification at the human adenosine deaminase (hADA) locus. We constructed IDLVs to deliver either one or two ZFN monomers and subsequently compared their efficiency. We investigated potential problems due to recombination in the vectors carrying contiguous ZFN monomers. We also constructed IDLVs to deliver donor templates and tested them for efficiency. Our results underscore potential challenges to using IDLVs for ZFN and donor delivery and provide solutions for these concerns. These results highlight the promise of using IDLVs for gene modification in human somatic and pluripotent cells.
Results
Site-specific genome modification of hADA
For this proof-of-principle study, we developed ZFNs to target the hADA gene for site-specific modification. We designed the ZFNs to recognize a 36 bp sequence in intron 6 of hADA, comprising two ZFN monomer-binding sites with a 5 bp spacer, 70 bp away from a known severe combined immunodeficiency-causing mutation in exon 7 (Figure 1a). The ZFN pair included ELD/KKR obligate heterodimeric FokI domains.18 We nucleofected K562 cells with ZFN plasmids and determined the extent of DNA cleavage and imperfect DNA repair by nonhomologous end joining flanking the target site using the Surveyor nuclease assay.19 We found that the ZFNs could induce up to 18% allelic disruption in K562 cells at 4 days following nucleofection of ZFN plasmids (Figure 1b).
Figure 1.

Zinc finger nucleases (ZFNs) for site-specific modification of human adenosine deaminase (hADA). (a) Schematic of hADA indicating the ZFN-binding site (top). The two half-binding sites are shown with thick lines. The EagI site is indicated by the arrow. The primer pair used for Surveyor nuclease assay is shown as well. Schematic of the donor template showing the silent mutations resulting in creation of an SstII site (middle). The extent of homology between the donor template and the genomic locus is indicated by broken lines. Schematic of the modified hADA locus showing the SstII site. The primer pairs for the restriction fragment length polymorphism (RFLP) assay as well as quantitative PCR (qPCR) assay are indicated (bottom). (b) Surveyor nuclease assay demonstrating ZFN activity in K562 cells 4 days post-nucleofection. The numbers indicate %allelic disruption. Mock: K562 cells nucleofected without DNA, PC: positive control, a sample with known allelic disruption, NC: no template reaction. (c) Quantification of site-specific genome modification in K562 cells 4 days post-nucleofection by qPCR assay. Y-axis shows frequency of gene modification, X-axis shows the plasmid combinations used in nucleofection. ZFN: 2.5 μg of ZFN plasmid, Donor: 25 μg of donor plasmid, ZFN + Donor: 2.5 μg of ZFN plasmid + 25 μg of donor plasmid, Mock: no DNA control. Bars indicate mean ± SEM; n = 4.
To achieve homology-directed repair in proximity of the ZFN-binding site, we amplified and cloned into a plasmid a 1.3 kb fragment from the hADA gene spanning the ZFN target site to serve as a homologous donor template (Figure 1a). The donor template was designed to contain two silent base pair changes in exon 7, 75 bp downstream of the ZFN-binding site, resulting in the introduction of an SstII site and thus enabling restriction fragment length polymorphism-based monitoring of gene modification. We nucleofected K562 cells with ZFN and donor plasmids and determined the gene modification levels by digestion of PCR amplicons with SstII. Although we demonstrated successful gene modification, the frequency was outside the limits of accurate quantification by densitometry. Hence, we developed a quantitative PCR-based assay to allow accurate quantification of gene modification frequencies (Figure 1a). In the first round of the assay, we amplified a 2 kbp region surrounding the ZFN-binding site using primers binding outside of the donor region to avoid amplification of co-purified donor templates. Using this PCR product as a template, we amplified gene modification events using a primer specific for changes introduced to form the SstII site. We normalized for the input template amount using a primer pair binding in intron 9. We determined the frequency of gene modification by quantifying the second round amplification using SYBRGreen. Using the quantitative PCR assay, we quantified the gene modification frequencies at 1% (Figure 1c). Throughout this study, we used the ZFNs and donor templates for gene modification of hADA as a model set of reagents for IDLV-mediated delivery.
IDLVs to deliver ZFNs
Using a previously established lentiviral backbone (CCL),20 we constructed Single-IDLVs expressing either the left or the right ZFN monomer under the control of truncated, intron-less human elongation factor 1α promoter (EFS).21 We linked a fluorescent reporter, mCherry, to the ZFN monomers using the self-cleaving 2A peptide from the porcine teschovirus-1 (P2A),22 to enable monitoring of gene transfer and expression from the IDLVs by flow cytometry. The IDLVs encoded the woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) to increase the stability of genomic as well as internal transcripts (Figure 2a). We packaged the vectors with a gag/pol plasmid carrying a D64V mutant integrase14 (EFS-Single-IDLVs), and packaged and concentrated the vectors using tangential flow filtration. The vector titers as well as average vector copy number (VCN) were determined using a multiplexed TaqMan quantitative PCR assay as previously described.23
Figure 2.

Integrase-defective lentiviral vectors (IDLVs) to deliver zinc finger nucleases (ZFNs). (a) Self-inactivating lentiviral constructs for delivery of ZFN monomers. ΔU3-R-U5: HIV-1–derived self-inactivating long terminal repeat, ψ: HIV-1 packaging signal, EFS: truncated, intron-less human elongation factor 1α promoter. ZFN-L and ZFN-R are left and right ZFNs, respectively, P2A: 2A peptide from porcine teschovirus-1, WPRE: woodchuck hepatitis virus post-transcriptional regulatory element. (b) Delivery of ZFNs to K562 cells by EFS-Single-IDLVs. Expression of mCherry in K562 cells transduced with EFS-Single-IDLVs at a range of concentrations from 2 × 107 to 1.6 × 108 transducing unit (TU)/ml (corresponding with multiplicity of infections of 200–1,600). Y-axis shows levels of transduction indicated by mCherry expression, X-axis shows days post-transduction (left). Average vector copy number (VCN) in transduced K562 cells at 4 days post-transduction (center). Bars and data points represent mean ± SD. (c) Allelic disruption in K562 cells transduced with EFS-Single-IDLVs (left). Representative Surveyor nuclease assay results from K562 cells transduced with various concentrations of EFS-Single-IDLVs are shown. The smaller products, corresponding to the cut bands arising from allelic disruption are indicated by the arrow. Quantified %allelic disruption is indicated by the numbers under the bands on the gel. (d) Transduction of CB-CD34+ cells with EFS-Single-IDLVs. Expression of mCherry in CB-CD34+ cells transduced with various concentrations of the EFS-Single-IDLVs, measured at 5 days post-transduction (left). Y-axis shows %mCherry+ cells, X-axis shows the vector concentrations. Average VCN in CB-CD34+ cells transduced with EFS-Single-IDLVs, measured at 4 days post-transduction (right). Y-axis indicates average VCN, X-axis indicates vector concentrations. Data points represent mean ± SD.
We cotransduced K562 cells with EFS-Single-IDLVs and monitored the cells for VCN, mCherry expression, and ZFN-induced allelic disruption. The IDLVs transduced K562 cells robustly, resulting in up to 100% cells expressing mCherry as determined by flow cytometry 4 days later. These vectors also demonstrated a vector dose-dependent transduction, reaching VCN of up to 40 at day 4 post-transduction (Figure 2b). K562 cells cotransduced with the two EFS-Single-IDLVs demonstrated allelic disruption at the ZFN target site measured by Surveyor nuclease assay, indicating that EFS-Single-IDLVs were able to successfully co-deliver and express both ZFN monomers in K562 cells (Figure 2c).
We tested the EFS-Single-IDLVs in primary CD34+ hematopoietic stem/progenitor cells isolated from human umbilical cord blood (CB-CD34+ cells). We screened the CB-CD34+ cells for the lack of sequence heterozygosity in the target region as well as for lack of single-nucleotide polymorphisms which might alter the ZFN-binding site. The EFS-Single-IDLVs demonstrated robust transduction of CB-CD34+ cells as shown by mCherry expression and VCN (Figure 2d). However, allelic disruption was below the detection limit of the Surveyor nuclease assay. One of the potential causes for this could be inefficient cotransduction with the two vectors, resulting in cells receiving unequal amounts of the two ZFNs.
Co-delivery of two ZFN monomers with one IDLV
A potential limitation to the use of Single-IDLVs to produce site-specific DNA modification is the requirement to achieve simultaneous cotransduction of cells with two vectors, needed for 1:1 stoichiometry of the two ZFN monomers. To improve delivery of both the ZFNs to the same cell in equal amounts, we constructed an IDLV encoding both the left and right ZFN monomers linked by the 2A peptide from Thosea asigna (T2A)22 and linked to mCherry by P2A (EFS-Double-IDLV) (Figure 3a). We transduced K562 cells with EFS-Double-IDLV and showed efficient transduction as evident from dose-dependent mCherry expression and VCN. However, these cells did not exhibit allelic disruption (Figure 3b). We hypothesized that this functional failure could be due to vector rearrangements during reverse transcription of the vector genome, because the vector contains repeated regions, including FLAG-tags, nuclear localization signals (NLSs), and the FokI endonuclease domains present in the two tandem ZFN sequences (summarized in Supplementary Table S1).
Figure 3.

Co-delivery of zinc finger nucleases (ZFN) monomers with one integrase-defective lentiviral vector (IDLV). (a) Schematic of the EFS-Double-IDLV construct. ΔU3-R-U5: HIV-1–derived self-inactivating long terminal repeat, ψ: HIV-1 packaging signal, EFS: truncated, intron-less human elongation factor 1α promoter. ZFN-L and ZFN-R are left and right ZFNs, respectively, P2A: 2A peptide from porcine teschovirus-1, T2A: 2A peptide from Thosea asigna, WPRE: woodchuck hepatitis virus post-transcriptional regulatory element. Detailed arrangement of various domains in the ZFN monomers is shown. 3xFLAG: triple FLAG-tag, NLS: nuclear localization signal, ZF motifs, ELD/KKR: obligate heterodimeric FokI domains. (b) Transduction of K562 cells by EFS-Double-IDLV. Expression of mCherry in K562 cells transduced with EFS-Double-IDLV at a range of concentrations from 2 × 107 to 1.6 × 108 transducing units (TU)/ml (corresponding with multiplicity of infectionss of 200–1,600). Y-axis shows levels of transduction indicated by mCherry expression; X-axis shows days post-transduction (left). Average vector copy number (VCN) in transduced K562 cells at days post-transduction (right). Bars and data points represent mean ± SD. (c) Primer pairs for testing integrity of proviral genome of EFS-Double-IDLV. Arrows connected by broken lines indicate primer pairs for reactions, A, B, C, D, E. (d) Vector integrity analysis on EFS-IDLV constructs. Gel electropherograms from reaction B performed on K562 cells transduced with IDLVs are shown. L100: 100 bp DNA ladder, Single: cells transduced with EFS-Single-IDLVs, Double: cells transduced with EFS-Double-IDLV, PC: EFS-Double-IDLV construct plasmid, NC: no template control, L1000: 1,000 bp DNA ladder. Product sizes of 1,300 bp and 400 bp are indicated by arrows.
To diagnose this phenomenon, we performed PCR analysis to determine the vector integrity post-transduction. We used primer pairs recognizing different overlapping regions of the vector genome as indicated (Figure 3c) and performed PCRs on proviral DNA from cells treated with the EFS-Double-IDLV or cotransduced with EFS-Single-IDLVs. Gel electrophoresis revealed that in one of the reactions, reaction B, which amplifies across the left ZFN, the major product was ~400 bp long, shorter than the expected size of 1.3 kbp (Figure 3d), whereas the other reactions revealed no differences (Supplementary Figure S1). This smaller PCR product indicated the occurrence of a deletion in the left ZFN region, the existence of which was subsequently confirmed by sequence analysis. We hypothesized that the sequence identity between the two contiguously arranged ZFN monomers resulted in recombination events leading to deletion of one of the ZFN monomers. The stretches of homology in the FLAG-tag, NLS, and FokI domains could potentially make the vector prone to undergoing recombination. Such recombination events pose a significant limitation for delivering two ZFNs from the same IDLV, as most of the ZFN architectures consist of highly similar monomers forming a functional nuclease dimer.24,25
Improvements in Double-IDLV constructs
To avoid rearrangements in the EFS-Double-IDLV, we modified the construct in two different ways (Figure 4a). In one of the constructs, EFS-Double-dF-IDLV, we deleted the N-terminal FLAG-tag from the left ZFN to eliminate one region of sequence homology; however, the NLS was retained. In the other construct, EFS-Double-CoOp-IDLV, we codon-optimized the left ZFN, including its FLAG-tag, NLS, ZF motifs, and the FokI domain. This resulted in a decrease in the DNA sequence identity between the two ZFNs (summarized in Supplementary Table S1), potentially reducing the possibility of recombination. We tested these two constructs by transducing K562 cells with the corresponding integrating LVs. The EFS-Double-dF-LV did not induce allelic disruption as assessed by the Surveyor nuclease assay. However, the EFS-Double-CoOp-LV construct did successfully induce allelic disruption (Figure 4b).
Figure 4.

Improvements in Double-integrase–defective lentiviral vector (IDLV) constructs. (a) Modified Double-IDLV constructs. EFS-Double-dF-IDLV construct (top) and Double-CoOp-IDLV constructs (bottom). ΔU3-R-U5: HIV-1–derived self-inactivating long terminal repeat, ψ: HIV-1 packaging signal, EFS: truncated, intron-less human elongation factor 1α promoter, MND: modified myeloproliferative sarcoma virus long terminal repeat. ZFN-L and ZFN-R are left and right ZFNs, respectively, P2A: 2A peptide from porcine teschovirus-1, T2A: 2A peptide from Thosea asigna, WPRE: woodchuck hepatitis virus post-transcriptional regulatory element. ZFN-LdF indicates the left ZFN without N-terminal FLAG-tag. ZFN-LCoOp indicates the codon-optimized left ZFN. (b) Comparison of ability of EFS-Double-LV constructs to induce allelic disruption in K562 cells. Surveyor nuclease assays on K562 cells transduced with EFS-Double-LV, EFS-Double-dF-LV, and EFS-Double-CoOp-LV are shown, demonstrating successful allelic disruption only with the Double-CoOp-LV construct. Numbers indicated quantified %allelic disruption. (c) Head-to-head comparison of EFS-Single-IDLVs, EFS-Double-CoOp-IDLV, and MND-Double-CoOp-IDLV. Expression of mCherry in K562 cells transduced with the ZFN IDLVs at 4 days post-transduction (top). Y-axis represents percentage of mCherry+ cells and X-axis represents vector concentrations. Average vector copy number (VCN) in K562 cells transduced with the ZFN IDLVs at 4 days post-transduction (bottom). Y-axis represents average VCN cells and X-axis represents vector concentrations. Data points represent mean ± SD. (d) Comparison of the ability of IDLVs to induce allelic disruption in K562 cells. Representative surveyor nuclease assay results from K562 cells transduced with EFS-Single-IDLVs (upper), EFS-Double-CoOp-IDLV (middle), and MND-Double-CoOp-IDLV (lower), at 4 days post-transduction are shown. PC: positive control, previously used sample with detectable allelic disruption, NC: no template control. Vector concentrations used for transduction are indicated on top. Numbers indicate quantified %allelic disruption. TU, transducing unit.
To determine whether the restoration of ZFN activity is due to reduction in recombination, we performed PCR analysis (reaction B) for vector integrity of the proviral genomes. We quantified the relative proportions of the two bands via densitometry and found that the expected 1.3 kbp product was tenfold to 15-fold more abundant in the EFS-Double-CoOp construct compared with the unmodified construct (Supplementary Figure S2). The increase in relative abundance of the 1.3 kbp product suggested a reduction in recombination levels in both the constructs. These data suggest strongly that the DNA sequence homology between two ZFN monomers in a given pair can be detrimental to their co-delivery by the same IDLV. However, by introducing DNA sequence changes to reduce homology, we successfully overcame this barrier and achieved delivery of both the ZFN monomers by one IDLV.
Upon successful restoration of delivery of two ZFNs from the same construct, we compared the EFS-Double-CoOp-IDLV with EFS-Single-IDLVs for transduction capacity as well as ability to deliver ZFNs to K562 cells. We found that despite achieving efficient transduction, EFS-Double-CoOp-IDLV was not able to induce detectable allelic disruption. We hypothesized that this was due to EFS being a weak promoter in hematopoietic cells as compared with retroviral long terminal repeat-derived promoters. Hence, we replaced EFS with a modified myeloproliferative sarcoma virus long terminal repeat containing a deletion in the negative regulatory region (MND-Double-CoOp-IDLV) (Figure 4a). The MND promoter was previously shown to be a strong promoter in hematopoietic cells.26 We compared the ability of the MND-Double-CoOp-IDLV to deliver ZFNs to K562 cells (Figure 4c). We found that K562 cells transduced with MND-Double-CoOp-IDLV did exhibit allelic disruption indicating that the IDLV was successful in delivering both the ZFNs, although activity was still reduced compared with Single-IDLVs (Figure 4d).
Delivery of ZFNs to primary hematopoietic cells using MND-Double-CoOp-IDLV
Based on evidence from K562 cells, we tested the MND-Double-CoOp-IDLV for ability to deliver ZFNs to human hematopoietic cells. We activated and expanded T-lymphocytes from peripheral blood mononuclear cells, and transduced them with increasing concentrations of the MND-Double-CoOp-IDLV. We monitored transduced T-lymphocytes for mCherry expression, VCN, and allelic disruption. MND-Double-CoOp-IDLV demonstrated robust transduction of T-lymphocytes based on mCherry expression and average VCN, with minimal cytotoxicity. However, in spite of successful transduction, the T-lymphocytes did not exhibit detectable allelic disruption (Figure 5a). We also tested the efficacy of the MND-Double-CoOp-IDLV in human umbilical cord blood CD34+ cells. We found that it was able to transduce CB-CD34+ cells efficiently, as indicated by mCherry expression and VCN, but did not induce detectable allelic disruption (Figure 5b).
Figure 5.
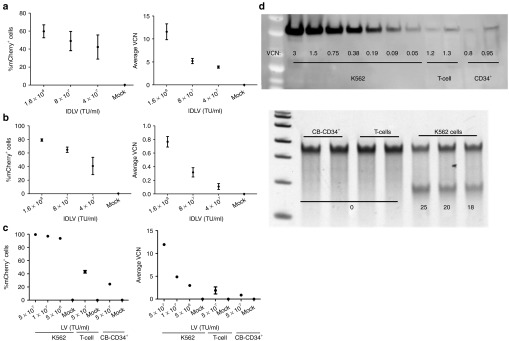
Delivery of zinc finger nucleases (ZFNs) to primary hematopoietic cells using integrase-defective lentiviral vectors (IDLVs). (a) Transduction of human T-lymphocytes with MND-Double-CoOp-IDLV. Expression of mCherry in T-lymphocytes transduced with the MND-Double-CoOp-IDLV at 4 days post-transduction (left). Y-axis represents percentage of mCherry+ cells and X-axis represents vector concentrations. Average vector copy number (VCN) in T-lymphocytes cells transduced with the MND-Double-CoOp-IDLV at 4 days post-transduction (right). Y-axis represents average VCN cells and X-axis represents vector concentrations. Data points represent mean ± SD. (b) Transduction of human CB-CD34+ cells with MND-Double-CoOp-IDLV. Expression of mCherry in CB-CD34+ cells transduced with the MND-Double-CoOp-IDLV at 4 days post-transduction (left). Y-axis represents percentage of mCherry+ cells and X-axis represents vector concentrations. Average VCN in CB-CD34+ cells transduced with the MND-Double-CoOp-IDLV at 8 days post-transduction (right). Y-axis represents average VCN cells and X-axis represents vector concentrations. Data points represent mean ± SD. (c) Transduction of K562 cells and primary hematopoietic cells with EFS-Double-CoOp-LV. Expression of mCherry in K562 cells, T-lymphocytes, and CB-CD34+ cells transduced with the EFS-Double-CoOp-LV at 4 days post-transduction (left). Y-axis represents percentage of mCherry+ cells and X-axis represents vector concentrations. Average VCN in K562 cells, T-lymphocytes, and CB-CD34+ cells transduced with the EFS-Double-CoOp-LV at 4 days post-transduction (right). Y-axis represents average VCN cells and X-axis represents vector concentrations. Data points represent mean ± SD. (d) Comparison of ZFN expression from EFS-Double-CoOp-LV in hematopoietic cells. Western blot against FLAG-tag (top) are shown. Equal amounts (50 μg each) of lysates from K562 cells, T-lymphocytes, and CB-CD34+ cells transduced with EFS-Double-CoOp-LV were used for the western blot. VCN corresponding to the lysates is indicated by the numbers. Surveyor nuclease assay results from K562 cells, T-lymphocytes, and CB-CD34+ cells transduced with EFS-Double-CoOp-LV (bottom). The numbers indicate quantified %allelic disruption. TU, transducing unit.
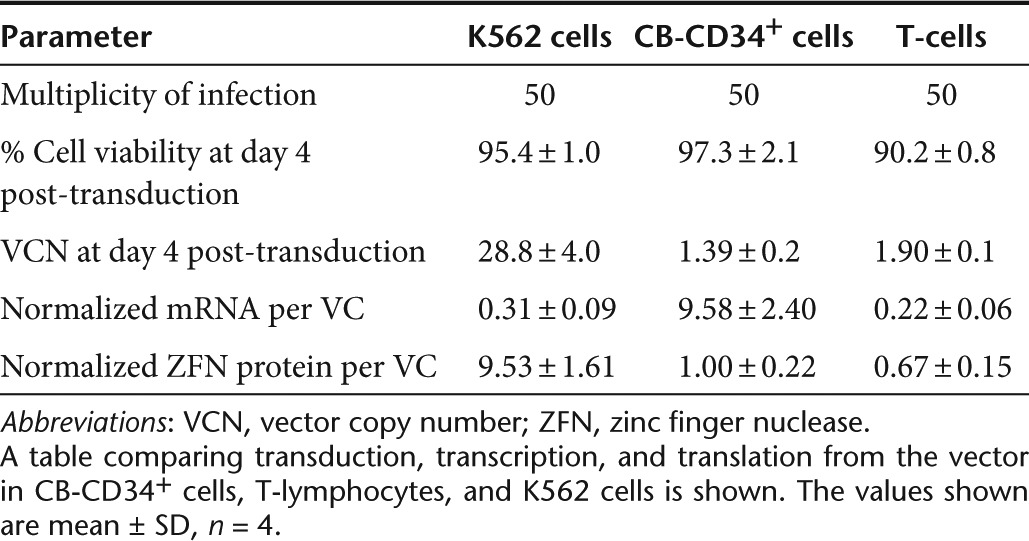
We hypothesized that the failure of Double-CoOp-IDLV to induce allelic disruption in primary cells could potentially be due to low expression per vector genome. To investigate this phenomenon, we transduced K562 cells, T-lymphocytes, and CB-CD34+ cells with an integrating EFS-Double-CoOp vector and monitored them for mCherry expression and VCN (Figure 5c). We also determined the level of expression of ZFNs by western blotting for the FLAG-tag present on both the ZFN monomers. We found that the expression of ZFNs at equivalent VCN was lower in T-lymphocytes and in CB-CD34+ cells by up to 25-fold (Figure 5d). This corroborated with the lack of detectable allelic disruption in either of these cell types. This would indicate that one of the major reasons for lower efficiency of IDLVs in primary cells is the expression per VC. Therefore, we measured the levels of mRNA from the LV by quantitative real-time PCR. Surprisingly, we found that mRNA produced per VC was higher in CB-CD34+ cells than in K562 cells. However, in T-lymphocytes, the mRNA levels per vector were similar to those in K562 cells. A comparison of transduction, transcription, and translation from the vector in CB-CD34+ cells, T-lymphocytes, and K562 cells is shown in (Table 1). This indicates different restrictions on the vectors in these different cell types. Further measures to increase expression from IDLVs are needed to make them efficient in primary cells.
Table 1. Comparison of EFS-Double-CoOp-LV in hematopoietic cells.
Delivery of donor templates by IDLVs
In addition to ZFN delivery, efficient delivery of homologous donor templates is required to achieve high levels of homology-directed repair. We showed previously that nucleofection of K562 cells with ZFN and donor plasmids yielded gene modification frequencies of up to 1%. However, we used a 10:1 molar ratio of donor plasmid to ZFN plasmid for nucleofection. At such high amounts of DNA, the nucleofection process can be cytotoxic. Therefore, we investigated the use of IDLVs for donor delivery with potentially reduced cytotoxicity.
We constructed the Donor-IDLV to deliver the aforementioned 1.3 kbp donor template with the SstII site (Figure 6a). The donor template was cloned along with the EFS promoter and WPRE to increase titers. Since the donor fragment contains introns, we cloned it in the reverse direction relative to the direction of lentiviral genome transcription during packaging to avoid potential splicing of the vector. We nucleofected K562 cells with ZFN plasmids, transduced them with Donor-IDLV 24 hours later, and monitored them for VCN as well as genome modification. The Donor-IDLV was able to transduce K562 cells efficiently as demonstrated by VCN (Figure 6b). The Donor-IDLV genomes were also able to act as donor templates as demonstrated by a vector dose-dependent increase in genome modification frequency when the ZFN plasmid was nucleofected (Figure 6b). With increasing amounts of the ZFN plasmid, we achieved increasing genome modification frequencies, suggesting ZFN expression as a limiting factor for this approach (Figure 6c).
Figure 6.

Integrase-defective lentiviral vectors (IDLVs) for delivery of donor templates. (a) Schematic of the donor-IDLV construct. Donor-IDLV construct representing the 1.3 kbp donor fragment arranged in reverse between EFS and WPRE. The SstII site which enables monitoring of gene modification is indicated. ΔU3-R-U5: HIV-1–derived self-inactivating long terminal repeat, ψ: HIV-1 packaging signal, EFS: truncated, intron-less human elongation factor 1α promoter, WPRE: woodchuck hepatitis virus post-transcriptional regulatory element. (b) Gene modification in K562 cells using Donor-IDLV. Gene modification frequencies in K562 cells nucleofected with 2.5 μg of ZFN plasmid and followed by transduction with various concentrations of Donor-IDLV 24 hours later (left). Gene modification frequencies are measured at 4 days post-transduction. Y-axis represents % gene modification, X-axis represents vector concentrations. Average vector copy number (VCN) in K562 cells nucleofected with 2.5 μg of ZFN plasmid and followed by transduction with various concentrations of Donor-IDLV 24 hours later (right). Y-axis represents average VCN at 4 days post-transduction, X-axis represents vector concentrations. Bars and data points represent mean ± SD. (c) Gene modification in K562 cells using Donor-IDLV. Gene modification frequencies in K562 cells nucleofected with various amounts of ZFN plasmid and followed by transduction with 3 × 107 transducing unit (TU)/ml of Donor-IDLV 24 hours later (left). Gene modification frequencies are measured at 4 days post-transduction. Y-axis represents % gene modification, X-axis represents nucleofection conditions. Average VCN in K562 cells nucleofected with various amounts of ZFN plasmid and followed by transduction with 3 × 107 TU/ml of Donor-IDLV 24 hours later (right). Y-axis represents average VCN at 4 days post-transduction, X-axis represents nucleofection conditions. Bars and data points represent mean ± SD. GFP, green fluorescent protein.
Co-delivery of ZFNs and donor templates by IDLVs
To determine whether gene modification could be achieved using a completely viral delivery approach, we investigated co-delivery of ZFNs and donor templates by IDLVs. We transduced K562 cells with varying doses of the pairs of Single-IDLVs while keeping the Donor-IDLV dose constant (3 × 107 transducing unit/ml) and monitored them for VCN and gene modification (Figure 7a). Gene modification frequencies exhibited a ZFN dose-dependent increase; however, the absolute levels were lower than with nucleofected plasmids (Figure 7b). To increase the cotransduction efficiency, we delivered the ZFNs by Double-CoOp-IDLV and delivered the donor templates by Donor-IDLV. Cotransduction of K562 cells with the two IDLVs exhibited efficient transduction and a similar dose-dependent response in gene modification (Figure 7b).
Figure 7.

Co-delivery of zinc finger nucleases (ZFNs) and donor templates by integrase-defective lentiviral vectors (IDLVs). (a) Gene modification in K562 cells with ZFN and donor delivery by IDLVs. Gene modification frequencies in K562 cells cotransduced with various concentrations of EFS-Single-IDLVs and 3 × 107 transducing unit (TU)/ml of Donor-IDLV, measured at 4 days post-transduction (left). Y-axis represents gene modification frequencies, X-axis represents concentrations of EFS-Single-IDLVs. Average vector copy number (VCN) in K562 cells cotransduced with various concentrations of EFS-Single-IDLVs and 3 × 107 TU/ml Donor-IDLV, measured at 4 days post-transduction (right). Y-axis represents average VCN, X-axis represents concentrations of EFS-Single-IDLVs. Bars and data points represent mean ± SD. (b) Gene modification in K562 cells with ZFN and donor delivery by IDLVs. Gene modification frequencies in K562 cells cotransduced with various concentrations of MND-Double-CoOp-IDLV and 3 × 107 TU/ml of Donor-IDLV, measured at 4 days post-transduction (left). Y-axis represents gene modification frequencies, X-axis represents concentrations of MND-Double-CoOp-IDLV. Average VCN in K562 cells cotransduced with various concentrations of MND-Double-CoOp-IDLV and 3 × 107 TU/ml Donor-IDLV, measured at 4 days post-transduction (right). Y-axis represents average VCN, X-axis represents concentrations of MND-Double-CoOp-IDLV. Bars and data points represent mean ± SD.
Discussion
In this study, we focused on IDLV-mediated delivery as this method has multiple potential advantages over other delivery methods: (i) IDLVs pseudotyped with vesicular stomatitis virus-G protein can efficiently transduce virtually every mammalian cell type,27 (ii) IDLVs exhibit low cytotoxicity, (iii) IDLVs can deliver transgenes in a transient manner. One of the most important advantages of IDLVs is that LVs have already been used in clinical trials for gene therapy.28 Methods to scale-up production of LVs are well established for vector preparations required at clinical scale29 and IDLVs production would use similar methods. In summary, IDLVs potentially offer advantages over other methods of gene delivery.
IDLVs were previously shown to deliver ZFNs and donor templates successfully, although at relatively low efficiency in primary cells. Studies using IDLVs have reported efficacy delivering one ZFN per vector, requiring cotransduction by two vectors to ensure ZFN delivery.10,11 The use of IDLVs for delivering two ZFNs in the same vector has shown lack of efficacy, although the reasons for it have not been investigated thoroughly. In this study, we identified potential concerns with using IDLVs to deliver ZFNs and donor templates and demonstrated a significant improvement in the design and usage of IDLVs for ZFN delivery. We tested IDLVs to deliver either one or two ZFNs. The IDLVs delivering one ZFN per vector, EFS-Single-IDLVs, were able to deliver ZFNs to cell lines. However, they exhibited lower efficiency of activity in primary cells, corroborating previous reports using IDLVs.10,11
Transduction with two IDLVs has potential drawbacks as a ZFN delivery system, especially in the stoichiometry of the two ZFN monomers. The optimal stoichiometry of ZFN monomers is 1:1, which might not be attained with cotransduction of two IDLVs. To ensure the production of equivalent amounts of both ZFN monomers, we linked them with 2A peptides. We constructed an IDLV to co-deliver two ZFNs (EFS-Double-IDLV). However, we found that despite comparable rates of transduction, EFS-Double-IDLV failed to deliver functional ZFNs. We further investigated this phenomenon and found that the IDLV underwent recombination due to regions of sequence identity between the two ZFNs. The presence of repeat regions was reported previously to induce recombination in the HIV genome.30 The mechanism of recombination mediated by genomic repeats is thought to occur during reverse transcription in an intermolecular fashion,31,32,33 often giving rise to deletion of the regions between the two repeats. We found that in the EFS-Double-IDLV construct, the recombination events were potentially due to the FLAG-tags and NLSs present in both ZFN constructs. This intravector recombination is clearly an important factor limiting the use of IDLVs for ZFN delivery, as most of the ZFN architectures include the FLAG-tag as part of the open-reading frame and all have incorporated NLS into the ZFN protein to mediate their nuclear import for access to the genome as well as the FokI endonuclease sequences. The ZF motifs within a ZFN monomer could also have repeat regions based on the target sequence, potentially facilitating rearrangements. Occurrence of repeat sequences in vectors is also a potential concern for viral delivery of transcription activator-like effector nucleases. Transcription activator-like effector nucleases are comprised of 33–35 amino acids long repeat-variable di-residue repeats and would be expected to be highly prone to recombination if used in viral vectors. To deliver either ZFNs or transcription activator-like effector nucleases via viral vectors, it is essential to develop measures to reduce recombination.
To minimize recombination, we modified the EFS-Double-IDLV construct in two ways. One of the modifications, codon optimization of the sequence of one left ZFN in the EFS-Double-CoOp construct, minimized the recombination, leading to successful ZFN delivery from the same vector. The EFS-Double-CoOp construct, when packaged as IDLV, did not exhibit allelic disruption in K562 cells. We replaced the EFS promoter with the MND promoter to boost expression in hematopoietic cells. The resulting construct, MND-Double-CoOp-IDLV, was able to induce allelic disruption in K562 cells. The MND-Double-CoOp-IDLV was tested in primary T-lymphocytes and CB-CD34+ cells, but failed to induce allelic disruption. This could potentially be due to insufficient expression of the ZFNs from the IDLV genomes.
Gene expression from IDLVs is lower than other delivery methods and might not be sufficient for every system. The main reasons for low gene expression could be: (i) low VCN per cell, (ii) limited persistence of VCs, and (iii) reduced expression per VC. We observed that we could get VCNs of up to 50 per cell. In the experiments with the integrating vectors, an average VCN of 1 per cell was sufficient to achieve detectable allelic disruption by ZFNs. However, since the expression from IDLVs is 10–100 times lower, we anticipate that VCNs of 10–100 per cell would be sufficient for ZFN delivery. Nevertheless, in primary hematopoietic cells, even at VCN values in that range, we did not detect allelic disruption. VCN can be increased by sequential repeated transductions. Unfortunately, certain primary cell types, especially hematopoietic stem/progenitor cells begin to differentiate if cultured ex vivo for longer than 2–3 days. At equivalent numbers of VCs, the expression of ZFNs was markedly lower in T-lymphocytes and CB-CD34+ cells as compared with K562 cells. Analysis of mRNA levels per VC in CB-CD34+ cells indicated that the block to expression from the vector genomes is not at a transcriptional level, as the levels were higher than those in K562 cells and T-lymphocytes. At higher mRNA levels per VC, protein levels were lower than those in K562 cells. This would indicate that the restriction to lentiviral expression in CB-CD34+ cells could potentially be post-transcriptional. This could be due to many reasons, such as, short mRNA half-life, reduced translational efficiency, short protein half-life. On the other hand, in T-lymphocytes, the transcript and protein levels were lower than in K562 cells. This suggests that the restriction in T-cells could be at both transcriptional and post-transcriptional stages. Deeper analysis of this phenomenon will need to be performed. We can speculate that multiple remedies could be applied to increase gene expression from IDLVs. A recent study reported that the use of histone deacetylase inhibitors could increase transcription from IDLVs.34 Stability of ZFN protein could be enhanced by treating transduced cells with proteasome inhibitors.35 Combining multiple such ways to increase gene expression from IDLVs can potentially increase their efficacy as a delivery method for ZFNs.
In addition to delivering ZFNs, we demonstrated that IDLVs could also deliver donor templates efficiently. We used an IDLV to deliver donor templates in conjunction with ZFN delivery by either nucleofection or IDLVs. Using nucleofection to deliver ZFNs, we could achieve levels of gene modification with Donor-IDLV comparable to those obtained by nucleofection of donor plasmid. We showed that the use of IDLVs for donor delivery could be advantageous as it permits use of high amounts of ZFN plasmids if nucleofection is used for ZFN delivery. However, co-delivery of ZFNs and donors by IDLVs is not as efficient as using nucleofection for ZFN delivery. Analysis of VCNs indicates that when a fixed amount of ZFN plasmid is nucleofected, there is an optimal value of Donor-IDLV VCN, which maximized gene modification, whereas, at the same amount of Donor-IDLV, with increasing ZFN plasmids, it is possible to increase the efficiency of gene modification. We also demonstrated gene modification using IDLV delivery of both ZFNs and donor templates. However, the absolute values were much lower than those obtained by ZFN delivery by nucleofection. This could be a compound effect of the lower efficiency of the IDLVs combined with the requirement of cotransduction of two or three vectors. Hence, based on our results, we can speculate that ZFN expression is the greater limiting factor in the context of IDLV delivery.
Although we demonstrate successful use of IDLVs for ZFNs, their efficiency is sub-optimal in primary hematopoietic cells. Further improvements in vector constructs may be necessary to increase the efficiency of IDLVs for ZFN delivery. We also showed that IDLVs can work efficiently as donor templates. These results highlight the promise of IDLV-based gene delivery of ZFN-mediated site-specific genome modification.
Materials and Methods
Cell lines and culture. K562 cells (CRL-243; ATCC, Manassas, VA) were cultured in RPMI1640 (Cellgro, Manassas, VA) supplemented with 10% Fetal Bovine Serum (Gemini Bio-products, West Sacramento, CA) and Penicillin/Streptomycin/L-glutamine (Gemini Bio-products). Human umbilical cord blood samples were collected from University of California, Los Angeles (UCLA) Ronald Reagan Medical Center and have been deemed exempt from review from institutional review board as anonymous medical waste. Mononuclear cells were isolated from cord blood by Ficoll-Paque (GE Healthcare Biosciences, Piscataway, NJ)-based separation. CD34+ cells were isolated from the mononuclear cells using MACS CD34+ enrichment kit (Miltenyi Biotec, Auburn, CA).
Adult human peripheral blood mononuclear cells were obtained from the Center for AIDS Research Virology Core Lab that is supported by the National Institutes of Health award AI-28697 and by the UCLA AIDS Institute and the UCLA Council of Bioscience Resources. Peripheral blood mononuclear cells were activated using Dynabeads Human T-Activator CD3/CD28 (Life Technologies, Green Islands, NY) for 3 days and cultured subsequently with RPMI1640 (Cellgro) supplemented with 10% Fetal Bovine Serum (Gemini Bio-products), Penicillin/Streptomycin/L-glutamine (Gemini Bio-products), and 5 ng/ml Interleukin-2 (R&D Systems, Minneapolis, MN). HEK293T (CRL-11268; ATCC) and HT-29 (ATCC HTB-38) cells were maintained in Dulbecco's modified Eagle's medium (Cellgro) supplemented with 10% Fetal Bovine Serum (Gemini Bio-products) and Penicillin/Streptomycin/L-glutamine (Gemini Bio-products). Genomic DNA from cells was extracted using PureLink Genomic DNA mini kit (Life Technologies) and quantified using NanoVue (GE Healthcare Biosciences). Cells were monitored for mCherry expression on BD-LSRFortessa or BD-LSRII flow cytometers (Becton-Dickinson, Franklin lakes, NJ). Cell counts were measured using a Beckman-Coulter ViCell-XR automated cell counter (Beckman-Coulter, Indianapolis, IN).
Vector constructs and molecular cloning. The ZFNs targeting hADA intron 6 were produced at Sangamo Biosciences (Richmond, CA). Single ZFN IDLV-coding sequence was constructed using spliced overlap PCR. ZFN-P2A was amplified from pVAX-ZFN using primers Flag-forward (5′-ATGGACTACAAAGACCATGACGG-3′) and P2A-Fok-reverse (5′-CTCCGCTTCCGGATCTCTTTGC TCGGAAGTTGATCTCGCCGTTGTT-3′). P2A-mCherry was amplified from pmCherry (Clontech Laboratories, Mountain View, CA) using primers P2A-mCh-forward (5′-CATGTTATCCTCCTCGCCCTTGCTCACCATGGATCCCCGGGTACCG-3′) and mCh-reverse (5′-CGCGGAATTCCGACTTGTACAGCTCGTCCATGC-3′). ZFN-P2A-mCherry was amplified using the Flag-forward and mCh-reverse primers using an equimolar mixture of ZFN-P2A and P2A-mCherry amplicons as a template. ZFN-P2A-mCherry amplicons were cloned into the EcoRI site of pCCL-EFS-x-WPRE. For Double-IDLV, ZFN-L-T2A was amplified using Flag-forward and T2A-Fok-reverse (5′-CCTAGGGCCGGGATTCTCCTCCACGTCACCGCATGTTAGAAGACT TCCTCTGCCCTCTCCGCCGCCAGATCTGAAGTTGATCTCGCCGTTGTT-3′); T2A-ZFN-R-P2A-mCherry was amplified from pCCLc-EFS-ZFN-R-P2A-mCherry-WPRE using T2A-Flag-forward (5′-AGAATCCCGGCCCTAGGATGGACTACAAAGACCATGACGG-3′) and mCh-reverse. ZFN-L-T2A and T2A-ZFN-R-P2A-mCherry were cloned into pCCLc-EFS-x-WPRE using EcoRI and AvrII. Double-dF-IDLV constructs were made from Double-IDLV constructs using InFusion HD cloning system (Clontech Laboratories). The 3xFLAG-tag (5′-DYKDHDGDYKDHDIDYKDDDDK-3′) was deleted from the left ZFN, but was kept intact in the right ZFN. Double-CoOp constructs were designed using GeneArt gene synthesis system (Life Technologies). The left ZFN, including 3xFLAG-tag, NLS, ZF motifs, and FokI domain was codon-optimized. The codon-optimized region was synthesized in overlapping 500 bp gBlocks fragments (Integrated DNA Technologies, Coralville, IA). The gBlocks were fused using spliced overlap PCR using primers Frag1-forward (5′-AGGTGTCGTGACGCGGGATCTCGA-3′) and Frag3-reverse (5′-CCTAGGGCCAGGGTTCTCTTCCAC-3′) and cloned into Double-IDLV constructs using XhoI-BmgBI (New England Biolabs, Ipswich, MA).
For the donor construct, the 1.3 kb region surrounding the ZFN-binding site of the hADA gene intron 6 (33133-34488 in NG_007385) was amplified using X7-F1 (5′-AGGGACTCCTGCTTCCTATGCG-3′) and X7-R2 (5′-CTGCTTCTGGCTGTGATTTGC-3′) and cloned in pCR4-TOPO (Life Technologies). The EagI site was mutated to SstII site using QuickChange Site-directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). For Donor-IDLV constructs, the 1.3 kb donor template was amplified using X7-F1-Eco (5′-CGCGGAATTCAGGGACTCCTGCTTCCTATGCG-3′) and X7-R2-Eco (5′-CGCGGAATTCCTGCTTCTGGCTGTGATTTGC-3′) and cloned in the reverse orientation into pCCLc-EFS-x-WPRE using EcoRI. LVs were packaged and their titers were determined as per published protocols.23
Nucleofection of K562 cells. K562 cells were nucleofected using Nucleofector 4D (Lonza, Walkersville, MD) as per the recommended protocol. Briefly, 1 × 106 cells per sample were centrifuged at 90g for 10 minutes. The pellets were resuspended in 100 μl of Cell Line SF (Lonza, Walkersville, MD) solution. Appropriate amounts of DNA were added to the cells and the mixture was nucleofected using the recommended program. The cells were allowed to recover at room temperature for 10 minutes and then supplemented with culture medium and transferred to 6-well tissue culture-treated plates (Corning, Corning, NY).
Transduction of K562 cells. K562 cells, 1 × 105 per sample, were centrifuged at 90g for 10 minutes and resuspended in 50 μl of culture medium per sample. Appropriate dilutions of viruses were made in culture medium in 50 μl. The diluted virus was added to cells in 48-well tissue culture-treated plates (Corning). Two days post-transduction, the cells were transferred to 6-well tissue culture-treated plates (Corning) with 2 ml of culture medium.
Transduction of human cord blood CD34+ cells. CB-CD34+ cells were prestimulated on Retronectin (Clontech Laboratories, Mountain View, CA)-coated 48-well non-tissue culture-treated plates (Corning) with X-Vivo15 (Lonza) supplemented with 50 ng/ml Stem Cell Factor (Amgen, Thousand Oaks, CA), 50 ng/ml Flt3-Ligand (R&D Systems), and 50 ng/ml Thrombopoietin (R&D Systems) for 18 hours at 1 × 105 cells/ml. CB-CD34+ cells were transduced with appropriate dilutions of the virus in X-Vivo15. At 48 hours post-transduction, cells were transferred to 24-well tissue culture-treated plates (Corning) and maintained in Iscove's modified Dulbecco's medium (Cellgro) supplemented with 20% Fetal Bovine Serum (Gemini Bio-products), 25 ng/ml Stem Cell Factor (Amgen), 10 ng/ml Interleukin-6 (R&D Systems), and 5 ng/ml Interleukin-3 (R&D Systems).
Transduction of human T-lymphocytes. Activated T-lymphocytes were resuspended to disrupt clumps and 1 × 105 cells per sample were centrifuged at 300g for 5 minutes. The cell pellets were resuspended in 50 μl culture medium with 10 ng/ml rhIL2. Virus dilutions were made in culture medium without interleukin-2 and added to the cells to achieve a final rhIL2 concentration of 5 ng/ml. The transductions were performed in 48-well tissue culture-treated plates (Corning). Two days post-transduction, the cells were transferred to 12-well tissue culture-treated plates (Corning) in 1 ml of culture medium with 5 ng/ml rhIL2.
Surveyor nuclease assay. Surveyor nuclease assay was used to determine ZFN-induced site-specific allelic disruption. A 332 bp region surrounding the ZFN-binding site was amplified from 200 ng of genomic DNA using 474F (5′-CTACCTGACACATGGTAACTGGCTAATGAG-3′) and 474R (5′-AATAGAGCCAAGTATGGGAGGAGGCAGTGAGGAGGG-3′) using Accuprime Taq Hi-Fi (Life Technologies) under the following conditions: 94 °C for 2 minutes followed by 30 cycles of 94 °C for 30 seconds, 62 °C for 30 seconds, and 68 °C for 1 minute, followed by a final extension of 68 °C for 5 minutes. The PCR product was diluted to 1:2 in 6 μl of 1X Accuprime buffer and subjected to denaturation and reannealing under the following conditions: 94 °C for 10 minutes, followed by cooling to 85 °C at −2 °C per second and cooling to 25 °C at −0.1 °C per second. The reaction mixture was then digested with 1 μl of Surveyor Nuclease (Transgenomic, Omaha, NE) at 42 °C for 15 minutes. The reactions were resolved on 8% TBE polyacrylamide gels (Life Technologies) at 120 V for 45 minutes. The gels were removed from the cassettes and stained with 5 μl of Gel-Green (Phenix Research Products, Candler, NC) in 50 ml of 1X TBE for 15 minutes and imaged on a Kodak Molecular Imaging Station (Kodak, Rochester, NY). The gel images were analyzed by densitometry and allelic disruption was determined using the following formula:
% Allelic disruption = 100 × (1 − √(intensity of the uncut band/total intensity of cut and uncut bands))
Determination of frequency of gene modification. Site-specific gene modification was detected by restriction fragment length polymorphism. A 2 kbp region surrounding the ZFN-binding site was amplified using primers X7-2 kb-F (5′-AGACCGTGGTAGCCATTGAC-3′) and X7-2 kb-R (5′-GCCAGGTGTCAGGAAGAGAG-3′) and Accuprime Taq Hi-Fi (Life Technologies) under the following conditions: 94 °C for 2 minutes followed by 30 cycles of 94 °C for 30 seconds, 62 °C for 30 seconds, and 68 °C for 2 minutes, followed by a final extension of 68 °C for 5 minutes. The PCR product was purified using PureLink PCR cleanup kit (Life Technologies) and digested using 10 units of SstII (Life Technologies) for 2 hours at 37 °C. The digestion products were separated on 0.8% TBE-Agarose gel pre-stained with Gel-Green and imaged on a Kodak Molecular Imaging Station.
To quantify gene modification, a quantitative PCR-based assay was used. A set of two PCR reactions were performed using the 2 kb PCR product as a template. The first PCR was performed to amplify modified genomes, using primers X7-forward (5′-tgacacatggtaactggctaatgag-3′) and SstII-reverse (5′-ctcacctcttttactacttccgcg-3′). The second PCR was performed to normalize the input template using primers i9-forward (5′-ttcccctgggcctgtt-3′) and i9-reverse (5′-tggaagcttccctcagagtaatg-3′). Both of these PCRs were made quantitative using SYBRGreen PCR master mix (Life Technologies) and acquired on ViiA7 Real time PCR system (Life Technologies). Frequency of gene modification was determined using the Ct (cycles to threshold) difference between the two PCRs and using a plasmid standard curve.
Western blot for detecting ZFN expression. ZFN expression was monitored by western blotting for FLAG-tag. Lysates from cells transduced with the ZFN vectors were produced using Denaturing cell extraction buffer (Life Technologies) supplemented with Complete Mini protease inhibitor tablets (Roche Applied Science, Indianapolis, IN). Cells were centrifuged at 500g for 10 minutes at 4 °C and the supernatant was aspirated. The cell pellets were resuspended in 50 μl lysis buffer per 1 × 106 cells and incubated on ice for 45 minutes with frequent vortexing. The lysate was centrifuged at 16,000g for 20 minutes at 4 °C. The supernatant was transferred to fresh tubes and stored at −20 °C until further use. The protein quantities were estimated using BCA assay (Thermo scientific, Rockford, IL) and read on a Tecan Infinite M1000 Microplate Reader (Tecan, Morrisville, NC). Equal amounts of protein lysate (40 μg per lane) combined with Laemmli sample buffer (Bio-Rad, Hercules, CA) were loaded on 4–12% bis-tris SDS-PAGE gels (Life Technologies). The gel was run at 150 V for 1 hour in 1X MOPS-SDS running buffer (Life Technologies) in a Novex gel electrophoresis unit (Life Technologies). Prism Ultra (Abcam, Cambridge, MA) protein standards were loaded for size determination. The proteins were transferred from the gel to a PVDF membrane (Life Technologies) in 1X Novex transfer buffer with 10% methanol (Life Technologies). The transfer was carried out at 30 V for 1 hour at 25 °C. After the transfer, the membrane was blocked using 5% non-fat milk in phosphate-buffered saline-Tween 20 for 30 minutes at 25 °C. After blocking, the membrane was incubated with 1:1,000 diluted Mouse anti-FLAG primary antibody (Sigma-Aldrich, St Louis, MO) overnight at 4 °C. The membrane was then washed three times with phosphate-buffered saline-Tween 20 and incubated with 1:5,000 dilution of Goat anti-mouse IgG-HRP secondary antibody (Sigma-Aldrich) for 3 hours at 25 °C. The membrane was washed again three times with phosphate-buffered saline-Tween 20. The membrane was incubated at 25 °C with ECL2 reagent (Thermo scientific) for 5 minutes and imaged on Typhoon FLA 9500 phosphorimager (GE Healthcare Biosciences, Pittsburgh, PA).
Quantitative real-time PCR for mRNA expression from IDLVs. Transcription from vectors was monitored by quantitative real-time PCR on RNA extracted from cells transduced with the EFS-Double-CoOp-LV. Total RNA was extracted from transduced cells using RNeasy Mini kit (Qiagen, Valencia, CA) and eluted in water. Residual DNA contamination was removed by Turbo DNA-free (Life Technologies). The RNA was reverse-transcribed using oligo-dT primer and Superscript II first strand synthesis kit (Life Technologies), with appropriate “no reverse transcriptase” controls. Transcripts from the vectors were amplified using mCherry-forward (5′-AGATCGAGGGCGAGGGCGAG-3′) and WPRE-reverse (5′-CGTCCCGCGCAGAATCCAGG-3′). The transcripts were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) amplified with GAPDH-forward (5′-CACCAGGGCTGCTTTTAACTCTG-3′) and GAPDH-reverse (5′-ATGGTTCACACCCATGACGAAC-3′). The amplification was performed using SYBRGreen PCR master mix (Life Technologies) and acquired on ViiA7 Real time PCR system (Life Technologies).
SUPPLEMENTARY MATERIAL Figure S1. Analysis of vector integrity by PCR, reactions A, C, D, and E as indicated in Figure 3c are shown. Figure S2. Analysis of vector integrity by PCR, gel electropherograms from reaction B performed on K562 cells transduced with IDLVs are shown. Table S1. Comparison of repeat sequences in Double-IDLV and Double-CoOp-IDLV.
Acknowledgments
We thank Arineh Sahaghian and Deborah Anisman-Posner for supplying and processing of human umbilical cord blood and human peripheral blood. We thank Andreas Reik, Colin Flinders, Fyodor Urnov, Michael Holmes, and Philip Gregory at Sangamo BioSciences, Inc. for expert advice, and the Sangamo design and production team for generation of zinc finger nucleases. This work was supported by a research award from the National Heart, Lung, and Blood Institute to D.B.K. (2P01 HL073104), as well as by the University of California, Los Angeles (UCLA) Jonsson Comprehensive Cancer Center and the Eli & Edythe Broad Center of Regenerative Medicine & Stem Cell Research at UCLA. The Flow Cytometry Core of the UCLA Broad Stem Cell Research Center and the UCLA CFAR Virology Core were used for this research. A.V.J. was the recipient of a UCLA CIRM Research Training Program II award (TG2-01169) from the California Institute of Regenerative Medicine and the Eli & Edythe Broad Center of Regenerative Medicine & Stem Cell Research at UCLA predoctoral fellowship in stem cell research. The authors declared no conflict of interest.
Supplementary Material
References
- Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- Klug A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem. 2010;79:213–231. doi: 10.1146/annurev-biochem-010909-095056. [DOI] [PubMed] [Google Scholar]
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- Carroll D. Progress and prospects: zinc-finger nucleases as gene therapy agents. Gene Ther. 2008;15:1463–1468. doi: 10.1038/gt.2008.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Sweeney CL, Chou BK, Choi U, Pan J, Wang H, et al. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood. 2011;117:5561–5572. doi: 10.1182/blood-2010-12-328161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–221. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, Reik A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. 2012;18:807–815. doi: 10.1038/nm.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asokan A, Schaffer DV, Samulski RJ. The AAV vector toolkit: poised at the clinical crossroads. Mol Ther. 2012;20:699–708. doi: 10.1038/mt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann A, Kessler B, Ewerling S, Kabermann A, Brem G, Wolf E, et al. Epigenetic regulation of lentiviral transgene vectors in a large animal model. Mol Ther. 2006;13:59–66. doi: 10.1016/j.ymthe.2005.07.685. [DOI] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gage FH, Trono D, Verma IM. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci USA. 1996;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas J, Jr, Gusella GL, Najfeld V, Klotman ME, Cara A. Novel integrase-defective lentiviral episomal vectors for gene transfer. Hum Gene Ther. 2004;15:361–372. doi: 10.1089/104303404322959515. [DOI] [PubMed] [Google Scholar]
- Nightingale SJ, Hollis RP, Pepper KA, Petersen D, Yu XJ, Yang C, et al. Transient gene expression by nonintegrating lentiviral vectors. Mol Ther. 2006;13:1121–1132. doi: 10.1016/j.ymthe.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Cornu TI, Cathomen T. Targeted genome modifications using integrase-deficient lentiviral vectors. Mol Ther. 2007;15:2107–2113. doi: 10.1038/sj.mt.6300345. [DOI] [PubMed] [Google Scholar]
- Doyon Y, Vo TD, Mendel MC, Greenberg SG, Wang J, Xia DF, et al. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods. 2011;8:74–79. doi: 10.1038/nmeth.1539. [DOI] [PubMed] [Google Scholar]
- Guschin DY, Waite AJ, Katibah GE, Miller JC, Holmes MC, Rebar EJ. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Donello JE, Trono D, Hope TJ. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J Virol. 1999;73:2886–2892. doi: 10.1128/jvi.73.4.2886-2892.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zychlinski D, Schambach A, Modlich U, Maetzig T, Meyer J, Grassman E, et al. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol Ther. 2008;16:718–725. doi: 10.1038/mt.2008.5. [DOI] [PubMed] [Google Scholar]
- Kim JH, Lee SR, Li LH, Park HJ, Park JH, Lee KY, et al. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE. 2011;6:e18556. doi: 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AR, Patel S, Senadheera S, Plath K, Kohn DB, Hollis RP. Highly efficient large-scale lentiviral vector concentration by tandem tangential flow filtration. J Virol Methods. 2011;177:1–9. doi: 10.1016/j.jviromet.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Thibodeau-Beganny S, Sander JD, Voytas DF, Joung JK. Oligomerized pool engineering (OPEN): an ‘open-source' protocol for making customized zinc-finger arrays. Nat Protoc. 2009;4:1471–1501. doi: 10.1038/nprot.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DA, Thibodeau-Beganny S, Sander JD, Winfrey RJ, Hirsh AS, Eichtinger M, et al. Standardized reagents and protocols for engineering zinc finger nucleases by modular assembly. Nat Protoc. 2006;1:1637–1652. doi: 10.1038/nprot.2006.259. [DOI] [PubMed] [Google Scholar]
- Papadakis ED, Nicklin SA, Baker AH, White SJ. Promoters and control elements: designing expression cassettes for gene therapy. Curr Gene Ther. 2004;4:89–113. doi: 10.2174/1566523044578077. [DOI] [PubMed] [Google Scholar]
- Robbins PB, Yu XJ, Skelton DM, Pepper KA, Wasserman RM, Zhu L, et al. Increased probability of expression from modified retroviral vectors in embryonal stem cells and embryonal carcinoma cells. J Virol. 1997;71:9466–9474. doi: 10.1128/jvi.71.12.9466-9474.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser J, Harmison G, Kluepfel-Stahl S, Brady RO, Karlsson S, Schubert M. Transduction of nondividing cells using pseudotyped defective high-titer HIV type 1 particles. Proc Natl Acad Sci USA. 1996;93:15266–15271. doi: 10.1073/pnas.93.26.15266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- Leath A, Cornetta K. Developing novel lentiviral vectors into clinical products. Meth Enzymol. 2012;507:89–108. doi: 10.1016/B978-0-12-386509-0.00005-3. [DOI] [PubMed] [Google Scholar]
- Basu VP, Song M, Gao L, Rigby ST, Hanson MN, Bambara RA. Strand transfer events during HIV-1 reverse transcription. Virus Res. 2008;134:19–38. doi: 10.1016/j.virusres.2007.12.017. [DOI] [PubMed] [Google Scholar]
- Fuentes GM, Palaniappan C, Fay PJ, Bambara RA. Strand displacement synthesis in the central polypurine tract region of HIV-1 promotes DNA to DNA strand transfer recombination. J Biol Chem. 1996;271:29605–29611. doi: 10.1074/jbc.271.47.29605. [DOI] [PubMed] [Google Scholar]
- Schambach A, Bohne J, Chandra S, Will E, Margison GP, Williams DA, et al. Equal potency of gammaretroviral and lentiviral SIN vectors for expression of O6-methylguanine-DNA methyltransferase in hematopoietic cells. Mol Ther. 2006;13:391–400. doi: 10.1016/j.ymthe.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Leuci V, Mesiano G, Gammaitoni L, Cammarata C, Capellero S, Todorovic M, et al. Transient proteasome inhibition as a strategy to enhance lentiviral transduction of hematopoietic CD34(+) cells and T lymphocytes: implications for the use of low viral doses and large-size vectors. J Biotechnol. 2011;156:218–226. doi: 10.1016/j.jbiotec.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Pelascini LP, Janssen JM, Gonçalves MA. Histone deacetylase inhibition activates transgene expression from integration-defective lentiviral vectors in dividing and non-dividing cells. Hum Gene Ther. 2013;24:78–96. doi: 10.1089/hum.2012.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.